

Abstract
K+ channels play a key role in regulating cellular excitability. It was thought that the strong K+-selectivity of these channels was static, only altered by mutations in their selectivity filter, which can cause severe genetic disorders. Recent studies demonstrate that selectivity of K+ channels can also exhibit dynamic changes. Under acidic conditions or in low extracellular K+ concentrations, the two-pore domain K+ channel (K2P) TWIK1 becomes permeable to Na+, shifting from an inhibitory role to an excitatory role. This phenomenon is responsible for the paradoxical depolarization of human cardiomyocytes in pathological hypokalemia, and therefore may contribute to cardiac arrhythmias. In other cell types, TWIK1 produces depolarizing leak currents under physiological conditions. Dynamic ion selectivity also occurs in other K2P channels. Here we review evidence that dynamic selectivity of K2P channels constitutes a new regulatory mechanism of cellular excitability, whose significance is only now becoming appreciated.
Keywords: channelopathies, resting potential, cationic channels, ionic pore
Ion selectivity of K+ channels
Rapid diffusion of ions through ion channels is essential for electrolyte homeostasis, cell development/differentiation and excitability. In physiological conditions, open K+ channels allow K+ to exit the cell, creating an excess of negative charges inside the cell and driving the membrane potential to hyperpolarized values. The opening of Na+ channels has the opposite effect, allowing an inward rush of positive charges, which promote depolarization. Thus, at any moment, the excitability of the cell relies directly on the selectivity of the activated ion channels, with Na+ influx favouring excitability and K+ efflux opposing it. Genetic mutations leading to permanent alterations in the ion selectivity of K+ channels are associated with severe disorders, further demonstrating how crucial K+-selectivity is to cellular function [1–8].
Expressed in everything from viruses and bacteria to plants and animals, K+ channels form the largest ion channel family. Despite their diversity, all these ion channels share the property of exhibiting highly selective transport of K+ [9–11]. The signature sequence TxGY(F/L)G, which is conserved in the pore-forming loop (P-loop) of all K+ channels, constitutes the core of the selectivity filter, which mainly determine the K+ selectivity (Box 1) [10,11]. Mutations in the P-loop and its surrounding regions can affect ion selectivity [9,12]. This K+ selectivity was originally thought to be static, independent of regulation by external stimuli. Recent studies have challenged this dogma by showing that K+ channels exhibit dynamic ion selectivity in response to physiological and pathophysiological stimuli [13–15]. In the subsequent sections, we review the recent findings that have contributed to this new view that ion selectivity of K+ channels can be both dynamic and regulated.
Box 1. Ion selectivity in K+ channels.
Four TVGYG signature sequences form the selectivity filter of the KcsA channel (Figure IA). In the conduction pathway, dehydrated K+ ions are stabilized by negatively charged groups and translocate in a single file [11]. Side-chains of the first threonines and the backbone’s carbonyl groups point toward the central axis of the pore, creating cage-like K+ binding sites, which are noted S1 to S4. This unique environment stands in for the hydration shells of solvated K+ ions (Figure IB) [10] and establishes favorable structural constraints for both selectivity and fast translocation. In the selectivity filter, other ion binding sites located in the planes of the carbonyls have been suggested, in which smaller ions, such as Na+ or Li+, would be more efficiently coordinated [48–50]. K+ within the conduction pathway is required to preserve a high rate and selective conductive conformation more favorable for K+ than for Na+ [51,52]. The large energy barrier formed by the concomitant presence of two K+ ions at their binding sites would prevent Na+ to access their own binding positions in the carbonyl planes (Figure IC). The structures of KcsA channels at low K+ concentrations exhibit changes in the conduction pathway and nearby vicinity, showing a collapse of the pore [10]. This rearrangement results in a non-conductive conformation of the filter. However, some K+ channels exhibit Na+ permeability in the absence of K+ [53–55], suggesting that perturbations in the selectivity filter geometry can also generate pores permeable to smaller ions. In KcsA, residues in the selectivity filter and the adjacent pore helix domain form, with water molecules, a complex network of hydrogen bonds, salt-bridges and electrostatics interactions [10,56,57] (Figure ID) that determine the stability of the selectivity filter [57,58]. Similar interactions exist in or are predicted for other K+ channels [40,59,60]. Depending on their nature and strength, the channel would be susceptible to collapse in a non-conductive state or in a state permeable to smaller ions.
Gene mutations alter ion selectivity in K+ channels and result in disorders
A point mutation in the selectivity filter of Kir3.2 subunits permanently changes the ion selectivity and accounts for the phenotype of the weaver mouse. The neurological mutant mouse weaver has been studied for decades because of its particular neuronal development and neurologic disorders. The weaver mouse is profoundly ataxic because of the loss of granule cell neurons during cerebellar development [16]. This loss is due to a point mutation occurring in the gene encoding the G-protein-activated, inwardly rectifying K+ channel protein, Kir3.2 (Figure 1). Kir3.2 (GIRK2, KCNJ6) forms functional heterotetramers with another member of the same channel family, Kir3.1 (GIRK1, KCNJ3) (reviewed in [17]). The weaver mutation (G156S) exchanges the first glycine residue for a serine in the selectivity sequence TxGYG of Kir3.2 [1,2]. Channels containing Kir3.2•G156S subunits lose K+ selectivity, become nonselective cation channels, and constitutively conduct inward leak Na+ currents. The Na+ to K+ relative permeability (PNa/PK) increases from ~0.03 for heteromeric Kir3.1/Kir3.2 wild type channels to ~0.94 for homomeric Kir3.2•G156S mutant channels and ~0.74 for heteromeric Kir3.1/Kir3.2•G156S mutant channels [2]. Kir3.1 and Kir3.2 are highly expressed in cerebellar granule cells during development, when weaver mouse granule cells are lost. The constitutive inward leak Na+ conductance in weaver Kir3 mutant channels leads to excessive Na+ influx, producing marked changes in intracellular Na+ concentrations and increasing granule cell death [16].
Figure 1. Somatic and inherited mutations alter ion selectivity of G protein-gated K+ channels.
