

Abstract
Complex regional pain syndrome - type I (CRPS-I; Reflex Sympathetic Dystrophy) is a chronic pain condition that usually follows a deep-tissue injury such as fracture or sprain. The cause of the pain is unknown. We have developed an animal model (chronic post-ischemia pain; CPIP) that creates CRPS-I –like symptomology. The model is produced by occluding the blood flow to one hind paw for 3 hr under general anesthesia. Following reperfusion, the treated hind paw exhibits an initial phase of hyperemia and edema. This is followed by mechano-hyperalgesia, mechano-allodynia, and cold-allodynia that last for at least one month. Light- and electron microscopic analyses of the nerves at the site of the tourniquet show that the majority of these animals have no sign of injury to myelinated or unmyelinated axons. However, electron microscopy shows that the ischemia-reperfusion (I-R) injury produces a microvascular injury, slow-flow/no-reflow, in the capillaries of the hind paw muscle and digital nerves. We propose that the slow-flow/no-reflow phenomenon initiates and maintains deep tissue ischemia and inflammation, leading to the activation of muscle nociceptors, and the ectopic activation of sensory afferent axons due to endoneurial ischemia and inflammation. These data, and a large body of clinical evidence, suggest that in at least a subset of CRPS-I patients, the fundamental cause of the abnormal pain sensations is ischemia and inflammation due to microvascular pathology in deep tissues, leading to a combination of inflammatory and neuropathic pain processes. Moreover, we suggest a unifying idea that relates the pathogenesis of CRPS-I to that of CRPS-II. Lastly, our hypothesis suggests that the role of the sympathetic nervous system in CRPS-I is a factor that is not fundamentally causative, but may have an important contributory role in early stage disease.
Symptomology of CRPS-I
CRPS-I is a chronic pain syndrome that occurs following injuries such as sprains, fractures and crush injuries that are not accompanied by a clinically-verified nerve injury [1]. CRPS-I is a relatively rare disorder, with an estimated incidence of 26.2 per 100,000 [2]. Nevertheless, this condition has fascinated, perplexed, and frustrated clinicians for well over a century. Symptoms of CRPS-I include spontaneous pain (“burning” pain referred to the skin, and “aching” pain referred to deep tissues), and a variety of stimulus-evoked abnormal pain sensations, including mechano-hyperalgesia, mechano-allodynia, cold-allodynia and sometimes heat-hyperalgesia. Other symptoms include disorders of vasomotor and sudomotor regulation; trophic changes in skin, hair, nails, and bone; and dystonia and other motor abnormalities [3–9]. CRPS-II (causalgia) is similar in all respects except that a clinically-verified nerve injury is present [3,4].
Until recently, it has been generally accepted that most, if not all, the ancillary symptoms of CRPS-I are due to a hyperactive sympathetic outflow to the painful region, and that the sympathetic outflow is somehow causally related to the pain. This belief gained credence from the therapeutic benefit that is achieved in at least some patients following sympathectomy or local anaesthetic block of the sympathetic ganglia [10]. However, it is well known that not all CRPS-I patients benefit from sympathetic interventions, and those that do sometimes receive only partial or temporary pain relief. Not surprisingly, the role of the sympathetic innervation in the CRPS-I syndrome is not well understood [11].
It is generally believed that there is a temporal progression of symptoms in CRPS-I, resulting in three broadly defined stages of disease, with the first typified by hyperemia and edema, the second by cold, hyperhydrosis and cyanosis, and the third by dystonia and dystrophic changes [5,9]. There is evidence that this temporal progression is not present in all patients [8]. Clinical experience and animal experiments suggest that the sympathetic innervation may contribute in the early stage of CRPS-I, but that the underlying pathology evolves from sympathetically-maintained pain (SMP) to sympathetically-independent pain (SIP) [5,10,12,13].
I. A new animal model of CRPS-I
We have developed an animal model (chronic post-ischemia pain; CPIP) that creates CRPS-I-like symptomology [14]. The CPIP model is produced in rats under general anesthesia with a tourniquet placed around the ankle for 3 hr. Following reperfusion, the hind paw exhibits an initial phase of hyperemia and edema lasting for 2–12 hrs, followed by neuropathic pain (mechano-hyperalgesia, mechano-allodynia, and cold-allodynia) that lasts for at least one month. Light- and electron microscopic analyses of the hind paw nerves from CPIP rats show that nearly all have no sign of nerve injury due to the tourniquet [15]. Some of the animals have a small number of degenerating myelinated axons, which would be difficult or impossible to verify if present in a human patient. In addition, there are no changes in conduction velocity of the sural nerve at 5 and 7 days post-reperfusion [16]. However, there is a reduced density of the sensory fibers’ terminal arbors in the epidermis in the injured hind paw, as determined by PGP9.5 immunohistochemistry [16], similar to that observed in CRPS-I patients [17].
Electron microscopic analysis of the hind paw digital muscle and digital nerves reveals microvascular pathology that is indicative of the capillary slow-flow/no-reflow phenomenon [15] – a well known consequence of I-R injury in cardiac and skeletal muscle. The hind paw muscle also exhibits poor perfusion and a reduced density of viable capillaries for a week following reperfusion [16]. Single fiber recordings from primary afferent axons in CPIP rats at intervals of 2–9 days after tourniquet release have found spontaneously discharging Aβ, Aδ, and C fibers in every case examined [15]. Normal sensory fibers have little or no spontaneous discharge.
Slow flow/no-reflow in deep tissue microvasculature would be expected to produce a persistent inflammatory state. Data from the CPIP animals is consistent with this idea. Malondialdehyde, a product of free radical-induced lipid peroxidation, was significantly elevated in the CPIP rat hind paw, and CPIP allodynia was dose-dependently attenuated by the anti-oxidant N-acetyl-L-cysteine, and the free radical scavenger 4-hydroxy-2,2,6,6-tetramethyl-piperydine-1-oxyl, suggesting a key role of free radicals [16]. Furthermore, the pro-inflammatory cytokines, tumor necrosis factor alpha (TNFα), interleukin-1 (IL-1) and interleukin-6 (IL-6), and the related transcription factor, nuclear factor kappa B (NFκB), are all elevated in the CPIP rat hind paws early after reperfusion, and both an IL-1 receptor antagonist and an NFκB inhibitor, pyrrolidine dithiocarbamate, dose-dependently reduce CPIP allodynia, suggesting that an NFκB-dependent generation of pro-inflammatory cytokines plays a role in CPIP. Lactate is also increased in the muscle tissue of CPIP rat hind paws, and the levels of lactate are further elevated when the rats exercise. Exercise also decreases mechanical paw withdrawal thresholds in CPIP rats, and there is a direct correlation between mechanical allodynia and lactate levels in unexercised and exercised CPIP rats [16].
CPIP rats also develop a hypersensitivity to norepinephrine (NE), reflected by enhanced arterial responsiveness to NE, as well as enhanced painful responses to hind paw injections of NE [18]. Mice also develop CPIP after I-R injury, and CPIP mice have an up-regulation of endothelin A (ET-A) receptors in their hind paw muscles. Accordingly, CPIP mice exhibit enhanced sustained nociceptive behaviours following hind paw injections of ET-1[19].
II. A hypothesis for the pathogenesis CRPS-I
Our findings in CPIP animals lead us to propose a hypothesis that we believe to be applicable to both the CPIP animals and to at least a subset of CRPS-I patients. We propose that the fundamental cause of the pain is a persistent deep-tissue (muscle, bone, and nerve) ischemia and consequent inflammatory reaction produced by microvascular pathology subsequent to an I-R injury. We propose that the following processes (summarized in Fig. 1) are critical to the initiation, development, and maintenance of the CPIP and CRPS-I syndromes.
Fig. 1.
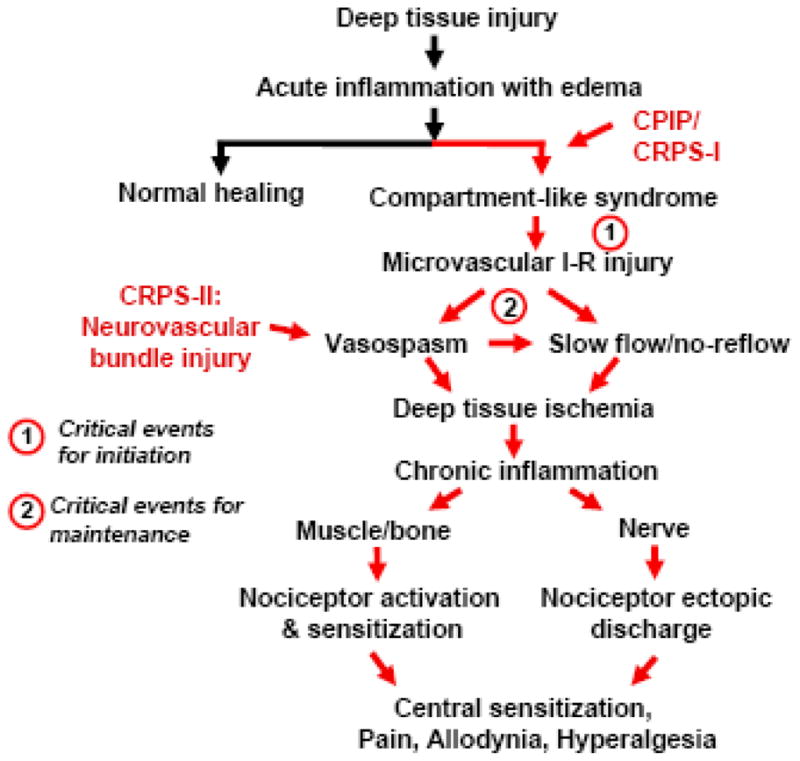
Schematic of the hypothesized pathophysiological mechanisms in the CPIP model and at least a subset of CRPS-I patients. In CRPS-I patients, deep tissue injury leads to edema and a compartment-like syndrome. Reperfusion (release of the tourniquet in the animal model) leads to injury of the microvascular endothelial cells induced by free radicals. These events lead to arterial vasospasms and slow flow/no-reflow in deep tissue microvasculature, which produces persistent ischemia that both spreads the I-R injury, and causes chronic inflammation. In muscle and bone, the resulting ischemia and inflammation (including generation of lactate) activates and sensitizes nociceptors. In nerve, the ischemia and inflammation cause ectopic nociceptor discharge. Injury to a neurovascular bundle may also evokes arterial vasospasm, and this may initiate the same vicious cycle in patients with CRPS-II (causalgia).
