

Abstract
Atomistic level understanding of interaction of α,β-unsaturated carbonyls with late transition metals is a key prerequisite for rational design of new catalytic materials with the desired selectivity toward C=C or C=O bond hydrogenation. The interaction of this class of compounds with transition metals was investigated on α,β-unsaturated ketone isophorone on Pd(111) as a prototypical system. In this study, infrared reflection–absorption spectroscopy (IRAS), near-edge X-ray absorption fine structure (NEXAFS) experiments, and density functional theory calculations including van der Waals interactions (DFT+vdW) were combined to obtain detailed information on the binding of isophorone to palladium at different coverages and on the effect of preadsorbed hydrogen on the binding and adsorption geometry. According to these experimental observations and the results of theoretical calculations, isophorone adsorbs on Pd(111) in a flat-lying geometry at low coverages. With increasing coverage, both C=C and C=O bonds of isophorone tilt with respect to the surface plane. The tilting is considerably more pronounced for the C=C bond on the pristine Pd(111) surface, indicating a prominent perturbation and structural distortion of the conjugated π system upon interaction with Pd. Preadsorbed hydrogen leads to higher tilting angles of both π bonds, which points to much weaker interaction of isophorone with hydrogen-precovered Pd and suggests the conservation of the in-plane geometry of the conjugated π system. The results of the DFT+vdW calculations provide further insights into the perturbation of the molecular structure of isophorone on Pd(111).
1. Introduction
Chemo- and enantioselectivity in hydrogenation of α,β-unsaturated carbonyls on transition metals is a topic of ongoing research in the field of heterogeneous catalysis. Particularly important is the possibility to tune surface chemistry of multiunsaturated hydrocarbons, such as α,β-unsaturated ketones and aldehydes, and their derivatives since they represent a broad class of valuable intermediates for practically important processes.1−3 For molecules containing both a C=C and a C=O π-bond, such as, e.g., the α,β-unsaturated ketone isophorone, hydrogenation can yield either a saturated ketone (3,3,5-trimethylcyclohexanone), an unsaturated alcohol (isophorol), or a saturated alcohol (trimethylcyclohexanol). To avoid the formation of undesired products and thereby an often difficult and cost-intense separation of the different products, a high selectivity in hydrogenating either the C=C or the C=O bond is desirable. Over Pd catalysts, the hydrogenation of the C=C double bond of isophorone is strongly favored, yielding the saturated ketone in high selectivity (∼100%) and essentially no alcohols.4−6 The origin of this chemoselectivity is, however, not fully understood.
Not only a high chemoselectivity but even high enantioselectivities in hydrogenation reactions can be achieved over heterogeneous catalysts using a chiral modifier7−10 that renders the surface asymmetric. Compared to homogeneous catalysts traditionally applied in enantioselective synthesis, the use of heterogeneous catalysts has operational, economical, and often environmental advantages. A number of different modifiers have been tested for the enantioselective hydrogenation of isophorone, which is a benchmark test molecule for enantioselective hydrogenation reactions of enones.7,11−18 Even though the exact origin of the enantioselectivity during hydrogenation of isophorone is not well understood so far, the specific adsorption geometry of isophorone, particularly the tilting angles of the unsaturated C=C and C=O bonds with respect to the surface plane, in combination with the adsorption geometry of the chiral modifier are usually discussed to play a decisive role in rendering the surface chemistry chiral.16,17 The effects of isophorone coverage and presence of hydrogen on the isophorone adsorption geometry have not been thoroughly investigated so far.
In this work, we investigate the adsorption of isophorone on Pd(111). Infrared reflection absorption spectroscopy (IRAS) and NEXAFS studies were combined to investigate the effects of isophorone coverage and hydrogen coadsorption on the orientations of the C=C and C=O π bonds with respect to the Pd(111) surface plane. Near-edge X-ray absorption fine structure (NEXAFS) has been demonstrated to be a powerful tool that provides electronic and structural information on adsorbed molecules. In the past two decades it was shown that the application of NEXAFS can be extended from small to large organic molecules.19,20 The determination of the orientation of large molecules by C 1s → π* resonances is employed to study the adsorption of organic molecules on metal surfaces,21−24 Langmuir–Blodgett monolayers,25 self-assembly of long-chain alkanes,26 or aromatic molecules.22 More recently high-resolution beamlines enabled spectroscopy with highly resolved additional fine structures, such as vibronic coupling and local vibronic excitations. NEXAFS data of organic molecules that show vibrational fine structure in superposition with the electronic excitation were obtained.27 With regard to catalytic conversions, several NEXAFS studies focus on the interaction of unsaturated hydrocarbons with metal surfaces. Attempts were made to correlate chemoselectivity in hydrogenation of multiple unsaturated compounds with the conformation of the adsorbates on the catalyst surface as determined by NEXAFS.28,29 In this study, infrared spectroscopy is used to complement NEXAFS in determining the adsorbate molecular structure as well as to provide additional information on the geometry of chemical bonds with respect to the substrate. While NEXAFS probes electronic states with very high sensitivity to small adsorbate coverages, IR spectroscopy is a very established tool to study the vibrations of chemical bonds. Complementary density functional theory studies including van der Waals interaction (DFT+vdW) were performed to rationalize the experimental observations.
We show that isophorone adsorbs on Pd(111) in a flat-lying geometry at low coverages preserving the in-plane geometry of the conjugated π system. The tilting angle of both double bonds changes with increasing coverage, however, to a different extent. Preadsorbed hydrogen leads to high tilting angles of both π bonds, which points to the conservation of the in-plane geometry of the conjugated π system and weaker interaction with Pd. DFT+vdW calculations confirm the flat-lying adsorption geometry of isophorone at low coverages. Both experimental and computational results suggest that the C=C bond of isophorone is significantly perturbed by the interaction with Pd, in terms of both the electronic and geometric structure, while the carbonyl bond appears to be hardly affected by the interaction with the metal.
2. Experimental Details and Methods
IRAS experiments were performed at the Fritz-Haber-Institut, Berlin, in an UHV apparatus that has been described in detail before.30 In brief, either normal or deuterium-labeled isophorone (d5-isophorone) was dosed onto the sample cooled to 100 K through a doubly differentially pumped multichannel array molecular beam controlled by valves and shutters. The source was operated at room temperature, and the beam diameter was chosen to exceed the sample size. The Pd(111) single crystal was cleaned prior to use by repeated cycles of Ar+ ion bombardment at room temperature, annealing at 1000 K, and oxidation in 1 × 10–6 mbar O2 at 750 K to remove residual carbon. The final cleaning cycle was stopped after annealing. The flatness and cleanliness of the Pd(111) single-crystal surface were checked by low-energy electron diffraction (LEED) and IRAS of adsorbed CO. IRAS data were acquired using a vacuum Fourier-Transform infrared (FT-IR) spectrometer (Bruker IFS 66v/S) with a spectral resolution of 2 cm–1 and using a mid-infrared (MIR) polarizer and p-polarized IR light. The surface was precovered with hydrogen by dosing 100 L of H2. Isophorone (Acros Organics, 98%) or d5-isophorone (Quotient Bioresearch (Radiochemicals) Limited, 90%) were purified prior to the experiments by repeated freeze–pump–thaw cycles. The stated coverages for IRAS experiments are given in fractions of a monolayer (ML) that were determined by TPD experiments (1 ML is defined as the surface coverage where the multilayer desorption feature begins to appear in the temperature-programmed desorption of isophorone from Pd(111)). The upper limit of a monolayer of isophorone is estimated to approximately 7 × 1014 molecules/cm2.