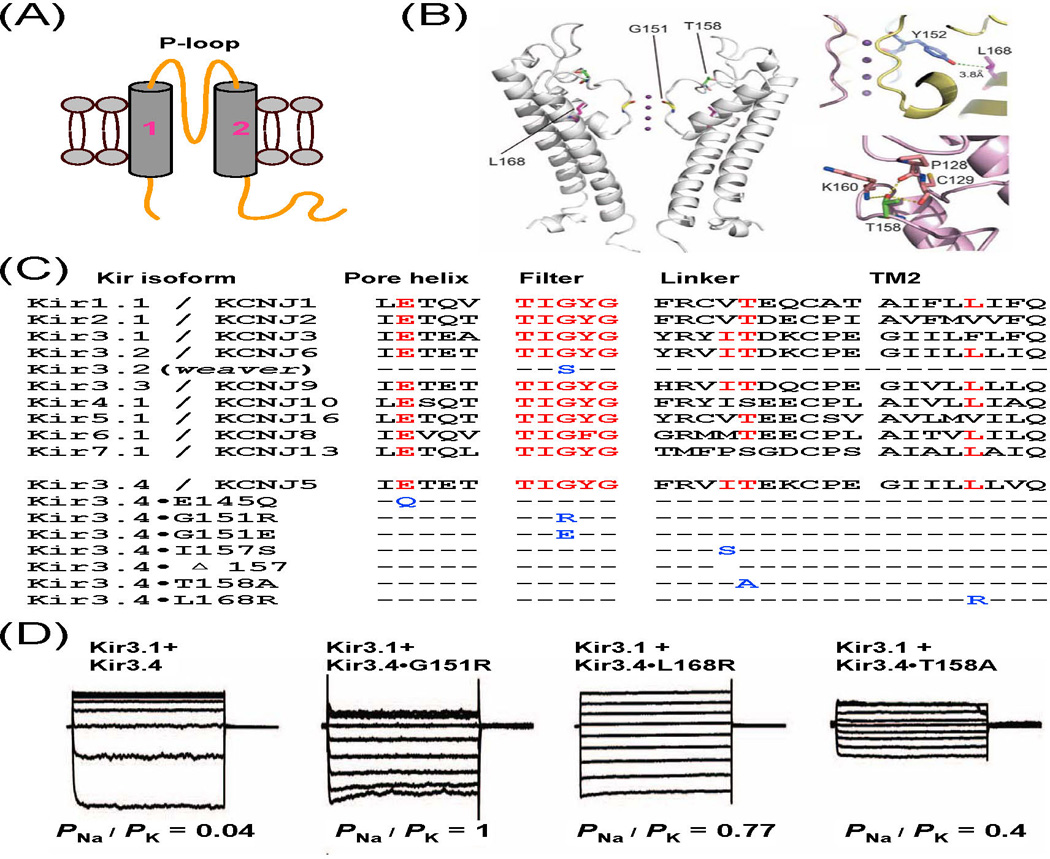
(A) Topology of Kir subunits with two transmembrane domains (TM1 and TM2) and one P-loop. (B) Residues G151, T158, and L168 of Kir3.4 are located respectively in the selectivity filter, the linker and TM2. (C) Sequence alignment of P-loops and upper parts of TM2 of human Kir channels and mouse Kir3.2 channel. Conserved residues are labelled in red, and mutations in Kir3.2 and Kir3.4 in blue. (D) Whole-cell currents recorded in HEK293T cells and corresponding PNa/PK values. (B and D adapted from [3]).
Mutations in the selectivity filter and surrounding regions of Kir3.4 subunits also change the K+ selectivity and are associated with human disorders. Kir3.4 (GIRK4, KCNJ5) is closely related to Kir3.2. In the heart, the heteromer Kir3.1/Kir3.4 underlies the cardiac muscarinic-gated channel (KACh) that contributes to the decrease in heart rate triggered by the activity of the parasympathetic nervous system [18]. Although the normal function of Kir3.4 in the adrenal gland is poorly understood, recent studies have shown that mutations in Kir3.4 cause aldosterone-producing adenomas (APAs) and inherited primary aldosteronism [3,4]. Two somatic mutations (G151R and L168R) in Kir3.4 were first identified by exome sequencing in APAs [3]. Further sequencing has found additional somatic mutations (E145Q; ΔI157, deletion of the residue I157) and four inherited mutations (G151R, G151E, I157S, and T158A) that are associated with APAs and familial primary aldosteronism [3–7]. E145 is located in the pore helix and G151 is the first glycine in the selectivity sequence TxGYG [9], the same glycine replaced by a serine in weaver Kir3.2 [1,2]. I157 and T158 are conserved among other Kir3 subunits and lie in the extracellular linker between the P-loop and the second transmembrane domain (TM2). L168 lies in the TM2, and its side chain abuts the highly conserved tyrosine side chain of the TxGYG motif (Figures 1B and 1C). All these mutations alter the ion selectivity of Kir3 channels. The PNa/PK values of heteromeric channels comprising Kir3.1 and Kir3.4•G151R, Kir3.4•T158A, and Kir3.4•L168R subunits are ~1, ~0.4, and ~0.77, respectively, as compared with ~0.04 for Kir3.1/Kir3.4 wild type channels (Figure 1D) [3]. Channels containing Kir3.1 and Kir3.4•E145Q (or Kir3.4•G151E or Kir3.4•ΔI157 or Kir3.4•I157S) also conduct inward leak Na+ currents [4,5,12,19], although their precise PNa/PK values have not been determined. These inward leak Na+ currents cause cellular depolarization. Such depolarization is expected to activate voltage-gated Ca2+ channels, resulting in Ca2+ entry into the cells, which in turn increases aldosterone production and cell proliferation of adrenal glomerulosa cells. Most interestingly, both G151E and G151R mutations occur at the same position, but G151E generates larger Na+ conductance than G151R, sufficient to induce mammalian cell death [4]. Thus, G151E and G151R could cause different phenotypes in inherited aldosteronism, with severe hyperplasia in the one case and APAs in the other.
