

Abstract
Cardiomyopathies, diseases of the heart muscle, are major causes of morbidity and mortality. A significant percentage of patients with cardiomyopathies have genetic-based, inheritable disease and, over the past two decades the genetic causes of these disorders have been increasingly discovered. The genes causing these disorders when they are mutated appear to encode proteins that frame a “final common pathway” for that specific disorder but the specifics of the phenotype, including age of onset, severity, and outcome is variable for reasons not yet understood. The “final common pathways” for the classified forms of cardiomyopathy include the sarcomere in the primarily diastolic dysfunction disorders hypertrophic cardiomyopathy (HCM) and restrictive cardiomyopathy (RCM), the linkage of the sarcomere and sarcolemma in the systolic dysfunction disorder dilated cardiomyopathy (DCM), and the desmosome in arrhythmogenic cardiomyopathy (AVC). Left ventricular noncompaction cardiomyopathy (LVNC) is an overlap disorder and appears that any of these “final common pathways” can be involved depending on the specific form of LVNC. The genetics and mechanisms responsible for these clinical phenotypes will be described.
INTRODUCTION
Cardiomyopathies are major causes of morbidity and mortality and over the past twenty-five years, limited improvements in outcome have been reported [1, 2]. However, improvement in the understanding of the major forms of cardiomyopathy has occurred over that time, in large part due to advances in imaging, genetics and genomics [3, 4]. A new classification scheme was recently developed for the cardiomyopathies in which five forms of disease were formally classified as distinct forms of cardiomyopathy: DCM, HCM, RCM, AVC, and LVNC [5]. These were further classified into genetic/inherited forms and acquired/non-inherited forms. All forms occur at all ages except AVC, which is almost always identified in teenagers and young adults.
Our understanding of the genetic causes of cardiomyopathy has expended significantly over the last decade. In many cases, the genetic and mechanistic causes of these disorders follow a disturbance in a particular, disease-specific “final common pathway” [6]. For instance, HCM is now viewed as predominantly an inherited abnormality of contractile protein function (“disease of the sarcomere”). Progress in understanding the genetics of familial DCM has been complicated by its heterogeneous etiologies but a genetic cause is thought to occur in approximately 50% of subjects, higher in children [3-5]. The genes identified to date as causative appear to disturb the functional link between the cytoskeleton and sarcomere most commonly [4]. Multiple genes have been identified for AVC, most resulting in disturbed desmosome/intercalated disk function [4, 7]. Although understanding the genetic basis for the development of RCM and LVNC has been more elusive, genes for both have been identified and appear to include sarcomere dysfunction as critical factors [8].
In this review, we will review the current genetic knowledge of each cardiomyopathy phenotype and will describe how disruption of these “final common pathways” leads to DCM (sarcolemma-sarcomere link), HCM (sarcomere), RCM (sarcomere), AVC (desmosome), and LVNC (sarcolemma and sarcomere).
FINAL COMMON PATHWAYS
Over the past two decades, many of the genes responsible for the development of the different cardiomyopathies have been identified. In an initial attempt to target genes for genetic study, we developed the “final common pathway hypothesis” which stated that genes encoding proteins with similar functions or involved in the same pathway are responsible for a particular disease or syndrome phenotype [6]. As a result, we and others identified structure-function similarities of proteins encoded by genes that, when disrupted, led to a somewhat predictable gross clinical phenotype. For instance, the genes identified a causative for inherited arrhythmia disorders tend to encode for ion channels, those for HCM encode for sarcomeric proteins, and AVCs by genes encoding cell-cell junction proteins, as examples (Figure 1). In addition, it appears that the proteins disturbed by the mutated gene directly disrupts the normal function of the structures in which it is integrated (such as the sarcomere in HCM when the mutated gene encodes a sarcomeric protein) most commonly but, in some instances, disrupt a binding partner protein that causes downstream disturbance of the “final common pathway” (such as a Z-disk protein disrupting the cell-cell junction via a maladaptive binding to desmin which negatively interacts with a desmosomal protein causing AVC) (Figure 1).
Figure 1. Final Common Pathway Hypothesis.
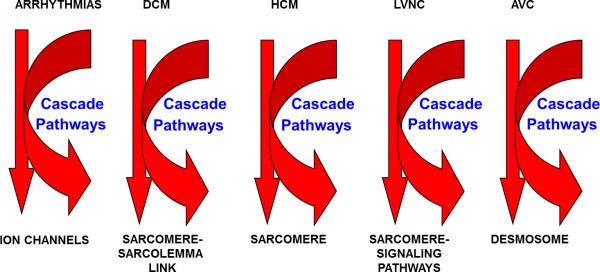
Illustration outlining the proposed “final common pathways” for arrhythmia disorders, dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM) or restrictive cardiomyopathy (RCM), left ventricular noncompaction (LVNC), or arrhythmic ventricular cardiomyopathy (AVC).
DILATED CARDIOMYOPATHY
Dilated cardiomyopathy (DCM) is the most common cardiomyopathy, accounting for approximately 55% of cardiomyopathies [5]. The annual incidence is 2 to 8/100,000 (0.57/100 000/year in children) with an estimated prevalence of 1/2,500 population [5, 9]. The percentage of cases with a genetic etiology is approximately 30-50% based on the presence of a family history [10-12]. DCM is characterized by LV dilation and systolic dysfunction (a reduction in myocardial force generation) and is the most common indication for cardiac transplantation (Figure 2) [5, 9-12]. Clinical manifestations include heart failure (HF), thromboembolism, and sudden cardiac death (SCD). The age at onset includes newborn through late adulthood, although most patients are diagnosed between 20-50 years of age [12].
Figure 2. Echocardiographic features of cardiomyopathies.

Panel A. 4-Chamber echocardiographic view of dilated cardiomyopathy (DCM). Note the dilated left ventricle (LV; arrow); Panel B. 4-Chamber echocardiographic view of hypertrophic cardiomyopathy (HCM). Note the thickened mid-portion of the interventricular septum (arrow); Panel C. 4-Chamber echocardiographic view of restrictive cardiomyopathy (RCM). Note the dilated atria (RA, LA; arrows); Panel D. 4-Chamber echocardiographic view of left ventricular noncompaction cardiomyopathy (LVNC). Note the hypertrabaeculation in the LV (arrow); Panel E. 4-Chamber echocardiographic view of arrhythmogenic right ventricular cardiomyopathy (ARVC). Note the dilated, trabeculated right ventricle (RV; arrow); Commonly, an aneurysm of the RV or RV outflow tract can be seen, particularly by MRI.
The genetic forms of DCM usually result from mutated genes encoding two major subgroups of proteins, cytoskeletal and sarcomeric proteins [3, 4, 6, 13]. DCM can also present with muscular involvement and may be the presenting or primary clinical feature of several multi-system conditions, including Emery-Dreifuss muscular dystrophy (EDMD), Barth syndrome, myofibrillar myopathy, limb-girdle muscular dystrophy (LGMD), and Duchenne or Becker muscular dystrophy (DMD/BMD) [14].
GENETICS OF DILATED CARDIOMYOPATHY
Inherited, familial DCM (FDCM) has been shown to occur in 30-50% of cases of DCM with autosomal dominant inheritance being the predominant pattern of transmission; X-linked, autosomal recessive, and mitochondrial inheritance is less common [10-12, 15, 16]. Over the past 20+ years, substantial progress has been made in the understanding of the genetic etiology of FDCM (Table 1). Initial progress was made in the early 1990s studying families with X-linked forms of DCM, with the autosomal dominant forms unraveling shortly thereafter.
