

Abstract
The existence of specific cellular subpopulations within primary tumors with increased tumorigenic potential and chemotherapy resistance (tumor-initiating cells, TICs) holds great therapeutic implications. Resistant cells can remain quiescent for long periods and be responsible for local relapses and metastasis. We and others have previously described in non–small-cell lung cancer the presence of cisplatin-resistant CD133+ cells with tumor-initiating potential and co-expression of CXCR4 as possible indicator of TICs with disseminating potential. In this study, we report, by in vitro cell fate tracing systems, heterogeneity within the TIC compartment with a highly quiescent pool and a slowly dividing subpopulation, both containing CD133+ cells but respectively enriched for CD133+/CXCR4− and CD133+/CXCR4+ cells. Pretreatment with differentiating agent all-trans retinoic acid counteracts cisplatin resistance specifically of the slowly dividing compartment indicating effect on CD133+/CXCR4+ cells. The same effects are appreciable also in vivo in patient-derived xenografts, where several cycles of all-trans retinoic acid and cisplatin treatment are able to stably reduce this fraction of TICs and tumor dissemination. Thus, partially affecting the heterogeneous TICs compartment, differentiating therapy has promising effects in counteracting cisplatin resistance of CD133+ cells, reducing both local tumor growth and dissemination. In addition, our approach discloses a further level of complexity of chemotherapy-resistant CD133+ TICs, revealing phenotypical and functional heterogeneity of the cancer stem cell compartment in lung cancer.
Keywords: Non–small-cell lung cancer, Retinoic acid, Cancer stem cells dynamics, Cisplatin resistance
Non–small-cell lung cancer (NSCLC), representing approximately 80% of all lung cancers, is the main cause of cancer-related deaths worldwide. Even though recently developed targeted therapies have been shown to provide some benefit in predefined subclasses of patients with tumors carrying specific mutations, platinum-based chemotherapy still represent the standard systemic treatment for NSCLC. However, 5-year survival rates have not raised substantially in the past decades, remaining as low as 20% for late stage disease (III–IV).1 Cisplatin resistance is the consequence of a multifactorial event which involves a combination of features, such as drug inactivation by detoxifying factors, alterations in checkpoint and apoptotic proteins, and variation in intracellular drug accumulation.2–4 However a rising number of observations also indicates that, in several tumor types, distinct cellular subpopulations persist after treatment and that these cells own intrinsic characteristics associated with a stem-like phenotype5 and can be indicated as cancer stem cells or tumor-initiating cells (TICs). TICs characterized by increased tumorigenicity, self-renewal ability, and multipotency6,7 have been described for several tumor types, such as myeloid leukemia,8 glioblastoma,9–11 melanoma,12 and several epithelial cancers13–18 including lung cancer.19,20 Particularly, lung cancer TICs are detected by the expression of cell surface markers as CD133 or by the high activity of aldehyde dehydrogenase and finally by increased capacity to efflux the DNA-binding dye Hoechst 33342 that define the so-called side population cells.21 Even if some controversies are still open about the choice of the most appropriate marker to enrich for TICs, almost all published studied demonstrating the drug resistance properties of lung TICs reported the enrichment for a fraction of CD133+ cells after chemotherapy, indicating the leading role of such cell subset in chemoresistance.22
Furthermore, the stem-like and tumorigenic phenotype has been associated to a slow proliferating fraction of cells within primary lines or cancer cells.23,24 The peculiar characteristics of TICs, and in particular their capability to remain in a quiescent state,25 are consistent with a lower susceptibility to replication-related drugs as previously described also in NSCLC, where CD133+ TICs have been reported as being spared by Cisplatin treatment.20 Moreover, in vitro CDDP treatment of NSCLC-derived spheres enriched for TICs induces a replication block (G2/M) and a more efficient DNA damage repair that can be prevented by treatment with inhibitor of checkpoint protein kinase (Chk1).26 Recently, cancer stem cells (CSC) from NSCLC have also been shown to be susceptible to Bcl-XL inhibition indicating potential therapeutic strategies targeting TICs.27
We reasoned that an efficient strategy to target TICs could also rely on their mobilization from the quiescence state inducing a chemo-susceptible phenotype, as already proposed and substantiated for leukemic stem cells.28,29 Altogether, this would deplete the tumor of its CSCs reservoir preventing relapse, metastasis formation and, eventually, increasing the efficacy of chemotherapy.
In this study, to overcome CDDP resistance of CD133+ NSCLC CSCs, we tested the capacity of all-trans retinoic acid (ATRA), in vitro and in vivo, to force the TICs fraction to differentiate to a more CDDP susceptible phenotype. ATRA has been reported to be effective in the cure and prevention of many types of cancer (reviewed in 30), however, its differentiating role against solid tumors CSC’s compartment is still poorly explored.31–33 Using in vitro cell fate tracing systems and in vivo models of patient-derived xenografts (PDXs),34 we show here that ATRA is able to interfere with the dynamics of the CSC compartment and prevents chemotherapy-related TICs increase.
MATERIALS AND METHODS
Cell Cultures
LT73 cells were derived in our laboratory from a primary lung tumor of a 68-year-old Caucasian male patient with lung adenocarcinoma. Cells were grown in RPMI-1640 (Lonza, Basel, Switzerland), supplemented with 10% fetal bovine serum (Lonza) and penicillin/streptomycin (Invitrogen, Carlsbad, CA) at 37°C with 5% CO2.
PKH Labeling
LT73 cells were labeled with PKH67 Fluorescent cell linker (Sigma-Aldrich, St. Louis, MO) according to the manufacturer’s instruction. In brief, cells were incubated 5 minutes at room temperature with PKH67 Dye Solution. Staining was stopped adding fetal bovine serum (Lonza) and labeled cells were analyzed by flow cytometry at different time points.