The last example of a genetic mutation known to cause an altered selectivity of the K+ channel leading to a human syndrome relates to the HERG1 K+ channel (KCNH2). The N629D mutation, which lies adjacent to the GFG signature sequence (GFGN), is associated with the cardiac long QT syndrome (LQTS). LQTS results from defects in K+ and Na+ channels that cause prolongation of cardiac repolarization and arrhythmias [20]. The tetrameric HERG1•N629D channel is non-selective among monovalent cations. Its PNa/PK value is ~0.65, as compared with 0.01 for the wild type channel. The N629D mutant channels replace the wild type outward repolarizing K+ tail current with an inward depolarizing Na+ current, which is expected to delay later stages of repolarization and contribute to arrhythmogenesis [8].
Hypokalemia induces changes in ion selectivity of TWIK1 that result in paradoxical depolarization in human cardiomyocytes
Hypokalemia is a common electrolyte disorder that occurs when blood K+ concentrations are lower than the normal values of 3.5 to 4.8 mM. Whereas moderate hypokalemia (2.5–3 mM K+) may lead to cardiac arrhythmias and other muscular disorders, more severe hypokalemia (less than 2.5 mM K+) may cause cardiac arrest and sudden death [21,22]. The Na+-K+ pumps generate the physiological K+ gradients across the cell membrane, which produce the hyperpolarized resting membrane potentials of cardiac cells. Background K+ channels maintain the resting membrane potentials of cardiac cells at around −80 mV, close to the K+ equilibrium potential [23]. Under hypokalemic conditions, in which the extracellular K+ concentrations ([K+]o) are lower than normal physiological levels, many cardiac cells, including human cardiomyocytes and Purkinje fibers, exhibit three well-known aberrant behaviors [24–28]. First, their resting membrane potentials become depolarized rather than hyperpolarized, which is inconsistent with the predictions made by the Nernst equation for K+; second, their resting membrane potentials can become bi-stable; third, the recovery of the hyperpolarized resting membrane potential from the paradoxical depolarization after [K+]o is restored to normal levels exhibits a kinetic hysteresis. Although some previous studies had implied that inward Na+ currents might cause the paradoxical depolarization of cardiac cells [27,28], it was more generally believed that the paradoxical depolarization arose from the strong inward rectifier K+ channels (Kir2), because these channels exhibit non-linear conductance near the K+ equilibrium potential. They pass current more easily in the inward direction (into the cell) than in the outward direction (out of the cell). Because decreases in [K+]o cause a progressive decline in the Kir2 conductance, Kir2 channels cannot fully compensate for the influence of the depolarizing inward currents [29,30]. However, this explanation could not account for several key observations related to the paradoxical depolarization of cardiac cells in lowered [K+]o, including bi-stability and the hysteresis of the recovery [30].
Data from our labs has now demonstrated that TWIK1 is involved in this phenomenon [13]. TWIK1 belongs to the family of two-pore domain K+ channels (K2P) that mediate background or leak K+ conductance, and they participate in maintaining the resting membrane potential of many cell types. Each K2P subunit contains two P-loops (P1 and P2) and 4 membrane-spanning helices (TM1 to TM4). K2P channels are active as dimers (for a recent review on K2P channels see [31]). TWIK1 mRNA is highly expressed in human atrium and Purkinje fibers [32,33], where paradoxical depolarization is often observed. In normal [K+]o, TWIK1 channels are highly selective for K+, and their reversal potentials shift by ~53 mV in the depolarizing direction for every 10-fold increase in [K+]o, which is in agreement with the Nernst equation for K+. In pathological hypokalemic conditions (when [K+]o falls to lower than 3 mM), the reversal potentials are more depolarized than the values predicted by the Nernst equation (Figures 2B–2E). Thus, TWIK1 changes ion selectivity, becomes permeable to Na+, and conducts inward leak Na+ current under hypokalemic conditions. TWIK1 has a unique threonine residue (T118) in the selectivity sequence TT118GYG of the first P-loop, where other K2P channels have a valine or isoleucine. Replacement of the threonine by isoleucine (T118I) eliminates the dynamic ion selectivity of TWIK1 channels (Figure 2D) [13].
Figure 2. In hypokalemic conditions, TWIK1 K+ channels conduct inward leak Na+ currents and induce cardiac paradoxical depolarization.
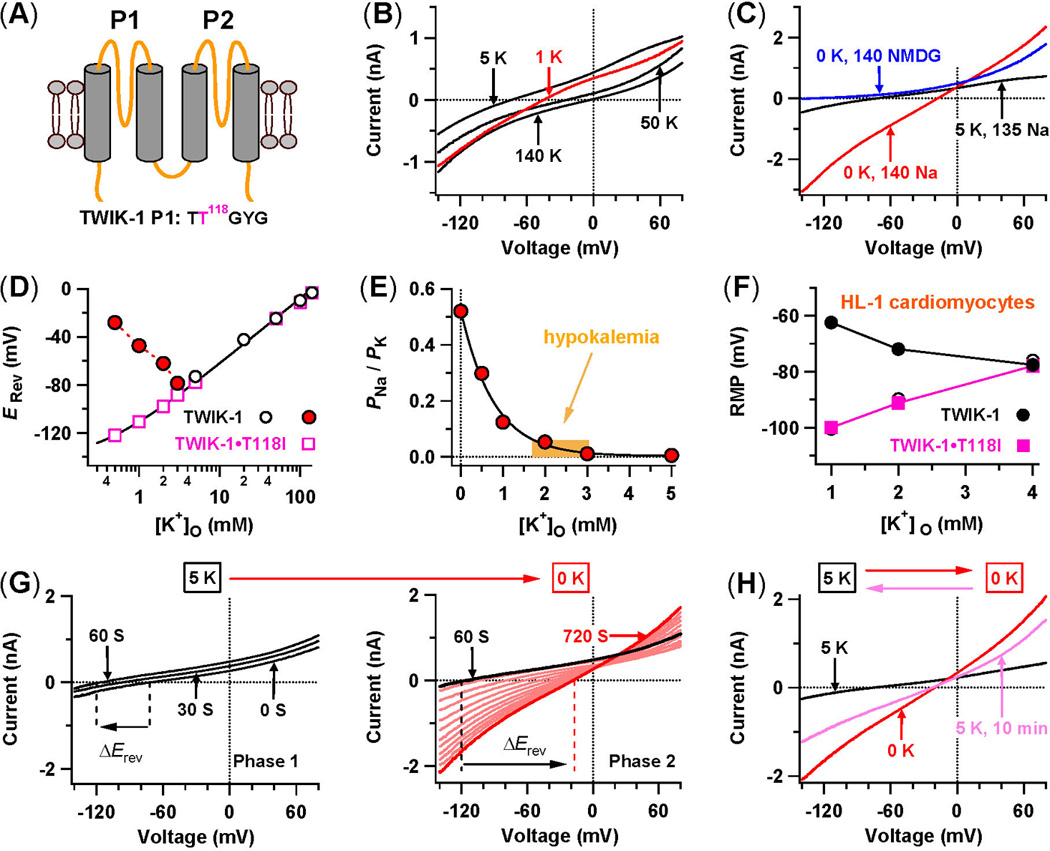