Table 1.
Dilated Cardiomyopathy (DCM) Genetics
| GENE | GENE NAME |
|---|---|
| ABCC9 | ATP-Binding cassette, sub-family C, ,ember 9 |
| ACTC | α-Cardiac Actin |
| ACTN2 | α-Actinin2 |
| ANKRD1 | Cardiac Ankyrin Repeat, Domain 1 |
| BAG3 | BCL2-associated athanogene 3 |
| CSRP3 | Cysteine and Glycine-rich Protein 3 |
| CTF1 | Cardiotrophin 1 |
| DES | Desmin |
| DYS | Dystrophin |
| DNAJC19 | DnaJ (Hsp40) homolog, subfamily C, member 19 |
| DSC2 | Desmocollin 2 |
| DSG2 | Desmoglin 2 |
| DSP | Desmoplakin |
| EMD | Emerin |
| EYA4 | Eyes absent homolog 4 |
| FHL2 | Four and a half LIM domains 2 |
| FKTN | Fukutin |
| FOXD4 | Forkhead box D4 |
| LAMA4 | α4-Laminin |
| LAMP2 | Lysosomal-associated membrane protein 3 |
| LDB3 | LIM-domain binding 3 |
| LMNA | Lamin A/C |
| MYBPC3 | Myosin Binding Protein C |
| MYH6 | β-Myosin Heavy Chain 6 |
| MYH7 | β-Myosin Heavy Chain 7 |
| MYPN | Myopalladin |
| NEBL | Nebulette |
| NEXN | Nexilin (F actin binding protein) |
| PLN | Phospholamban |
| PSEN1 | Presenilin 1 |
| PSEN2 | Presenilin 2 |
| RBM20 | RNA binding motif protein 20 |
| SCN5A | Voltage-gated sodium channel, α subunit |
| SDHA | Succinate dehydrogenase complex, subunit A, flavoprotein |
| SGCD | δ-Sarcoglycan |
| SYNE1 | Spectrin repeat containing nuclear protein 1 |
| SYNE2 | Spectrin repeat containing nuclear protein 2 |
| TAZ | Tafazzin |
| TCAP | Titin-cap (Telethonin) |
| TMPO | Thymopoeitin |
| TNNC1 | Cardiac Troponin C, type 1 |
| TNNI3 | Cardiac Troponin I, type 3 |
| TNNT2 | Cardiac Troponin T, type 2 |
| TPM1 | α-Tropomyosin 1 |
| TTN | Titin |
| TTR | Transthyretin |
| VCL | Vinculin |
X-linked DCM (XLCM), occurring in males in the teen years and early twenties with rapid progression from HF to death due to arrhythmias or transplantation, is distinguished by elevated serum creatine kinase muscle isoforms (CK-MM) [16, 17]. Female carriers develop milder, slowly progressive DCM (fifth decade). Towbin et al (1993) identified the disease-causing gene as dystrophin and demonstrated significant reduction or absence of dystrophin protein in the heart [17]. These findings were later confirmed by Muntoni et al and others, with mutations generally occurring in the 5’ end of the gene [18, 19]. Dystrophinlinks the sarcomere to the sarcolemma and extracellular matrix (ECM) (Figure 3)[16]. When mutated, dystrophin causes DMD/BMD, skeletal myopathies that present in males early in life with elevated CK-MM and lead to the early need for wheelchairs (DMD, before age 12 years; BMD, older than 16 years of age) [20-22] with DCM usually occurring between10-25 years [16-22].
Figure 3. Cardiac myocyte cytoarchitecture.

Schematic illustration of key proteins of the extracellular matrix (ECM), sarcolemma (SL), sarcomere (SM), and nucleus.and some of their protein-protein interactions. Note the sarcolemmal proteins include ion channels such as SCN5A, L-type calcium channels and others, as well as the dystrophin associated binding proteins that interact with dystrophin and other cytoplasmic cytoskeletal proteins(dystroglycans, sarcoglycans, syntrophins, dystrobrevin, sarcospan, caveolin), and cadherins that bind with desmosomal proteins (desmocollin, plakophillin, desmoplakin, plakoglobin). The integral sarcolemmal membrane proteins interact with the extracellular matrix via α-dystroglycan-laminin α2 connections. The sarcomere includes thick and thin filament contractile proteins and Z-disk proteins. The amino-terminus of dystrophin binds actin and connects dystrophin with the sarcomere intracellularly, the sarcolemma and extracellular matrix. The nucleus includes important membrane proteins lamin A/C and emerin. The intermediate filament protein desmin is another important and prominent linker protein. MLP=muscle LIM protein.
The most common form of inherited DCM has autosomal dominant inheritance [6, 13, 15], presenting either as classic “pure” DCM or DCM associated with early-onset conduction system disease (CDDC). Genetic heterogeneity exists for autosomal dominant DCM with more than 40 genes identified for DCM and CDDC [3, 4, 13, 23, 24], with most genes encoding either cytoskeletal, sarcomeric, or Z-disk proteins, although mutations in a small number of ion channel-encoding and desmosome-encoding genes have also been identified (Table 1). These include genes primarily encoding cytoskeletal (δ-sarcoglycan (SGCD), β–sarcoglycan (SGCB), desmin (DES), lamin A/C (LMNA), vinculin (VCL)), sarcomeric/myofibrillar (α-cardiac actin (ACTC), troponin T (TNNT2), troponin I (TNNI3), β–myosin heavy chain (MYH7), myosin binding protein C (MBPC3), and α-tropomyosin (TPM1)), and Z-disk proteins (muscle LIM protein (MLP)/ cysteine and glycine-rich protein 3 (CSRP3), titin (TTN), telethonin/TCAP, α-actinin-2 (ACTN2), nebulette (NEBL), myopalladin (MYPN), ANKRD1/CARP, and ZASP/ LIM-domain binding 3 (LBD3) (Table 1, Figure 3). Ion channel-encoding genes identified to date include the cardiac sodium channel gene SCN5A and calcium homeostasis regulator phospholamban (PLN) [23, 24] while associated desmosome-encoding genes include desmoplakin (DSP), desmoglein-2 (DSG2), and desmocolin-2 (DSC2) have also been shown to result in a DCM phenotype (Table 1, Figure 3) [25]. In the case of CDDC, LMNA is the most common cause, but other causes include DES and SCN5A (Table 1, Figure 3) [26, 27]. Mechanistically, cytoskeletal proteins are thought to cause defects of force transmission resulting in the DCM phenotype, while defects of force generation have been speculated to be associated with sarcomere protein-induced DCM [28, 29]. The altered desmosomal proteins appear to disrupt the links between the intercalated disk, Z-disk, and sarcomere [30,31]. Purevjav et al recently showed that a gene mutation may disrupt protein binding partners and, depending on this interaction, differential phenotypes and severity may occur [32, 33].
Mutations in the sarcomere may produce variable forms of cardiomyopathy (Tables 1-4, Figure 3). As previously noted, abnormalities in force generation or transmission are thought to contribute to the development of DCM [28, 29, 34, 35]. α-Cardiac actin, a sarcomeric thin filament protein that interacts with tropomyosin and the troponin complex, links the sarcomere to the sarcolemma via its binding to the N-terminus of dystrophin. DCM-causing actin mutations disturb dystrophin-actin binding [36, 37], causing force transmission abnormalities and DCM. Further, actin interacts in the sarcomere with Troponin T and β-myosin heavy chain; mutations in these genes cause DCM or HCM, depending on mutation position [37] via development of force generation abnormalities. For MYH7, this probably occurs by perturbing the actin-myosin interaction or altering cross-bridge movement during contraction [34-37]. TNNT2 mutations disrupt calcium-sensitive troponin C binding [34-37]. Mutations in PLN have also been identified which further support calcium handling as a potentially important mechanism in DCM development [34-37]. α-tropomyosin (TPM1) mutations alter its surface charge, causing impaired interaction with actin and potentially changing its secondary structure, its binding to actin and position on the thin filament, and alternating actin-myosin interactions and myofilament Ca(2+) sensitivity.