Culture of Cancer Tissue-Originated Spheroids
Cancer tissue-originated spheroids (CTOS) culture method was adapted from Kondo et al.35
In brief, tumor tissue was mechanically and then enzymatically digested in a solution of collagenase IV (5 mg/ml) and DNAse (100 U/ml; Sigma Aldrich, St. Louis, MO) in DMEM/F12 (Lonza, Verviers, Belgium) for 1 hour at 37°C and subsequently filtered through 100-μm and 40-μm cell strainers (Becton Dickinson, Franklin Lakes, NJ). The tumor tissue organoids retained in the strainer were washed in 30 ml of DMEM/F12 and centrifuged at 100g for 5 minutes. Tumor organoids were plated in stem cell medium SCM (described in 20) and in 60 mm Petri dishes. Spheroids appeared in about 3 days. For culture expansion, spheroids were centrifuged at 100g for 5 minutes and incubated with a mild digestion solution of DMEM/F12 + collagenase IV 5 mg/ml at 37°C for 5 minutes.
Flow Cytometry Analysis
Single-cell suspensions were washed and incubated in staining solution containing 1% BSA and 2 mM ethylenediaminetetraacetic acid with specific antibodies at appropriate dilutions. For CD133 and CXCR4 staining, 106 cells were incubated with phycoerythrin-conjugated anti-CD133/1 (Miltenyi Biotec, Bergish Gladbach, Germany) and allophycocyanin-conjugated anti-CXCR4 (Becton Dickinson). Samples were acquired by FACS Calibur and analyzed with FlowJo_V10 software.
For lung dissemination analysis, a morphological gate allowing the identification of the highest percentage of human tumor cells in murine lungs was identified36 and subsequent exclusion for 7-AAD+ dead cells and mouse H2K+ cells was performed. This method was able to specifically detect as few as 103 single tumor cells in murine lungs.
Patient-Derived Xenograft Tumor Growth
All experiments were carried out with female SCID mice, 7–10 weeks old (Charles River Laboratories, Calco, Italy). Mice were maintained in laminar flow rooms, with constant temperature and humidity. Mice had free access to food and water. Experiments were approved by the Ethics Committee for Animal Experimentation of the Fondazione IRCCS Istituto Nazionale dei Tumori, according to institutional guidelines. PDXs were established as described.34 PDX111 (EGFRwt, KRASwt, LKB1wt, HER2wt, PIK3wt, BRAFwt) and PDX73 (EGFRwt, KRASwt, LKB1K287X, HER2wt, PIK3wt, BRAFwt) were derived from a 77-year-old female and a 68-year-old Caucasian male patient, respectively, both with lung adenocarcinoma. For pharmacological experiments, mice were randomly distributed into equal groups (five mice per group, grafted in both flanks). Mice were treated with All-Trans Retinoic Acid (Sigma-Aldrich; 10 mg/kg gavage, qd × 5 × 3 weeks) and/or with Cisplatin (Teva, Petach Tikva, Israel; 5 mg/kg i.v. q7d × 3).
Immunofluorescence
104 LT73 cells were grown on Lab-Tek (ThermoFisher, Waltham, MA) slides and incubated with BSA 2% + NGS 5% blocking solution for 30 minutes, incubated with anti-human CD133/1 (Miltenyi; Biotec) for 1 hour at RT, then 30' at RT with AlexaFluor 488 goat anti human IgG (H+L) (Invitrogen) washed in tween 1× and mounted with the VECTASHIELD Mounting Medium, containing DAPI (Vector Laboratories, Burlingame, CA).
Statistical Analysis
All data are shown as mean value ± standard error. t tests and Fisher exact test have been performed with GraphPad Prism 4 Software. p values are represented as follows: *: p < 0.05; **: p < 0.01, ***: p < 0.001.
RESULTS
Identification of a Slow Proliferating Fraction of NSCLC Cells Enriched for CD133+ TICs with High In Vivo Tumorigenic Potential
To investigate the compartment of slow proliferating cells and its dynamics in NSCLC, we exploited a general cell membrane labeling system (Fluorescent Cell Linker Kit PKH67, Sigma-Aldrich, St. Louis, MO), previously reported as a useful tool to identify slow proliferating cells37,38 because the labeling is progressively diluted through repeated cell divisions whereas not proliferating cells maintain it for weeks. LT73 primary cell line, established from patient’s lung adenocarcinoma, was labeled with PKH67 and label retaining was monitored by flow cytometry during serial population doublings (PD). After 10 PDs (PD10) a small amount of labeled cells (PKH+) was still appreciable (0.22 ± 0.11%, n = 5 replicates) as confirmed also by immunofluorescence analysis (Fig. 1A, Supplementary 1a, SDC 1, http://links.lww.com/JTO/A843). Interestingly, a remarkable enrichment of CD133+ TICs was appreciable in the PKH+ subpopulation (200-fold average: 24.7 ± 4.8% versus 0.12 ± 0.05% in total population, n = 5 replicates; Fig. 1B). Within this subpopulation of quiescent cells, a