(A) TWIK1 topology with four transmembrane domains (TM1 to TM4) and two P-loops (P1 and P2). The sequence of its K+ selectivity sequence TTGYG in P1 is shown. (B) Whole-cell TWIK1 currents in transfected CHO cells in different external K+ concentrations. (C) Direct recordings of inward leak Na+ currents. Whole-cell TWIK1 currents are shown while Na+-based bath solutions with 5 mM K+ are shifted to 0 mM K+, and finally to an NMDG+-based bath solution with 0 mM K+. (D) Reversal potentials (Erev) measured in Na+-based bath solutions with various [K+]o. The continuous line is a fit for open squares with the Goldman-Hodgkin-Katz voltage equation. (E) PNa/PK values in normal and low [K+]o. (F) Resting membrane potentials (RMP) of mouse HL-1 cardiomyocytes with and without over-expression of TWIK1 and TWIK-1•T118I in 1, 2, and 4 mM [K+]o. (G) Two-phase changes of whole-cell TWIK-1 currents are shown while Na+-based bath solutions are changed from 5 mM to 0 mM K+. (H) Whole-cell TWIK1 currents are shown when a Na+-based bath solution is reversibly changed from 5 mM to 0 mM K+, and then back to 5 mM K+ for 10 minutes. (adapted from [13]).
The inward leak Na+ currents through Na+-permeable TWIK1 have a large impact on the resting membrane potentials. Mouse atrial-like HL-1 cardiomyocytes, which lack TWIK1, do not exhibit the paradoxical depolarization in sub-physiological [K+]o. However, when these same cardiomyocytes ectopically express TWIK1, they acquire the ability to undergo paradoxical depolarization in lowered [K+]o (Figure 2F). Moreover, TWIK1 knockdown in human spherical cardiac myocytes significantly decreases their chances of showing the paradoxical depolarization in lowered [K+]o [13]. These observations indicate that TWIK1 could indeed be responsible for triggering the paradoxical depolarization in human cardiomyocytes, influencing cardiac excitability through dynamic changes of ion selectivity under pathological, hypokalemic conditions.
The kinetics of the conformational changes between the K+-selective and Na+-permeable states of TWIK1 are very slow. The time to reach the maximal Na+ permeability state takes ~10 min, with a time constant of ~373 s, and the time to recovery of the K+ selectivity from the maximal Na+ permeability is ~76 min (Figures 2G and 2H). Such a slow recovery of K+ selectivity in TWIK1 suggests that these channels remain Na+-permeable and able to conduct inward leak Na+ currents for a prolonged period of time after [K+]o is restored to normal levels. This behavior actually provides a biophysical basis for the hysteresis of restoring hyperpolarization from paradoxical depolarization and for bi-stable resting membrane potentials seen in sub-physiological [K+]o. Therefore, dynamic changes in ion selectivity of TWIK1 provide a reasonable explanation for three well-known behaviours of cardiac cells in lowered [K+]o.
TWIK1 ion selectivity is dynamically modulated by the external pH
The changing nature of the ion selectivity of TWIK1 also manifests under physiological conditions. In renal principal cells, for example, loss of TWIK1 expression leads to a decrease in cationic Na+/K+ conductance [34]. In pancreatic β cells, however, inactivation of TWIK1 leads to a hyperpolarization of the membrane potential rather than the expected depolarization, which is puzzling [15]. How could TWIK1 produce depolarizing currents at physiological K+ concentrations?
In heterologous expression systems, the exogenous, newly synthesized TWIK1 is targeted to the plasma membrane, then becomes constitutively internalized and stored in recycling endosomes [35]. Mutation of a di isoleucine motif in the cytoplasmic Cterminal of TWIK1 prevents this endocytosis, resulting in more channels at the plasma membrane and measurable currents [36]. We took advantage of this mutant to reassess the regulation of TWIK1 by extracellular pH, a physiological parameter that modulates the activity of many other K2P channels [37]. An initial report had suggested that TWIK1 was blocked by external acidification [38]. We found that although TWIK1 indeed displays a marked sensitivity to acidification, this sensitivity is biphasic (Figure 3A). TWIK1 is stimulated when pH falls from 7.5 to pH 6.5, but becomes inhibited when pH falls below 6.5. In TASK1/3 channels, which are related to TWIK1, which are related to TWIK1, the pH-sensor is a histidine residue following the TxGYG motif (TIGYGH) in the first P-loop [15]. This histidine is conserved in TWIK1, and its replacement by a neutral residue (H122N), which cannot be protonated upon acidification, completely abolishes TWIK1 channel sensitivity to acidic pH (Figure 3A). This result shows that histidine 122 is an essential component of the pH sensor.
Figure 3. TWIK1 ion selectivity is regulated by extracellular pH.

(A). Sensitivity of TWIK1 and TWIK1 mutants to acidification. The shift of the reversal potential (Erev) at pH 5.5 is indicated by an arrowhead. (B). Erev as a function of the extracellular K+ concentration at pH 7.4 and 6.0. (C). PNa/PK values of different TWIK1 mutants at pH 7.4 and 6.0. (D). Kinetics of the ion selectivity change. TWIK1 currents were recorded at −80mV (adapted from [15]).