Table 4.
LVNC Genetics
| GENE | GENE NAME |
|---|---|
| ACTC1 | α-Cardiac Actin |
| CASQ2 | Cardiac Calsequestrin 2 |
| DTNA | α-Dystrobrevin |
| DYS | Dystrophin |
| GLA | α-Galactosidase |
| LDB3 | LIM-domain binding 3 |
| LMNA | Lamin A/C |
| MYBPC3 | Myosin Binding Protein C |
| MYH7 | β-Myosin Heavy Chain 7 |
| TAZ | Tafazzin |
| TNNT2 | Cardiac Troponin T, type 2 |
Another molecular level target for DCM is the Z-disk [38-40]. Knoll et al identified mutations in MLP/ CSPR3, which disturbs its interaction with TCAP [39] and its stretch sensor activity. Mutations in CSPR3 cause abnormalities in the T-tubule system and Z-disk architecture as well [41]. Mutations in ACTN2, which binds CSPR3, is involved in crosslinking actin filaments, and shares a common actin-binding domain with dystrophin, disrupts CSPR3 binding and stretch sensing [38-41]. Mutations in ZASP/ LBD3 cause DCM and LVNC, as well as myofibrillar myopathy [38-40, 42]. This protein, which interacts with ACTN2, the FATZ/calsarcin/myozenin protein family, the myotilin (myotilin, myopalladin, palladin) family, and TCAP that together forms a complex with the cardiac sodium channel, NEBL, and phosphoglucomutase 1, disrupts the actin cytoskeleton when mutated [43]. TTN, encoding a giant sarcomeric cytoskeletal protein, contributes to the maintenance of sarcomere organization and myofibrillar elasticity, and interacts with these proteins at the Z disk/I band transition zone and causes DCM or AVC when mutated [44].
Due to significant locus and allelic heterogeneity, genetic testing for DCM has been of limited utility, with pathogenic variants identified in 17-40% of cases using current 40 gene panels, with most genes contributing only a small percentage of pathogenic variants [4, 13, 23, 24]. Clear genotype-phenotype correlations are rare.
MUSCLE IS MUSCLE: CARDIOMYOPATHY AND SKELETAL MYOPATHY GENES OVERLAP
Interestingly, nearly all of the genes identified for inherited DCM are also known to cause skeletal myopathy in humans and/or mouse models: dystrophin mutations cause DMD/BMD, SGCD causes LGMD2F, LMNA causes autosomal dominant EDMD and LGMD1B, ACTC and DES cause nemaline myopathy. In addition, taffazin (TAZ), α-dystrobrevin, LBD3, CSRP3, ACTN2, TTN, and SGCB mutations also cause skeletal myopathy, suggesting that cardiac and skeletal muscle function is interrelated; skeletal muscle fatigue seen in patients with DCM with and without HF may be due to primary skeletal muscle disease and not only related to cardiac dysfunction. It also suggests that striated muscle dysfunction results from disturbed “final common pathways” and both sets of muscles should be evaluated in DCM.
HYPERTROPHIC CARDIOMYOPATHY
Hypertrophic cardiomyopathy (HCM) is characterized by asymmetric or concentric wall thickening (Figure 2) in the absence of an underlying systemic condition or other cardiac disease [5, 6, 13, 16, 45]. The major impact of this disorder on human health is its predilection to be inherited, its reputation as the most common cause of SCD in young, healthy individuals, and its potential to develop HF due to diastolic factors or development of systolic dysfunction [5, 46].
Gene Identification in Familial Hypertrophic Cardiomyopathy
HCM is among the most common inherited cardiac disorders, with a prevalence of 1/500 in young adults [5]. During the last 20+ years, molecular genetic studies have given important insights into the pathogenesis of HCM and provided new perspectives for the diagnosis and management of affected patients. Genetic causes for HCM have been described with over 20 genes discovered to date, most affecting the sarcomere; however, mutations in genes encoding proteins of the Z-disk or intracellular calcium modulators have also been identified (Table 2). Eight causative genes encode sarcomere proteins, with most (~80%) mutations identified in the MYH7 and MYBPC3 genes (Figure 3) [4, 5, 13, 45]. Mutations identified in sarcomeric genes are typically single nucleotide substitutions and in most instances the mutant protein is thought to incorporate into the sarcomere, exerting a poison peptide effect (dominant negative mutations), with the only exception being MYBPC3, where deletions or insertions leading to a frameshift, a truncated protein with no function, resulting in haploinsufficiency occur [47]. In addition to defects in the sarcomere-encoding genes, HCM patients have been identified hosting mutations in Z-disk and other non-sarcomere encoding genes [4, 13, 32, 48]. The giant protein titin and its interactive Z-disc proteins, including MLP, ZASP, telethonin, nexilin, myopalladin, myozenin-2, α-actinin 2, CARP, and vinculin have been identified as causes of HCM when their gene is mutated [48] (Table 2, Figure 3). For Z-disk and calcium modulator genes, the specific mechanism has not been clearly elucidated.
Table 2.
HCM Genetics
| GENE | GENE NAME |
|---|---|
| ACTC | α-Cardiac Actin |
| ACTN2 | α-Actinin2 |
| ANKRD1 | Cardiac Ankyrin Repeat, Domain 1 |
| CAV3 | Caveolin 3 |
| COX15 | COX 15 homolog, cytochrome C oxidase assembly protein |
| CRYAB | Crystallin αB |
| CSRP3 | Cysteine and Glycine-rich Protein 3 |
| GLA | α-Galactosidase |
| LAMP-2 | Lysomal-Associated Membrane Protein 2 |
| MYBPC3 | Myosin Binding Protein C |
| MYH6 | β-Myosin Heavy Chain 6 |
| MYH7 | β-Myosin Heavy Chain 7 |
| MYL2 | Myosin Regulatory Light Chain 2, slow |
| MYL3 | Myosin Light Chain 3, slow |
| MYLK2 | Myosin Light Chain kinase 2 |
| MYO6 | Unconventional Myosin VI |
| MYOZ2 | Myozenin 2 |
| NEXN | Nexilin (F actin binding protein) |
| PLN | Phospholamban |
| PRKAG2 | AMP=activated Protein kinase, γ2, non-catalytic subunit |
| TNNC1 | Cardiac Troponin C, type 1 |
| TAZ | Tafazzin |
| TCAP | Titin-cap (Telethonin) |
| TNNI3 | Cardiac Troponin I, type 3 |
| TNNT2 | Cardiac Troponin T, type 2 |
| TPM1 | α-Tropomyosin 1 |
| TTN | Titin |
| TTR | Transthyretin |
| VCL | Vinculin |
Recent clinical guidelines for HCM recommend comprehensive testing for five HCM genes (MYBPC3, MYH7, TNNI3, TNNT2, and TPM1) although current Nexgen sequencing approaches have facilitated much broader testing panels to be widely available from commercial laboratories [19;http://www.genetests.org and http://www.ncbi.nlm.nih.gov/gtr]. Currently, genetic testing for HCM is primarily used to identify families with a detectable genetic cause of disease and to screen at-risk family members. In our clinic, mutations are seen in approximately 65-70% of all subjects. The use of genetic test results in guiding clinical management is limited due to limited genotype-phenotype correlations. Although almost 1000 variants for HCM have been identified to date [4, 13, 32, 48, 49], most are private and can, therefore, be detected only through comprehensive genetic testing.