PKH brightest subgroup was recognized (long-term quiescent cells, PKHBRIGHT) and analysis revealed a further enrichment of CD133+ cells (400-fold average: 48.3 ± 7.7 versus 0.12 ± 0.05 in total population, n = 5 replicates). We previously demonstrated that CD133+ cells spared by CDDP were particularly enriched for the subset co-expressing the chemokine receptor CXCR420 that could be involved in dissemination of TICs.36,39 Interestingly, the observed heterogeneity within the slow proliferating PKH+ cells is mirrored by different distribution of CXCR4 co-expressing cells, indeed PKHBRIGHT fraction was mainly composed by CD133+/CXCR4− cells (36.1 ± 4.61% CD133+/CXCR4− versus 12.22 ± 2.803% CD133+/CXCR4+, n = 5, Supplementary 1B, SDC 1, http://links.lww.com/JTO/A843), conversely an enrichment for CD133+/CXCR4+ cells was appreciable in the PKHDIM fraction (short-term quiescent, 4.1 ± 0.75% CD133+/CXCR4− versus 11.7 ± 2.3% CD133+/CXCR4+, n = 5, Supplementary 1B, SDC 1, http://links.lww.com/JTO/A843). PKHBRIGHT cells showed enhanced tumorigenic potential when injected subcutaneously in SCID mice. In this assay, 102 PKHBRIGHT, but not 102 PKHDIM cells successfully gave rise to fully developed tumors (4/4 take rate, average tumor volume of 1000 mm3 reached 60 days after injection versus 0/4 take rate; Fig. 1C). Moreover, injection of 103 PKHDIM cells led to a delayed tumor growth (3/4 take rate and tumor growth delayed by 30 days, Fig. 1C). We previously reported the increased tumorigenicity of CD133+ compared with CD133− cells. As the estimated amount of CD133+ TICs (and CD133+/CXCR4+ fraction) was higher in 103 PKHDIM than in 102 PKHBRIGHT injected cells, we speculate that the amount of CD133+/CXCR4− cells in the PKHBRIGHT subset was responsible for the observed tumor growth (Table 1). A subset of PKH retaining, CD133+-enriched population was also detectable ex vivo in PDX models. Cells obtained from disaggregation of PDX samples (PDX111, Fig. 2A and PDX73, Fig. 2B) were stained with PKH67 and reinjected subcutaneously in the flank of immunocompromised mice. In tumors analyzed at different volumes (V1 = 250 mm3, V2 = 1000 mm3), a small fraction of PKH-labeled cells was still detectable and showed an enrichment of CD133+ TICs (three- to 50-fold depending on the model, Fig. 2), more evident in the PKHBRIGHT fraction (up to 115-fold). Thus, PKH labeling led to the identification of the heterogeneity of TICs compartment, with more or less quiescent CD133+ cells, and, interestingly, suggested heterogeneity also within the quiescent fraction. Indeed, the PKHBRIGHT fraction, few cells which underwent very few replications, was mainly composed by CD133+/CXCR4− cells, whereas in the PKHDIM fraction, a larger number of cells lying across the quiescence–nonquiescence border, CD133+/CXCR4+ cells were more represented. The high tumorigenicity of PKHBRIGHT cells suggests that quiescent CD133+/CXCR4− cells are the main responsible for the observed in vivo growth after subcutaneous implantation.
FIGURE 1.
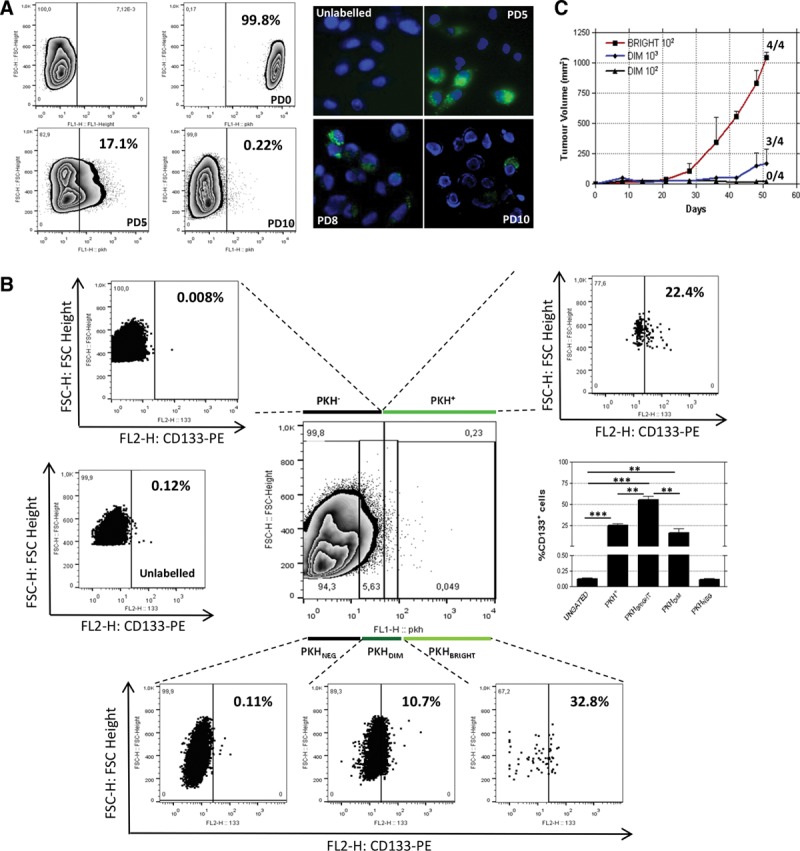
In vitro identification of a slow proliferating fraction enriched for CD133+ TICs in a NSCLC-derived cell line (LT73). PKH67 (Sigma) labeled cells were almost completely labeled at day 0 and progressively lost the labeling. At PD10, a small amount of positive cells were still appreciable, both by fluorescence activated cell sorting and immunofluorescence (representative FACS and immunofluorescence analysis out of three replicates are shown) (A); CD133+ tumor initiating cells were 200-fold enriched in the PKH retaining fraction (24.7 ± 2.8% versus 0.12 ± 0.05%). A representative FACS analysis out of five replicates shows this enrichment (upper right box) compared with basal level of CD133+ cells in this cell line (middle left box); the low box shows that CD133+ cells enrichment is higher in the most quiescent fraction (PKHBRIGHT, 450-fold enrichment, 55.1 ± 4.5 versus 0.12 ± 0.05), intermediate in the PKHDIM fraction (130-fold enrichment, 15.8 ± 3.1 versus 0.12 ± 0.05) and absent in the PKHNEG fraction, data are shown as percentage of CD133+ cells, t test p values are represented as: *p < 0.05, **p < 0.01, ***p < 0.001 (B). In vivo injection of PKH-labeled cells shows that 100 PKHBRIGHT cells injected subcutaneously in SCID mice (n = 4) are able to give rise to a completely developed tumor (1000 mm3). Same tumor growth are not achieved injecting 100 PKHDIM cells, where a delayed tumor growth is appreciable after injection of 1000 PKHDIM cells. Tumor take is represented as “number of developed tumors/numbers of injection” (C).
TABLE 1.