NEXAFS experiments were performed at the undulator beamline UE52-PGM at the BESSY-II synchrotron facility in Berlin. The Pd(111) single crystal was cleaned as described above by repeated sputtering–annealing–oxidizing cycles. Isophorone was purified by freeze–pump–thaw cycles prior to the exposure. Isophorone was deposited onto the Pd(111) at 100 K by placing the crystal in front of the gas doser. The monolayer coverage of isophorone was determined by the position of the C 1s peak in X-ray photoelectron spectroscopy that was found to change at the onset of a multilayer formation (data not shown) in agreement with literature data.29 To deposit any defined submonolayer coverage of isophorone, correspondingly lower exposure times were used. Following deposition of isophorone onto the Pd(111) crystal, the sample was transferred to a separate analysis chamber for NEXAFS spectra collection. The analysis chamber was equipped with a channeltron detector with a retarding field of 150 V for partial electron yield NEXAFS measurements. The energy of the incident X-ray beam was scanned from 250 to 350 eV with a resolution of 0.1 eV in the range of 280–300 and 0.5 eV elsewhere. Spectra were taken at incident beam angles of 70° and 80° with respect to the sample normal, both with horizontal and vertical polarization with respect to the Pd(111) surface. The spectra were normalized by their C K-edge. The pre-edge peaks were fitted by Gaussian functions. The absorption edge was fitted by a Gaussian onset and a linear decrease toward high energies.19,25 The orientations of the molecular orbitals were calculated from the ratio of the corresponding peak areas in the spectra measured with horizontally and vertically polarized light (the upper estimate of accuracy is ±5°).19
The DFT calculations were carried out using the recently developed PBE+vdWsurf method,31 as implemented in the FHI-aims all-electron code.32 The PBE+vdWsurf method extends pairwise vdW approaches to modeling of adsorbates on surfaces by a synergetic combination of the PBE+vdW method33 for intermolecular interactions with the Lifshitz–Zaremba–Kohn theory34,35 for the nonlocal Coulomb screening within the bulk. We employed the “tight” settings for integration grids and standard numerical atom-centered orbitals basis sets in FHI-aims code. We used the FHI-aims “tier2” basis set for light elements (H, C, and O) and “tier1” for Pd. The scaled zeroth-order regular approximation (ZORA)36 was used to treat relativistic effects for Pd atoms. We built up four-layer Pd slabs with a (4 × 4) unit cell, and each slab was separated by a 20 Å vacuum. The bottom two metal layers were constrained, whereas the uppermost two metal layers and the adsorbate were allowed to fully relax during geometry relaxations. For slab calculations, we used a 3 × 3 × 1 k-points mesh. On the basis of the most stable geometries, infrared vibrational spectra were calculated by a second derivative of the energy from the numerical change of the forces arising from small finite displacements. Six finite displacements were applied to each of the atoms with a value of 0.005 Å.
3. Results and Discussion
The adsorption of isophorone on Pd(111) at 100 K was experimentally studied under well-defined UHV conditions by IRAS and NEXAFS experiments. DFT+vdW simulations of the vibration modes of isophorone were employed to help the interpretation of the IR spectra. In this section, we discuss the adsorption geometry of isophorone, specifically focusing on tilting angles of both unsaturated bonds—C=C and C=O—with respect to the Pd(111) surface. The adsorption geometry of isophorone was investigated as a function of coverage on both pristine and hydrogen-precovered Pd(111).
3.1. Unsaturated C=C and C=O Bonds/Pd(111)
The structural formulas of normal and deuterium-substituted (d5) isophorone are shown in Figure 1. In the latter molecule, all five hydrogen atoms directly attached to the C6 ring were substituted by five deuterium atoms. The IR spectra of isophorone ice, providing a reference for a nonperturbed molecular structure, are displayed in Figure 2 for normal and d5-isophorone. For both molecules, three main spectral regions can be distinguished, which are characteristic of C–H stretching vibrations (2800–3200 cm–1), C=C and C=O stretching (1550–1850 cm–1), as well as for C–H, C–D, and C–C deformation vibrations (<1500 cm–1). While the exact assignment of the stretching and deformation vibrations of C–H, C–D, and C–C bonds is rather complex and will be the subject of a forthcoming publication, the vibrational features corresponding to the C=C and C=O bonds can be clearly identified. In the normal isophorone molecule, the most intense vibration is the C=O stretching mode at 1665 cm–1, which lies very close to the vibrational frequency 1655 cm–1 characteristic for the C=C stretching vibration. As can be clearly seen from the spectrum, these vibrational features strongly overlap making the distinction of both bonds rather difficult. In order to overcome this problem, the ring-substituted d5-isophorone can be used, where the C=C vibrational feature appears at 1620 cm–1, showing a shift by 35 cm–1 as compared to its nonsubstituted counterpart. The C=O bond in the d5-isophorone appears to be hardly affected by the substitution in the ring and remains essentially at the same position as in the nonsubstituted isophorone.
Figure 1.

Molecular structure of isophorone (a) and d5-isophorone (b).
Figure 2.

IR spectra of isophorone ice for the regular (upper trace) and d5-substituted (lower trace) forms formed at 100 K on Pd(111). Three main spectral regions can be distinguished characteristic for C–H stretching (2800–3200 cm–1), C=C and C=O stretching (1550–1850 cm–1), and C–H, C–D, and C–C deformation vibrations (<1500 cm–1).