In both wild-type TWIK1 and TWIK1•H122K, a marked shift of the reversal potential is observed upon acidification, suggesting a reduced selectivity for K+. TWIK1•T118I. TWIK1•T118I remains K+ selective in hypokalemic conditions and is insensitive to extracellular acidification, which suggests a link between TWIK1 inhibition by acidic pH and altered ion selectivity. To confirm the loss of K+-selectivity at acidic pH, TWIK1 currents were recorded in different [K+]o and the reversal potentials were plotted against [K+]o (Figure 3B). At pH 7.4, the relationships for TWIK1 and TWIK1•T118I were linear, as expected for K+-selective currents. At low [K+]o, however, only a slight deviation was observed for TWIK1, which corresponds to its altered selectivity in these conditions. At pH 6, the relationship for TWIK1 was strongly affected, even in high [K+]o, confirming the loss of K+-selectivity. Upon acidification, the PNa/PK value shifted from 0.03 to 0.19, confirming that TWIK1 conducts leak Na+ currents upon acidification. The shift from the K+-selective to the Na+-permeable state was reversible but very slow (Figure 3D), exhibiting kinetics similar to those seen with decreases in [K+]o (~373 s) [13] or with acidification (~266 s) [15], suggesting that they use similar mechanisms.
Residues in TWIK1 are important for its dynamic selectivity (Figure 3C and Box 1). TWIK1 has a unique motif, LFF, in the pore helix of the first P-loop, whereas other K2P channels have FFF (TREK1, KCNK0) or FYF (TASK1) in the corresponding position. Whereas a TWIK1 mutant containing a FFF triplet behaves as the wild type channel, TWIK1 with FYF (TWIK1•L108F•F109Y) displays altered ion selectivity at both pH 6 and pH 7.4. TWIK1 also contains a leucine residue (L228) in the selectivity sequence TIG(F/L)G of the second P-loop, which is unique to the TWIK channel subfamily that also comprises TWIK2 and KCNK7 (Figure 4). However, mutating this leucine to a phenylanine, which reconstitutes the “classical” signature sequence found in the other K2P channels, fails to abolish the dynamic selectivity of TWIK1. Stimuli modulating TREK channel activity converge on a common molecular gate comprising elements of the pore domains and the N-terminus of TM4 [39]. This gating element is conserved among K2P channels and is employed regardless of whether the gating stimuli are inhibitory or stimulatory. In TWIK1, this element is a threonine (T250). In terms of selectivity TWIK1•T250L is more similar to TWIK1•T118I than it is to TWIK1 and retains K+ selectivity at pH 6 (Figure 3C). The various types of K+ channels, including K2P channels, share a common molecular gate called the “C-type inactivation gate” or “upper gate”. Originally described in voltage-gated K+ channels (Kv), which spontaneously inactivate upon depolarization, this gating involves conformational rearrangements of the upper pore region leading to a closed state [40]. In many K+ channels, the absence of external K+ leads to a collapse of the pore in a closed state that is believed to be similar to the C-type inactivated state of Kv channels. For instance, before to enter in a stable non-conducting state, KV2.1 switches from K+-selective in the open state to Na+-permeable during C-type inactivation [41]. Our results show that residues in P-loops and TM4 that are important for K+ channel pore conformations and upper gating are also involved in the pH-dependent change of ion selectivity in TWIK1. Upon upper gate closure, TWIK1 would reach a Na+-permeable state rather than a strictly closed state.
Figure 4. Human and nematode K2P channels show unusual sequences in their Ploops.

Upper panel: Regions constituting the pore helix and the selectivity filter are shown in the crystal structure of TWIK1. Extracellular and intracellular domains of TWIK1 are not included. Pore helices (PH) are in blue and selectivity filters (SF) are in green. The side chains of residues affecting ion selectivity (L108, F109, T118, H122, L228, T250) are in red. Lower panel: sequence alignment of the pore domains of human and Caenorhabditis elegans K2P channels. Unusual residues are in dark bold.
Taken together, these results indicate that TWIK1 can conduct Na+ upon acidification. However, why would TWIK1 in renal principal cells and in pancreatic β cells behave as a Na+/K+ cationic channel at extracellular pH 7.4? The recycling rate of TWIK1 between acidic intracellular compartments and the cell surface may underlie its depolarizing or hyperpolarizing behaviour. During its transport along biosynthetic and recycling pathways, TWIK1 is exposed to intravesicular acidic pH. This condition is expected to promote a conformational arrangement of the selectivity filter of TWIK1 toward the non-selective state. When TWIK1 arrives at the cell surface it acts as a depolarizing Na+/K+ channel rather than as a hyperpolarizing K+-selective channel. If TWIK1 spends several tens of minutes at the plasma membrane before endocytosis, it would eventually acquire K+ selectivity and produce K+ currents. This is certainly the case in both human cardiomyocytes and Xenopus oocytes, explaining the original description of TWIK1 as a K+-selective channel [42]. However, if TWIK1 is rapidly internalized, it would have insufficient time in the membrane to recover its K+ selectivity. Its influence would then be depolarizing, which is what is observed in renal and pancreatic cells [15,34]. If true, this hypothesis would imply that the degree of influence of TWIK1 in the many tissues and cell types that express this unique channel depends on its recycling rate between endosomes and the plasma membrane.
Other K+ channels with permanently or dynamically-altered ion selectivity?
An alternative translation initiation site generates a truncated form of TREK1 that is permeable to Na+ [43]. Recently, the TASK1/3 channels have been shown to change their ion selectivity upon acidification [14]. Sequence comparisons against TWIK1 suggest that many other K2P channels should exhibit dynamically or permanently altered ion selectivity (Figure 4). A polymorphism occurring at high frequency (60%) in the human gene encoding TASK5 has the unusual sequence EYG in P1 instead of the classical GYG motif (Figure 4) [44,45]. Also the human KCNK7 channel contains a GLE sequence in P2, whereas the mouse KCNK7 contains GLG at the equivalent position. These variants, EYG and GLE, are incompatible with a K+-selective pore (Box 1). Thus, depending on the expressed polymorphism, TASK5 channels might behave as depolarizing channels in some individuals and as hyperpolarizing channels in others, which is rather puzzling. Equally puzzling is the possibility that the ion selectivity KCNK7 be radically different in the two mammalian lineages. This rather suggests that all these channels either are inactive or active but with a permanently altered ion selectivity. Unfortunately this prediction has not yet been tested, since when expressed in heterologous systems, TASK5 and KCNK7 are mainly detected in intracellular compartments rather than in the plasma membrane, where their electrophysiological properties could be tested. When the GLE mutation is introduced in TWIK1, which is highly homologous to KCNK7, TWIK1 becomes permeable to Na+, suggesting that KCNK7 is a non-selective Na+/K+ channel (unpublished result).