Sarcomere variants are identified in up to 60-70% of patients with HCM who also have a family history and in 40-60% with sporadic HCM [13, 49]. Genotype-phenotype correlations for HCM, however, are incompletely defined, in part due to the private nature of the mutations.Further, suggestions that a specific mutation in a specific sarcomeric gene will lead to a specific phenotype and specific outcome have been questioned. The pattern and extent of LVH in patients with HCM, as well as outcomes, is heterogeneous even in first-degree relatives. This heterogeneity has made it difficult to correlate genotype and phenotype, including outcomes. A systematic review and meta-analysis of genotype-phenotype correlations in 2459 patients and 53 families with sarcomeric gene mutations reported in 18 publications showed that presence of any sarcomere gene mutation was associated with younger age at presentation (38.4 vs 46.0 years), family history of HCM (50.6% vs 23.1%), family history of SCD (27.0% vs 14.9%), and greater LV wall thickness (21.0 vs 19.3 mm) [51]. The relationship between cardiac morphology, mutated gene, and outcome has also been evaluated [52-54]. Bos et al studied 359 patients with 34% being genotype-positive for a putative HCM-associated mutation in 1 or more genes. Echocardiographic reverse curve morphological subtype (Figure 4), age at diagnosis younger than 45 years, maximum LV wall thickness of 20 mm or greater, family history of HCM, and family history of SCD were identified as positive predictors of mutation identification; hypertension was a negative predictor [52, 53]. A score, based on the number of predictors of a positive genetic test result, predicted mutation identification in 6% when only hypertension was present to 80% when all 5 positive predictor markers were present.
Figure 4. Septal morphologies in hypertrophic cardiomyopathy (HCM) and the relationship to sarcomeric myofilament-encoding gene mutations.
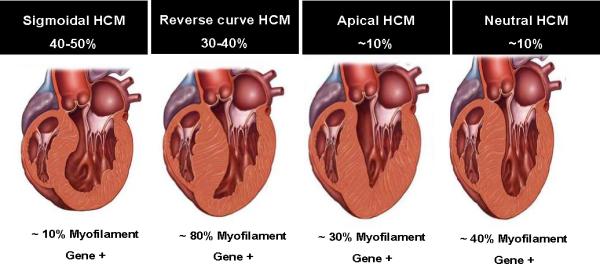
The four subgroups of septal morphology in HCM (sigmoidal, reverse curve, apical, neutral) are shown, along with the percentage of HCM cases represented by each morphology and the likelihood of identifying a mutation in a myofilament-encoding gene. With permission from Bos JM et al. J Am Coll Cardiol 2009, 54: 201-211 (reference 52).
RESTRICTIVE CARDIOMYOPATHY
Restrictive cardiomyopathy (RCM) is the rarest form of cardiomyopathy, characterized by dialted atria, increased ventricular stiffness with normal ventricular wall thickness and systolic function (Figure 2) [5, 8]. SCD or a life-threatening cardiac event due to ventricular arrhythmias or heart block is common, as is HF; death occurs within a few years of diagnosis [5, 8]. Some reports suggest clinical overlap between RCM and HCM [55, 56].
Genetics of Restrictive Cardiomyopathy
RCM usually has autosomal dominant inheritance, but autosomal recessive, X-linked and mitochondrial-transmitted disease occurs. Most identified genes encode sarcomere or Z-disk proteins, such as TNNI3, TNNT2, MYH7, ACTC1, TPM1, MYL3, and MYL2 [8, 57] (Table 3). Z-disk protein-encoding genes, including MYPN, TTN, and Bag3 have also been identified [4, 5, 8, 13, 33]. Missense variants in DES have been identified in several families with desmin-related myopathy, which can present with RCM, with or without skeletal myopathy and/or atrioventricular block [8]. Transthyretin (TTR) is associated with amyloid-related RCM [8].
Table 3.
RCM Genetics
| GENE | GENE NAME |
|---|---|
| ACTC1 | α-Cardiac Actin |
| BAG3 | BCL2-associated athanogene 3 |
| DES | Desmin |
| GLA | α-Galactosidase |
| MYH7 | β-Myosin Heavy Chain 7 |
| MYL2 | Myosin Regulatory Light Chain 2, slow |
| MYL3 | Myosin Light Chain 3, slow |
| MYPN | Myopalladin |
| TNNI3 | Cardiac Troponin I, type 3 |
| TNNT2 | Cardiac Troponin T, type 2 |
| TPM1 | α-Tropomyosin 1 |
| TTN | Titin |
| TTR | Transthyretin |
LEFT VENTRICULAR NONCOMPACTION
Left ventricular noncompaction cardiomyopathy (LVNC) is a heterogeneous myocardial disorder characterized by prominent trabeculae most evident in the LV apex, intra-trabecular recesses, and LV myocardium having two distinct layers: compacted and noncompacted myocardium (Figure 2) [5, 13, 16, 58]. Although LVNC primarily affects the LV, isolated RV and biventricular noncompaction also occurs [58]. Multiple forms of LVNC occur and these include primary myocardial forms, a form associated with arrhythmias, and LVNC associated with congenital heart disease (CHD), including septal defects (ASD, VSD), hypoplastic left heart syndrome (HLHS), and right heart obstructive abnormalities including pulmonic stenosis (PS) and Ebstein's anomaly (Figure 5), among others [58]. Outcomes appear to differ based on the specifics of the phenotype.
Figure 5. Echocardiographic features of the left ventricular noncompaction phenotype heterogeneous clinical characteristics.

Panel A: Four-chamber view demonstrates normal atrial and ventricular sizes and thickness with a hypertrabeculated left ventricular wall and apex (arrow); Panel B: Four-chamber view demonstrates normal atrial sizes with a dilated and hypertrabeculated left ventricular wall and apex (arrow); Panel C: Four-chamber view demonstrates hypertrophic and hypertrabeculated ventricular walls and apex with a hypertrophic septum (arrow); Panel D: Four-chamber view demonstrates normal to small ventricular sizes with dilated atria bilaterally (arrows). The LV is hypertrabeculated. Panel E: Four-chamber view demonstrates normal atrial sizes with severely hypertrabeculated left and right ventricular wall and apices (arrows); Panel F: Four-chamber view demonstrates normal atrial sizes with tricuspid atresia (no tricuspid valve) and a hypertrabeculated left ventricular wall and apex (arrow); Panel G: Four-chamber view of Ebstein's anomaly with LVNC which demonstrates a normal left atrial size with a large right atrium, displaced tricuspid valve towards the right verntricle (closed arrowhead), and a severely hypertrabeculated left ventricular wall and apex (open arrowhead).
LVNC Genetics
LVNC most commonly has X-linked recessive or autosomal dominant inheritance, with reports suggesting 70% having autosomal dominant and 30% X-linked inheritance, at least in childhood [58-60]. Autosomal recessive and mitochondrial inheritance also occurs [58]. When LVNC is associated with CHD, the congenital cardiac defect may be heterogeneous in families but this form of LVNC is transmitted as an autosomal dominant trait along with the CHD; affected members may have no CHD at initial evaluation because the cardiac defects include “minor” forms of CHD (small VSDs, ASDs, patent ductus arteriosus), which have spontaneously normalized or penetrance may be reduced, while others may have severe forms of CHD (HLHS, Ebstein's, etc [58, 61]. Ichida et al. reported that 44% of her patients had inherited LVNC, with 70% having autosomal dominant and 30% X-linked inheritance [59]. In addition, patients with chromosomal abnormalities are associated with LVNC.