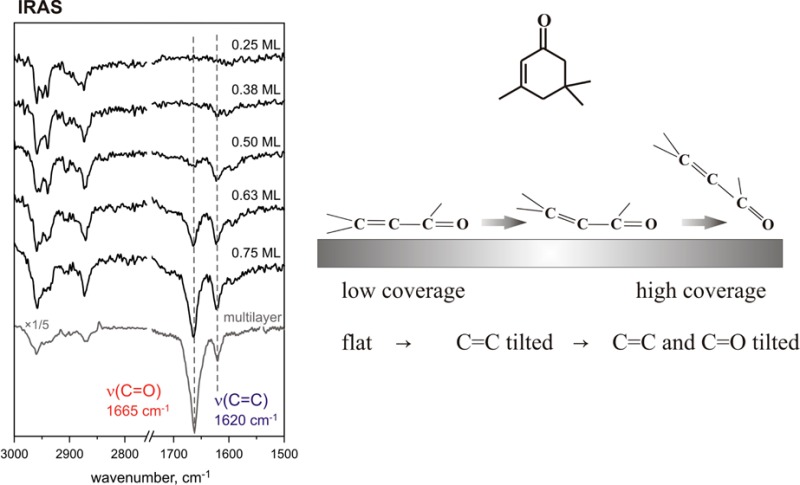
The adsorption geometry of molecular species adsorbed on the metal surface can be deduced from their IRAS spectra based on the metal surface selection rule (MSSR).37,38 According to the MSSR, only vibrations having a projection of the dynamic dipole moment perpendicular to the surface are visible in IRA spectra, while the vibrations parallel to the surface are strongly attenuated due to formation of an image dipole moment in the underlying metal substrate. To determine the orientation of isophorone, particularly the C=C and C=O bonds, a series of IR spectra were obtained at different isophorone coverages spanning the range from 0.25 ML up to 2.40 ML.
Figure 3a shows the IR spectra of d5-isophorone adsorbed on Pd(111) at 100 K at different coverages and the spectrum of isophorone ice for comparison with an unperturbed molecule. In these spectra, two vibrational regions are displayed for simplicity: the region of the C–H stretching vibrations (2800–3000 cm–1) and the region of the C=O and C=C stretching vibrations (1665–1620 cm–1). At the lowest coverage of d5-isophorone (0.25 ML), there is a significant signal in the C–H stretching region; however, there is essentially no signal for the double bonds: neither for the C=O stretching mode nor for the C=C stretching mode. This intensity distribution is in a sharp contrast to the situation found for isophorone ice (see Figure 2 and the last spectrum shown in this series), where the most intense vibrations are that of the C=O and C=C bonds. The absence of absorption bands in the C=C and C=O stretching region indicates that these bonds are either oriented parallel to the surface and therefore cannot be seen because of MSSR or strongly perturbed (e.g., dissociated to form bidentate species) by the interaction with Pd(111). With increasing d5-isophorone coverage (0.38–0.75 ML), the intensity of the C–H stretching region increases only slightly, but the intensity of the absorption features in the region characteristic for C=C and C=O stretching vibrations changes very strongly. For an exposure of 0.5 ML d5-isophorone, a pronounced C=C stretching signal centered around 1620 cm–1 is observed, while the C=O stretching peak around 1665 cm–1 is hardly visible. With increasing d5-isophorone coverage, the intensity of the C=O stretching signal at 1665 cm–1 increases rapidly and becomes the most intense peak in the spectra from 0.75 ML and higher. The ratio of the C=O to C=C peak intensities at 0.75 ML amounts to approximately IC=O/IC=C ≈ 4 and is roughly similar to the intensity ratio in the isophorone multilayer (e.g., at 2.4 ML) IC=O/IC=C ≈ 6–8. The intensities IC=O and IC=C are determined by integration of the IRAS peak areas.
Figure 3.

(a) IR spectra of d5-isophorone adsorbed at 100 K on pristine Pd(111) for different exposures displayed for two main vibration regions: from 3000 to 2750 cm–1 (C–H stretching vibrations) and from 1750 to 1500 cm–1 (C=O and C=C stretching vibrations). (b) NEXAFS spectra of isophorone/Pd(111) obtained at 100 K show coverage-dependent orientations of both π bonds.
NEXAFS experiments were performed under the same experimental conditions to obtain quantitative information on the coverage-dependent adsorption geometry of the C=C and C=O bonds in isophorone on Pd(111). In the present experiments, two different ways of determining the molecular tilting angles were used. The first method is based on the use of vertically polarized light; the NEXAFS spectra were obtained as a function of the incidence angle of the photon beam.39 In the second method the incidence angle of the X-ray beam was kept constant, and the polarization was changed from vertically to horizontally polarized light.40 The second method has the advantage that the sample geometry remains unchanged, and therefore the spectra become less susceptible to experimental uncertainties (e.g., the illuminated area of the sample is the same). Both methods were applied, and the results showed very good qualitative agreement; however, the spread of the experimental data was considerably larger for the method one. For this reason, we will discuss only the results of the experiments based on the changing polarization while keeping the sample geometry constant.
Figure 4 shows two example NEXAFS curves in horizontal and vertical polarization corresponding to 0.7 ML of isophorone on Pd(111) with the fitted C K-edge and indication of the most pronounced excitations around the edge.
Figure 4.

(a) C K-edge NEXAFS of 0.7 ML isophorone/Pd(111) at 100 K in vertical and horizontal polarization. The spectra are normalized to the C K edge, which was fitted and is indicated with the dashed line. The most pronounced transitions around the edge are indicated with numbers 1 to 6. The assignment is given in Table 1. (b) Schematic representation of the NEXAFS experimental geometry.
Table 1. Assignment of NEXAFS Peaks of Isophorone.
The pre-edge peak 1 around 284.9 eV and peak 2 around 286.6 eV appear much sharper than the postedge transitions. According to previous studies, peaks 1 and 2 are assigned to C 1s → π*(C=C) and C 1s → π*(C=O) excitations.23,24,29,41,42 It should be noted that what permits the differentiation between the C=O and C=C resonances is not the final state (LUMO) but the initial state, i.e., the C 1s states of the C=O carbon and the C=C carbons having different binding energies due to different chemical shifts. As long as the chemical nature of the molecule is preserved, i.e., both C=C and C=O bonds are not dissociated as in the case of this study, both resonances C 1s → π*(C=C) and C 1s → π*(C=O) can be observed as separate peaks.