Singular sequences in pore domains are not restricted to mammalian K2P channels. Caenorhabditis elegans has more than 40 genes encoding K2P channels, making this class of K+ channels the most abundant in the nematode [46]. Indeed, a dozen worm K2P channels have uncommon motifs. Figure 4 shows an alignment of pore helices and selectivity filters of these channels. Most of them are mainly expressed in excitable cells, including neurons (TWK-3, TWK-4, TW-17), interneurons (TWK-16, TWK-32) and excretory cells (TWK-36) [47]. With a threonine equivalent to TWIK1 T118 in P1 and a GLG triplet in P2, the nematode TWK-25, TWK-34 and TWK-36 channels are very similar to mammalian TWIK1, making them very good candidates for channels with dynamic ion selectivity. However, it should be noted that they lack the histidine in the first P-loop, which is the pH-sensor in TWIK1. TWK-17, which is related to the channels mentioned above, does have a histidine at this position, but also displays a very unusual first pore domain with a GYD motif instead of the classical GYG, suggesting that ion selectivity is permanently altered. TWK-3, TWK-4, TWK-10, TWK-32 on the one hand, and TWK-6, TWK-16, TWK-29 on the other hand, are also quite unusual among K2P channels. Instead of having the strictly conserved threonine located two residues upstream of the signature sequence in one of their P-loops (TXGXG), both groups of channels have another amino acid, either a serine or a proline, for the first group and an alanine or a leucine for the second group. Given the role of the threonine side chain in K+ coordination (Box 1), its substitution by other residues is expected to alter ion conduction. In addition, the second group of channels displays uncommon signature sequences in P2, suggesting also a non-selective state. TWK-16 and TWK-29 also have substitutions in their signature sequences (GYN in P1 and DFG in P2 for TWIK-16 and EYG in P2 for TWK-29) that indicate a possibly altered ion selectivity. Possessing a combination of two GYG motifs within the same subunit, TWK-44 is also very interesting. This combination is never seen in mammalian K2P channels, and when introduced into mammalian TWIK1, it produces a non-functional channel [15]. Cloning and heterologous expression of Caenorhabditis elegans channels should allow these predictions to be tested through electrophysiological characterization and determination of their ion selectivity.
Concluding remarks
K+-selectivity is a hallmark that characterizes a wide class of inhibitory ion channels present from virus and bacteria to plants and animals. Recent data have revealed that an alteration of this property is not only related to deleterious gene mutations but also seen in both pathological and physiological conditions. Future work will address the physiological relevance of the dynamic ion selectivity of TWIK1, and particularly its role in inhibitory and excitatory processes in the many tissues and cell types that express this channel. Cloning and characterization of the ion selectivity of channels related to TWIK1 in mammals and Caenorhabditis elegans should help to verify that dynamic ion selectivity is not a unique event in evolution but rather a property shared by multiple K+ channels that has been overlooked. Ion channels are major targets of the current pharmaceutical drugs. These drugs that modulate channel activity and cell excitability have proven to be useful as anxiolytics, neuroleptics, anticonvulsants, local and general anesthetics, pain killer as well as cardiac antiarrhythmics. If dynamic selectivity proves to be more common than expected, designing drugs that modulate ionic selectivity may constitute a new avenue in therapeutic intervention.
Figure I. Ion conduction pathway in K+ channels.
(A) Coordination of K+ in the conduction pathway of the KcsA channel. (B) Hydratation of a K+ ion in solution. (C) Selectivity filter sites suggested by Molecular Dynamics (MD) simulations. (D) Network of hydrogen bonds, salt-bridges and electrostatic interactions between the selectivity filter and the adjacent pore helix domain. (adapted from [49,56,61]).

Highlights.
K+ channels produce K+ influx that decreases cell excitability
Gene mutations altering K+-selectivity of K+ channels produce disorders in mouse and human
TWIK1 K+ channels become permeable to Na+ upon hypokalemia
TWIK1 K+ channels become permeable to Na+ upon acidification
This dynamic selectivity relies on specific residues in the pore domain
Sequence conservation suggests that dynamic selectivity has been conserved during evolution
ACKNOWLEDGMENTS
We thank Ben G. Szaro and David Tieman for comments on the manuscript.
This work was supported by the Fondation pour la Recherche Médicale (Equipe labellisée FRM 2011 to FL) and by the Agence Nationale de la Recherche (Laboratory of Excellence “Ion Channel Science and Therapeutics“, grant ANR-11-LABX-0015-01 to FL) as well as by National Institutes of Health (1R01GM102943-01A1 to HC) through the NIGMS, and American Heart Association (11GRNT7270014 to HC).