In Vivo Injection of PKHBRIGHT or PKHDIM-Sorted Cells
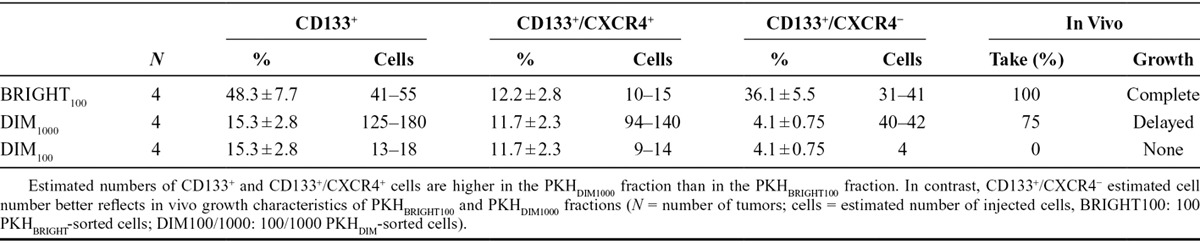
FIGURE 2.
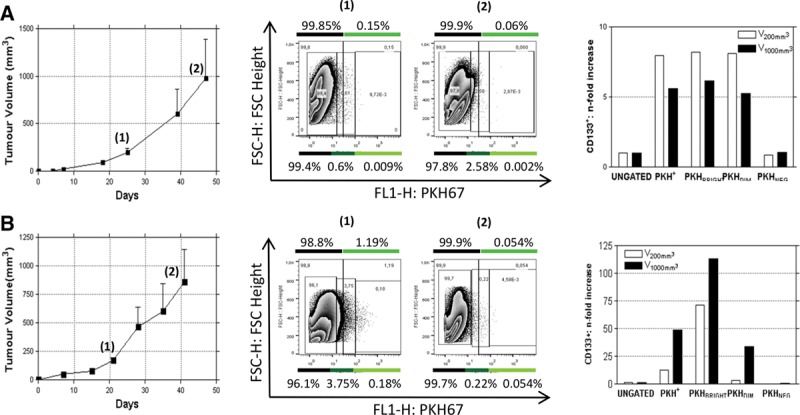
The slow proliferating fraction is appreciable also ex vivo in NSCLC PDX. Cells derived from disaggregation of PDX were PKH labeled and reinjected subcutaneously in SCID mice (n = 4). PKH retaining CD133+ enriched cells are appreciable in tumors analyzed at different volumes (V1 = 250 mm3, V2 = 1000 mm3) in both PDX tested; PDX111 (A) and PDX73 (B).
CDDP-Resistant Subpopulation is Highly Enriched in Slow Proliferating CD133+ Cells
To further demonstrate the link between quiescence and chemoresistance, LT73PKH (PD10) cells were treated with 10 µM CDDP (LD50) for 72 hours. The PKH-positive fraction showed resistance to CDDP treatment, increasing by a 15-fold average enrichment (n = 5 replicates; 2.7 ± 0.7% compared with 0.22 ± 0.11% PKH+ cells in untreated controls; Fig. 3A). CDDP was preferentially active against proliferating PKHNEG cells as expected but interestingly also on PKH+/CD133− cells. Indeed, after CDDP treatment, both PKHBRIGHT and PKHDIM fractions were almost entirely composed by CD133+ cells (Fig. 3B) confirming our previous data on resistance of CD133+ cells to CDDP treatment.20 Notably, a relevant enrichment of CD133+/CXCR4+ cells after CDDP treatment was observed in the PKHDIM fraction (Fig. 3B). Taken together, these data indicate that the quiescent PKH+/CD133+ subpopulation overlaps with the already reported cisplatin-resistant CD133+ TICs and that different dynamics within PKH subpopulations after CDDP treatment are appreciable.
FIGURE 3.
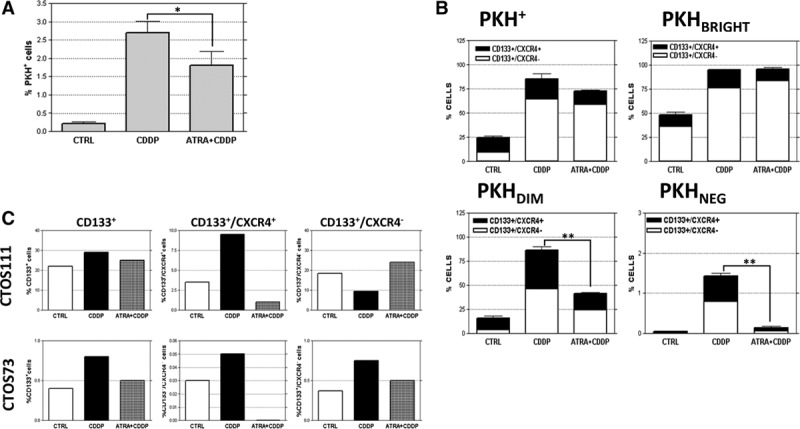
In vitro ATRA pretreatment counteracts CDDP-derived TICs enrichment, except for the PKHBRIGHT CD133+/CXCR4− fraction. PKH-labeled LT73 NSCLC cell line was treated with 10 µM CDDP alone or after ATRA (10 µM) pretreatment. ATRA pretreatment was able to counteract CDDP-induced PKH+ cells enrichment (A). A general enrichment of PKH+/CD133+ cells is appreciable after CDDP treatment (B, upper left box plot), this enrichment is striking for CD133+/CXCR4− cells. The increase of CD133+ cells is partially reverted by ATRA pretreatment especially in PKHDIM (lower left box plot) and PKHNEG (lower right box plot) fraction. Interestingly, CD133+/CXCR4+ cells are affected by CDDP and ATRA also in the PKHBRIGHT (upper right box plot) fraction, whereas CD133+/CXCR4− cells only in PKHDIM and PKHNEG fraction., t test p values are represented as: *p < 0.05, **p < 0.01, ***p < 0.001. Also in ex vivo PDX-derived CTOS, the enrichment of CD133+ cells after CDDP is reverted by ATRA cotreatment, especially in the CD133+/CXCR4+ fraction (C).