In more detail, peak 1 consists of two features at 284.8 and 285.0 eV, which we correlate to excitations of C 1s electrons located at the two different C atoms of the C=C bond. However, they cannot be resolved as two peaks in every measurement and might appear as one feature located around 284.9 eV (±0.1 eV). Peak 2 has its maximum at 286.6 eV (±0.1 eV); in every recorded spectrum two shoulders appear around 286.9 eV (±0.1 eV) and 287.2 eV (±0.1 eV). The energy difference between the three features amounts to approximately 0.3 eV (±0.2 eV), which we tentatively assign to coupled excitations of C 1s → π*(C=O) electronic transitions and C=O vibronic excitations. Previously such coupled electronic and vibronic excitations have been observed for various organic molecules.27 From IRAS the energy of the C=O bond vibration is calculated to be 0.21 eV. However, the resolution limit of the NEXAFS experiments was 0.1 eV, which is not ideal to determine vibration frequencies. According to previous assignments, the broad features 3 and 4 around 288.8 and 290.7 eV are assigned to C 1s → π*2(C=C) and C 1s → π*2(C=O) transitions.29,42 Both features show identical dependence on the polarization of the incident beam as the corresponding C 1s → π*(C=C) and C 1s → π*(C=O). The very broad excitation features 5 and 6 around 293.1 and 299 eV are to a large extend independent of the polarization and can be attributed mainly to several C 1s → σ* transitions, as reported elsewhere.29,42
C K-edge NEXAFS was measured for isophorone coverages of 0.2, 0.4, and 0.7 ML, each with horizontally and vertically polarized X-ray beam and incident angles of 70° and 80° with respect to the surface normal. A series of representative coverage-dependent, step-edge normalized C K-edge NEXAFS spectra with incident angle of 70° are shown in Figure 3b. The two most important features in the spectra are the pre-edge peaks that are assigned to the C 1s → π*(C=C) resonance at 284.9 eV and the C 1s → π*(C=O) resonance at 286.6 eV. These peaks can be already clearly seen at the lowest coverages of isophorone (0.2 ML), indicating thus that both C=C and C=O bonds are not dissociated upon interaction with Pd(111). At the lowest coverage (0.2 ML), a flat-lying geometry of the π(C=C) and the π(C=O) bond is evident by the strong dependence of the intensity of both pre-edge peaks on the polarization of the incident beam. While there is a strong resonance of both π bonds with vertically polarized light (black), almost no absorption is detectable with horizontally polarized light (gray) suggesting a flat-lying adsorption geometry of isophorone. With increasing coverage, the intensity of both π resonance peaks with horizontally polarized light increases relative to the resonance with vertically polarized light, indicating that the tilting of isophorone increases. The coverage-dependent angles of the C=C and C=O bonds with respect to the Pd(111) crystal plane were calculated and are summarized in Table 2. It should be pointed out that in a general case the directions of specific bonds are not accessible with NEXAFS. Only the angle between the substrate surface normal and the direction of the transition dipole moments of the respective C 1s → π* transitions can be determined. In this study, we make an assumption that the geometry of the π* orbital with respect to the corresponding double bond remains fairly constant upon adsorption, an assumption that is supported by theoretical calculations. Therefore, we assume that the changes of the C 1s → π* transitions corresponding to the C=C and C=O double bonds reflect the tilting of the conjugated fraction of the molecule. An excellent agreement between the NEXAFS results and IRAS data as well as the results of theoretical calculations, which will be discussed in the following, support a good validity of this assumption.
Table 2. Inclination Angles of the Unsaturated Bonds in Isophorone with Respect to the Pd(111) Surface for Different Coverages of Isophorone and for 0.7 ML Isophorone with Coadsorbed Hydrogen.
| isophorone coverage/ML | tilting C=C bond/° | tilting C=O bond/° |
|---|---|---|
| 0.2 | <13 (±6) | <13 (±6) |
| 0.4 | 33 (±2) | 28 (±2) |
| 0.7 | 41 (±2) | 37 (±2) |
| H/Pd(111)+0.7 | 45 (±2) | 40 (±2) |
The strong polarization dependence of the pre-edge peaks at a coverage of 0.2 ML allows us to conclude that isophorone adopts essentially a flat adsorption geometry on Pd(111) at low coverages. Since NEXAFS becomes relatively inaccurate at very small tilting angles of the π bonds, the formally determined tilting angle of 13° with respect to the Pd(111) should be rather considered as an upper limit of the inclination angle. The possible source of the error can also be the uncertainty of the experimental alignment, e.g., difficulty of setting the sample surface precisely parallel to the horizontal electric vector or the polarization factor of the incident X-ray. At an intermediate coverage (0.4 ML), the C=C bond (33° ± 2°) is slightly more tilted than the C=O bond (28° ± 2°). At high coverage (0.7 ML) both unsaturated bonds are strongly inclined. Still, the C=C bond (41° ± 2°) takes a tentatively more upright position than the C=O bond (37° ± 2°). The inclination angles obtained for the highest isophorone coverages are in a good agreement with the recent NEXAFS results from Lambert et al., where the C=C–C=O framework in isophorone was found to be tilted by 42° with respect to the surface at high isophorone coverages.29
The IRAS and NEXAFS results for the coverage-dependent adsorption geometry of isophorone on clean Pd(111) are in qualitative agreement. At low coverages isophorone lays flat on the Pd(111) surface, which results in a complete attenuation of the vibrational features characteristic for C=O and C=C bonds, while the dynamic dipole moments of C–H stretching vibrations are at least partly inclined and therefore the corresponding bond vibrations visible. As observed by NEXAFS, both C=C and C=O bonds are present in a nondissociated form, which rules out the hypothesis on scission of these bonds as a reason for the missing IR bands at the lowest isophorone coverages. The estimated upper limit for the inclination angle of both π bonds (13°) is in a good agreement with the flat adsorption geometry of the conjugated C=C and C=O bonds deduced from the IRAS data. This observation suggests that isophorone essentially preserves the in-plane configuration of the conjugated π system in the low coverage limit.
With increasing coverage, the inclination angles of both the C=C and C=O bonds increase, with the tilting of the C=C bond being considerably more pronounced than that of the C=O bond according to IRAS. In fact, at the coverage of 0.5 ML the intensity of the IRAS absorption of the C=C bond is already comparable with the intensity of this bond on the isophorone-saturated surface, while the intensity of C=O vibration is still very close to zero. This observation indicates that the conjugated π system of C=C and C=O bonds is very strongly distorted. In a gas-phase molecule, these two bonds are lying in the same plane. If the molecule would uniformly lift up, one would expect identical tilting angles and, hence, the ratio of the absorptions in IRAS IC=O/IC=C close to the ice value (IC=O/IC=C ≈ 6–8). The observation of the intense C=C vibration and the absence of the C=O vibration in IRAS indicate that at intermediate coverages the C=O bond is still nearly lying flat on the surface, while the C=C bond lifts up resulting in a strong distortion of the original in-plane molecular structure of isophorone. The higher tilting of isophorone at intermediate coverages can also be observed in the NEXAFS data; however, this trend is somewhat less pronounced, probably because of an absolute difference in coverage. The pronouncedly tilted adsorption geometry at high coverages most likely originates from steric constraints on the surface. Since the intensity ratio in IRAS IC=O/IC=C ≈ 4 at this coverage becomes closer to the ice value of 6–8, diminishing distortion of the isophoron molecular structure can be concluded for the high coverage limit. Note that the IRAS data allow more reliable conclusions on the adsorption geometry of both double bonds since the relative orientation of the C–C and C–O axes with respect to the metal surface plane is determined. In NEXAFs, only the angle between the metal surface plane and the direction of the transition dipole moments of the respective C 1s → π* transitions can be obtained; possible rotation of the π bonds with respect to the C–C or C–O axes might complicate the interpretation of the NEXAFS spectra.