REFERENCES
- 1.Kofuji P, et al. Functional analysis of the weaver mutant GIRK2 K+ channel and rescue of weaver granule cells. Neuron. 1996;16(5):941–952. doi: 10.1016/s0896-6273(00)80117-8. [DOI] [PubMed] [Google Scholar]
- 2.Navarro B, et al. Nonselective and G betagamma-insensitive weaver K+ channels. Science. 1996;272(5270):1950–1953. doi: 10.1126/science.272.5270.1950. [DOI] [PubMed] [Google Scholar]
- 3.Choi M, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science. 2011;331(6018):768–772. doi: 10.1126/science.1198785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scholl UI, et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc Natl Acad Sci U S A. 2012;109(7):2533–2538. doi: 10.1073/pnas.1121407109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charmandari E, et al. A novel point mutation in the KCNJ5 gene causing primary hyperaldosteronism and early-onset autosomal dominant hypertension. The Journal of clinical endocrinology and metabolism. 2012;97(8):E1532–E1539. doi: 10.1210/jc.2012-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azizan EA, et al. Somatic mutations affecting the selectivity filter of KCNJ5 are frequent in 2 large unselected collections of adrenal aldosteronomas. Hypertension. 2012;59(3):587–591. doi: 10.1161/HYPERTENSIONAHA.111.186239. [DOI] [PubMed] [Google Scholar]
- 7.Akerstrom T, et al. Comprehensive re-sequencing of adrenal aldosterone producing lesions reveal three somatic mutations near the KCNJ5 potassium channel selectivity filter. PloS one. 2012;7(7):e41926. doi: 10.1371/journal.pone.0041926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lees-Miller JP, et al. Novel gain-of-function mechanism in K(+) channel-related long-QT syndrome: altered gating and selectivity in the HERG1 N629D mutant. Circ Res. 2000;86(5):507–513. doi: 10.1161/01.res.86.5.507. [DOI] [PubMed] [Google Scholar]
- 9.Heginbotham L, et al. Mutations in the K+ channel signature sequence. Biophys J. 1994;66(4):1061–1067. doi: 10.1016/S0006-3495(94)80887-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou Y, et al. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 2001;414(6859):43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 11.Doyle DA, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280(5360):69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 12.Dibb KM, et al. Molecular basis of ion selectivity, block, and rectification of the inward rectifier Kir3.1/Kir3.4 K(+) channel. J Biol Chem. 2003;278(49):49537–49548. doi: 10.1074/jbc.M307723200. [DOI] [PubMed] [Google Scholar]
- 13.Ma L, et al. TWIK-1 two-pore domain potassium channels change ion selectivity and conduct inward leak sodium currents in hypokalemia. Science signaling. 2011;4(176):ra37. doi: 10.1126/scisignal.2001726. [DOI] [PubMed] [Google Scholar]
- 14.Ma L, et al. Acid-sensitive TWIK and TASK Two-pore Domain Potassium Channels Change Ion Selectivity and Become Permeable to Sodium in Extracellular Acidification. J Biol Chem. 2012;287(44):37145–37153. doi: 10.1074/jbc.M112.398164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chatelain FC, et al. TWIK1, a unique background channel with variable ion selectivity. Proc Natl Acad Sci U S A. 2012;109(14):5499–5504. doi: 10.1073/pnas.1201132109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hess EJ. Identification of the weaver mouse mutation: the end of the beginning. Neuron. 1996;16(6):1073–1076. doi: 10.1016/s0896-6273(00)80133-6. [DOI] [PubMed] [Google Scholar]
- 17.Hibino H, et al. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90(1):291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 18.Bettahi I, et al. Contribution of the Kir3.1 subunit to the muscarinic-gated atrial potassium channel IKACh. J Biol Chem. 2002;277(50):48282–48288. doi: 10.1074/jbc.M209599200. [DOI] [PubMed] [Google Scholar]
- 19.Murthy M, et al. Characterization of a novel somatic KCNJ5 mutation delI157 in an aldosterone-producing adenoma. J Hypertens. 2012;30(9):1827–1833. doi: 10.1097/HJH.0b013e328356139f. [DOI] [PubMed] [Google Scholar]
- 20.Lu JT, Kass RS. Recent progress in congenital long QT syndrome. Current opinion in cardiology. 2010;25(3):216–221. doi: 10.1097/HCO.0b013e32833846b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zaza A. Serum potassium and arrhythmias. Europace. 2009;11(4):421–422. doi: 10.1093/europace/eup005. [DOI] [PubMed] [Google Scholar]
- 22.Rastegar A, Soleimani M. Hypokalaemia and hyperkalaemia. Postgrad Med J. 2001;77(914):759–764. doi: 10.1136/pmj.77.914.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gurney A, Manoury B. Two-pore potassium channels in the cardiovascular system. Eur Biophys J. 2009;38(3):305–318. doi: 10.1007/s00249-008-0326-8. [DOI] [PubMed] [Google Scholar]
- 24.McCullough JR, et al. Two stable levels of diastolic potential at physiological K+ concentrations in human ventricular myocardial cells. Circ Res. 1990;66(1):191–201. doi: 10.1161/01.res.66.1.191. [DOI] [PubMed] [Google Scholar]
- 25.Christe G. Effects of low [K+]o on the electrical activity of human cardiac ventricular and Purkinje cells. Cardiovasc Res. 1983;17(4):243–250. doi: 10.1093/cvr/17.4.243. [DOI] [PubMed] [Google Scholar]
- 26.McCullough JR, et al. Intra- and extracellular potassium activities and the potassium equilibrium potential in partially depolarized human atrial cells. J Mol Cell Cardiol. 1987;19(5):477–486. doi: 10.1016/s0022-2828(87)80399-1. [DOI] [PubMed] [Google Scholar]
- 27.Sheu SS, et al. Intra- and extracellular K+ and Na+ activities and resting membrane potential in sheep cardiac purkinje strands. Circ Res. 1980;47(5):692–700. doi: 10.1161/01.res.47.5.692. [DOI] [PubMed] [Google Scholar]
- 28.Gadsby DC, Cranefield PF. Two levels of resting potential in cardiac Purkinje fibers. J Gen Physiol. 1977;70(6):725–746. doi: 10.1085/jgp.70.6.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geukes Foppen RJ, et al. Effects of chloride transport on bistable behaviour of the membrane potential in mouse skeletal muscle. J Physiol. 2002;542(Pt 1):181–191. doi: 10.1113/jphysiol.2001.013298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Struyk AF, Cannon SC. Paradoxical depolarization of BA2+-treated muscle exposed to low extracellular K+: insights into resting potential abnormalities in hypokalemic paralysis. Muscle Nerve. 2008;37(3):326–337. doi: 10.1002/mus.20928. [DOI] [PubMed] [Google Scholar]
- 31.Enyedi P, Czirjak G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev. 2010;90(2):559–605. doi: 10.1152/physrev.00029.2009. [DOI] [PubMed] [Google Scholar]