(1) CRPS-I is initiated when an inflammatory response to a deep-tissue injury produces a compartment-like syndrome that impairs blood flow to muscle, nerve and bone
At its onset, CRPS-I is often characterized by significant regional edema that is sometimes described as “exaggerated” [20–22]. Oedema develops due to the extravasation and accumulation of plasma in the interstitial space [23]. In the early stage of CRPS-I, there is plasma extravasation [20], increased density of perfused vessels, and higher capillary filtration capacity (an index of microvascular permeability) [24,25].
The pressure exerted by the interstitial accumulation of extravasated plasma within a relatively confined anatomical space occludes the capillaries of adjacent tissues and causes a compartment syndrome [26,27]. Pressures within a myofascial compartment of as little as 30–40 mm Hg will occlude capillary flow [28,29]. Compartment syndrome is a well-known complication of fractures, joint trauma and joint surgery [26,30–32]. CRPS-I also often follows fractures, joint injuries, and wrist, elbow, and knee surgery [33,34]. We think that it is extremely important to note that CRPS-I nearly always follows injury to deep tissues (fractures, sprains, surgeries, crush injuries, etc.). CRPS-I initiated by a strictly cutaneous injury (e.g., a laceration or burn) is exceedingly rare [35]. Oedema in the subcutaneous space does not lead to a compartment syndrome.
In its most severe form, musculoskeletal compartment syndrome leads to ischemia that is severe enough to cause tissue necrosis, but subtotal or episodic (exertional) ischemia lead to compartment-like syndromes that do not progress to tissue necrosis [36–38]. We propose that in the CPIP animal, a compartment-like syndrome is created by the period of extensive edema that follows release of the tourniquet. In man, both compartment syndrome and CRPS-I are known risks of excessive tourniquet exposure [39,40].
(2) The compartment syndrome leads to an I-R injury and persistent deep-tissue microvascular pathology
While the proposed compartment-like syndrome does not produce tissue necrosis, it does produce microvascular injury. Ischemic tissues accumulate oxidative enzymes, primarily xanthine oxidase and NADPH oxidase. Upon reperfusion, the accumulated oxidases reduce the returning molecular oxygen and the cells composing the microvessels are exposed to high levels of oxygen free radicals that damage both vascular endothelial and smooth muscle cells [41–48]. The onset of reperfusion is obviously well defined when a tourniquet is removed, but less clearly demarcated when the ischemia is due to a compartment-like syndrome. In the latter case, it is probable that reperfusion is episodic or partial, and may migrate from one part of the tissue’s capillary bed to another.
I-R injury is a multifactorial phenomenon characterized by several pathological mechanisms affecting arterioles, capillaries, and venules [48–55]. These mechanisms interact and generally contribute to positive-feedback loops that perpetuate and worsen the I-R injury. Although most studied as a consequence of ischemic insult to heart muscle, I-R injury phenomena are also known to occur in the microvasculature of skeletal muscle [52,56]. We propose here that I-R injury also occurs in the microvasculature of bone and peripheral nerve.
I-R injury, aterioles, vasospasm and the sympathetic nervous system
Following I-R injury, the arteriole’s endothelial cells release less nitric oxide, and the reduced amount of nitric oxide that is released is converted to toxic nitrogen free radicals after interaction with oxygen free radicals. The result is a deficit in the nitric oxide-mediated vasodilatation that normally modulates the vasoconstriction that is evoked by the sympathetic nervous system [49,57,58]. Recent data show that CRPS-I patients have abnormal vasodilatation responses after sympathetically-evoked vasoconstriction [59], and decreased levels of nitric oxide have been found in blister fluid from the affected region [60]. Endothelin-1 (ET-1) is a potent vasoconstrictor derived from vascular endothelial cells that produces it’s pressor effects by acting on ET-A receptors on vascular smooth muscle cells [61]. Following I-R injury, ET-1 production and release are increased, and so is the vasoconstrictor response that it evokes [62]. Increased levels of ET-1 are found in blister fluid from the affected extremity in early stage CRPS-I patients [60]. As we described above, CPIP mice have up-regulated ET-A receptors in hind paw muscle, and show enhanced painful responses to intraplantar ET-1 injections [19].
I-R injury also evokes an up-regulation of the expression of α-adrenoceptors on arterial smooth muscle cells, resulting in a three-fold increase in the contractile response to NE [63]. As a result, arteries will spasm in response to normal levels of sympathetic discharge (vasoconstrictor “tone”), to the myriad of sympathetic reflexes that are evoked by daily activity, and perhaps to catecholamines (NE and epinephrine) that arrive via the circulation. As discussed above, CPIP rats exhibit enhanced arterial vasoconstrictor responses to NE. Both CRPS-I patients and CPIP rats display abnormal pain responses to intraplantar injections of NE [18,64]. There is clear evidence that there is dramatically reduced sympathetic reflex activity in early CRPS [65]. There is also evidence that this reduced sympathetic outflow itself results in a hypersensitivity of the smooth muscle adrenoceptors, i.e., functional denervation super-sensitivity [66]. There is evidence for increased vascular α-adrenoceptor responsiveness in CRPS-I patients [67–69]. The situation is likely to be very complex and variable from patient-to-patient, or from time-to-time in a given patient: increased vascular α-adrenoceptor responsiveness that might be driven by circulating catecholamines and/or the NE released in the context of decreased activity at the postganglionic sympathetic fiber’s synapse. In any case, the result is that sympathetic activity would be contributory, rather than fundamentally causative, and exacerbate ischemia, inflammation and consequent pain.
Nerve injury evokes sympathetic fiber sprouting in the dorsal root ganglion. However, there is evidence that this sprouting may not contribute to CRPS-II pain (and, by implication, CRPS-I pain) [70]. There is also evidence that nerve injury evokes the de novo expression of α-adrenoceptors on primary afferent nociceptors, which suggests a direct link between NE and epinephrine (via the circulation or via the sympathetic postganglionic synapse) and nociceptor activation [71]. The presence of such a mechanism is not incompatible with the hypothesis that we propose.
Vasopasm in pre-capillary arterioles exacerbates the ischemia and leads to further I-R injury, and may contribute to the spread of microvascular dysfunction. The resulting vicious cycle contributes to the maintenance and worsening of the ischemic state. Such spreading dysfunction may account for the “contiguous” spread of CRPS-I symptoms, i.e., the tendency for edema and pain to gradually spread outwards from the initially symptomatic region [72]. There is extensive evidence for such a spreading phenomenon in the case of ischemic heart disease [73]; we propose here that the same thing occurs in skeletal muscle. A long forgotten paper by Foisie [74] suggested that arterial vasospasm might contribute to causalgic-like pain after crush injuries and soft tissue injuries that did not injury a nerve (i.e., CRPS-I). Arterial vasospasm after I-R injury is sometimes relieved by a brief tourniquet application [75]. CRPS-I patients sometimes report temporary pain relief after brief tourniquet application [76,77].
I-R injury to capillaries
Free radicals also damage capillary endothelial cells and stimulate them to release various pro-inflammatory mediators. Free radicals increase the expression of selectins, intracellular and extracellular adhesion molecules [78], complement factors [79], leukotriene B4 [80,81], and platelet activating factor [82,83]. Many of these molecules are chemotaxic and recruit monocytes, leukocytes, and platelets which accumulate and occlude the capillary lumen, and release TNFα, IL-1 and IL-6, which produce toxic effects that might spread tissue injury to adjacent regions [84,85].
Occlusion of capillaries generates the phenomenon known as slow-flow/no-reflow, which we believe to be a key feature of both the CRPS-I and CPIP syndromes. Slow-flow/no-reflow is characterized by the swelling of microvascular endothelial cells, platelet aggregation, and plugging of the capillary lumen by leukocytes and erythrocytes [45,48,49,51,52,55,86–95]. “Slow-flow” refers to the condition where the lumen is partly occluded; complete occlusion is “no-reflow”. In cardiac muscle, the onset of slow-flow/no-reflow is detectable immediately after reperfusion, but it worsens significantly during the following hours and days [55,96]. It is important to recall how easy it is to block capillary flow. The smallest capillaries are formed from a single endothelial cell, and their lumen diameters are nearly the same as the shortest diameter of a red blood cell. The lumens’ diameter is restricted further at the level of the endothelial cell’s nucleus. Red blood cells must deform and squeeze through the capillary lumen. The pain of a sickle cell crisis is due to ischemia that occurs because of an abnormality of the erythrocye’s membrane that makes it too stiff to deform and squeeze through the capillary lumen.
As well as occurring in skeletal muscle, we have evidence from the CPIP animals that slow-flow/no-reflow also occurs in the nerve’s capillaries, and we think it likely that it also occurs in periosteal and intramedullary bone capillaries. There is no logical reason to suppose that slow-flow/no-reflow does not also occur in cutaneous capillaries. However, because of their important role in thermoregulation, the cutaneous capillary beds in the distal extremities of man are extremely dense and highly anastomotic; this renders the skin’s capillary bed relatively resistant to slow-flow/no-reflow [97].
I-R injury to venules
Free radical damage to the endothelial cells of venules resembles that seen in capillaries. However, venules have lumens that have a greater diameter than that of capillaries and venules are thus relatively resistant to slow-flow/no-reflow. However, I-R injury causes damage at post-capillary venules, which causes leakage of plasma through resultant gaps between adjacent endothelial cells [98]. It is significant that plasma extravasation occurs mostly at the level of the venules, not the capillaries. Free radical damage to venules is thus the immediate cause of edema formation. As we discuss below, it is significant to note that a leaky venule has nothing to leak if its upstream capillary supply is blocked.
Compensatory reactions to microvascular pathology
Although arterial vasospasms and slow flow/no-reflow would produce areas of tissue ischemia, the system responds with attempts to compensate via reactive hyperemia and arteriovenous shunting [52,56]. Limbs in early stages of CRPS-I often have high arterial flow, yet at the same time have elevated venous pressure and arteriovenus shunting [24,25]. These phenomena are likely to be due to slow-flow/no-reflow in the affected capillary bed. The alternation between ischemia and reperfusion provides a way to spare tissue from lethal injury, but it also creates a vicious cycle of I-R injury which maintains microvascular dysfunction.
(3) Microvascular pathology leads to persistent ischemia which in turn leads to persistent inflammation
We propose that microvascular pathology leads to an ischemic state that evokes persistent inflammatory responses.
Microvascular pathology and ischemia
Several observations are consistent with the presence of impaired blood flow in CRPS-I patients. Skin capillary hemoglobin oxygenation (HbO2) is lowered and skin lactate is increased in CRPS-I limbs, suggesting that there is both impaired nutritive blood flow and enhanced anaerobic glycolysis [99,100]. Nail bed capillary flow is decreased [101], and reactive hyperemia in the cutaneous vasculature is impaired in CRPS-I patients [66]. Additionally, there is an impairment of high-energy phosphate metabolism in CRPS-I muscles [21,102], consistent with an impaired blood supply. Experimentally-evoked plasma extravasation is exaggerated in CRPS-I patients [103,104].
