

Abstract
We describe a developmentally normal Amish child who has a karyotype with 47 chromosomes, including a supernumerary ring-shaped chromosome 18 in each metaphase studied. The only phenotypic findings in the patient were hemivertebrae and rib anomalies. Further analysis of interphase cells revealed an additional, less frequent mosaic, apparently normal cell population. Genes in the triplicated region that possibly are contributing to her skeletal phenotype include GATA6, MC2R, MC5R, RBBP8, ESCO1, and ROCK1, among others. By studying such patients with abnormal genetic dosage, genotype–phenotype correlations can be used to refine gene function.
Keywords: trisomy 18, scoliosis, hemivertebrae, ring chromosome 18, Edwards syndrome
INTRODUCTION
We report on a developmentally normal Amish child with hemivertebrae and a mosaic de novo supernumerary r(18), resulting in pericentromeric partial 18 triplication. Of the genes in the triplicated region we describe those with known or possible effects on skeletal function.
CLINICAL REPORT
The child was born at 36 weeks to her then, 40-year-old G14 P12 mother via emergency cesarean for placental previa/abruption. There were no other pregnancy problems, such as maternal diabetes or hypertension. Her birth weight was 2.95 kg (60th centile). Relevant family history includes a paternal grandmother with scoliosis of unclear presentation and etiology, a maternal first cousin with spina bifida, and a maternal first cousin with a cardiomyopathy of unclear etiology. There is no other reported scoliosis in the family. There is no known consanguinity; however both parents are Amish and they think there may have been consanguinity many generations back.
During an admission at 1-year-of-age for a significant respiratory syncytial virus infection requiring intubation and ventilation, severe thoracic dextroscoliosis of 60° and kyphosis were noted. Chest X-ray revealed multiple thoracic hemivertebrae, absent left-sided ribs, and a right-sided bifid rib (Fig. 1). The patient had no other known medical issues or previous hospitalizations.
FIG. 1.
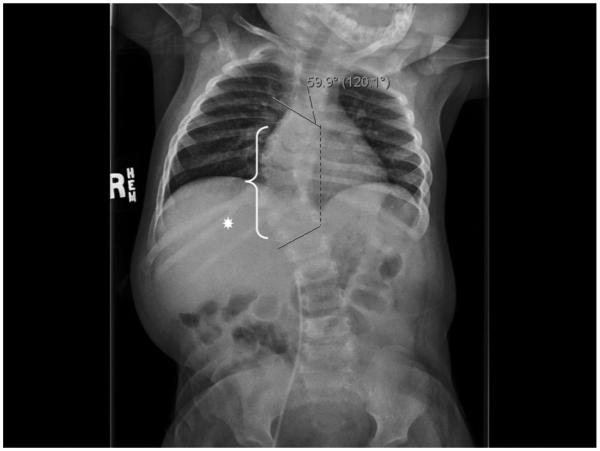
Patient plain skeletal film displaying her severe 59.9° thoracic dextroscoliosis. Based on 3-D CT reconstruction (not shown) the dextroscoliosis appears to have its apex at the T10/11 level. Multiple vertebral anomalies were also identified (white bracket), including right hemivertebrae at levels T8 and T11. The right T8 hemivertebra was partially fused to the right aspect of T9. Additionally, there was clefting of vertebral bodies T6 and T9 and fusion of the posterior elements of T11 and T12. There also appeared to be a spina bifida occulta at both the T11 and T12 levels. There are only nine ribs on the left side. Note is made of a bifid right rib at the T11 level (white asterisk).
At a diagnostic evaluation at 14 months she had growth parameters of: height, 72.5 cm (10th centile); weight, 10.6 kg (70th centile); head circumference, 46.8 cm (60th centile). A physical exam showed a well appearing child without facial dysmorphic features. Her chest and back exam revealed a significant dextroscoliotic curvature with the right shoulder higher than the left, more pronounced when she was standing. Her physical exam was otherwise normal, without other minor abnormalities. Furthermore, all other systems were reviewed and were negative. A full skeletal survey showed no additional bony abnormalities. Echocardiogram was normal and renal ultrasound showed mild fullness of the left kidney. Developmental screening at the time of our initial consult was normal based on Denver II Developmental screening guidelines.
An additional Denver II Developmental Screen performed at 30 months of age by telephone with her father demonstrated the child to be still completely on track for both fine and gross motor skills, as well as personal and social skills. In fact, her father thought she was cognitively advanced for her age compared to his other 11 children. She had not developed kidney problems and her only medical issues remained to be a result of her scoliosis, for which she underwent spinal fusion of T10–T12, with placement of a rod and pedicle screws. Additionally, she was given a Risser body cast for 3 months. At 34 months of age, her height was 93 cm (50th centile) and radiography showed 22° of scoliosis of the left thoracic curve (T1–T6), 47° of the right lower thoracic curve (T6–T11), and 21° of the left lumbar curve (T11-L4). This was interpreted as a small amount of further progression through the main thoracic curve. Her weight at that time was 13 kg (40th centile).