The experimental results pointing to the flat-lying adsorption geometry of isophorone at the lowest coverage were corroborated by the theoretical calculations. In this study, the DFT+vdWsurf method with the Perdew–Burke–Ernzerhof (PBE) functional43 was applied to computationally investigate the details of the electronic structure of isophorone adsorbed on Pd. The PBE+vdWsurf method is an accurate and efficient vdW-inclusive approach that allows quantitative treatment of both weakly and strongly adsorbed molecules on metal surfaces.31,44−46
Figure 5a illustrates the most stable adsorption structure found in PBE+vdWsurf geometry relaxations for isophorone. In agreement with the experimental observations by IRAS and NEXAFS, isophorone was computed to adsorb in a flat-lying adsorption geometry with the C=C and C=O bonds oriented parallel to the substrate surface plane.47 The O–Pd and C–Pd distances for the C=O and C=C bonds are in the range of 2.14–2.29 Å, close to typical covalent bond lengths. In contrast, carbon atoms in the three methyl groups attached to the ring, which are highly affected by the vdW forces, are lifted above the surface by 2.87–4.98 Å. The C=C bond in the adsorbed isophorone was found to be elongated by 0.1 Å as compared to the isolated molecule (from 1.35 to 1.45 Å), suggesting a change of the effective bond order of the C=C bond. The carbonyl bond is less elongated than the C=C bond (from 1.23 to 1.29 Å).
Figure 5.
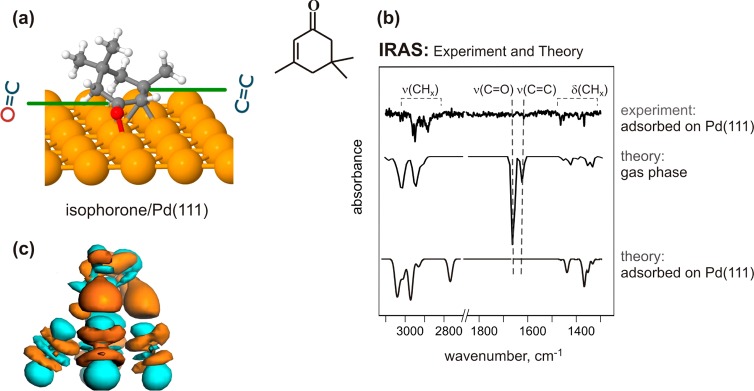
(a) Calculated structures of isophorone adsorbed on the Pd(111) surface. (b) Experimental and theoretical IR spectra of gas-phase and surface-adsorbed isophorone on Pd(111); the uppermost trace is the experimental spectrum measured at 100 K; the intermediate and the lowest traces are the calculated harmonic IR spectra for the gas-phase and the adsorbed molecules, correspondingly. The peak at 2753 cm–1 does not appear in the experimental spectrum due to substantial broadening of this vibrational band because of a very short lifetime of the C–H–Pd bond. (c) A side view of the electron density difference upon isophorone adsorption on Pd(111) at its equilibrium adsorption structure, using the value of the isosurface of 0.25 Å–3. Cyan and orange indicate electron depletion and accumulation, respectively.
To examine the feasibility of the calculated adsorption structure, the isophorone IR spectra were calculated both for gas-phase and adsorbed on Pd(111) molecules using the harmonic approximation (Figure 5b, two lowest curves). The calculated spectra were found to reproduce the most essential features of the experimentally measured IR spectra. The most intense vibrational features for the isophorone gas-phase molecule were computed in the range of C=O and C=C vibrations, which are in good agreement with the experimental observations obtained for isophorone ice (Figure 2) that can be considered as a proxy for a nonperturbed molecular form of isophorone. For the adsorbed molecule, a vanishing of C=O and C=C stretching modes was computed as expected for a flat-lying molecule. This observation is in excellent agreement with the experimental IR spectra at the lowest measured isophorone coverage (the upper curve in Figure 5b and Figure 3a). For the high frequency region of C–H stretching vibrations (2200–2900 cm–1), there is good qualitative agreement between the calculated and the measured spectra of adsorbed isophorone. The only visible difference appears for the calculated isophorone spectrum at 2573 cm–1, which is the stretch mode of the C–H–Pd vibration. This discrepancy arises from substantial broadening of this vibrational band due to a very short lifetime of the C–H–Pd bond precluding its experimental observation. To check the latter possibility, an ab initio molecular dynamics simulation was carried out at 130 K, and the anharmonic IR spectra for isophorone/Pd(111) through the Fourier transform of the dipole autocorrelation function were calculated.48 The explicit inclusion of anharmonic effects through dipole–dipole autocorrelation function leads to the disappearance of the peak at 2573 cm–1 in the PBE+vdW IR spectra, in very good agreement with experimental spectra. Further details will be presented in an upcoming publication.
The vdW forces were found to significantly contribute to the adsorption of isophorone. The standard PBE functional predicts 0.58 eV binding energy for isophorone, while the inclusion of the vdW interaction increases the binding energy to 1.80 eV. It should be noted that the most stable configuration (the flat-lying molecule) could not be located by PBE calculations when starting from a random isophorone configuration, while it was readily obtained using PBE+vdW. The large contribution of the vdW interaction to the final binding energy can be traced back to the fact that the methyl groups and the ring of isophorone lie very close to Pd(111).
Having obtained the adsorption geometries correctly reproducing the experimental observations from IRAS and NEXAFS, we performed an analysis of its electronic structure and the degree of its perturbation by the interaction with the metal surface. Figure 5c shows the side view of the electron density difference for isophorone on the Pd(111) surface, which serves to visualize the electron density redistribution upon adsorption. Cyan and orange colors indicate electron depletion and accumulation, respectively. The observed strong charge redistributions clearly indicate a substantial charge transfer between the adsorbate and the substrate.
As a next step, the electron density redistribution between the molecule and the substrate was analyzed by projection of the density of states (DOS) of the full adsorption system onto selected molecular orbitals of the free molecule.49 The full details of this analysis will be discussed in a forthcoming publication; here, we will only briefly discuss the main results. Three particular molecular orbitals of isophorone were found to be mostly perturbed by the interaction with the metal: the former HOMO–1, former HOMO, and former LUMO. While former HOMO and HOMO–1 of isophorone were computed to be rather localized orbitals in the full adsorption system (mainly located on the C=O and C=C bonds, respectively) the former LUMO is distributed over the entire π-system. The former LUMO of isophorone was found to be partially filled with 0.656 electrons and largely shifted below the Fermi level. The occupancy of the former HOMO–1, mainly located on the C=C bond, is reduced from 2 to 1.57. In contrast, the occupation number of the former HOMO, located on the C=O bond, remains hardly changed. In total, in the scope of this analysis about 1.27 electrons are donated from the molecule (HOMO and below) to the empty band of the metal, and 1.08 electrons are back-donated from metal to the empty bands of the molecule (LUMO and above). Combining these observations, we conclude that the electronic structure of isophorone is strongly perturbed by interaction with Pd(111), with notably larger charge transfer from the C=C bond than from the C=O bond.