Several lines of evidence suggest that at least some CRPS-I patients have a persistent inflammatory condition. Necropsy studies of amputated limbs from patients with severe CRPS-I find lipofuscin deposits, atrophic fibers, and severely thickened capillary basal membranes in muscle and subcutaneous tissue [105,106], consistent with the presence of ischemia-evoked inflammation. Serum levels of calcitonin-gene related peptide and bradykinin are elevated in the venous drainage of CRPS-I limbs, consistent with the presence of an ongoing ischemic and inflammatory state in deep-tissues [107,108].
Eisenberg et al. [109] have detected greatly elevated levels of inflammatory by-products (e.g., malondialdeyhde) and cellular anti-oxidants in the serum and saliva of CRPS-I patients with typical disease. We suggest that these markers originate in tissues made ischemic and inflamed by slow-flow/no-reflow. There are several reports of increased levels of pro-inflammatory cytokines in CRPS-I patients [60,110–112], as would be expected if there was an ongoing inflammatory reaction. These findings are consistent with our results that malondialdeyhde and pro-inflammatory cytokines are increased in the hind paw muscle of CPIP rats [16], as well as numerous reports that these mediators are elevated after skeletal I-R injury (see above).
(4) The ischemic and inflammatory state in muscle results in nociceptor activation
In muscle, an ischemic state would be expected to be a direct cause of nociceptor discharge and nociceptor sensitization due to acidosis [113]. It has long been known that tissue acidosis associated with increased lactate causes muscle pain in ischemic tissue [114]. It is significant that mechanical allodynia in CPIP rats is directly correlated with lactate levels in CPIP hind paw muscle [16]. These findings are consistent with that the reports of CRPS-I patients that the pain produced by infusing a low pH solution into muscle in the affected limb is far more painful than the same infusion into a contralateral muscle [99]. Moreover, they report that the pain produced by an infusion into the normal, contralateral limb is qualitatively similar to their ipsilateral CRPS-I pain, i.e., their CRPS-I pain feels like inflammatory muscle pain [99]. Differential activation of sympathetic vasoconstrictor reflex responses to muscle and skin shows that the response in muscle, rather than skin, causes the greatest pain [115], consistent with ischemic muscle being a primary cause of CRPS-I pain.
Muscle lactate has also long been known to increase following exercise [116], so it is not surprising that lactate increases in exercised CPIP rats. Importantly, mechanical allodynia is also increased after exercise in CPIP rats, and mechanical allodynia is directly correlated with muscle lactate levels in CPIP rats [16]. This could explain the often reported phenomenon that the pain of CRPS-I is increased during exercise [20,117]. In sum, these mechanisms would be expected to cause a persistent deep, aching pain sensation. CRPS-I patients frequently (perhaps always) report the presence of a deep, aching pain, and they also commonly report that movement worsens their pain, as would be expected if their muscles were ischemic and inflamed.
(5) The effects of an ischemic and inflammatory state in bone
Prolonged ischemia of bone has been shown to produce bone inflammation and injury [118,119]. Three-phase bone scans reveal that CRPS-I patients often have a late-phase periarticular accumulation of tracer [33], consistent with an I-R injury-induced injury to venules leading to intramedullary plasma extravasation. The abnormality may not be confined to periarticular bone, as is generally believed, but may affect the bone shafts as well [120]. Using scintigraphical studies of CRPS-I patients over time reveals an early hyper-perfusion of bone that is followed by later hypo-perfusion [121].
Some CRPS-I patients also have radiographic evidence of osteoporosis, although this may be transient and migratory [33,120,122–126]. Osteoporosis is an expected consequence of impaired medullary perfusion. Clinical reports of pain relief in at least some CRPS-I patients treated with bisphosphonates or calcitonin are consistent with the idea of pain arising from ischemic and inflamed bone [127–130]. The bone pathology of CRPS-I was originally attributed to an inflammatory process [122]. All of the observations noted above are likely results of edema, ischemia, and inflammation secondary to slow-flow/no-reflow in periosteal and intramedullary microvessels. Ischemia in bone would cause deep, aching pain. It is our experience that CRPS-I patients often insist that their bones hurt.
(6) The effects of an ischemic and inflammatory state in peripheral nerves
There is no reason to doubt that endoneurial capillaries are also prone to the slow-flow/no-reflow phenomenon. Small, distal nerves would be especially vulnerable, simply because they have few capillaries. We have shown evidence of slow-flow/no-reflow in endoneurial microvessels in hind paw digital nerves in CPIP rats [15]. An ischemic and inflammatory state within a peripheral nerve should produce spontaneous ectopic discharge in sensory fibers. We have shown that ectopic discharge is present in A-fibers and C-fibers in CPIP rats [15].
Oaklander et al. [17] have shown degeneration of intraepidermal nerve fibers (IENF) in CRPS-I patients, and we have shown similar reduction of IENF density in the skin of CPIP rats [16]. No-flow in the capillaries of small, distal nerves might account for such degeneration.
Endoneurial ischemia and inflammation would be associated with increased levels of pro-inflammatory cytokines that are known to produce neuropathic (“neuritic”) pain [131,132]. Ectopic discharge in nociceptors due to endoneurial microvascular pathology would cause spontaneous pain referred to the tissue innervated by the nociceptors within the nerve. Such pain is properly categorized as neuropathic, while the pain from ischemic and inflamed muscle and bone is not. We thus propose that both inflammatory and neuropathic pain mechanisms are present in CRPS-I.
(7) Nociceptor activation leads to central sensitization
Deep-tissue C-fiber nociceptor discharge and the ectopic discharge of C-fibers traveling within ischemic and inflamed nerves will initiate and maintain N-methyl-D-aspartate receptor-mediated central sensitization [133]. Importantly, input from muscle C-fiber nociceptors is a much more potent initiator of central sensitization than input from cutaneous C-fiber nociceptors [134]. Also, while injecting inflammatory agents into skin produces allodynia that lasts at most hours, the same injections into muscle induce central sensitization and cutaneous allodynia lasting up to several weeks [135,136].
We hypothesize that the central sensitization evoked by deep-tissue nociceptor discharge is the primary cause of cutaneous allodynia and hyperalgesia. The presence or absence, severity, and spatial extent of cutaneous hypersensitivity might thus be expected to vary from patient-to-patient and from time-to-time, depending on fluctuations in the intensity of deep-tissue pain. Fluctuating cutaneous hypersensitivity is a common observation in CRPS-I patients [137,138], and cutaneous allodynia can be suppressed with the persistence of ongoing deep-tissue pain [12,139]. Cutaneous allodynia and hyperalgesia might thus be categorized as epiphenomena.
III. The role of the sympathetic nervous system and the stages of CRPS-I may depend on a progression from slow-flow to no-reflow
The duration of the initial, edematous stage of CRPS-I is highly variable. Some patients remain in this state, have episodes of exacerbated edema, or progress to a non-edematous stage characterized by cyanosis and/or atrophy of subcutaneous tissue. Patients with slow-flow would be expected to exhibit edema, but there can be no plasma extravasation from leaky venules if there is no-reflow in their upstream capillaries. Thus, at later times, when no-reflow predominates, the patient would be in the cyanotic/atrophic phase. We propose that the progression from early-stage to late-stage CRPS-I reflects the evolving dominance of no-reflow over slow-flow in deep tissue capillaries. The variability in the progression from predominately slow-flow to predominately no-reflow is likely to be the basis of the argument over whether CRPS-I is a staged condition [140].
Ischemia in the edematous stage of CRPS-I is likely to be due primarily to slow-flow and arteriole vasospasm. There is evidence that both phenomena may persist long after an I-R injury [55]. The exacerbation of slow-flow by arteriole vasospasm is likely to significantly worsen ischemia in the edematous stage of CRPS-I. Sympathetic activity (whether normal, increased, or decreased) would significantly reinforce arterial vasospasm and worsen slow-flow, thereby increasing ischemia and pain. Interruption of the sympathetic outflow to the affected region would relieve the arterial vasospasm, increase capillary flow and decrease pain; such patients would be classified as having sympathetically-maintained pain (SMP) [141]. In patients with relatively mild I-R injury, the absence of sympathetic exacerbation might allow microvascular recovery. In those patients with more severe I-R injury, the pain relief would be expected to be partial and/or temporary. Prolonged ischemia would likely lead to the gradual accumulation of capillaries with actual no-reflow. Sympathetically-mediated arteriole vasospasm would make little or no contribution to downstream blood flow if the capillaries are already occluded. The patient would thus be unlikely to benefit from interruption of sympathetic outflow to the affected region; i.e, the patient would have sympathetically-independent pain (SIP). Thus, we propose that the transition from microvascular slow-flow to no-reflow is also the basis for the transition from SMP to SIP.
IV. In causalgia (CRPS-II), injury to the neurovascular bundle induces an arterial vasospasm that produces I-R injury similar to that of CRPS-I
Vasospasm is a likely consequence of a traumatic injury to a major nerve because major nerves travel alongside a large artery and vein (i.e., the neurovascular bundle). Interestingly, Mitchell’s publications describing causalgia [142,143] also include the first detailed descriptions of severe long-lasting nerve injury-evoked arterial vasospasm. However, Mitchell made no mention of a possible link between vasospasm and causalgia. In CRPS-II, we suggest that injury to the neurovascular bundle evokes arterial vasospasm and initiates tissue ischemia and subsequent slow-flow/no-reflow in deep-tissue microvessels. The end result is the same as the I-R injury evoked by a compartment-like syndrome in CRPS-I.
Injuries to the brachial plexus at the level of the thoracic outlet sometimes lead to CRPS-like symptoms in the hand and arm. In such cases it is possible that irritation to the subclavian artery or to the sympathetic postganglionic fibers in adjacent nerves evoke vasospasm and the I-R phenomena discussed above. Relief of arterial and neural irritation via decompression of the scalene triangle is beneficial in these patients [144,145].
V. Clinical implications and novel therapeutic avenues
Our hypothesis suggests new treatment options for the pain of CRPS-I, and suggests possible mechanisms of action for therapies that are in use.
α-Adrenoceptor antagonists like phenoxybenzamine [69,146] may be more effective than sympathectomy or ganglionic blocks, especially in early stage CRPS-I, since they would prevent the enhanced vasoconstriction evoked by NE from the sympathetic postganglionic neuron, or NE and epinephrine from the circulation, from acting on up-regulated smooth muscle cell α-adenoreceptors. Arterial vasospasms, reduced vasodilatation, and ischemia associated with slow-flow/no-reflow may also be alleviated by treatment with nitric oxide donors or other vasodilators. The nitric oxide donors, nitroglycerine [147] and isosorbide dinitrate [148], have been used to alleviate CRPS-I.