ATRA Pretreatment Partially Reverts CDDP-Induced TICs Enrichment In Vitro Acting on CD133+/CXCR4+ Cells
To verify the effect of differentiating agent on TICs subset, LT73 cell line was treated in vitro with ATRA 10 µM (every 48 hours, from PD8 to PD10); no effect on cell proliferation and viability was observed during treatment (Supplementary 3A, SDC 1, http://links.lww.com/JTO/A843), and a general decrease in CD133+ percentages was induced, even though the only significant effect was observed within PKH+/CD133+/CXCR4+ fraction (15.4 ± 1.6% in untreated versus 13.6 ± 1.7% in ATRA treated cells, n = 5; p = 0.0192, Supplementary 3B, SDC 1, http://links.lww.com/JTO/A843). CDDP treatment in cells previously exposed to ATRA led to a less pronounced increase of PKH+ cells compared with CDDP-only treated cells (1.8 ± 0.85% versus 2.7 ± 0.7% in CDDP-only treated, corresponding to an average prevention of 36.3% of CDDP-induced enrichment, n = 5, Fig. 3A). In details, ATRA pretreatment was able to prevent increase of CD133+ cells due to CDDP exposure, completely in the PKHNEG fraction and partially in the PKHDIM fraction, without affecting the more quiescent PKHBRIGHT fraction (Fig. 3B and Supplementary Table 1, SDC 2, http://links.lww.com/JTO/A844). These in vitro data suggest that the differentiating agent is able to sensitize proliferating PKHNEG CD133+ cells to CDDP and is also partially able to push a fraction of quiescent PKHDIM CD133+ into a cisplatin responsive state, seemingly acting on CD133+/CXCR4+ cells, whose increase is totally prevented. The same experiment was carried out on ex vivo CTOS established from two PDX models. Similarly to in vitro results, ATRA treatment induced a slight decrease of TICs (from 22% to 19%, corresponding to a 14% decrease compared with untreated CTOS111, whereas a higher effect was appreciable in CTOS73, from 0.4% to 0.05%, corresponding to a 87.5% decrease compared with untreated) associated with a down regulation of stemness genes (Supplementary 3B–D, SDC 1, http://links.lww.com/JTO/A843). Moreover, CD133+ cells enrichment after CDDP treatment was partially prevented by ATRA also in these ex vivo models (from 29% to 25% corresponding to a prevention of 57% of CDDP-induced enrichment in CTOS111, and from 0.8% to 0.5% corresponding to a prevention of 75% of CDDP-induced enrichment in CTOS73, Fig. 3C). Taken together, these in vitro and ex vivo data indicate that within the TIC compartment the differentiating treatment preferentially sensitizes PKHDIM cells, enriched in CD133+/CXCR4+ TICs, to CDDP treatment without affecting PKHBRIGHT cells.
In Vivo Differentiation Therapy Partially Depletes CD133+ TICs, Leading to a Reduced Tumor Regrowth after Cisplatin Treatment
To investigate if ATRA-driven mobilization of a specific TICs compartment was also effective in vivo, we treated two NSCLC PDXs models (PDX73 and PDX111). ATRA alone slightly decreased the percentage of CD133+ cells (Supplementary 3D, SDC 1, http://links.lww.com/JTO/A843) without affecting tumor growth (Fig. 4A). Moreover, in both models, the combination of ATRA and CDDP had no synergistic effect on tumor growth, but CD133+ enrichment due to CDDP treatment was strongly counteracted by combined treatment in PDX73 (prevention of 100% of the CDDP-induced enrichment) and partially in PDX111 (prevention of 30% of the CDDP-induced enrichment, Fig. 4A). Interestingly, in both models, effects of combination therapy were mostly linked to a variation in the CD133+/CXCR4+ fraction (prevention of 40% of the CDDP-induced enrichment in PDX73 and of 125% in PDX111, Fig. 4B). This effect was mirrored by a decreased dissemination of human cells to lungs in mice receiving ATRA pretreatment (Fig. 4B). On the other hand, CD133+/CXCR4− cell levels were not affected by combined treatment (PDX73: untreated: 0.15%, CDDP: 0.3%. ATRA pretreatment: 0.35%; PDX111: untreated: 16%, CDDP: 20%. ATRA pretreatment: 22%, Fig. 4B) and this was mirrored by a similar regrowth of CDDP only or combined-treated tumors after suspension of treatment (Fig. 4B). These findings indicate functional heterogeneity in the CD133+ TICs fraction with CD133+/CXCR4− cells (enriched within the PKHBRIGHT fraction) mainly supporting local tumor growth, whereas CD133+/CXCR4+ cells (highly represented in the PKHDIM fraction and susceptible to ATRA pretreatment) related to lung dissemination. Thus, differentiating therapy shows promising effects also in vivo, confirming the capability of ATRA to partially affect the TICs compartment by sensitizing CD133+/CXCR4+ cells to CDDP treatment resulting in a decreased lung dissemination, whereas local tumoral (re-)growth and CD133+/CXCR4− cells are less affected.
FIGURE 4.