The first genetic cause of isolated LVNC was initially described by Bleyl et al. when they identified mutations in the X-linked TAZ gene, the gene also responsible for Barth syndrome, in patients and carrier females (Table 3) [62]. Multiple genes causing autosomal dominant LVNC have now been identified, including mutations in genes causing CHD with LVNC. In patients with HLHS and LVNC, the cytoskeletal gene α-dystrobrevin was identified while mutations in Nkx-2.5 in children with LVNC and ASD and MYH7 in patients with LVNC and Ebstein's anomaly have also been reported (Table 3) [58, 63-66]. In LVNC without CHD, mutations in the Z-line protein-encoding ZASP/LDB3 gene and the sarcomere-encoding genes (MYH7, ACTC, TNNT2, MYBPC3, TMP1, and TNNI3) appear to account for 20% or more of LVNC [58, 67, 68]. Hoedemaekers et al. additionally demonstrated an association of LVNC with genetic variants in 2 calcium handling genes, as well as TAZ and LMNA [68], while Probst et al. further showed that sarcomere gene mutations are important in LVNC, showing a prevalence of 29%, with MYH7 and MYBPC3 most frequently mutated (13% and 8%, respectively) [67]. Dellefave et al. also identified sarcomere mutations in LVNC, including those presenting with HF in infancy with and without compound heterozygosity [69]. In addition to sarcomere-encoding genes and the cytoskeleton, mutations in the sodium channel gene, SCN5A, is associated wtih LVNC and rhythm disturbance [63]. Diagnostic testing in patients with LVNC appear to have a detection rate of clinically significant variants in 35-40% of individuals, with sarcomere-encoding genes most commonly found to be mutated [4, 70].
ARRHYTHMOGENIC CARDIOMYOPATHIES
Arrhythmogenic ventricular cardiomyopathy (AVC), previously referred to as arrhythmogenic right ventricular dysplasia (ARVD) or cardiomyopathy (ARVC) and recently re-named in a consensus statement [5], constitutes a hereditary cardiomyopathy usually with an autosomal dominant inheritance pattern, although a recessive cardiocutaneous disorder also occurs [71]. The RV was initially thought to solely be affected in AVC, and therefore any associated disease of the LV was considered exclusionary for the diagnosis in the original “Task Force criteria” for diagnosis but,more recently, it has been recognized that the LV can also be involved and actually is often associated with severe disease and a worse prognosis [72]. Classically, however, the RV is dilated with fibro-fatty infiltration with no or only minimal LV involvement, [5, 7]. Clinically, progressive disease is characterized by systolic impairment and biventricular dilation (with or without a ventricular aneurysm) with clinical features of HF (Figure 2). In most parts of the world, phenotypic expression is more common in men than in women (2-3:1) [73]. AVC commonly manifests during late childhood or adolescence but can also emerge in the elderly and has a prevalence of 1/2000, which can be higher in certain geographical regions such as the Veneto region or the Greek island Naxos [74]. AVC is recognized as a leading cause of SCD in young adults ≤ 35 years of age and may account for up to 10% of cardiovascular deaths in the < 65 age group [5, 7].
Genetics of Arrhythmogenic Cardiomyopathy
Analyses of first- and second-degree relatives of patients with AVC suggest that up to 50% of AVC cases are familial [5, 7,75]. AVC is most commonly inherited as an autosomal dominant trait with incomplete penetrance [5, 7, 75], although two autosomal recessive forms have been described [71]. To date, 15 genes have been reported to cause AVC (Table 5) [4, 5, 7, 13, 76]. We and others demonstrated that compound and digenic heterozygosity is involved in AVC pathogenesis in up to 20% of cases and leads to a more severe disease [77, 78]. As penetrance is incomplete, genetically affected relatives often demonstrate variable and mild (or no) phenotype and the prevalence of familial disease is often underestimated in clinical practice [13, 73].
Table 5.
AVC Genetics
| GENE | GENE NAME |
|---|---|
| CTNNA3 | αT-Catenin |
| DES | Desmin |
| DNAJC19 | DnaJ (Hsp40) homolog, subfamily C, member 19 |
| DSC2 | Desmocollin 2 |
| DSG2 | Desmoglin 2 |
| DSP | Desmoplakin |
| JUP | Junctional Plakoglobin |
| LMNA | Lamin A/C |
| PKP2 | Plakophilin 2 |
| PKP4 | Plakophilin 4 |
| RYR2 | Ryanodine receptor 2 |
| STRN | Striatin |
| TGFβ3 | Transforming growth factor-β3 |
| TMEM43 | Transmembrane 43 |
| TTN | Titin |
The first gene to be identified in AVC was junctional plakoglobin (JUP) when a homozygous 2-nucleotide deletion in JUP was reported in patients with Naxos disease [7, 71, 73]. This gene encodes the plakoglobin protein, a key protein of the desmosome in the intercalated disk. In autosomal dominant AVC, the desmosomal protein-encoding DSP gene was identified as disease-causing [7, 71, 73].
Three groups of desmosomal proteins are known: (1) transmembrane desmosomal cadherins including DSC2 and DSG2; (2) DSP, a plakin family protein that attaches directly to intermediate filaments (desmin in the myocardium); and (3) linker proteins such as armadillo family proteins including JUP and plakophilin 2 (PKP2) that mediate interactions between the desmosomal cadherin tails and DSP [91]. In about 80% of cases with confirmed pathogenic mutations, PKP2, DSP and DSG2 are altered [74]. Besides desmosomal gene mutations, mutations in genes encoding proteins that interact with desmosomal proteins were found as well. These include: (1) the transforming growth factor β3 that conveys cytokine-stimulating fibrosis and modulates cell adhesion and growth ; (2) the transmembrane protein 43 (TMEM43), which functions as a PPAR-γ response element, an adipogenic transcription factor ; (3) DES, which binds desmoplakin ; and (5) TTN, that bridges the sarcomere along its longitudinal axis and forms a continuous filament along the myofibril (Table 5) [4, 7, 13, 73, 79, 80].
Genetically-determined disruption of intercalated-disk integrity is a key factor promoting the development of AVC and sudden cardiac death. Recent data indicates that loss of desmosomal integrity can substantially affect gap junctions, sodium channel function and electrical propagation, thereby promoting ventricular arrhythmias in the absence of overt structural damage [81, 82], thus providing an overlapping phenotype (cardiomyopathy plus arrhythmias) due to disruption of two “final common pathways” (desmosome and ion channel) [80, 83].