CYTOGENETICS AND MOLECULAR STUDIES
Metaphase chromosomes were prepared from peripheral blood samples using standard procedures for high-resolution chromosome studies [Yunis, 1976; Ikeuchi, 1984]. Digital images were collected using an Olympus microscope controlled by CytoVision software manufactured and distributed by Genetix Corp., San Jose, CA.
Metaphase fluorescence in situ hybridization (FISH) studies were performed to determine the chromosomal origin of the marker chromosome, using commercially available centromere enumeration probes (Abbott Molecular Laboratories, Inc., Des Plaines, IL), and then, to delineate the deletion breakpoints, using homebrew BAC probes. BACs were selected for use on the basis of their physical location along chromosome 18. This information was obtained from the UCSC Human Genome Browser (March 2006 freeze), available at http://genome.ucsc.edu/. Isolated BAC DNA from a Human BAC filter library (RPCI-11) was obtained from the Roswell Park Cancer Institute DNA Microarray Facility (Rochester NY, USA). The DNA hybridization was performed in accordance with the technique described by Pinkel et al. [1986], with minor modifications [Sullivan et al., 1996]. The slides ranged in age from a few days to several months and were stored at −20°C prior to hybridization. The hybridization sequence strategy was patterned after a method previously described by Wandstrat et al. [1998]. Essentially, BACs were differentially labeled with fluorochromes [Spectrum Orange-dUTP or Spectrum Green-dUTP (Abbott Molecular Laboratories, Inc., Des Plaines, IL)] using a standard nick-translation reaction (PerkinElmer Life Sciences, Inc., Boston, MA) and two-color FISH was performed. A probe known to be located outside the region under investigation was co-hybridized as a control. For each hybridization, at least 5 metaphases were analyzed for the presence and number of probe signals on the homologues of interest. To confirm the metaphase analysis, interphase cells were studied for the number of probe signals. Digital images were collected using a Leitz DMRB microscope controlled by CytoVision ChromoFluor software manufactured and distributed by Applied Imaging Corporation.
Using the same approach, interphase FISH analysis was performed using a chromosome 18-specific centromere enumeration probe (CEP18, Abbott Molecular, Inc.). A chromosome 8-specific centromere enumeration probe (CEP8, Abbott Molecular, Inc.) was included in the hybridization mixture to serve as an internal control.
RESULTS
High-resolution chromosomal analysis of a peripheral blood sample from the patient showed the presence of a small supernumerary ring-shaped marker chromosome in each of the metaphase cells that was studied. The child’s initial karyotype was reported as 47,XX,+r.
To identify the origin of the marker chromosome and to delineate its breakpoints, a series of FISH assays was performed on metaphase cells from the blood sample, utilizing chromosome enumeration probes (Abbott Molecular, Inc.). These assays revealed that the ring-shaped chromosome was derived from chromosome 18 and included material from both the short (p) and long (q) arms. The breakpoint on the p-arm was refined to within an approximately 280 kb region in 18p11.21, bounded by BACs RP11-431B10(−) and RP11-48I17(+). The breakpoint in the q-arm was refined to within an approximately 90 kb region in 18q11.2 bounded by BACs RP11-780G2(+) and RP11-198L17(−). The physical length of the region included in the marker is estimated to be between 3.52 and 3.89 Mb, of which an estimated 1.4 Mb is alpha satellite sequences, devoid of unique DNA sequences. Parental blood karyotypes were normal, 46, XX and 46, XY.
Interphase FISH identified 368 of 491 cells (74.9%; range 74.4–75.5%) with three CEP18 (18cen) signals, consistent with the presence of the ring chromosome 18. The remaining 123 cells (~25%), however, had two CEP18 (18cen) signals, suggesting the presence of a second, normal cell line. The final karyotype report was 47, XX, +r.ish r(18)(p11.21q11.2).nuc ish 18cen(CEP18x3)-[368/491]/(CEP18x2)[123/491] (Fig. 2).
FIG. 2.