In the next section, we investigate the effect of preadsorbed hydrogen on the adsorption geometry of isophorone on Pd(111).
3.2. Effect of Hydrogen on the Geometry of C=C and C=O Bonds
The effect of preadsorbed H was investigated by IRAS and NEXAFS measurements that were conducted at 100 K on Pd(111). Prior to the isophorone exposure, Pd(111) was exposed to 100 L of H2. At this exposure, hydrogen forms a saturated layer of surface-adsorbed H species with a formal stoichiometry H:Pd 1:1.50 The corresponding IR spectra for isophorone exposures ranging from 0.25 to 0.75 ML are depicted in Figure 6a (black traces). For comparison, the IR spectra of isophorone adsorbed on clean Pd(111) are also displayed (gray traces).
Figure 6.

(a) IR spectra of d5-isophorone adsorbed at 100 K on hydrogen-precovered (black traces) and pristine (gray traces) Pd(111) for different exposures. Displayed are two main vibration regions: from 3000 to 2750 cm–1 (C–H stretching vibrations) and from 1750 to 1500 (C=O and C=C stretching vibrations). (b) NEXAFS spectra obtained at 100 K for 0.7 ML isophorone coverage on pristine Pd(111) (upper traces) and H-precovered Pd(111) (lower traces).
The total intensities of the C–H stretching features (2800–3000 cm–1) were found to be similar on both clean and hydrogen-precovered Pd(111) at different isophorone exposures. This observation might be considered as an indication that similar isophorone coverages are formed on both surfaces. In contrast, IR absorption due to C=C and the C=O vibrations strongly changes when hydrogen was preadsorbed and is most pronounced around the coverage of 0.5 ML. At the coverage of 0.5 ML, the C=O vibration is clearly visible on H-precovered Pd(111), while the C=C vibration cannot be identified yet. On the pristine Pd(111) surface, the situation was found to be reverse—the most intense vibrational feature corresponds to the C=C bond, while the C=O bond vibration is hardly visible. At 0.75 ML, the ratio of the C=O to the C=C vibration peak intensities (IC=O/IC=C ≈ 5) is similar to that of multilayer isophorone on clean Pd(111) ( IC=O/IC=C ≈ 6–8). At the lowest coverage of 0.25 ML the spectra are essentially identical on both surfaces pointing to the flat-lying adsorption geometry of isophorone also on the hydrogen-precovered surface.
The observed coverage dependence indicates that the interaction of isophorone with Pd is considerably affected by preadsorbed hydrogen. While at the lowest coverage isophorone adopts—similarly to the pristine Pd surface—a flat adsorption geometry, the interaction changes at the intermediate surface coverages. Particularly, the ratio IC=O/IC=C becomes close to the ice (or gas-phase) value, indicating that isophorone lifts up rather uniformly and that the conjugated π system of two double bonds preserves to a large extent its in-plane geometry characteristic for ice or the gas-phase molecule. This behavior is in sharp contrast for the coverage dependence of isophorone adsorption geometry obtained on the pristine Pd(111) surface, where the C=C bond was concluded to be strongly tilted with respect to the flat-lying C=O bond and the total conjugated π system is significantly geometrically distorted. The diminished distortion of the molecule in the presence of hydrogen most likely originates from the weaker interaction of isophorone with the hydrogen-containing Pd surface, a phenomenon which was discussed also for other hydrocarbon–metal systems.
NEXAFS studies were performed to gain quantitative information on the changes in tilting angle of the two unsaturated C=C and C=O bonds in the presence of preadsorbed hydrogen. In Figure 6b, representative C K-edge normalized NEXAFS spectra of 0.7 ML isophorone adsorbed on clean Pd(111) (top) and on hydrogen-precovered Pd(111) (bottom) are displayed. The angles of the C=C and the C=O bonds relative to the Pd(111) substrate were calculated and are given in Table 2. Our results indicate that coadsorbed hydrogen slightly increases the inclination angle of the C=C bond (from 41 ± 2° to 45 ± 2°) and the C=O bond (from 37 ± 2° to 41 ± 2°) with respect to Pd(111). It should be noted that the isophorone coverages used in these experiments are close to the saturation, where the IRAS data do not indicate any strong differences between the adsorption geometries of isophorone. NEXAFS data at lower coverage, where IRAS point to a stronger influence of hydrogen precovering on the adsorption of isophorone, are not available. However, the NEXAFS data are consistent with the observations of the IRAS experiments showing in general higher inclination angles of unsaturated bonds on the hydrogen-precovered Pd(111).
It should be noted that under the reaction conditions some of the isophorone molecules might undergo chemical transformations, e.g., partial dissociation, resulting in different surface species with presumably different adsorption geometries. Whether these eventual species could be produced and be involved in the reactive pathway is a question that needs to be answered in future studies. At the present time, we cannot exclude that higher variety of surface species might be present on Pd during isophorone partial hydrogenation.
4. Conclusions
Interaction of isophorone with Pd was investigated experimentally by the combination of NEXAFS and IRAS and theoretically with DFT+vdW. Particularly, the adsorption geometry of the two unsaturated bonds (C=C and C=O) in isophorone on pristine and hydrogen-precovered Pd(111) surfaces as well as the coverage dependence of the adsorbate structures were determined. At low coverages, both NEXAFS and IRAS data point to a flat-lying adsorption geometry of isophorone on Pd(111) with the C=C and C=O bonds being oriented parallel to the surface plane. This observation suggests that isophorone preserves the in-plane configuration of the conjugated π-system in the low coverage limit. For intermediate coverages, the structure of isophorone adsorbed on pristine Pd(111) was observed to be strongly distorted. The C=C bond becomes noticeably tilted with respect to the surface plane, while the C=O bond is still oriented flat on the surface. Close to saturation, both the C=O and the C=C bonds lift up, and the inclination angles of the entire conjugated π system increase to around 40° with slightly more pronounced tilting of the C=C bond. High tilting angles of the entire molecule at higher coverages most likely originate from steric constraints on the surface.
DFT+vdW calculations confirmed the flat-lying adsorption geometry of isophorone at low coverages and provided further microscopic insights into interaction of isophorone with Pd. It was found that the C=C bond of isophorone is significantly perturbed by the interaction with Pd, in terms of both the electronic and geometric structure, showing a strong elongation of the bond and a large extent of the electron density redistribution. In contrast, the carbonyl bond in isophorone was found to be hardly affected by the interaction with the metal. The computed IR spectra are in good agreement with the experimentally measured ones.
The IR spectra show that the adsorption of isophorone is significantly affected by the presence of coadsorbed hydrogen, particularly pronounced at the intermediate coverage. While at the lowest coverages isophorone exhibits a flat-lying geometry, similarly to pristine Pd(111), both unsaturated bonds strongly tilt already at intermediate coverages. In this case, the intensity ratio IC=O/IC=C of the main vibrational peaks on hydrogen-precovered Pd(111) indicates a rather unperturbed molecular structure of adsorbed isophorone that is similar to the gas-phase molecule. Higher tilting angles of both π bonds point to much weaker interaction of isophorone with hydrogen-precovered Pd and suggest the conservation of the in-plane geometry of the conjugated π system.