- 32.Gaborit N, et al. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol. 2007;582(Pt 2):675–693. doi: 10.1113/jphysiol.2006.126714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ordog B, et al. Gene expression profiling of human cardiac potassium and sodium channels. Int J Cardiol. 2006;111(3):386–393. doi: 10.1016/j.ijcard.2005.07.063. [DOI] [PubMed] [Google Scholar]
- 34.Nie X, et al. Expression and insights on function of potassium channel TWIK-1 in mouse kidney. Pflugers Arch. 2005;451(3):479–488. doi: 10.1007/s00424-005-1480-9. [DOI] [PubMed] [Google Scholar]
- 35.Decressac S, et al. ARF6-dependent interaction of the TWIK1 K+ channel with EFA6, a GDP/GTP exchange factor for ARF6. EMBO Rep. 2004;5(12):1171–1175. doi: 10.1038/sj.embor.7400292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feliciangeli S, et al. Potassium channel silencing by constitutive endocytosis and intracellular sequestration. J Biol Chem. 2010;285(7):4798–4805. doi: 10.1074/jbc.M109.078535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lesage F, Barhanin J. Molecular physiology of pH-sensitive background K(2P) channels. Physiology (Bethesda) 2011;26(6):424–437. doi: 10.1152/physiol.00029.2011. [DOI] [PubMed] [Google Scholar]
- 38.Rajan S, et al. Sumoylation silences the plasma membrane leak K+ channel K2P1. Cell. 2005;121(1):37–47. doi: 10.1016/j.cell.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 39.Bagriantsev SN, et al. Multiple modalities converge on a common gate to control K2P channel function. EMBO J. 2011;30(17):3594–3606. doi: 10.1038/emboj.2011.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCoy JG, Nimigean CM. Structural correlates of selectivity and inactivation in potassium channels. Biochimica et biophysica acta. 2012;1818(2):272–285. doi: 10.1016/j.bbamem.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kiss L, et al. Contribution of the selectivity filter to inactivation in potassium channels. Biophys J. 1999;76(1):253–263. doi: 10.1016/S0006-3495(99)77194-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lesage F, et al. TWIK-1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. Embo J. 1996;15(5):1004–1011. [PMC free article] [PubMed] [Google Scholar]
- 43.Thomas D, et al. Alternative translation initiation in rat brain yields K2P2.1 potassium channels permeable to sodium. Neuron. 2008;58(6):859–870. doi: 10.1016/j.neuron.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim D, Gnatenco C. TASK-5, a new member of the tandem-pore K(+) channel family. Biochem Biophys Res Commun. 2001;284(4):923–930. doi: 10.1006/bbrc.2001.5064. [DOI] [PubMed] [Google Scholar]
- 45.Karschin C, et al. Expression pattern in brain of TASK-1, TASK-3, and a tandem pore domain K(+) channel subunit, TASK-5, associated with the central auditory nervous system. Molecular and cellular neurosciences. 2001;18(6):632–648. doi: 10.1006/mcne.2001.1045. [DOI] [PubMed] [Google Scholar]
- 46.Salkoff L, et al. Potassium channels in C. elegans. WormBook. 2005:1–15. doi: 10.1895/wormbook.1.42.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salkoff L, et al. Evolution tunes the excitability of individual neurons. Neuroscience. 2001;103(4):853–859. doi: 10.1016/s0306-4522(01)00079-3. [DOI] [PubMed] [Google Scholar]
- 48.Kim I, Allen TW. On the selective ion binding hypothesis for potassium channels. Proc Natl Acad Sci U S A. 2011;108(44):17963–17968. doi: 10.1073/pnas.1110735108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nimigean CM, Allen TW. Origins of ion selectivity in potassium channels from the perspective of channel block. J Gen Physiol. 2011;137(5):405–413. doi: 10.1085/jgp.201010551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thompson AN, et al. Mechanism of potassium-channel selectivity revealed by Na(+) and Li(+) binding sites within the KcsA pore. Nat Struct Mol Biol. 2009;16(12):1317–1324. doi: 10.1038/nsmb.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Egwolf B, Roux B. Ion selectivity of the KcsA channel: a perspective from multi-ion free energy landscapes. J Mol Biol. 2010;401(5):831–842. doi: 10.1016/j.jmb.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou Y, MacKinnon R. The occupancy of ions in the K+ selectivity filter: charge balance and coupling of ion binding to a protein conformational change underlie high conduction rates. J Mol Biol. 2003;333(5):965–975. doi: 10.1016/j.jmb.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 53.Ye S, et al. Novel insights into K+ selectivity from high-resolution structures of an open K+ channel pore. Nat Struct Mol Biol. 2010;17(8):1019–1023. doi: 10.1038/nsmb.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Korn SJ, Ikeda SR. Permeation selectivity by competition in a delayed rectifier potassium channel. Science. 1995;269(5222):410–412. doi: 10.1126/science.7618108. [DOI] [PubMed] [Google Scholar]
- 55.Shin N, et al. Sodium permeability of a cloned small-conductance calcium-activated potassium channel. Biophys J. 2005;89(5):3111–3119. doi: 10.1529/biophysj.105.069542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cordero-Morales JF, et al. A multipoint hydrogen-bond network underlying KcsA C-type inactivation. Biophys J. 2011;100(10):2387–2393. doi: 10.1016/j.bpj.2011.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cordero-Morales JF, et al. Molecular driving forces determining potassium channel slow inactivation. Nat Struct Mol Biol. 2007;14(11):1062–1069. doi: 10.1038/nsmb1309. [DOI] [PubMed] [Google Scholar]
- 58.Sauer DB, et al. Protein interactions central to stabilizing the K+ channel selectivity filter in a four-sited configuration for selective K+ permeation. Proc Natl Acad Sci U S A. 2011;108(40):16634–16639. doi: 10.1073/pnas.1111688108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brohawn SG, et al. Crystal structure of the human K2P TRAAK, a lipid- and mechano-sensitive K+ ion channel. Science. 2012;335(6067):436–441. doi: 10.1126/science.1213808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller AN, Long SB. Crystal structure of the human two-pore domain potassium channel K2P1. Science. 2012;335(6067):432–436. doi: 10.1126/science.1213274. [DOI] [PubMed] [Google Scholar]
- 61.Alam A, Jiang Y. Structural studies of ion selectivity in tetrameric cation channels. J Gen Physiol. 2011;137(5):397–403. doi: 10.1085/jgp.201010546. [DOI] [PMC free article] [PubMed] [Google Scholar]