Relief of deep-tissue ischemia may underlie the beneficial effects of hyperbaric oxygen therapy [149], spinal cord stimulation [150], and physical therapy [151,152] in CRPS-I patients. The key role of free radicals and cytokines after I-R injury suggests that antioxidants, free radical scavengers and anti-cytokine drugs maybe useful as therapeutic agents in CRPS-I. There are reports of successful treatment of CRPS-I patients with free radical scavengers and anti-oxidants [95,153–156] and anti-TNFα therapies [110,157,158]. Proposed damage to endothelial cells and associated vascular leakage through post-capillary venules, and the role of leukocytes and neutrophils in plugging capillaries leading to no-reflow, suggests that pain associated with CRPS-I may be alleviated both by anti-inflammatory drugs, and by novel agents that inhibit endothelin, leukocyte, and neutrophil attraction and migration, and platelet aggregation.
VI. Limitations of the hypothesis
We propose a hypothesis that we believe is relevant to at least a subset CRPS-I patients, and perhaps also to CRPS-II patients. We do not claim that the pain mechanisms that we describe here are present in all CRPS-I patients. CRPS-I may be a term that encompasses pathophysiologically distinct conditions [1,140]. We are not the first to emphasize that CRPS-I might be an abnormal inflammatory condition [21,22,122,159], but we present a mechanism that would initiate and maintain such a state – deep-tissue ischemia and inflammation due to persistent microvascular pathology subsequent to an IR injury. Moriwaki et al. [160] presented the idea that early-stage CRPS-II pain might have both neuropathic and inflammatory pain mechanisms. We agree and think that this is also true for CRPS-I, and we suggest that the neuropathic component is due to ectopic nociceptor discharge subsequent to endoneurial microvascular pathology.
We have not extended our hypothesis to account for the motor abnormalities that occur in CRPS-I [7]. We have also not accounted for the deficits in somatosympathetic reflexes that characterize CRPS-I [65,161]. Although, it may be that decreased sympathetic activity is a homeostatic reflex that increases perfusion to ischemic tissue [162]. We have also not accounted for the occurrence of CRPS-I following stroke [163]. Our hypothesis does not address the question of why CRPS-I symptoms sometimes spread to the contralateral extremity [71,72,164,165]. However, we do see contralateral allodynia in CPIP rats [14], and contralateral symptoms are common in response to muscle afferent activation [134] or muscle inflammation [135,136].
The incidence of CRPS-I is 3–5 times higher in women than in men. Our hypothesis suggests that this may be related to the increased prevalence of other vascular conditions in women (e.g., varicosis and Raynaud’s disease). Muscle mass in women is typically less than in men, and lower muscle mass is likely to be associated with a less extensive and less anastomotic capillary bed that is more susceptible to the ischemia produced by slow-flow/no-reflow. There is evidence that smoking is a significant risk factor in CRPS-I [166], and this may be related to nicotine-evoked damage to microvessels. There is accumulating evidence that suggests that CRPS-I is accompanied by changes in CNS regions that regulate somatosensation, motor control, and the autonomic nervous system [71,167–169]. A role for CNS mechanisms as consequences of the peripheral pathology described here is not a contradiction.
VII. Conclusions
For many years now, the cutaneous manifestations of CRPS-I have been the primary focus of investigation. We propose that as an underlying mechanism cutaneous pathology may be of decidedly secondary importance. We propose instead that deep tissue pathology (endothelial dysfunction and persistent ischemia and inflammation) in muscle, bone, and peripheral nerve is the key to the syndrome’s pathophysiology, and we suggest that the deep-tissue pathology is driving the cutaneous abnormalities. We are not the first to suggest that deep tissue pathology may be of fundamental importance, but we have proposed a distinct and testable hypothesis for its cause. Our hypothesis grew from observations of an animal model of CRPS-I. However, our ideas can be tested in CRPS-I patients, and are consistent with many reported clinical observations and investigations. The recent discovery of markers of inflammation in the serum and saliva of CRPS-I patients with typical disease [109], suggests a likely selection criterion for studies seeking to detect abnormal deep tissue perfusion, i.e., the patients with the highest suspicion of inflammation. It is relatively easy to measure the perfusion of capillary beds in skin (and the nail bed), but difficult to measure perfusion in deep tissues. However, new methods like near-infrared spectroscopy [170] may allow direct measurements muscle capillary function.
Acknowledgments
This research was supported by grants from CIHR, NSERC, FRSQ and The Louise and Allan Edwards Foundation to T.J.C. and NIH to G.J.B.
References
- 1.Stanton-Hicks M, Jänig W, Hassenbusch S, Haddox JD, Boas R, Wilson P. Reflex sympathetic dystrophy: changing concepts and taxonomy. Pain. 1995;63:127–33. doi: 10.1016/0304-3959(95)00110-E. [DOI] [PubMed] [Google Scholar]
- 2.de Mos M, de Bruijn AG, Huygen FJ, Dieleman JP, Stricker BH, Sturkenboom MC. The incidence of complex regional pain syndrome: a population-based study. Pain. 2007;129:12–20. doi: 10.1016/j.pain.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 3.Bonica JJ. The management of pain. Philadelphia: Lea & Febiger; 1953. [Google Scholar]
- 4.Bonica JJ. Management of intractable pain in general practice. Gen Prac. 1966;33:107–23. [PubMed] [Google Scholar]
- 5.Schwartzman RJ, McLellan TL. Reflex sympathetic dystrophy: a review. Arch Neurol. 1987;44:555–61. doi: 10.1001/archneur.1987.00520170081028. [DOI] [PubMed] [Google Scholar]
- 6.Bhatia KP, Bhatt MH, Marsden CD. The causalgia-dystonia syndrome. Brain. 1993;116:843–51. doi: 10.1093/brain/116.4.843. [DOI] [PubMed] [Google Scholar]
- 7.Schwartzman RJ, Kerrigan J. The movement disorder of reflex sympathetic dystrophy. Neurology. 1990;40:57–61. doi: 10.1212/wnl.40.1.57. [DOI] [PubMed] [Google Scholar]
- 8.Veldman PH, Reyen HM, Arnt IE, Goris RJ. Signs and symptoms of reflex sympathetic dystrophy: prospective study of 829 patients. Lancet. 1993;342:1012–6. doi: 10.1016/0140-6736(93)92877-v. [DOI] [PubMed] [Google Scholar]
- 9.Schwartzman RJ. Reflex sympathetic dystrophy. In: Loscalzo J, Creager MA, Dzau VJ, editors. Vascular Medicine: A textbook of Vascular Biology and Disease. 2. Boston: Little, Brown; 1996. pp. 1209–21. [Google Scholar]
- 10.Bonica JJ. Causalgia and other reflex sympathetic dystrophies. In: Bonica JJ, editor. The Management of Pain. Vol. 2. Philadelphia: Lea & Febiger; 1990. pp. 220–43. [Google Scholar]
- 11.Baron R, Levine JD, Fields HL. Causalgia and reflex sympathetic dystrophy: does the sympathetic nervous system contribute to the generation of pain? Muscle Nerve. 1999;22:678–95. doi: 10.1002/(sici)1097-4598(199906)22:6<678::aid-mus4>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 12.Schwartzman RJ, Liu JE, Smullens SN, Hyslop T, Tahmoush AJ. Long-term outcome following sympathectomy for complex regional pain syndrome type 1 (RSD) J Neurol Sci. 1997;150:149–52. doi: 10.1016/s0022-510x(97)00078-6. [DOI] [PubMed] [Google Scholar]
- 13.Wakisaka S, Kajander KC, Bennett GJ. Abnormal skin temperature and abnormal sympathetic vasomotor innervation in an experimental painful peripheral neuropathy. Pain. 1991;46:299–313. doi: 10.1016/0304-3959(91)90113-C. [DOI] [PubMed] [Google Scholar]
- 14.Coderre TJ, Xanthos DN, Francis L, Bennett GJ. Chronic post-ischemia pain (CPIP): a novel animal model of complex regional pain syndrome-type I (CRPS-I; reflex sympathetic dystrophy) produced by prolonged hindpaw ischemia and reperfusion in the rat. Pain. 2004;112:94–105. doi: 10.1016/j.pain.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 15.Laferrière A, Millecamps M, Xanthos DN, Xiao W-H, Bennett GJ, Coderre TJ. Chronic post-ischemia pain: A novel animal model suggests that ischemia-reperfusion (I-R) injury, no-reflow and chronic tissue ischemia contribute to CRPS-I. Eur J Pain. 2007;11:S61–2. [Google Scholar]
- 16.Laferrière A, Millecamps M, Xanthos DN, Xiao W-H, Siau C, Vrolijk-de Mos M, Sachot C, Huygen FJPM, Bennett GJ, Coderre TJ. A novel animal model suggests complex regional pain syndrome (CRPS) depends on microvascular pathology and chronic deep tissue ischemia. Nature. 2008 [submitted] [Google Scholar]