With respect to the gas-phase structure, the molecules appear to be more perturbed on the clean Pd(111) surface and considerably less on the hydrogen-precovered surface. These coverage- and coadsorbate-dependent changes in the adsorption geometry of the C=C and/or the C=O bond are expected to have a decisive influence on the selectivity in hydrogenation reactions.
Acknowledgments
The authors thank Matthias Peter, Sergey Adamovski, José Manuel Flores Camacho, Helmut Kuhlenbeck, and the technical staff of BESSY-II for the scientific and technical support during the beam time. A.T. acknowledges support from the European Research Council (ERC Starting Grant VDW-CMAT). S.S. acknowledges support from the Fonds der Chemischen Industrie for the Chemiedozentenstipendium and the European Research Council (ERC Starting Grant 335205-ENREMOS).
The authors declare no competing financial interest.
References
- Pirnot M. T.; Rankic D. A.; Martin D. B. C.; MacMillan D. W. C. Photoredox activation for the direct β-arylation of ketones and aldehydes. Science 2013, 339, 1593–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Qian H.; Drake B. A.; Jin R. Atomically precise Au25 (SR) 18 nanoparticles as catalysts for the selective hydrogenation of α,β-unsaturated ketones and aldehydes. Angew. Chem. 2010, 122, 1317–1320. [DOI] [PubMed] [Google Scholar]
- Fleischer S.; Zhou S.; Junge K.; Beller M. General and highly efficient iron-catalyzed hydrogenation of aldehydes, ketones, and α,β-unsaturated aldehydes. Angew. Chem., Int. Ed. 2013, 52, 5120–5124. [DOI] [PubMed] [Google Scholar]
- Hitzler M. G.; Smail F. R.; Ross S. K.; Poliakoff M. Selective catalytic hydrogenation of organic compounds in supercritical fluids as a continuous process. Org. Process Res. Dev. 1998, 2, 137–146. [Google Scholar]
- Sato T.; Rode C. V.; Sato O.; Shirai M. Hydrogenation of isophorone with noble metal catalysts in supercritical carbon dioxide. Appl. Catal., B 2004, 49, 181–185. [Google Scholar]
- Enache D. I.; Hutchings G. J.; Taylor S. H.; Stitt E. H. The hydrogenation of isophorone to trimethyl cyclohexanone using the downflow single capillary reactor. Catal. Today 2005, 105, 569–573. [Google Scholar]
- Tungler A.; Fodor K.; Mathe T.; Sheldon R. A.. Enantioselective hydrogenation of ethyl pyruvate and isophorone over modified Pt and Pd catalysts. In Heterogeneous Catalysis and Fine Chemicals Iv; Blaser H. U., Baiker A., Prins R., Eds.; Elsevier Science Bv: Amsterdam, 1997; Vol. 108, pp 157–165. [Google Scholar]
- Studer M.; Blaser H. U.; Exner C. Enantioselective hydrogenation using heterogeneous modified catalysts: an update. Adv. Synth. Catal. 2003, 345, 45–65. [Google Scholar]
- Blaser H. U.; Pugin B.; Spindler F. Progress in enantioselective catalysis assessed from an industrial point of view. J. Mol. Catal. A: Chem. 2005, 231, 1–20. [Google Scholar]
- Mallat T.; Orglmeister E.; Baiker A. Asymmetric catalysis at chiral metal surfaces. Chem. Rev. 2007, 107, 4863–4890. [DOI] [PubMed] [Google Scholar]
- Tungler A.; Fogassy G. Catalysis with supported palladium metal, selectivity in the hydrogenation of C=C, C=O and C=N bonds, from chemo- to enantioselectivity. J. Mol. Catal. A: Chem. 2001, 173, 231–247. [Google Scholar]
- Tarnai T.; Tungler A.; Mathe T.; Petro J.; Sheldon R. A.; Toth G. A new chiral auxiliary in enantioselective hydrogenations: (−)-dihydrovinpocetine. Part I. Hydrogenation of isophorone. J. Mol. Catal. A: Chem. 1995, 102, 41–47. [Google Scholar]
- Farkas G.; Fodor K.; Tungler A.; Mathe T.; Toth G.; Sheldon R. A. New chiral auxiliaries in enantioselective heterogeneous catalytic hydrogenations: (−) and (+)-dihydro-apovincaminic acid. Comparison with (−)-dihydro-apovincaminic acid ethyl ester. III. J. Mol. Catal. A: Chem. 1999, 138, 123–127. [Google Scholar]
- Tungler A.; Nitta Y.; Fodor K.; Farkas G.; Mathe T. Comparison of chiral modifiers in the Pd catalysed hydrogenation of phenylcinnamic acid and isophorone. J. Mol. Catal. A: Chem. 1999, 149, 135–140. [Google Scholar]
- Sipos E.; Tungler A. Effect of pretreatment of the catalyst and catalyst-modifier system in the enantioselective hydrogenation of isophorone. React. Kinet. Catal. Lett. 2003, 80, 365–373. [Google Scholar]
- Sipos E.; Tungler A.; Fogassy G. New substrates and modifiers in the enantio selective heterogeneous catalytic hydrogenation of the C=C double bond. J. Mol. Catal. A: Chem. 2004, 216, 171–180. [Google Scholar]
- Fodor M.; Tungler A.; Vida L. Heterogeneous catalytic asymmetric hydrogenations with modifiers of axial chirality. React. Kinet. Catal. Lett. 2007, 90, 413–418. [Google Scholar]
- Li S.; Zhan E. S.; Li Y.; Xu Y. D.; Shen W. J. Enantio selective hydrogenation of isophorone and kinetic resolution of 3,3,5-trimethylcyclohexanone over Pd catalysts in the presence of (S)-proline. Catal. Today 2008, 131, 347–352. [Google Scholar]
- Stöhr J.NEXAFS spectroscopy; Springer-Verlag: Berlin Heidelbger New York, 1992. [Google Scholar]
- Hahner G. Near edge X-ray absorption fine structure spectroscopy as a tool to probe electronic and structural properties of thin organic films and liquids. Chem. Soc. Rev. 2006, 35, 1244–1255. [DOI] [PubMed] [Google Scholar]
- Lee H. K.; Han J. H.; Kim K. J.; Kang T. H.; Kim B. Configuration of pentacene (C22H14) films on Si(100)-2 × 1 studied by NEXAFS. Surf. Sci. 2007, 601, 1456–1460. [Google Scholar]
- Taborski J.; Vaterlein P.; Dietz H.; Zimmermann U.; Umbach E. NEXAFS investigations on ordered adsorbate layers of large aromatic molecules. J. Electron Spectrosc. Relat. Phenom. 1995, 75, 129–147. [Google Scholar]