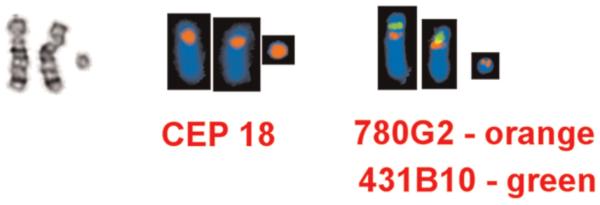
A partial karyotype of the patient’s chromosomes 18 following (from left) G-banding, CEP 18 FISH and BAC FISH. In the BAC FISH assay, the marker chromosome 18 binds the CEP 18 probe and BAC probe RP11-780G2 (in orange) (which localizes to G-band region 18q11.2) but does not bind BAC probe RP11-431B10 (in green) (which localized to G-band region 18p11.21). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
DATABASE SEARCH
We used the University of California Santa Cruz [UCSC, 2006] genome browser, March 2006 assembly, to evaluate the genes included in her trisomic segment. We expanded our search to include the regions 1 Mb in either direction of the triplicated segment in consideration of a possible epigenetic effect of a breakpoint and to include genes that may be within the breakpoint regions. Gene identification was limited to UCSC genes and excluded non-coding sequences and splice variants (UCSC). We evaluated 22 genes in total.
DISCUSSION
The differential diagnosis of congenital scoliosis is large [Burgoyne and Fairbanks, 2001]. In this patient it was quickly narrowed by radiography to be due to a structural bony abnormality, specifically hemivertebrae and other vertebral anomalies (Fig. 1). The multiple disrupted vertebrae likely caused the patient’s scoliosis, as well as three absent ribs on the left side, and her right bifid rib, which appears to be from a partial fusion of two initial individual ribs. The patient’s mild kidney fullness witnessed on her initial renal ultrasound has not developed into a kidney disorder, although ultrasound scans have not yet been repeated. The patient appears developmentally normal, however, formal cognitive testing has not been completed.
Although, hemivertebrae, fused vertebrae, rib anomalies, and scoliosis are found in 10% of patients with complete trisomy 18 or Edwards syndrome, this patient’s findings represent a significant deviation from the characteristic Edwards syndrome phenotype [Jones, 2006]. Rings involving chromosome 18 are very common among autosomal ring chromosomes, however, most of the time the ring replaces a normal homologous chromosome, causing partial monosomy of chromosome 18 with a phenotype similar to that of 18p- or 18q-syndromes [Kaiser-Rogers and Rao, 2005]. Clinical reports of partial duplications, supernumerary markers, or supernumerary rings are rarer in the literature and the phenotypes vary significantly with the size and location of the triplicated segment [Mewar et al., 1993].
Reports of karyotypes involving pericentromeric region partial duplications, supernumerary markers, or supernumerary rings of chromosome 18 in the absence of other chromosomal anomalies are even scarcer in the literature [Callen et al., 1991; Jenderny et al., 1993; Timur et al., 2004]. Other than mild fullness of the left kidney, the patient did not have the same multisystem complications as the mos 47,XY,+r(18)[13]/46,XY[42] patient described in Timur et al. [2004], such as dysmorphic features, limb hypertrophy, arteriovenous malformations, and a learning disability. As she does not have developmental delay or mental retardation, her phenotype is closer to the phenotypically normal, 47,XX,+r(18)[2]/46,XX[98] (breakpoints approximately at p11 and q23) and 47,XX,+r(18)/46,XX [85% of the cells with marker; break points unknown]), patients previously described by Callen et al. [1991] and Jenderny et al. [1993], respectively.
Furthermore, her isolated bone abnormalities hint towards the presence of genes affecting skeletal remodeling that are sensitive to dosage effects within her small triplicated genetic segment. However, other possible diagnoses could include a coincidental VATER association, particularly if she has persistent renal disease, or a recessive condition unmasked by possible distant consanguinity.
Additionally, the patient’s overall phenotype may be mild, or apparently isolated to her vertebral malformations, due to variable levels of tissue mosaicism. As the cell undergoes mitosis, ring chromosomes produce unstable structures such as double-sized dicentric rings, interlocked rings, broken rings, or open rings due to abnormal sister chromatid exchange [Kaiser-Rogers and Rao, 2005; Kosztolanyi, 2009; Sodre et al., 2010]. The instability of ring chromosomes can eventually lead to the loss of the entire ring or the formation of novel ring structures in some cells, a process known as “dynamic mosaicism” [Kaiser-Rogers and Rao, 2005; Kosztolanyi, 2009; Sodre et al., 2010]. Previous studies have shown that 87% of chromosome 18 non-disjunction happens during a meiotic event. Furthermore, 91.5% of the supernumerary chromosomes 18s are of maternal origin [Nicolaidis and Petersen, 1998]. Therefore, we hypothesize that the r(18) in our patient originated from a meiotic event, most likely in her mother, forming a trisomic zygote. However, due to the instability of the ring chromosome, it was lost in certain cells during post-zygotic cell divisions, producing some cells with only two copies of chromosome 18. This can explain the presence of normal diploid cells in her peripheral blood. Unfortunately, we were unable to evaluate for the presence of mosaicism in other tissues due to difficulty obtaining patient follow-up.