- 17.Oaklander AI, Rissmiller JG, Gelman LB, Zheng L, Chang Y, Gott R. Evidence of focal small-fiber axonal degeneration in complex regional pain syndrome-I (reflex sympathetic dystrophy) Pain. 2006;120:235–43. doi: 10.1016/j.pain.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 18.Xanthos DN, Bennett GJ, Coderre TJ. Norepinephrine-induced nociception and vasoconstrictor hypersensitivity in rats with chronic post-ischemia pain. Pain. 2008 doi: 10.1016/j.pain.2007.10.031. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Millecamps M, Laferriere A, Coderre TJ. Role of peripheral endothelin receptors in an animal model of Complex Regional Pain Syndrome type 1. doi: 10.1016/j.pain.2010.07.003. Manuscript in preparation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oyen WJ, Arntz IE, Claessens RM, Van der Meer JW, Corstens FH, Goris RJA. Reflex sympathetic dystrophy of the hand: an excessive inflammatory response? Pain. 1993;55:151–7. doi: 10.1016/0304-3959(93)90144-E. [DOI] [PubMed] [Google Scholar]
- 21.Goris RJA. Reflex sympathetic dystrophy: model of a severe regional inflamatory response syndrome. World J Surg. 1998;22:197–202. doi: 10.1007/s002689900369. [DOI] [PubMed] [Google Scholar]
- 22.van der Laan L, Goris RJA. The role of an exaggerated regional inflammatory response in the pathophysiology of CRPS. In: Harden RN, Baron R, Jänig W, editors. Complex Regional Pain Syndrome. Progress in Pain Research and Management. Vol. 22. Seattle: IASP Press; 2001. pp. 183–91. [Google Scholar]
- 23.Little RC, Ginsburg JM. The physiologic basis for clinical edema. Arch Intern Med. 1984;144:1661–4. doi: 10.1001/archinte.144.8.1661. [DOI] [PubMed] [Google Scholar]
- 24.Matsumura H, Jimbo Y, Watanabe K. Haemodynamic changes in early phase reflex sympathetic dystrophy. Scand J Plast Reconstr Surg Hand Surg. 1996;30:133–8. doi: 10.3109/02844319609056395. [DOI] [PubMed] [Google Scholar]
- 25.Schurmann M, Zaspel J, Grandl G, Wipfel A, Christ F. Assessment of the peripheral microcirculation using computer-assisted venous congestion plethysmography in post-traumatic complex regional pain syndrome type I. J Vasc Res. 2001;38:453–61. doi: 10.1159/000051078. [DOI] [PubMed] [Google Scholar]
- 26.Mubarak SJ, Pedowitz RA, Hargens AR. Compartment syndromes. Curr Orthop. 1989;3:36–40. doi: 10.1016/0268-0890(89)90069-8. [DOI] [PubMed] [Google Scholar]
- 27.Lyden SP, Shortell CK, Illig KA. Reperfusion and compartment syndromes: strategies for prevention and treatment. Semin Vasc Surg. 2001;14:107–13. doi: 10.1053/svas.2001.23166. [DOI] [PubMed] [Google Scholar]
- 28.Hargens AR, Mubarak SJ. Current concepts in the pathophysiology, evaluation, and diagnosis of compartment syndrome. Hand Clin. 1998;14:371–83. [PubMed] [Google Scholar]
- 29.Fulkerson E, Razi A, Tejwani N. Review: acute compartment syndrome of the foot. Foot Ankle Int. 2003;24:180–7. doi: 10.1177/107110070302400214. [DOI] [PubMed] [Google Scholar]
- 30.Velmahos GC, Toutouzas KG. Vacular trauma and compartment syndromes. Surg Clin North Am. 2002;82:125–41. doi: 10.1016/S0039-6109(03)00145-2. [DOI] [PubMed] [Google Scholar]
- 31.Kim TK, Savino RM, McFarland EG, Cosgarea AJ. Neurovascular complications of knee arthroscopy. Am J Sports Med. 2002;30:619–29. doi: 10.1177/03635465020300042501. [DOI] [PubMed] [Google Scholar]
- 32.Joseph J, Giannoudis PV, Hinsche A, Cohen A, Matthews SJ, Smith RM. Compartment syndrome following isolated ankle fracture. Int Orthop. 2000;24:173–5. doi: 10.1007/s002640000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allen G, Galer BS, Schwartz L. Epidemiology of complex regional pain syndrome: a retrospective chart review of 134 patients. Pain. 1999;80:539–44. doi: 10.1016/S0304-3959(98)00246-2. [DOI] [PubMed] [Google Scholar]
- 34.Galer BS, Henderson J, Perander J, Jensen MP. Course of symptoms and quality of life measurement in Complex Regional Pain Syndrome: a pilot survey. J Pain Symptom Manage. 2000;20:286–92. doi: 10.1016/s0885-3924(00)00183-4. [DOI] [PubMed] [Google Scholar]
- 35.van der Laan L, Goris RJ. Reflex sympathetic dystrophy after a burn injury. Burns. 1996;22:303–6. doi: 10.1016/0305-4179(95)00139-5. [DOI] [PubMed] [Google Scholar]
- 36.Khan K, Brown J, Way S, Vass N, Crichton K, Alexander R, Baxter A, Butler M, Wark J. Overuse injuries in classical ballet. Sports Med. 1995;19:341–57. doi: 10.2165/00007256-199519050-00004. [DOI] [PubMed] [Google Scholar]
- 37.Johansen K, Watson J. Compartment syndrome: new insights. Semin Vasc Surg. 1998;11:294–301. [PubMed] [Google Scholar]
- 38.Brennan FH, Kane SF. Diagnosis, treatment, options, and rehabilitation of chronic lower leg exertional compartment syndrome. Curr Sports Med Rep. 2003;2:247–50. doi: 10.1249/00149619-200310000-00003. [DOI] [PubMed] [Google Scholar]
- 39.Miller SH, Price G, Buck D, Neeley J, Kennedy TJ, Graham WP, 3rd, Davis TS. Effects of tourniquet ischemia and postischemic edema on muscle metabolism. J Hand Surg. 1979;4:547–55. doi: 10.1016/s0363-5023(79)80008-8. [DOI] [PubMed] [Google Scholar]
- 40.Wakai A, Winter DC, Street JT, Redmond PH. Pneumatic tourniquets in extremity surgery. J Am Acad Orthop Surg. 2001;9:345–51. doi: 10.5435/00124635-200109000-00008. [DOI] [PubMed] [Google Scholar]
- 41.Kellog EW. Superoxide, hydrogen peroxide and singlet oxygen in lipid peroxidation by a xanthine oxydase system. J Biol Chem. 1975;250:8812–17. [PubMed] [Google Scholar]
- 42.McCord JM. Oxygen-derived radicals: a link between reperfusion injury and inflammation. Fed Proc. 1987;46:2402–6. [PubMed] [Google Scholar]
- 43.Bolli R, Jeroudi MO, Patel BS, DuBose CM, Roberts EK, McCay PB. Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proc Natl Acad Sci USA. 1989;86:4695–99. doi: 10.1073/pnas.86.12.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Inauen W, Suzuki M, Granger DN. Mechanisms of cellular injury: potential sources of oxygen free radicals in ischemia/reperfusion. Microcirc Endothelium Lymphatics. 1989;5:143–55. [PubMed] [Google Scholar]
- 45.Przyklenk K, Kloner RA. “Reperfusion injury” by oxygen-derived free radicals? Effect of superoxide dismutase plus catalase, given at the time of reperfusion, on myocardial infarct size, contractile function, coronary microvasculature, and regional myocardial blood flow. Circ Res. 1989;64:86–96. doi: 10.1161/01.res.64.1.86. [DOI] [PubMed] [Google Scholar]
- 46.Partrick DA, Moore FA, Moore EE, Barnett CC, Jr, Silliman CC. Neutrophil priming and activation in the pathogenesis of postinjury multiple organ failure. New Horiz. 1996;4:194–210. [PubMed] [Google Scholar]
- 47.Yassin MM, Barros D’Sa AA, Parks TG, McCaigue MD, Leggett P, Halliday MI, Rowlands BJ. Lower limb ischaemia-reperfusion injury alters gastrointestinal structure and function. Br J Surg. 1997;84:1425–9. [PubMed] [Google Scholar]
- 48.Blaisdell FW. The pathophysiology of skeletal muscle ischemia and the reperfusion syndrome: a review. Cardiovascular Surgery. 2002;10:620–30. doi: 10.1177/096721090201000620. [DOI] [PubMed] [Google Scholar]
- 49.Lefer AM, Tsao PS, Lefer DJ, Ma XL. Role of endothelial dysfunction in the pathogenesis of reperfusion injury after myocardial ischemia. FASEB J. 1991;5:2029–34. doi: 10.1096/fasebj.5.7.2010056. [DOI] [PubMed] [Google Scholar]
- 50.Brooks B. Pathologic changes in muscle as the result of disturbances of circulation. Arch Surg. 1922;5:188–216. [Google Scholar]
- 51.Harris AG, Steinbauer M, Leiderer R, Messmer K. Role of leukocyte plugging and edema in skeletal muscle ischemia-reperfusion injury. Am J Physiol. 1997;273:H989–96. doi: 10.1152/ajpheart.1997.273.2.H989. [DOI] [PubMed] [Google Scholar]
- 52.Menger MD, Rücker M, Vollmar B. Capillary dysfunction in striated muscle ischemia/reperfusion: on the mechanisms of capillary “no-reflow”. Shock. 1997;8:26–31. [PubMed] [Google Scholar]
- 53.Urbaniak JR, Seaber AV, Chen LE. Assessment of ischemia and reperfusion injury. Clin Orthop. 1997;334:30–6. [PubMed] [Google Scholar]
- 54.Gute DC, Ishida T, Yarimizu K, Korthuis RJ. Inflammatory responses to ischemia and reperfusion in sketetal muscle. Mol Cell Biochem. 1998;179:169–87. doi: 10.1023/a:1006832207864. [DOI] [PubMed] [Google Scholar]
- 55.Kloner RA. No-reflow phenomenon persists long-term after ischemia/reperfusion in the rat and predicts infarct expansion. Circulation. 2003;108:2911–7. doi: 10.1161/01.CIR.0000101917.80668.E1. [DOI] [PubMed] [Google Scholar]
- 56.Kennedy TJ, Miller SH, Nellis SH, Buck D, Flaim SF, Graham WP, 3rd, Davis TS. Effects of transient ischemia on nutrient flow and arteriovenous shunting in canine hindlimb. Ann Surg. 1981;193:255–63. doi: 10.1097/00000658-198103000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kurose I, Wolf R, Grisham MB, Granger DN. Modulation of ischemia/reperfusion-induced microvascular dysfunction by nitric oxide. Circ Res. 1994;74:376–82. doi: 10.1161/01.res.74.3.376. [DOI] [PubMed] [Google Scholar]
- 58.Wang WZ, Anderson G, Fleming JT, Peter FW, Franken RJ, Acland RD, Barker J. Lack of nitric oxide contributes to vasospasm during ischemia/reperfusion injury. Plast Reconstr Surg. 1997;99:1099–108. doi: 10.1097/00006534-199704000-00028. [DOI] [PubMed] [Google Scholar]