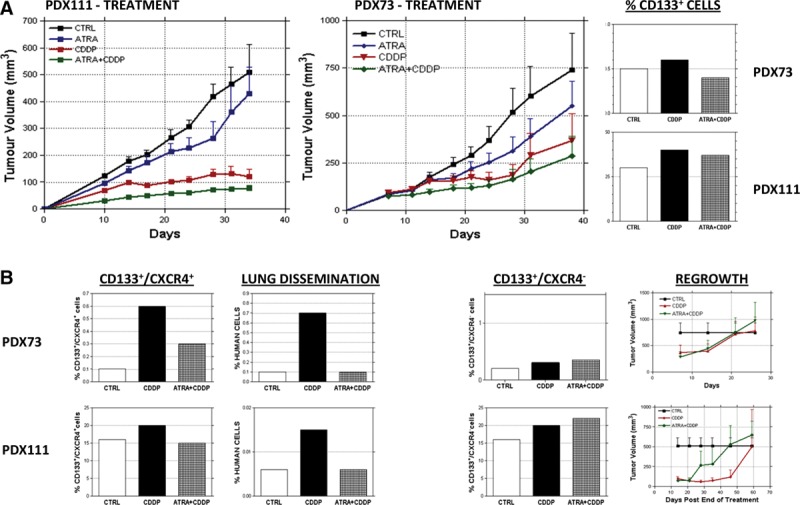
In vivo ATRA pretreatment counteracts CDDP-derived enrichment of CD133+/CXCR4+ cells and lung dissemination in NSCLC PDX. In vivo ATRA treatment produces no effects in terms of tumor growth for both models tested, PDX111 and PDX73. Pretreating both models with ATRA and CDDP gives no benefits in terms of tumor growth compared with CDDP only treatment (A). At the end of treatment, CD133+/CXCR4+ cell percentage in the residual tumors are lower compared with CDDP treated tumors, and this correlates with a lower lung dissemination of tumoral cells, whereas CD133+/CXCR4− cell levels are unaffected by ATRA pretreatment resulting in a similar regrowth rate of the treated tumors (B).
Multiple Cycles of In Vivo ATRA and CDDP Treatment Significantly Reduce Tumor Volume
To investigate if the effects of the double treatment in vivo were permanent after multiple treatment cycles and to achieve a progressive exhaustion of the TICs reservoir, a serial transplantation experiment was performed. PDX111 residual tumors after treatment were cut in small pieces (3 mm), injected subcutaneously in a set of new mice and treated with ATRA and/or CDDP. After four serial transplantations (S4), a significant decrease in tumor growth of double treated mice compared with CDDP treated counterpart was achieved (V = 467 ± 160 mm3 and 864 ± 252 in double-treated and CDDP-treated, respectively, Fig. 5A). This difference in tumor growth was anticipated (at S3) by a lower percentage of CD133+ cells (3.8 ± 0.9% and 13 ± 1.32% in double-treated and CDDP-treated, respectively, Fig. 5A), which is mainly ascribed to the reduction of CD133+/CXCR4+ subset. After five treatment cycles, the effects of double treatment on tumor growth were still appreciable with a decrease of tumor volume in double-treated PDX. Interestingly, CD133+/CXCR4+ cell levels remained low at each cycle of differentiating therapy and this resulted in an appreciable and long-lasting (up to five serial transplants) decrease of disseminating cells in lungs of mice carrying PDXs (Fig. 5C). When double-treated tumors were transplanted into new mice and not retreated, a progressive increase in tumor growth was observable (Fig. 5B), which was accompanied by the constant increase of CD133+/CXCR4− subset. Differentiating therapy with ATRA is therefore promising in controlling disseminating cells (CD133+/CXCR4+), whereas CD133+/CXCR4− cells appear not to be completely exhausted after five treatment cycles.
FIGURE 5.
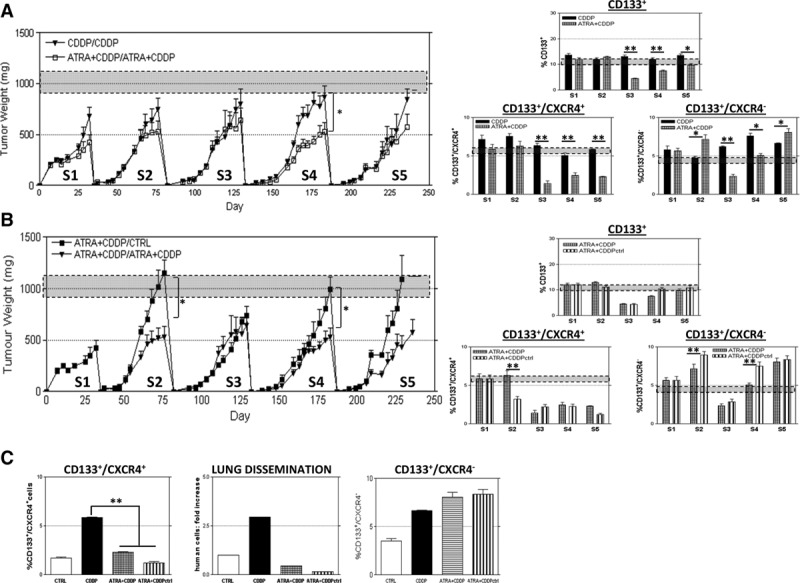
Multiple cycles of ATRA pretreatment in a serial transplantation assay leads to a long-lasting reduction of lung dissemination and CD133+/CXCR4+ cell fraction. In PDX111 tumors, explanted from mice after treatments and reimplanted subcutaneously in new SCID mice, a significant decrease of tumor growth is appreciable after four passages of serial transplants, but this effect attenuates in the following passage: S4,VCDDP = 864 ± 252 mm3, VATRA+CDDP = 467 ± 160 mm3, n = 8, p = 0.0272; S5, VCDDP = 991 ± 168 mm3, VATRA+CDDP = 658 ± 135 mm3, n = 8, p = 0.07. These effects are preceded by a reduction, already at S3, of CD133+ percentage in double treated tumor compared with CDDP treated (S3 %CD133+CDDP = 13 ± 1.32%, %CD133+ATRA+CDDP= 3.8 ± 0.9%). Interestingly, CD133+/CXCR4− cell levels reflect this trend of local growth throughout the serial passages, whereas CD133+/CXCR4+ cells levels are constantly low after third passage (A). If tumors were serially transplanted after double treatment but no more treated, tumor growth were significantly higher than in treated ones (S4,VNOT TREATED = 998 ± 228 mm3, VATRA+CDDP = 467 ± 160 mm3, n = 8, p = 0.0128; S5, VNOT TREATED = 1170 ± 174 mm3, VATRA+CDDP = 658 ± 135 mm3, n = 8, p = 0.10]. Again, these effects are mirrored by CD133+/CXCR4− cell levels and not by CD133+/CXCR4+ cells, whose levels remain low also when treatment is interrupted (B). t test p values are represented as: *p < 0.05, **p < 0.01, ***p < 0.001. At fifth serial passage, CD133+/CXCR4+ cell levels and lung dissemination of tumoral cells but not CD133+/CXCR4− cell levels are permanently controlled by ATRA pretreatment (C).