CONCLUSIONS
The cardiomyopathies appear to occur due to disruption of “final common pathways”. These disruptions may be due to purely genetic causes, such as mutations in a single gene that results in a dysfunctional protein, which leads to a domino effect of downstream protein interaction abnormalities and ultimately a phenotype. In other situations, multiple mutations in the same gene (compound heterozygosity) or in different genes (digenic heterozygosity) may lead to a phenotype that may be classic, more severe, or even overlapping with other disease forms. In some cases, different intersecting pathways may become disturbed, resulting in complex phenotypes. Further, acquired causes may play a role by causing to disruption of these functional pathways. For instance, dilated cardiomyopathy results from disruption in the “final common pathway” linking the sarcomere and sarcolemma and mutations in the affected genes are responsible for cardiac and skeletal muscle dysfunction. The mechanisms of disease, which include disruption of the linkage due to protein-protein interaction abnormalities that occur from dysfunctional proteins, as well as the interplay of other factors such as mechanical stress and stretch, are being elucidated in detail with the development of animal models of the human disease. Many of the genes identified are now clinically available in fee-for-service laboratories. Novel therapies have resulted from the improved understanding of this clinical phenotype, as noted above. Similarly, the genetic basis and mechanistic understanding of HCM as a disturbance of sarcomere function has occurred over the past two decades and genetic tests are clinically available. The development of novel targeted therapies has been somewhat slow in coming but is expected to develop in the near future. In the case of LVNC, the “new kid on the block”, genetic understanding is in the early phases. The future of cardiomyopathy care is poised to shift in the next decade due to these new developments, as well as the growing science of stem cell therapy. Since children have “pure” disease states, unfettered by comorbidities, the dream of “cures” of muscle disease (cardiac, skeletal muscle) will likely be realized more fully in this population. We must move toward that goal.
REFERENCES
- 1.Lee CS, Chien CV, Bidwell JT, Gelow JM, Denfeld QE, Creber RM, et al. Comorbidity profiles and inpatient outcomes during hospitalization for heart failure: an analysis of the U.S. Nationwide inpatient sample. BMC Cardiovasc Disord. 2014;14:73. doi: 10.1186/1471-2261-14-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shamszad P, Hall M, Rossano JW, Denfield SW, Knudson JD, Penny DJ, et al. Characteristics and outcomes of heart failure-related intensive care unit admissions in children with cardiomyopathy. J Card Fail. 2013;19:672–677. doi: 10.1016/j.cardfail.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 3.Cahill TJ, Ashrafian H, Watkins H. Genetic cardiomyopathies causing heart failure. Circ Res. 2013;113:660–675. doi: 10.1161/CIRCRESAHA.113.300282. [DOI] [PubMed] [Google Scholar]
- 4.Teekakirikul P, Kelly MA, Rehm HL, Lakdawala NK, Funke BH. Inherited cardiomyopathies: molecular genetics and clinical genetic testing in the postgenomic era. J Mol Diagn. 2013;15:158–167. doi: 10.1016/j.jmoldx.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 6.Bowles NE, Bowles KR, Towbin JA. The “final common pathway” hypothesis and inherited cardiovascular disease. The role of cytoskeletal proteins in dilated cardiomyopathy. Herz. 2000;25:168–175. doi: 10.1007/s000590050003. [DOI] [PubMed] [Google Scholar]
- 7.Ellinor PT, MacRae CA, Thierfelder L. Arrhythmogenic right ventricular cardiomyopathy. Heart Fail Clin. 2010;6:161–177. doi: 10.1016/j.hfc.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 8.Sen-Chowdhry S, Syrris P, McKenna WJ. Genetics of restrictive cardiomyopathy. Heart Fail Clin. 2010;6:179–186. doi: 10.1016/j.hfc.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 9.Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, et al. Incidence, causes and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867–1876. doi: 10.1001/jama.296.15.1867. [DOI] [PubMed] [Google Scholar]
- 10.Fatkin D. members of the CSANZ Cardiac Genetic Diseases Council Writing Group. Guidelines for the diagnosis and management of familial dilated cardiomyopathy. Heart Lung Circ. 2011;20:691–693. doi: 10.1016/j.hlc.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 11.Givertz MM, Mann DL. Epidemiology and natural history of recovery of left ventricular function in recent onset dilated cardiomyopathies. Curr Heart Fail Rep. 2013;10:321–330. doi: 10.1007/s11897-013-0157-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dec GW, Fuster V. Idiopathic dilated cardiomyopathy. N Engl J Med. 1994;331:1564–1575. doi: 10.1056/NEJM199412083312307. [DOI] [PubMed] [Google Scholar]
- 13.Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011;8:1308–1339. doi: 10.1016/j.hrthm.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 14.Finsterer J, Stöllberger C, Wahbi K. Cardiomyopathy in neurological disorders. Cardiovasc Pathol. 2013;22:389–400. doi: 10.1016/j.carpath.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 15.Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA. Heart Failure Society of America. Genetic evaluation of cardiomyopathy--a Heart Failure Society of America practice guideline. J Card Fail. 2009;15:83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Towbin JA, Bowles NE. The failing heart. Nature. 2002;415:227–233. doi: 10.1038/415227a. [DOI] [PubMed] [Google Scholar]
- 17.Towbin JA, Hejtmancik JF, Brink P, Gelb B, Zhu XM, Chamberlain JS, et al. X-linked dilated cardiomyopathy (XLCM): Molecular genetic evidence of linkage to the Duchenne muscular dystrophy gene at the Xp21 locus. Circulation. 1993;87:1854–1865. doi: 10.1161/01.cir.87.6.1854. [DOI] [PubMed] [Google Scholar]
- 18.Muntoni F, Cau M, Ganau A, Congiu R, Arvedi G, Mateddu A, et al. Brief report: Deletion of the dystrophin muscle-specific promoter region associated with X-linked dilated cardiomyopathy. N Engl J Med. 1993;329:921–925. doi: 10.1056/NEJM199309233291304. [DOI] [PubMed] [Google Scholar]
- 19.Diegoli M, Grasso M, Favalli V, Serio A, Gambarin FI, Klersy C, et al. Diagnostic work-up and risk stratification in X-linked dilated cardiomyopathies caused by dystrophin defects. J Am Coll Cardiol. 2011;58:925–934. doi: 10.1016/j.jacc.2011.01.072. [DOI] [PubMed] [Google Scholar]
- 20.Rahimov F, Kunkel LM. The cell biology of disease: cellular and molecular mechanisms underlying muscular dystrophy. J Cell Biol. 2013;201:499–510. doi: 10.1083/jcb.201212142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Constantin B. Dystrophin complex functions as a scaffold for signalling proteins. Biochim Biophys Acta. 2014;1838:635–642. doi: 10.1016/j.bbamem.2013.08.023. [DOI] [PubMed] [Google Scholar]
- 22.Chelly J, Desguerre I. Progressive muscular dystrophies. Handb Clin Neurol. 2013;113:1343–1366. doi: 10.1016/B978-0-444-59565-2.00006-X. [DOI] [PubMed] [Google Scholar]