- 59.Dayan L, Salman S, Norman D, Vatine JJ, Calif E, Jacob G. Exaggerated vasoconstriction in Complex Regional Pain Syndrome-1 is associated with impaired resistance artery endothelial function and local vascular reflexes. J Rheumatol. 2008 In press. [PubMed] [Google Scholar]
- 60.Groeneweg JG, Huygen FJ, Heijmans-Antonissen C, Niehof S, Zijlstra FJ. Increased endothelin-1 and diminished nitric oxide levels in blister fluids of patients with intermediate cold type complex regional pain syndrome type 1. BMC Musculoskelet Disord. 2006;7:91. doi: 10.1186/1471-2474-7-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rubanyi GM. Endothelium-derived relaxing and contracting factors. J Cell Biochem. 1991;46:27–36. doi: 10.1002/jcb.240460106. [DOI] [PubMed] [Google Scholar]
- 62.Pernow J, Wang QD. Endothelin in myocardial ischaemia and reperfusion. Cardiovasc Res. 1997;33:518–26. doi: 10.1016/s0008-6363(96)00265-9. [DOI] [PubMed] [Google Scholar]
- 63.Sapienza P, Edwards JD, Mingoli A, McGregor PE, Cavallari N, Agrawal DK. Ischemia-induced peripheral arterial vasospasm role of alpha 1- and alpha 2-adrenoceptors. J Surg Res. 1996;62:192–6. doi: 10.1006/jsre.1996.0194. [DOI] [PubMed] [Google Scholar]
- 64.Ali Z, Raja SN, Wesselmann U, Fuchs PN, Meyer RA, Campbell JN. Intradermal injection of NE evokes pain in patients with sympathetically maintained pain. Pain. 2000;88:161–168. doi: 10.1016/S0304-3959(00)00327-4. [DOI] [PubMed] [Google Scholar]
- 65.Wasner G, Heckmann K, Maier C, Baron R. Vascular abnormalities in acute reflex sympathetic dystrophy (CRPS I): complete inhibition of sympathetic nerve activity with recovery. Arch Neurol. 1999;56:613–20. doi: 10.1001/archneur.56.5.613. [DOI] [PubMed] [Google Scholar]
- 66.Kurvers HA, Jacobs MJ, Beuk RJ, Van den Wildenberg FA, Kitslaar PJ, Slaaf DW, Reneman RS. Reflex sympathetic dystrophy: evolution of microcirculatory disturbances in time. Pain. 1995;60:333–40. doi: 10.1016/0304-3959(94)00133-y. [DOI] [PubMed] [Google Scholar]
- 67.Arnold JMO, Teasell RW, Macleod AP, Brown JE, Carruthers SG. Increased venous alpha-adrenoceptors responsiveness in patients with reflex sympathetic dystrophy. Ann Intern Med. 1993;83:185–92. doi: 10.7326/0003-4819-118-8-199304150-00008. [DOI] [PubMed] [Google Scholar]
- 68.Drummond PD, Skipworth S, Finch PM. alpha 1-adrenoceptors in normal and hyperalgesic human skin. Clin Sci (Lond) 1996;91:73–7. doi: 10.1042/cs0910073. [DOI] [PubMed] [Google Scholar]
- 69.Teasell RW, Arnold JM. Alpha-1 adrenoceptor hyperresponsiveness in three neuropathic pain states: complex regional pain syndrome 1, diabetic peripheral neuropathic pain and central pain states following spinal cord injury. Pain Res Manag. 2004;9:89–97. doi: 10.1155/2004/150503. [DOI] [PubMed] [Google Scholar]
- 70.Häbler HJ, Jänig W. Neuropathy after spinal nerve injury in rats: A model for sympathetically-maintained pain? In: Harden RN, Baron R, Jänig W, editors. Complex Regional Pain Syndrome, Progress in Pain Research and Management. Vol. 22. Seattle: IASP Press; 2001. pp. 39–52. [Google Scholar]
- 71.Mailis-Gagnon A, Bennett GJ. Abnormal contralateral pain responses from an intradermal injection of phenylephrine in a subset of patients with complex regional pain syndrome (CRPS) Pain. 2004;111:378–84. doi: 10.1016/j.pain.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 72.Maleki J, LeBel AA, Bennett GJ, Schwartzman RJ. Patterns of spread in Complex Regional Pain Syndrome, Type I (Reflex Sympathetic Dystrophy) Pain. 2000;88:259–66. doi: 10.1016/S0304-3959(00)00332-8. [DOI] [PubMed] [Google Scholar]
- 73.Hellstrom HR. Coronary microvascular spasm in patients with vasospastic angina. J Am Coll Cardiol. 2002;40:573–4. doi: 10.1016/s0735-1097(02)02000-4. [DOI] [PubMed] [Google Scholar]
- 74.Foisie PS. Traumatic arterial vasospasm. N Engl J Med. 1947;237:295–302. doi: 10.1056/NEJM194708282370901. [DOI] [PubMed] [Google Scholar]
- 75.Hellstrom HR. Coronary artery stasis after induced myocardial infarction in the dog. Cardiovasc Res. 1971;5:371–75. doi: 10.1093/cvr/5.3.371. [DOI] [PubMed] [Google Scholar]
- 76.Blumberg H, Hoffmann U. Diagnosis of sympathetic reflex dystrophy. Comparison of ischemia test and modified guanethidine blockade. Nervenarzt. 1994;65:370–4. [PubMed] [Google Scholar]
- 77.Blanchard J, Ramamurthy S, Walsh N, Hoffman J, Schoenfeld L. Intravenous regional sympatholysis: a double-blind comparison of guanethidine, reserpine, and normal saline. J Pain Symptom Manage. 1990;5:357–61. doi: 10.1016/0885-3924(90)90030-n. [DOI] [PubMed] [Google Scholar]
- 78.Granger DN, Kvietys PR, Perry MA. Leucocyte-endothelial cell adhesion induced by ischemia and reperfusion. Can J Physiol Pharmacol. 1993;71:67–75. doi: 10.1139/y93-011. [DOI] [PubMed] [Google Scholar]
- 79.Lindsay TF, Hill J, Ortiz F, Rudolph A, Valeri CR, Hechtman HB, Moore FD., Jr Blockade of complement activation prevents local and pulmonary albumin leak after lower torso ischemia-reperfusion. Ann Surg. 1992;216:677–83. doi: 10.1097/00000658-199212000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lehr HA, Guhlmann A, Nolte D, Keppler D, Messmet K. Leukotrienes as mediators in ischemia-reperfusion injury in a microcirculation model in the hamster. J Clin Invest. 1991;81:2036–41. doi: 10.1172/JCI115233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Goldman G, Welbourn R, Paterson IS, Klausner JM, Kobzik L, Valeri CR, Shepro D, Hechtman HB. Ischemia-induced neutrophil activation and diapedesis is lipoxygenase dependent. Surgery. 1990;107:428–33. [PubMed] [Google Scholar]
- 82.Kubes P, Ibbotson G, Russell J, Wallace JL, Granger DN. Role of platelet-activating factor in ischemia/reperfusion-induced leukocyte adherence. Am J Physiol. 1990;259:G300–5. doi: 10.1152/ajpgi.1990.259.2.G300. [DOI] [PubMed] [Google Scholar]
- 83.Kubes P, Suzuki M, Granger DN. Platelet-activating factor-induced microvascular dysfunction: role of adherent leukocytes. Am J Physiol. 1990;258:G158–63. doi: 10.1152/ajpgi.1990.258.1.G158. [DOI] [PubMed] [Google Scholar]
- 84.Sternbergh WC, Tuttle TM, Makhoul RG, Bear HD, Sobel M, Fowler AA. Postischemic extremities exhibit immediate release of tumor necrosis factor. J Vasc Surg. 1994;20:474–81. doi: 10.1016/0741-5214(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 85.Yan SF, Tritto I, Pinsky D, Liao H, Huang J, Fuller G, Brett J, May L, Stern D. Induction of interleukin 6 (IL-6) by hypoxia in vascular cells. Central role of the binding site for nuclear factor-IL-6. J Biol Chem. 1995;270:11463–71. doi: 10.1074/jbc.270.19.11463. [DOI] [PubMed] [Google Scholar]
- 86.Strock PE, Manjo G. Microvascular changes in acutely ischemic rat muscle. Surg Gyn Obst. 1969;129:1213–24. [PubMed] [Google Scholar]
- 87.Kloner RA, Ganote CE, Jennings RB. The “no-reflow” phenomenon after temporary coronary occlusion in dogs. J Clin Invest. 1974;54:1496–508. doi: 10.1172/JCI107898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Engler RL, Schmid-Schonbein GW, Pavelec RS. Leukocyte capillary plugging in myocardial ischemia and reperfusion in the dog. Am J Pathol. 1983;111:98–111. [PMC free article] [PubMed] [Google Scholar]
- 89.Suval WD, Duran WN, Boric MP, Hobson RW, 3rd, Berendsen PB, Ritter AB. Microvascular transport and endothelial cell alterations preceding skeletal muscle damage in ischemia and reperfusion injury. Am J Surg. 1987;154:211–8. doi: 10.1016/0002-9610(87)90181-4. [DOI] [PubMed] [Google Scholar]
- 90.Menger MD, Pelikan S, Steiner D, Messmer K. Microvascular ischemia-reperfusion injury in striated muscle: significance of “reflow paradox”. Am J Physiol. 1992;263:H1901–6. doi: 10.1152/ajpheart.1992.263.6.H1901. [DOI] [PubMed] [Google Scholar]
- 91.Harris AG, Leiderer R, Peer F, Messmer K. Skeletal muscle microvascular and tissue injury after varying durations of ischemia. Am J Physiol. 1996;271:H2388–98. doi: 10.1152/ajpheart.1996.271.6.H2388. [DOI] [PubMed] [Google Scholar]
- 92.Jerome SN, Akimitsu T, Korthius RJ. Leukocyte adhesion, edema, and development of postischemic capillary no-reflow. Am J Physiol. 1994;267:H1329–36. doi: 10.1152/ajpheart.1994.267.4.H1329. [DOI] [PubMed] [Google Scholar]
- 93.Jerome SN, Kong L, Korthuis RJ. Microvascular dysfunction in postischemic skeletal muscle. J Invest Surg. 1994;7:3–16. doi: 10.3109/08941939409018278. [DOI] [PubMed] [Google Scholar]
- 94.Reffelmann T, Kloner RA. Microvascular reperfusion injury: rapid expansion of anatomic no-reflow during reperfusion in the rabbit. Am J Physiol. 2002;283:H1099–107. doi: 10.1152/ajpheart.00270.2002. [DOI] [PubMed] [Google Scholar]
- 95.Nanobashvili J, Neumayer C, Fuegl A, Blumer R, Prager M, Sporn E, Polterauer P, Malinski T, Huk I. Development of ‘no-reflow’ phenomenon in ischemia/reperfusion injury: failure of active vasomotility and not simply passive vasoconstriction. Eur Surg Res. 2003;35:417–24. doi: 10.1159/000072226. [DOI] [PubMed] [Google Scholar]