Refining the patient’s breakpoints with FISH to 18p11.21–q11.2 allowed us to examine the genes in the centromeric region for any known influence on bone modeling. Genes in the region that may have possible involvement in bone formation include: GATA binding protein 6 (GATA6), melanocortin-2 receptor (MC2R), melanocortin-5 receptor (MC5R), retinoblastoma binding protein 8 (RBBP8), establishment of cohesion 1 homolog 1 (ESCO1), and Rho-associated, coiled-coil containing protein kinase 1 (ROCK1).
GATA6 belongs to the GATA family of genes that encode transcriptional regulatory proteins. One study involving a zebrafish knock-out model suggested GATA6 may be a positive regulator of bone morphogenetic protein-4 (BMP-4) [Peterkin et al., 2003]. MC2R and MC5R are receptors for adrenocorticotropic hormone (ACTH) and melanocyte-stimulating hormones (MSH). A similar gene, melanocortin-4 receptor (MC4R), has been shown to be expressed in periosteum and in osteoblasts in the mouse and it is possible that MC2R and MC5R are likewise involved in the regulation of bone formation [Dumont et al., 2005]. RBBP8 is found among several proteins that bind directly to the retinoblastoma protein, which regulates cell proliferation. It also associates with BRCA1 and is thought to modulate the functions of BRCA1 in transcriptional regulation [Yu and Baer, 2000]. Therefore, it may globally influence cell cycle regulation in the body. ESCO1 belongs to a family of acetyltransferases involved in sister chromatid cohesion [Hou and Zou, 2005]. The cohensionopathies, Cornelia de Lange syndrome and Roberts phocomelia syndrome, have well described skeletal malformations and are due to similar defects in sister chromatid cohesion [Liu and Krantz, 2009]. ROCK1 induces the formation of actin-related structures and helps in cellular cytoskeleton regulation. Additionally, ROCK1 inhibition has been shown to suppress the proliferation of vascular smooth muscle cells and in one in situ study, led to osteoblastic cell proliferation [Tian et al., 2009].
Even though none of the discussed genes is known to be associated with skeletal malformations, over-expression of one or more of them may have the potential to cause improper cartilage and vertebral formation. Further studies to determine functions of the above genes, as well as other genes and non-coding segments in the 18p11.21–q11.2 region, may one day better explain the skeletal malformations in this patient. Also, to better define the breakpoints and the genes involved in the triplicated segment, a whole genome array would be the next step in this patient’s work-up, however we have been unable to obtain one to date.
In summary, we have molecularly defined a mosaic supernumerary r(18) in a patient with isolated congenital scoliosis and other minor vertebral malformations. Analysis of the 18p11.21–q11.2 triplicated region resulted in a list of genes that may be involved in skeletal formation and regulation. Further functional studies of these genes are needed to better understand their role in skeletal development.
ACKNOWLEDGMENTS
We would like to acknowledge the patient and her family for their kind cooperation, as well as all clinical and laboratory personnel involved in her care. We also thank Anne Matthews for review of this manuscript.