DISCUSSION
TICs have been described in many tumor types for specific features, such as potential to initiate and maintain tumor growth and to give rise to all observed heterogeneous lineages, high expression of stem-related genes and of xenobiotic transporter, and quiescence. These peculiar features suggest them as the prime suspect for tumor relapse or metastasis formation after chemotherapeutical treatment. We previously reported the presence of a cisplatin-resistant CD133+ cellular subpopulation with stem-like characteristics in NSCLC cell lines and in vivo xenograft models. The dynamics of CSCs compartment are beginning to be unraveled in the better understood leukemia models where hierarchy appears more delineated,29 whereas little is known of this complexity in solid cancers.40 However, resistance of CSC has already been reported to be linked to a slow rate of proliferation in ovarian,41 breast,42 and pancreatic43 cancer models and here, we demonstrated that the CD133+ residual fraction after CDDP treatment is mainly composed by slow proliferating label-(PKH67) retaining cells. The purpose of this study was to counteract the cisplatin resistance of CD133+ cells, inducing a mobilization of these TICs to a less resistant phenotype using ATRA. ATRA was the first differentiation agent found to be successful in the treatment of acute promyelocytic leukemia where it promotes differentiation of promyelocytic to myeloid cells, blocked by the chimeric protein product of the fusion gene PML/RARα.44 Retinoic acid has been reported to be involved in processes of lung embryogenesis with an accumulation within the lung during organ development and a temporally association with the process of alveolar septation. The importance of retinoids in the generation of alveoli is confirmed by studies involving retinoid receptor knock-out animals45 as well as by in vitro experiments on human cell lines demonstrating that retinoic acid can regulate an array of cellular and molecular pathways in lung remodeling.46 Moreover, a randomized phase II trial indicated that adding ATRA to chemotherapy could increase response rate and progression-free survival in patients with advanced (IIIb–IV) NSCLC.47 On the basis of these data, we hypothesized that a differentiating therapy with retinoids could play a role in lung tumor growth, promoting the differentiation to a CDDP susceptible state of CD133+ NSCLC TICs. A partial reversion of CDDP-induced enrichment of CD133+ cells in vitro was observed after exposure to the differentiating compound. However, ATRA does not completely affect CD133+ cells, suggesting that this subpopulation represents a nonhomogeneous TICs reservoir. We demonstrated TICs heterogeneity also in terms of quiescence; indeed, in PKH labeling experiments, CD133+ cells are represented in the entire PKH label spectrum, but PKHBRIGHT cells are highly enriched for CD133+/CXCR4− population, whereas CD133+/CXCR4+ cells are more represented in the PKHDIM fraction. Interestingly, the entire spectrum of PKH retaining CD133+ cells are CDDP resistant, but ATRA is able to nick in the PKHDIM group making CD133+/CXCR4+ cells more susceptible to chemotherapeutics in in vitro, ex vivo, and in vivo experiments. Moreover, ATRA treatment alone was able to induce a general but reduced decrease of CD133+ cell percentages as well as a slight decrease in expression levels of a panel of stem-related genes and these effects reached statistical significance in the PKH+/CD133+/CXCR4+ fraction, confirming a role of the differentiating agent ATRA in the mobilization of a specific subset of quiescent TICs. A functional heterogeneity of CD133+ cells, according to CXCR4 expression, has also been demonstrated in different tumor types; in particular, CD133+/CXCR4+ cells have been indicated as a subset of CD133+ TICs with high metastatic potential in pancreatic cancer48 and as a marker for lymph node metastasis in colon cancer models.49 Moreover, the functional role of CXCR4/CXLC12 axis in cell mobilization, dissemination, and metastasis formation has already been elucidated.50 Based on PKH retaining ability, we can hypothesize a precise hierarchy within CD133+ TICs, with CD133+/CXCR4− (PKHBRIGHT) cells representing the pluripotent CSCs and CD133+/CXCR4+ (PKHDIM) cells representing a pool of multipotent progenitors derived from them. In vivo experiments corroborate this hypothesis, indeed a low number of PKHBRIGHT, but not PKHDIM cells is able to give rise to a completely developed tumor if xenografted in immunocompromised mice. Interestingly, injection of 103PKHDIM cells (corresponding to about 40 CD133+/CXCR4− cells) led to a delayed tumor growth compared with 102 PKHBRIGHT cells (corresponding to a similar amount of CD133+/CXCR4− cells), suggesting a higher tumorigenic potential of the most quiescent subset of CD133+/CXCR4− cells compared with the same population in the PKHDIM fraction. However, in the PKHBRIGHT fraction CD133+/CXCR4− cells are represented at high purity (representing more than 30% of the fraction), whereas in the PKHDIM the same number of CD133+/CXCR4− cells is contained within a higher number of non-TICs. It is therefore also possible that interference of non-TICs with TICs in the early phases of tumor initiation could be responsible for the delayed and decreased tumorigenicity. High level of complexity within TICs niche was also confirmed by in vitro ATRA pretreatment which counteracted TICs enrichment after CDDP challenge differently in different compartments: increase of CD133+/CXCR4+ was in fact prevented in both PKH fractions, whereas increase of CD133+/CXCR4− cells was efficiently prevented in PKHDIM but not in PKHBRIGHT fraction. Moreover, in vivo ATRA pretreatment was not able to affect CD133+/CXCR4− cells, resulting in a continuous tumor growth at every passage in the serial transplantation assay. This suggests that quiescent CD133+/CXCR4− cells represent the TICs subset which is able, alone, to regenerate a heterogeneous tumor. Conversely, susceptibility of CD133+/CXCR4+ cells to ATRA pretreatment leads to a progressive eradication of this TICs fraction in the PDX serial transplantation assay, and this is correlated with a decrease of tumor dissemination to murine lungs, indicating a role of double positive cells in tumor spread from subcutaneous implants. Interestingly, CXCR4 expression has been already linked to a higher metastatic potential of NSCLC.51,52 Thus ATRA, interfering with TICs dynamics and therefore counteracting cisplatin resistance of CD133+/CXCR4+ cells, can have a therapeutic impact for lung cancers in terms of reduction of the metastatic potential. Moreover, differential activity of ATRA on specific subsets within the CSC compartment allows us to describe, for the first time, the heterogeneity of CD133+ TICs. It remains mandatory to better understand the TICs compartment’s dynamics, and in particular the equilibrium between CD133+/CXCR4− and CD133+/CXCR4+ cells, to specifically target the quiescent niche or at least to block its mobilization, whereas disseminating progenitors can be affected and controlled by ATRA treatment in combination with standard chemotherapy.