- 96.Ambrosio G, Weisman HF, Mannisi JA, et al. Progressive impairment of regional myocardial perfusion after initial restoration of postischemic blood flow. Circulation. 1989;80:1846–61. doi: 10.1161/01.cir.80.6.1846. [DOI] [PubMed] [Google Scholar]
- 97.Gorodkin R, Moore T, Herrick A. Assessment of endothelial function in complex regional pain syndrome type I using iontophoresis and laser Doppler imaging. Rheumatology. 2004;43:727–30. doi: 10.1093/rheumatology/keh158. [DOI] [PubMed] [Google Scholar]
- 98.McDonald DM, Thurston G, Baluk P. Endothelial gaps as sites for plasma leakage in inflammation. Microcirculation. 1999;6:7–22. [PubMed] [Google Scholar]
- 99.Birklein F, Weber M, Neundorfer B. Incresed skin lactate in complex regional pain syndrome: evidence for tissue hypoxia? Neurology. 2000;55:1213–5. doi: 10.1212/wnl.55.8.1213. [DOI] [PubMed] [Google Scholar]
- 100.Koban M, Leis S, Schultze-Mosgau S, Birklein F. Tissue hypoxia in complex regional pain syndrome. Pain. 2003;104:149–57. doi: 10.1016/s0304-3959(02)00484-0. [DOI] [PubMed] [Google Scholar]
- 101.Rosén L, Ostergren J, Fagrell B, Stranden E. Skin microvascular circulation in the sympathetic dystrophies evaluated by videophotometric capillaroscopy and laser Doppler fluxmetry. Eur J Clin Invest. 1988;18:305–8. doi: 10.1111/j.1365-2362.1988.tb01263.x. [DOI] [PubMed] [Google Scholar]
- 102.Heerschap A, den Hollander JA, Reynen H, Goris RJA. Metabolic changes in reflex sympathetic dystrophy: A 31P NMR spectroscopy study. Muscle Nerve. 1993;16:367–73. doi: 10.1002/mus.880160405. [DOI] [PubMed] [Google Scholar]
- 103.Weber M, Birklein F, Neundorfer B, Schmelz M. Facilitated neurogenic inflammation in complex regional pain syndrome. Pain. 2001;91:251–7. doi: 10.1016/S0304-3959(00)00445-0. [DOI] [PubMed] [Google Scholar]
- 104.Leis S, Weber M, Isselmann A, Schmelz M, Birklein F. Substance-P-induced protein extravasation is bilaterally increased in complex regional pain syndrome. Exp Neurol. 2003;183:197–204. doi: 10.1016/s0014-4886(03)00163-8. [DOI] [PubMed] [Google Scholar]
- 105.van der Laan L, ter Laak HJ, Gabreels-Festen A, Gabreels F, Goris RJA. Complex regional pain syndrome type I (RSD): pathology of skeletal muscle and peripheral nerve. Neurology. 1998;51:20–5. doi: 10.1212/wnl.51.1.20. [DOI] [PubMed] [Google Scholar]
- 106.Albrecht PJ, Hines S, Eisenberg E, Pud D, Finlay DR, Connolly MK, Pare M, Davar M, Rice FL. Pathologic alterations of cutaneous innervations and vasculature in affected limbs from patients with complex regional pain syndrome. Pain. 2006;120:244–266. doi: 10.1016/j.pain.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 107.Blair SJ, Chinthagada M, Hoppenstehdt D, Kijowski R, Fareed J. Role of neuropeptides in pathogenesis of reflex sympathetic dystrophy. Acta Orthop Belg. 1998;64:448–51. [PubMed] [Google Scholar]
- 108.Birklein F, Schmelz M, Schifter S, Weber M. The important role of neuropeptides in complex regional pain syndrome. Neurology. 2001;57:2179–84. doi: 10.1212/wnl.57.12.2179. [DOI] [PubMed] [Google Scholar]
- 109.Eisenberg E, Shtahl S, Geller R, Reznick AZ, Sharf O, Ravbinovich M, Erenreich A, Nagler RM. Serum and salivary oxidative analysis in Complex Regional Pain Syndrome. Pain. 2008 doi: 10.1016/j.pain.2008.04.019. (In press) [DOI] [PubMed] [Google Scholar]
- 110.Huygen FJ, De Bruijn AG, De Bruin MT, Groeneweg JG, Klein J, Zijistra FJ. Evidence for local inflammation in complex regional pain syndrome type 1. Mediators Inflamm. 2002;11:47–51. doi: 10.1080/09629350210307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Huygen FJ, Niehof S, Zijlstra FJ, van Hagen PM, van Daele PL. Successful treatment of CRPS 1 with anti-TNF. J Pain Symptom Manage. 2004;27:101–3. doi: 10.1016/j.jpainsymman.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 112.Alexander GM, van Rijn MA, van Hilten JJ, Perreault MJ, Schwartzman RJ. Changes in cerebrospinal fluid levels of pro-inflammatory cytokines in CRPS. Pain. 2005;116:213–9. doi: 10.1016/j.pain.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 113.Mense S. The pathogenesis of muscle pain. Curr Pain Headache Rep. 2003;7:419–25. doi: 10.1007/s11916-003-0057-6. [DOI] [PubMed] [Google Scholar]
- 114.Hagberg H. Intracellular pH during ischemia in skeletal muscle: relationship to membrane potential, extracellular pH, tissue lactic acid and ATP. Pflugers Arch. 1985;404:342–347. doi: 10.1007/BF00585346. [DOI] [PubMed] [Google Scholar]
- 115.Schattschneirder J, Binder A, Siebrecht D, Wasner G, Baron R. Complex regional pain syndromes: the influence of cutaneous and deep somatic sympathetic innervation on pain. Clin J Pain. 2006;22:240–4. doi: 10.1097/01.ajp.0000169672.49438.67. [DOI] [PubMed] [Google Scholar]
- 116.Sahlin K, Harris RC, Nylind B, Hultman E. Lactate content and pH in muscle obtained after dynamic exercise. Pflugers Arch. 1976;367:143–9. doi: 10.1007/BF00585150. [DOI] [PubMed] [Google Scholar]
- 117.Perez RS, Collins S, Marinus J, Zuurmond WW, de Lange JJ. Diagnostic criteria for CRPS I: differences between patient profiles using three different diagnostic sets. Eur J Pain. 2007;11:895–902. doi: 10.1016/j.ejpain.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 118.Skawina A, Wyczółkowski M, Radecki T. Studies of the effect of ischemia of the legs on the degree of osteoporosis of the femur and tibia using the cortex-shaft index. Przegl Lek. 1989;46:346–9. [PubMed] [Google Scholar]
- 119.Kälebo P, Johansson C, Albrektsson T. Temporary bone tissue ischemia in the hind limb of the rabbit. A vital microscopic study. Arch Orthop Trauma Surg. 1986;105:321–5. doi: 10.1007/BF00449935. [DOI] [PubMed] [Google Scholar]
- 120.Atkins RM, Tindale W, Bickerstaff D, Kanis JA. Quantitative bone scintigraphy in reflex sympathetic dystrophy. Br J Rheumatol. 1993;32:41–5. doi: 10.1093/rheumatology/32.1.41. [DOI] [PubMed] [Google Scholar]
- 121.Demangeat JL, Constantinesco A, Brunot B, Foucher G, Farcot JM. Three-phase bone scanning in reflex sympathetic dystrophy of the hand. J Nucl Med. 1988;29:26–32. [PubMed] [Google Scholar]
- 122.Sudeck P. Uber die acute (trophoneurotische) knochenatrophie nach entzundungen und traumen der extremitaten. Deustsche Med Wochenschrift. 1902;29:336–8. [Google Scholar]
- 123.Kozin F, Genant HK, Bekerman C, McCarty DJ. The reflex sympathetic dystrophy syndrome. II. Roentgenographic and scintigraphic evidence of bilaterality and of periarticular accentuation. Am J Med. 1976;60:332–43. doi: 10.1016/0002-9343(76)90748-8. [DOI] [PubMed] [Google Scholar]
- 124.Kozin F, McCarty DJ, Simms J, Genant H. The reflex sympathetic dystrophy syndrome. I. Clinical and histologic studies: evidence for bilaterality, response to corticosteriods and articular involvement. Am J Med. 1976;60:321–31. doi: 10.1016/0002-9343(76)90747-6. [DOI] [PubMed] [Google Scholar]
- 125.Mailis A, Inman R, Pham D. Transient migratory osteoporosis: a variant of reflex sympathetic dystrophy? Report of 3 cases and literature review. J Rheumatol. 1992;19:758–64. [PubMed] [Google Scholar]
- 126.Leitha T, Korpan M, Staudenherz A, Wunderbaldinger P, Fialka V. Five phase bone scintigraphy supports the patholphysiological concept of a subclinical inflammatory process in reflex sympathetic dystrophy. Q J Nucl Med. 1996;40:188–93. [PubMed] [Google Scholar]
- 127.Cortet B, Flipo RM, Coquerelle P, Duquesnoy B, Delcambre B. Treatment of severe, recalcitrant reflex sympathetic dystrophy: assessment of efficacy and safety of the second generation bisphosphonate pamidronate. Clin Rheumatol. 1997;16:51–6. doi: 10.1007/BF02238763. [DOI] [PubMed] [Google Scholar]
- 128.Kubalek I, Fain O, Paries J, Kettaneh A, Thomas M. Treatment of reflex sympathetic dystrophy with pamidronate: 29 cases. Rheumatol (Oxford) 2001;40:1394–97. doi: 10.1093/rheumatology/40.12.1394. [DOI] [PubMed] [Google Scholar]
- 129.Forouzanfar T, Koke AJ, van Kleef M, Weber WE. Treatment of complex regional pain syndrome type I. Eur J Pain. 2002;6:105–22. doi: 10.1053/eujp.2001.0304. [DOI] [PubMed] [Google Scholar]
- 130.Robinson JN, Sandom J, Chapman PT. Efficacy of pamidronate in complex regional pain syndrome type I. Pain Med. 2004;5:276–80. doi: 10.1111/j.1526-4637.2004.04038.x. [DOI] [PubMed] [Google Scholar]
- 131.Eliav E, Herzburg U, Ruda MA, Bennett GJ. Neuropathic pain from an experimental neuritis of the rat sciatic nerve. Pain. 1999;83:169–82. doi: 10.1016/s0304-3959(99)00102-5. [DOI] [PubMed] [Google Scholar]
- 132.Watkins LR, Maier SF. Beyond neurons: evidence that immune and glial cells contribute to pathological pain states. Physiol Rev. 2002;82:981–1011. doi: 10.1152/physrev.00011.2002. [DOI] [PubMed] [Google Scholar]
- 133.Woolf CJ, Thompson SW. The induction and maintenance of central sensitization is dependent on N-methyl-D-aspartic acid receptor activation; implications for the treatment of post-injury pain hypersensitivity states. Pain. 1991;44:293–9. doi: 10.1016/0304-3959(91)90100-C. [DOI] [PubMed] [Google Scholar]