REFERENCES
- Burgoyne W, Fairbanks J. The management of scoliosis. Curr Paediatr. 2001;11:323–331. [Google Scholar]
- Callen DF, Eyre HJ, Ringenbergs ML, Freemantle CJ, Woodroffe P, Haan EA. Chromosomal origin of small ring marker chromosomes in man: Characterization by molecular genetics. Am J Hum Genet. 1991;48:769–782. [PMC free article] [PubMed] [Google Scholar]
- Dumont LM, Wu CS, Tatnell MA, Cornish J, Mountjoy KG. Evidence for direct actions of melanocortin peptides on bone metabolism. Peptides. 2005;26:1929–1935. doi: 10.1016/j.peptides.2004.12.034. [DOI] [PubMed] [Google Scholar]
- Hou F, Zou H. Two human orthologues of Eco1/Ctf7 acetyltransferases are both required for proper sister-chromatid cohesion. Mol Biol Cell. 2005;16:3908–3918. doi: 10.1091/mbc.E04-12-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeuchi T. Inhibitory effect of ethidium bromide on mitotic chromosome condensation and its application to high resolution chromosome banding. Cytogenet Cell Genet. 1984;33:56–61. doi: 10.1159/000132030. [DOI] [PubMed] [Google Scholar]
- Jenderny J, Caliebe A, Beyer C, Grote W. Transmission of a ring chromosome 18 from a mother with 46,XX/47,XX,+r(18) mosaicism to her daughter, resulting in a 46,XX,r(18) karyotype. J Med Genet. 1993;30:964–965. doi: 10.1136/jmg.30.11.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KL. Smith’s recognizable patterns of human malformation. 6th Elsevier Saunders Company; Philadelphia: 2006. pp. 13–17. [Google Scholar]
- Kaiser-Rogers K, Rao K. Structural chromosome rearrangements. In: Gersen SL, Keagle MB, editors. The principles of clinical cytogenetics. Humana Press; Totowa: 2005. pp. 165–206. [Google Scholar]
- Kosztolanyi G. The genetics and clinical characteristics of constitutional ring chromosomes. J Assoc Genet Technol. 2009;35:44–48. [PubMed] [Google Scholar]
- Liu J, Krantz ID. Cornelia de Lange syndrome, cohesin, and beyond. Clin Genet. 2009;76:303–314. doi: 10.1111/j.1399-0004.2009.01271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mewar R, Kline AD, Harrison W, Rojas K, Greenberg F, Overhauser J. Clinical and molecular evaluation of four patients with partial duplications of the long arm of chromosome 18. Am J Hum Genet. 1993;53:1269–1278. [PMC free article] [PubMed] [Google Scholar]
- Nicolaidis P, Petersen MB. Origin and mechanisms of non-disjunction in human autosomal trisomies. Hum Reprod. 1998;13:313–319. doi: 10.1093/humrep/13.2.313. [DOI] [PubMed] [Google Scholar]
- Peterkin T, Gibson A, Patient R. GATA-6 maintains BMP-4 and Nkx2 expression during cardiomyocyte precursor maturation. EMBO J. 2003;22:4260–4273. doi: 10.1093/emboj/cdg400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkel D, Gray JW, Trask B, van den Engh G, Fuscoe J, van Dekken H. Cytogenetic analysis by in situ hybridization with fluorescently labeled nucleic acid probes. Cold Spring Harb Symp Quant Biol. 1986;51:151–157. doi: 10.1101/sqb.1986.051.01.018. [DOI] [PubMed] [Google Scholar]
- Sodre CP, Guilherme RS, Meloni VF, Brunoni D, Juliano Y, Andrade JA, Belangero SI, Christofolini DM, Kulikowski LD, Melaragno MI. Ring chromosome instability evaluation in six patients with autosomal rings. Genet Mol Res. 2010;9:134–143. doi: 10.4238/vol9-1gmr707. [DOI] [PubMed] [Google Scholar]
- Sullivan BA, Jenkins LS, Karson EM, Leana-Cox J, Schwartz S. Evidence for structural heterogeneity from molecular cytogenetic analysis of dicentric robertsonian translocations. Am J Hum Genet. 1996;59:167–175. [PMC free article] [PubMed] [Google Scholar]
- Tian YS, Kim HJ, Kim HM. Rho-associated kinase (ROCK) inhibition reverses low cell activity on hydrophobic surfaces. Biochem Biophys Res Commun. 2009;386:499–503. doi: 10.1016/j.bbrc.2009.06.087. [DOI] [PubMed] [Google Scholar]
- Timur AA, Sadgephour A, Graf M, Schwartz S, Libby ED, Driscoll DJ, Wang Q. Identification and molecular characterization of a de novo supernumerary ring chromosome 18 in a patient with Klippel– Trenaunay syndrome. Ann Hum Genet. 2004;68:353–361. doi: 10.1046/j.1529-8817.2004.00095.x. [DOI] [PubMed] [Google Scholar]
- University of California Santa Cruz (UCSC) Genome Browser, March 2006 assembly. 2006 Accessible at http://genome.ucsc.edu/cgi-bin/hgGateway. Last accessed February 4th, 2010. [Google Scholar]
- Wandstrat AE, Leana-Cox J, Jenkins L, Schwartz S. Molecular cytogenetic evidence for a common breakpoint in the largest inverted duplications of chromosome 15. Am J Hum Genet. 1998;62:925–936. doi: 10.1086/301777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Baer R. Nuclear localization and cell cycle-specific expression of CtIP, a protein that associates with the BRCA1 tumor suppressor. J Biol Chem. 2000;275:18541–18549. doi: 10.1074/jbc.M909494199. [DOI] [PubMed] [Google Scholar]
- Yunis JJ. High resolution of human chromosomes. Science. 1976;191:1268–1270. doi: 10.1126/science.1257746. [DOI] [PubMed] [Google Scholar]