- Yannoulis P.; Frank K. H.; Koch E. E. Electronic structure and orientation of anthracene on Ag(111). Surf. Sci. 1991, 241, 325–334. [Google Scholar]
- Horsley J. A.; Stohr J.; Hitchcock A. P.; Newbury D. C.; Johnson A. L.; Sette F. Resonances in the K shell excitation spectra of benzene and pyridine: gas phase, solid, and chemisorbed states. J. Chem. Phys. 1985, 83, 6099–6107. [Google Scholar]
- Outka D. A.; Stohr J. Curve fitting analysis of near-edge core excitation spectra of free, adsorbed, and polymeric molecules. J. Chem. Phys. 1988, 88, 3539–3554. [Google Scholar]
- Hahner G.; Hofer R.; Klingenfuss I. Order and orientation in self-assembled long chain alkanephosphate monolayers adsorbed on metal oxide surfaces. Langmuir 2001, 17, 7047–7052. [Google Scholar]
- Schoell A.; Zou Y.; Huebner D.; Urquhart S. G.; Schmidt T.; Fink R.; Umbach E.. A comparison of fine structures in high-resolution x-ray-absorption spectra of various condensed organic molecules. J. Chem. Phys. 2005, 123. [DOI] [PubMed] [Google Scholar]
- Urquhart A. J.; Williams F. J.; Vaughan O. P. H.; Cropley R. L.; Lambert R. M. Adsorbate conformation determines catalytic chemoselectivity: crotonaldehyde on the Pt(111) surface. Chem. Commun. 2005, 1977–1979. [DOI] [PubMed] [Google Scholar]
- Beaumont S. K.; Kyriakou G.; Watson D. J.; Vaughan O. P. H.; Papageorgiou A. C.; Lambert R. M. Influence of adsorption geometry in the heterogeneous enantioselective catalytic hydrogenation of a prototypical enone. J. Phys. Chem. C 2010, 114, 15075–15077. [Google Scholar]
- Libuda J.; Meusel I.; Hartmann J.; Freund H. J. A molecular beam/surface spectroscopy apparatus for the study of reactions on complex model catalysts. Rev. Sci. Instrum. 2000, 71, 4395–4408. [Google Scholar]
- Ruiz V. G.; Liu W.; Zojer E.; Scheffler M.; Tkatchenko A. Density-functional theory with screened van der Waals interactions for the modeling of hybrid inorganic-organic systems. Phys. Rev. Lett. 2012, 108, 146103–146108. [DOI] [PubMed] [Google Scholar]
- Blum V.; Gehrke R.; Hanke F.; Havu P.; Havu V.; Ren X.; Reuter K.; Scheffler M. Ab initio molecular simulations with numeric atom-centered orbitals. Comput. Phys. Commun. 2009, 180, 2175–2196. [Google Scholar]
- Tkatchenko A.; Scheffler M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 073005–073009. [DOI] [PubMed] [Google Scholar]
- Lifshitz E. M. The theory of molecular attractive froces between solids. Sov. Phys. JETP 1956, 2, 73–83. [Google Scholar]
- Zaremba E.; Kohn W. Van der Waals interaction between an atom and a solid surface. Phys. Rev. B 1976, 13, 2270–2285. [Google Scholar]
- van Lenthe E.; Baerends E. J.; Snijders J. G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar]
- Hollins P.; Pritchard J. Infrared studies of chemisorbed layers on single crystals. Prog. Surf. Sci. 1985, 19, 275–349. [Google Scholar]
- Hoffmann F. M. Infrared reflection-absorption spectroscopy of adsorbed molecules. Surf. Sci. Rep. 1983, 3, 107–192192. [Google Scholar]
- Stohr J.; Outka D. A. Determination of molecular orientations on surfaces from the angular dependence of near-edge x-ray-absorption fine-structure spectra. Phys. Rev. B 1987, 36, 7891–7905. [DOI] [PubMed] [Google Scholar]
- Casu M. B.; Scholl A.; Bauchspiess K. R.; Hubner D.; Schmidt T.; Heske C.; Umbach E. Nucleation in organic thin film growth: perylene on Al2O3/Ni3Al(111). J. Phys. Chem. C 2009, 113, 10990–10996. [Google Scholar]
- Ishii I.; Hitchcock A. P. The oscillator strengths for C1s and O1s excitation of some saturated and unsaturated organic alcohols, acids and esters. J. Electron Spectrosc. Relat. Phenom. 1988, 46, 55–84. [Google Scholar]
- Bournel F.; Laffon C.; Parent P.; Tourillon G. Adsorption of some substituted ethylene molecules on Pt(111) at 95 K 0.1. NEXAFS, XPS and UPS studies. Surf. Sci. 1996, 350, 60–78. [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [DOI] [PubMed] [Google Scholar]
- Bürker C.; Ferri N.; Tkatchenko A.; Gerlach A.; Niederhausen J.; Hosokai T.; Duhm S.; Zegenhagen J.; Koch N.; Schreiber F. Exploring the bonding of large hydrocarbons on noble metals: diindoperylene on Cu (111), Ag (111), and Au (111). Phys. Rev. B 2013, 87, 165443–165448. [Google Scholar]
- Liu W.; Carrasco J.; Santra B.; Michaelides A.; Scheffler M.; Tkatchenko A. Benzene adsorbed on metals: concerted effect of covalency and van der Waals bonding. Phys. Rev. B 2012, 86, 245405–245411. [Google Scholar]
- Egger D. A.; Ruiz V. G.; Saidi W. A.; Bučko T.; Tkatchenko A.; Zojer E. Understanding structure and bonding of multilayered metal-organic nanostructures. J. Phys. Chem. C 2013, 117, 3055–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Savara A.; Ren X.; Ludwig W.; Dostert K.-H.; Schauermann S.; Tkatchenko A.; Freund H.-J.; Scheffler M. Toward low-temperature dehydrogenation catalysis: isophorone adsorbed on Pd (111). J. Phys. Chem. Lett. 2012, 3, 582–586. [DOI] [PubMed] [Google Scholar]
- Gaigeot M.-P.; Martinez M.; Vuilleumier R. Infrared spectroscopy in the gas and liquid phase from first principle molecular dynamics simulations: application to small peptides. Mol. Phys. 2007, 105, 2857–2878. [Google Scholar]
- Rangger G. M.; Romaner L.; Heimel G.; Zojer E. Understanding the properties of interfaces between organic self-assembled monolayers and noble metals—a theoretical perspective. Surf. Int. Anal. 2008, 40, 371–378. [Google Scholar]
- Christmann K. Interaction of hydrogen with solid surfaces. Surf. Sci. Rep. 1988, 9, 1–163. [Google Scholar]