- 134.Wall PD, Woolf CJ. Muscle but not cutaneous C-afferent input produces prolonged increases in the excitability of the flexion reflex in the rat. J Physiol (Lond) 1984;356:443–58. doi: 10.1113/jphysiol.1984.sp015475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Radhakrishnan R, Moore SA, Sluka KA. Unilateral carrageenan injection into muscle or joint induces chronic bilateral hyperalgesia in rats. Pain. 2003;104:567–577. doi: 10.1016/s0304-3959(03)00114-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sluka KA. Stimulation of deep somatic tissue with capsaicin produces long-lasting mechanical allodynia and heat hypoalgesia that depends on early activation of the cAMP pathway. J Neurosci. 2002;22:5687–93. doi: 10.1523/JNEUROSCI.22-13-05687.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Moriwaki K, Yuge O. Topographical features of cutaneous tactile hypoesthetic and hyperesthetic abnormalities in chronic pain. Pain. 1999;81:1–6. doi: 10.1016/s0304-3959(98)00257-7. [DOI] [PubMed] [Google Scholar]
- 138.Treede RD, Davis KD, Campbell JN, Raja SN. The plasticity of cutaneous hyperalgesia during sympathetic ganglion blockade in patients with neuropathic pain. Brain. 1992;615:607–21. doi: 10.1093/brain/115.2.607. [DOI] [PubMed] [Google Scholar]
- 139.Mailis A, Amani N, Umana M, Basur R, Roe S. Effect of intravenous sodium amytal on cutaneous sensory abnormalities, spontaneous pain and algometric pain pressure thresholds in neuropathic pain patients: a placebo-controlled study. Pain. 1997;70:69–81. doi: 10.1016/s0304-3959(96)03300-3. [DOI] [PubMed] [Google Scholar]
- 140.Bruehl S, Harden RN, Galer BS, Saltz S, Backonja M, Stanton-Hicks M. Complex regional pain syndrome: are there distinct subtypes and sequential stages of the syndrome? Pain. 2002;95:119–24. doi: 10.1016/s0304-3959(01)00387-6. [DOI] [PubMed] [Google Scholar]
- 141.Roberts W. A hypothesis on the physiological basis for causalgia and related pains. Pain. 1986;24:297–311. doi: 10.1016/0304-3959(86)90116-8. [DOI] [PubMed] [Google Scholar]
- 142.Mitchell SW. Injuries of nerves and their consequences. London: Smith Elder; 1872. [Google Scholar]
- 143.Mitchell SW, Morehouse CR, Keen WW. Gunshot wounds and other injuries to nerves. Philadelphia: JB Lippincott; 1864. [Google Scholar]
- 144.Liu JE, Tahmoush AJ, Roos DB, Schwartzman RJ. Shoulder-arm pain from cervical bands and scalene muscle anomalies. J Neurol Sci. 1995;128:175–180. doi: 10.1016/0022-510x(94)00220-i. [DOI] [PubMed] [Google Scholar]
- 145.Wilhelm A. Surgical treatment of therapy refractory Sudeck’s dystrophy by transaxillary decompression of the neurovascular bundle and sympathectomy. On the pathogenesis of Sudeck’s disease. Handchir Mikrochir Plast Chir. 1997;29:60–72. [PubMed] [Google Scholar]
- 146.Muizelaar JP, Kleyer M, Hertogs IA, DeLange DC. Complex regional pain syndrome (reflex sympathetic dystrophy and causalgia): management with the calcium channel blocker nifedipine and/or the alpha-sympathetic blocker phenoxybenzamine in 59 patients. Clin Neurol Neurosurg. 1997;99:26–30. doi: 10.1016/s0303-8467(96)00594-x. [DOI] [PubMed] [Google Scholar]
- 147.Manahan AP, Burkman KA, Malesker MA, Benecke GW. Clinical observation on the use of topical nitroglycerin in the management of severe shoulder-hand syndrome. Nebr Med J. 1993;78:87–9. [PubMed] [Google Scholar]
- 148.Groeneweg G, Niehof S, Wesseldijk F, Huygen FJ, Zijlstra FJ. Vasodilative effect of isosorbide dinitrate ointment in complex regional pain syndrome type 1. Clin J Pain. 2008;24:89–92. doi: 10.1097/AJP.0b013e318156db3b. [DOI] [PubMed] [Google Scholar]
- 149.Kiralp MZ, Yildiz S, Vural D, Keskin I, Ay H, Dursun H. Effectiveness of hyperbaric oxygen therapy in the treatment of complex regional pain syndrome. J Int Med Res. 2004;32:258–62. doi: 10.1177/147323000403200304. [DOI] [PubMed] [Google Scholar]
- 150.Grabow TS, Tella PK, Raja SN. Spinal cord stimulation for complex regional pain syndrome: an evidence-based medicine review of the literature. Clin J Pain. 2003;19:371–83. doi: 10.1097/00002508-200311000-00005. [DOI] [PubMed] [Google Scholar]
- 151.Harden RN. The rationale for integrated functional restoration. In: Wilson PR, Stanton-Hicks M, Harden RN, editors. CRPS: Current diagnosis and therapy. Progress in pain research and management. Vol. 32. Seattle: IASP Press; 2005. pp. 163–72. [Google Scholar]
- 152.Kemler MA, Rijks CP, de Vet HC. Which patients with chronic reflex sympathetic dystrophy are most likely to benefit from physical therapy? J Manipulative Physiol Ther. 2001;24:272–8. doi: 10.1067/mmt.2001.114364. [DOI] [PubMed] [Google Scholar]
- 153.Zollinger PE, Tuinebreijer WE, Kreis RW, Breederveld RS. Effect of vitamin C on frequency of reflex sympathetic dystrophy in wrist fractures: a randomised trial. Lancet. 1999;354:2025–8. doi: 10.1016/S0140-6736(99)03059-7. [DOI] [PubMed] [Google Scholar]
- 154.Goris RJ, Dongen LM, Winters HA. Are toxic oxygen radicals involved in the pathogenesis of reflex sympathetic dystrophy? Free Radic Res Commun. 1987;3:13–8. doi: 10.3109/10715768709069764. [DOI] [PubMed] [Google Scholar]
- 155.Zuurmond WW, Langendijk PN, Bezemer PD, Brink HE, de Lange JJ, van Loenen AC. Treatment of acute reflex sympathetic dystrophy with DMSO 50% in a fatty cream. Acta Anaesthesiol Scand. 1996;40:364–7. doi: 10.1111/j.1399-6576.1996.tb04446.x. [DOI] [PubMed] [Google Scholar]
- 156.Perez RS, Zuurmond WW, Bezemer PD, Kuik DJ, van Loenen AC, de Lange JJ, Zuidhof AJ. The treatment of complex regional pain syndrome type I with free radical scavengers: a randomized controlled study. Pain. 2003;102:297–307. doi: 10.1016/S0304-3959(02)00414-1. [DOI] [PubMed] [Google Scholar]
- 157.Schwartzman RJ, Chevlen E, Bengtson K. Thalidomide has activity in treating complex regional pain syndrome. Arch Inten Med. 2003;163:1487–8. doi: 10.1001/archinte.163.12.1487. [DOI] [PubMed] [Google Scholar]
- 158.Manning DC. New and emerging pharmacological targets for neuropathic pain. Curr Pain Headache Rep. 2004;8:192–8. doi: 10.1007/s11916-004-0051-7. [DOI] [PubMed] [Google Scholar]
- 159.Goris RJA. Conditions associated with impaired oxygen extraction. In: Guitierrez G, Vincent JL, editors. Tissue Oxygen Utilization. Berlin: Springer Verlag; 1991. pp. 350–69. [Google Scholar]
- 160.Moriwaki K, Yuge O, Tanaka H, Sasaki H, Izumi H, Kaneko K. Neuropathic pain and prolonged regional inflammation as two distinct symptomatological components in complex regional pain syndrome with patchy osteoporosis--a pilot study. Pain. 1997;72:277–82. doi: 10.1016/s0304-3959(97)00029-8. [DOI] [PubMed] [Google Scholar]
- 161.Birklein F, Riedl B, Neundorfer B, Handwerker HO. Sympathetic vasoconstrictor reflex pattern in patients with complex regional pain syndrome. Pain. 1998;75:93–100. doi: 10.1016/S0304-3959(97)00209-1. [DOI] [PubMed] [Google Scholar]
- 162.Dornhorst AC, Whelan RF. The blood flow in muscle following exercise and circulatory arrest; the influence of reduction in effective local blood pressure, of arterial hypoxia and of adrenaline. Clin Sci (Lond) 1953;12:33–40. [PubMed] [Google Scholar]
- 163.Petchkrua W, Weiss DJ, Patel RR. Reassessmetn of the incidence of complex regional pain syndrome type 1 following stroke. Neurorehabil Neural Repair. 2000;14:59–63. doi: 10.1177/154596830001400107. [DOI] [PubMed] [Google Scholar]
- 164.Karacan I, Aydin T, Ozaras N. Bone loss in the contralateral asymptomatic hand in patients with complex regional pain syndrome type 1. J Bone Miner Metab. 2004;22:44–7. doi: 10.1007/s00774-003-0447-1. [DOI] [PubMed] [Google Scholar]
- 165.Leis S, Weber M, Schmelz M, Birklein F. Facilitated neurogenic inflammation in unaffected limbs of patients with complex regional pain syndrome. Neurosci Lett. 2004;359:163–6. doi: 10.1016/j.neulet.2004.02.025. [DOI] [PubMed] [Google Scholar]
- 166.An HS, Hawthorne KB, Jackson WT. Reflex sympathetic dystrophy and cigarette smoking. J Hand Sur. 1988;13:458–60. doi: 10.1016/s0363-5023(88)80032-7. [DOI] [PubMed] [Google Scholar]
- 167.Apkarian AV, Thomas PS, Krauss BR, Szeverenyi NM. Prefrontal cortical hyperactivity in patients with sympathetically mediated chronic pain. Neurosci Lett. 2001;311:193–7. doi: 10.1016/s0304-3940(01)02122-x. [DOI] [PubMed] [Google Scholar]
- 168.Jänig W, Baron R. Complex regional pain syndrome: mystery explained? Lancet Neurol. 2003;2:687–97. doi: 10.1016/s1474-4422(03)00557-x. [DOI] [PubMed] [Google Scholar]
- 169.Maihofner C, Forster C, Birklein F, Neundorfer B, Handwerker HO. Brain processing during mechanical hyperalgesia in complex regional pain syndrome: a functional MRI study. Pain. 2005;114:93–103. doi: 10.1016/j.pain.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 170.Harel F, Denault A, Ngo Q, Dupuis J, Khairy P. Near-infrared spectroscopy to monitor peripheral blood flow perfusion. J Clin Monit Comput. 2008;22:37–43. doi: 10.1007/s10877-007-9105-9. [DOI] [PubMed] [Google Scholar]