- 23.Morales A, Hershberger RE. Genetic evaluation of dilated cardiomyopathy. Curr Cardiol Rep. 2013;15:375. doi: 10.1007/s11886-013-0375-1. doi: 10.1007/s11886-013-0375-1. [DOI] [PubMed] [Google Scholar]
- 24.Mestroni L, Taylor MR. Genetics and genetic testing of dilated cardiomyopathy: a new perspective. Discov Med. 2013;15:43–49. [PMC free article] [PubMed] [Google Scholar]
- 25.Garcia-Pavia P, Syrris P, Salas C, Evans A, Mirelis JG, Cobo-Marcos M, et al. Desmosomal protein gene mutations in patients with idiopathic dilated cardiomyopathy undergoing cardiac transplantation: a clinicopathological study. Heart. 2011;97:1744–1752. doi: 10.1136/hrt.2011.227967. [DOI] [PubMed] [Google Scholar]
- 26.Wolf CM, Wang L, Alcalai R, Pizard A, Burgon PG, Ahmad F, et al. Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J Mol Cell Cardiol. 2008;44:293–303. doi: 10.1016/j.yjmcc.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor MR, Slavov D, Ku L, Di Lenarda A, Sinagra G, Carniel E, et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation. 2007;115:1244–1251. doi: 10.1161/CIRCULATIONAHA.106.646778. [DOI] [PubMed] [Google Scholar]
- 28.Debold EP, Schmitt JP, Patlak JB, Beck SE, Moore JR, Seidman JG, et al. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse alpha-cardiac myosin in the laser trap assay. Am J Physiol Heart Circ Physiol. 2007;293:H284–291. doi: 10.1152/ajpheart.00128.2007. [DOI] [PubMed] [Google Scholar]
- 29.Ehler E, Perriard JC. Cardiomyocyte cytoskeleton and myofibrillogenesis in healthy and diseased heart. Heart Fail Rev. 2000;5:259–269. doi: 10.1023/A:1009861504264. [DOI] [PubMed] [Google Scholar]
- 30.Basso C, Czarnowska E, Della Barbera M, Bauce B, Beffagna G, Wlodarska EK, et al. Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: an electron microscopy investigation on endomyocardial biopsies. Eur Heart J. 2006;27:1847–1854. doi: 10.1093/eurheartj/ehl095. [DOI] [PubMed] [Google Scholar]
- 31.Yang Z, Bowles NE, Scherer SE, Taylor MD, Kearney DL, Ge S, et al. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Res. 2006;99:646–655. doi: 10.1161/01.RES.0000241482.19382.c6. [DOI] [PubMed] [Google Scholar]
- 32.Purevjav E, Arimura T, Augustin S, Huby AC, Takagi K, Nunoda S, et al. Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. Hum Mol Genet. 2012;21:2039–2053. doi: 10.1093/hmg/dds022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Purevjav E, Varela J, Morgado M, Kearney DL, Li H, Taylor MD, et al. Nebulette mutations are associated with dilated cardiomyopathy and endocardial fibroelastosis. J Am Coll Cardiol. 2010;56:1493–1502. doi: 10.1016/j.jacc.2010.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopathies. Physiol Ref. 2002;82:945–980. doi: 10.1152/physrev.00012.2002. [DOI] [PubMed] [Google Scholar]
- 35.McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013;123:19–26. doi: 10.1172/JCI62862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olson TM, Michels VV, Thibodeau SN, Tai YS, Keating MT. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science. 1998;280:750–752. doi: 10.1126/science.280.5364.750. [DOI] [PubMed] [Google Scholar]
- 37.Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343:1688–1696. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- 38.Pyle WG, Solaro RJ. At the crossroads of myocardial signaling: The role of Z-discs in intracellular signaling and cardiac function. Circ Res. 2004;94:296–305. doi: 10.1161/01.RES.0000116143.74830.A9. [DOI] [PubMed] [Google Scholar]
- 39.Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen-Schmidt I, Bang ML, et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;11:943–955. doi: 10.1016/s0092-8674(02)01226-6. [DOI] [PubMed] [Google Scholar]
- 40.Vatta M, Sinagra G, Brunelli L, Faulkner G. Remodeling of dystrophin and sarcomeric Z-band occurs in pediatric cardiomyopathies: a unifying mechanism for force transmission defect. J Cardiovasc Med (Hagerstown) 2009;10:149–156. doi: 10.2459/JCM.0b013e328318954c. [DOI] [PubMed] [Google Scholar]
- 41.Mohapatra B, Jimenez S, Lin JH, Bowles KR, Coveler KJ, Marx JG, et al. Mol Genet Metab. 2003;80:207–215. doi: 10.1016/s1096-7192(03)00142-2. [DOI] [PubMed] [Google Scholar]
- 42.Lin X, Ruiz J, Bajraktari I, Ohman R, Banerjee S, Gribble K, et al. Z-disc-associated, Alternatively Spliced, PDZ Motif-containing Protein (ZASP) Mutations in the Actin-binding Domain Cause Disruption of Skeletal Muscle Actin Filaments in Myofibrillar Myopathy. J Biol Chem. 2014;289:13615–13626. doi: 10.1074/jbc.M114.550418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martinelli VC, Kyle WB, Kojic S, Vitulo N, Li Z, Belgrano A, et al. ZASP interacts with the mechanosensing protein Ankrd2 and p53 in the signalling network of striated muscle. PLoS One. 2014;9:e92259. doi: 10.1371/journal.pone.0092259. doi: 10.1371/journal.pone.0092259. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.LeWinter MM, Granzier HL. Cardiac titin and heart disease. J Cardiovasc Pharmacol. 2014;63:207–212. doi: 10.1097/FJC.0000000000000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381:242–255. doi: 10.1016/S0140-6736(12)60397-3. [DOI] [PubMed] [Google Scholar]
- 46.O'Mahony C, Elliott PM. Prevention of sudden cardiac death in hypertrophic cardiomyopathy. Heart. 2014;100:254–260. doi: 10.1136/heartjnl-2012-301996. [DOI] [PubMed] [Google Scholar]
- 47.Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, et al. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. 2009;105:219–222. doi: 10.1161/CIRCRESAHA.109.202440. [DOI] [PubMed] [Google Scholar]
- 48.Lopes LR, Zekavati A, Syrris P, Hubank M, Giambartolomei C, Dalageorgou C, et al. Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J Med Genet. 2013;50:228–239. doi: 10.1136/jmedgenet-2012-101270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ho CY. Genetics and clinical destiny: improving care in hypertrophic cardiomyopathy. Circulation. 2010;122:2430–2440. doi: 10.1161/CIRCULATIONAHA.110.978924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lopes LR, Rahman MS, Elliott PM. A systematic review and meta-analysis of genotypephenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart. 2013;99:1800–1811. doi: 10.1136/heartjnl-2013-303939. [DOI] [PubMed] [Google Scholar]
- 51.Bos JM, Will ML, Gersh BJ, Kruisselbrink TM, Ommen SR, Ackerman MJ. Characterization of a Phenotype-Based Genetic Test Prediction Score for Unrelated Patients with Hypertrophic Cardiomyopathy. Mayo Clin Proc. 2014;89:727–737. doi: 10.1016/j.mayocp.2014.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bos JM, Towbin JA, Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54:201–211. doi: 10.1016/j.jacc.2009.02.075. [DOI] [PubMed] [Google Scholar]
- 53.Theis JL, Bos JM, Bartleson VB, Will ML, Binder J, Vatta M, et al. Echocardiographic-determined septal morphology in Z-disc hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 2006;351:896–902. doi: 10.1016/j.bbrc.2006.10.119. [DOI] [PubMed] [Google Scholar]
- 54.Geske JB, Bos JM, Gersh BJ, Eidem BW, Ackerman MJ. Deformation patterns in genotyped patients with hypertrophic cardiomyopathy. Eur Heart J Cardiovasc Imaging. 2014;15:456–465. doi: 10.1093/ehjci/jet234. [DOI] [PubMed] [Google Scholar]
- 55.Rivenes SM, Kearney DL, Smith EO, Towbin JA, Denfield SW. Sudden death and cardiovascular collapse in children with restrictive cardiomyopathy. Circulation. 2000;102:876–882. doi: 10.1161/01.cir.102.8.876. [DOI] [PubMed] [Google Scholar]
- 56.Walsh MA, Grenier MA, Jefferies JL, Towbin JA, Lorts A, Czosek RJ. Conduction abnormalities in pediatric patients with restrictive cardiomyopathy. Circ Heart Fail. 2012;5:267–273. doi: 10.1161/CIRCHEARTFAILURE.111.964395. [DOI] [PubMed] [Google Scholar]
- 57.Caleshu C, Sakhuja R, Nussbaum RL, Schiller NB, Ursell PC, Eng C, et al. Furthering the link between the sarcomere and primary cardiomyopathies: restrictive cardiomyopathy associated with multiple mutations in genes previously associated with hypertrophic or dilated cardiomyopathy. Am J Med Genet A. 2011;155A:2229–2235. doi: 10.1002/ajmg.a.34097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Towbin JA. Left ventricular noncompaction: a new form of heart failure. Heart Fail Clin. 2010;6:453–469. doi: 10.1016/j.hfc.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 59.Ichida F, Hamamichi Y, Miyawaki T, Ono Y, Kamiya T, Akagi T, et al. Clinical features of isolated noncompaction of the ventricular myocardium: long-term clinical course, hemodynamic properties, and genetic background. J Am Coll Cardiol. 1999;34:233–240. doi: 10.1016/s0735-1097(99)00170-9. [DOI] [PubMed] [Google Scholar]
- 60.Sasse-Klaassen S, Gerull B, Oechslin E, Jenni R, Thierfelder L. Isolated noncompaction of the left ventricular myocardium in the adult is an autosomal dominant disorder in the majority of patients. Am J Med Genet Part A. 2003;119A:162–167. doi: 10.1002/ajmg.a.20075. [DOI] [PubMed] [Google Scholar]
- 61.Stahli BE, Gebhard C, Biaggi P, Klaassen S, Valsangiacomo Buechel E, et al. Left ventricular non-compaction: Prevalence in congenital heart disease. Intl J Cardiol. 2013;167:2477–2481. doi: 10.1016/j.ijcard.2012.05.095. [DOI] [PubMed] [Google Scholar]
- 62.Bleyl SB, Mumford BR, Brown-Harrison MC, Pagotto LT, Carey JC, Pysher TJ, et al. Xq28-linked noncompaction of the left ventricular myocardium: prenatal diagnosis and pathologic analysis of affected individuals. Am J Med Genet. 1997;72:257–265. [PubMed] [Google Scholar]
- 63.Udeoji DU, Philip KJ, Morrissey RP, Phan A, Schwarz ER. Left ventricular noncompaction cardiomyopathy: updated review. Ther Adv Cardiovasc Dis. 2013;7:260–273. doi: 10.1177/1753944713504639. [DOI] [PubMed] [Google Scholar]
- 64.Ouyang P, Saarel E, Bai Y, Luo C, Lv Q, Xu Y, et al. A de novo mutation in NKX2.5 associated with atrial septal defects, ventricular noncompaction, syncope and sudden death. Clin Chim Acta. 2011;412:170–175. doi: 10.1016/j.cca.2010.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Postma AV, van Engelen K, van de Meerakker J, Rahman T, Probst S, Baars MJ, et al. Mutations in the sarcomere gene MYH7 in Ebstein anomaly. Circulation Cardiovasc Genet. 2011;4:43–50. doi: 10.1161/CIRCGENETICS.110.957985. [DOI] [PubMed] [Google Scholar]
- 66.Vermeer AM, van Engelen K, Postma AV, Baars MJ, Christiaans I, De Haij S, et al. Ebstein anomaly associated with left ventricular noncompaction: an autosomal dominant condition that can be caused by mutations in MYH7. Am J Med Genet C Semin Med Genet. 2013;163C:178–184. doi: 10.1002/ajmg.c.31365. [DOI] [PubMed] [Google Scholar]
- 67.Probst S, Oechslin E, Schuler P, Greutmann M, Boyé P, Knirsch W, et al. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circulation Cardiovasc Genet. 2011;4:367–374. doi: 10.1161/CIRCGENETICS.110.959270. [DOI] [PubMed] [Google Scholar]
- 68.Hoedemaekers YM, Caliskan K, Michels M, Frohn-Mulder I, van der Smagt JJ, Phefferkorn JE, et al. The importance of genetic counseling, DNA diagnostics, and cardiologic family screening in left ventricular noncompaction cardiomyopathy. Circulation Cardiovasc Genet. 2010;3:232–239. doi: 10.1161/CIRCGENETICS.109.903898. [DOI] [PubMed] [Google Scholar]
- 69.Dellefave LM, Pytel P, Mewborn S, Mora B, Guris DL, Fedson S, et al. Sarcomere mutations in cardiomyopathy with left ventricular hypertrabeculation. Circulation Cardiovasc Genet. 2009;2:442–449. doi: 10.1161/CIRCGENETICS.109.861955. [DOI] [PubMed] [Google Scholar]
- 70.Shieh JT. Implications of genetic testing in noncompaction/hypertrabeculation. Am J Med Genet C Semin Med Genet. 2013;163C:206–211. doi: 10.1002/ajmg.c.31371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Protonotarios N, Tsatsopoulou A. Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol. 2004;13:185–194. doi: 10.1016/j.carpath.2004.03.609. [DOI] [PubMed] [Google Scholar]
- 72.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Azaouagh A, Churzidse S, Konorza T, Erbel R. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: a review and update. Clin Res Cardiol. 2011;100:383–394. doi: 10.1007/s00392-011-0295-2. [DOI] [PubMed] [Google Scholar]
- 74.Herren T, Gerber PA, Duru F. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: a not so rare “disease of the desmosome” with multiple clinical presentations. Clin Res Cardiol. 2009;98:141–158. doi: 10.1007/s00392-009-0751-4. [DOI] [PubMed] [Google Scholar]
- 75.Hamid MS, Norman M, Quraishi A, Firoozi S, Thaman R, Gimeno JR, et al. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol. 2002;40:1445–1450. doi: 10.1016/s0735-1097(02)02307-0. [DOI] [PubMed] [Google Scholar]
- 76.Te Rijdt WP, Jongbloed JD, de Boer RA, Thiene G, Basso C, van den Berg MP, van Tintelen JP. Clinical utility gene card for: arrhythmogenic right ventricular cardiomyopathy (ARVC). Eur J Hum Genet. 2014;22 doi: 10.1038/ejhg.2013.124. doi: 10.1038/ejhg.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu T, Yang Z, Vatta M, Rampazzo A, Beffagna G, Pilichou K, et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2010;55:587–597. doi: 10.1016/j.jacc.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bauce B, Nava A, Beffagna G, Basso C, Lorenzon A, Smaniotto G, et al. Multiple mutations in desmosomal proteins encoding genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm. 2010;7:22–29. doi: 10.1016/j.hrthm.2009.09.070. [DOI] [PubMed] [Google Scholar]
- 79.Patel DM, Green KJ. Desmosomes in the heart: a review of clinical and mechanistic analyses. Cell Commun Adhes. 2014;21:109–128. doi: 10.3109/15419061.2014.906533. [DOI] [PubMed] [Google Scholar]
- 80.Vatta M, Marcus F, Towbin JA. Arrhythmogenic right ventricular cardiomyopathy: a 'final common pathway' that defines clinical phenotype. Eur Heart J. 2007;28:529–530. doi: 10.1093/eurheartj/ehl530. [DOI] [PubMed] [Google Scholar]
- 81.Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 2010;107:700–714. doi: 10.1161/CIRCRESAHA.110.223412. [DOI] [PubMed] [Google Scholar]
- 82.Noorman M, Hakim S, Kessler E, Groeneweg JA, Cox MG, Asimaki A, et al. Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm. 2013;10:412–419. doi: 10.1016/j.hrthm.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Towbin JA, Lorts A. Arrhythmias and dilated cardiomyopathy common pathogenetic pathways? J Am Coll Cardiol. 2011;57:2169–2171. doi: 10.1016/j.jacc.2010.11.061. [DOI] [PubMed] [Google Scholar]