ACKNOWLEDGMENTS
The authors thank the help of AIRC (Associazione Italiana per la Ricerca sul Cancro: IG13403 to L.R.; IG11991 to G.S.), European Community Integrated Project 037665 “CHEMORES” (to G.S.) and the European Community Seventh Framework Programme (FP7/2007–2013) under Grant Agreement No. HEALTH-F2-2010–258677 (Collaborative Project CURELUNG to L.R.).
Supplementary Material
Footnotes
Disclosure: The authors declare no conflict of interest.
MM, LR, UP, and GS designed the research; MM and GB performed the research; MM, LC, GB, and GS analyzed the data; MM, LR, and GB wrote the paper; GS and LR gave the study supervision. All authors participated in the critical revision of the report. Massimo Moro, Giulia Bertolini, Luca Roz, and Gabriella Sozzi contributed equally to this study.
Samples of primary NSCLC were obtained from patients undergoing surgical resection, who gave their informed consent after approval from the Internal Review and the Ethics Boards of the Fondazione IRCCS Istituto Nazionale Tumori. Animal studies were performed according to the Ethics Committee for Animal Experimentation of the Fondazione IRCCS Istituto Nazionale Tumori, according to institutional guidelines.
REFERENCES
- 1.Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Kim ES, Lee JJ, He G, et al. Tissue platinum concentration and tumor response in non-small-cell lung cancer. J Clin Oncol. 2012;30:3345–3352. doi: 10.1200/JCO.2011.40.8120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stewart DJ. Tumor and host factors that may limit efficacy of chemotherapy in non-small cell and small cell lung cancer. Crit Rev Oncol Hematol. 2010;75:173–234. doi: 10.1016/j.critrevonc.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galluzzi L, Senovilla L, Vitale I, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 5.Creighton CJ, Li X, Landis M, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA. 2009;106:13820–13825. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 7.Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: Cancer stem cells versus clonal evolution. Cell. 2009;138:822–829. doi: 10.1016/j.cell.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 8.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 9.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 10.Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 12.Schatton T, Murphy GF, Frank NY, et al. Identification of cells initiating human melanomas. Nature. 2008;451:345–349. doi: 10.1038/nature06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bapat SA, Mali AM, Koppikar CB, Kurrey NK. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005;65:3025–3029. doi: 10.1158/0008-5472.CAN-04-3931. [DOI] [PubMed] [Google Scholar]
- 15.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946–10951. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 16.Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 17.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 18.Dalerba P, Dylla SJ, Park IK, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA. 2007;104:10158–10163. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eramo A, Lotti F, Sette G, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504–514. doi: 10.1038/sj.cdd.4402283. [DOI] [PubMed] [Google Scholar]
- 20.Bertolini G, Roz L, Perego P, et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci USA. 2009;106:16281–16286. doi: 10.1073/pnas.0905653106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koren A, Motaln H, Cufer T. Lung cancer stem cells: A biological and clinical perspective. Cell Oncol (Dordr) 2013;36:265–275. doi: 10.1007/s13402-013-0141-9. [DOI] [PubMed] [Google Scholar]
- 22.Freitas DP, Teixeira CA, Santos-Silva F, Vasconcelos MH, Almeida GM. Therapy-induced enrichment of putative lung cancer stem-like cells. Int J Cancer. 2014;134:1270–1278. doi: 10.1002/ijc.28478. [DOI] [PubMed] [Google Scholar]
- 23.Richichi C, Brescia P, Alberizzi V, Fornasari L, Pelicci G. Marker-independent method for isolating slow-dividing cancer stem cells in human glioblastoma. Neoplasia. 2013;15:840–847. doi: 10.1593/neo.13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pece S, Tosoni D, Confalonieri S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62–73. doi: 10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 25.Kleffel S, Schatton T. Tumor dormancy and cancer stem cells: Two sides of the same coin? Adv Exp Med Biol. 2013;734:145–179. doi: 10.1007/978-1-4614-1445-2_8. [DOI] [PubMed] [Google Scholar]
- 26.Bartucci M, Svensson S, Romania P, et al. Therapeutic targeting of Chk1 in NSCLC stem cells during chemotherapy. Cell Death Differ. 2012;19:768–778. doi: 10.1038/cdd.2011.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeuner A, Francescangeli F, Contavalli P, et al. Elimination of quiescent/slow-proliferating cancer stem cells by Bcl-XL inhibition in non-small cell lung cancer. Cell Death Differ. 2014;21:1877–1888. doi: 10.1038/cdd.2014.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Essers MA, Offner S, Blanco-Bose WE, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 29.Essers MA, Trumpp A. Targeting leukemic stem cells by breaking their dormancy. Mol Oncol. 2010;4:443–450. doi: 10.1016/j.molonc.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Connolly RM, Nguyen NK, Sukumar S. Molecular pathways: Current role and future directions of the retinoic acid pathway in cancer prevention and treatment. Clin Cancer Res. 2013;19:1651–1659. doi: 10.1158/1078-0432.CCR-12-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niu CS, Li MW, Ni YF, et al. Effect of all-trans retinoic acid on the proliferation and differentiation of brain tumor stem cells. J Exp Clin Cancer Res. 2010;29:113. doi: 10.1186/1756-9966-29-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Guan DX, Shi J, et al. All-trans retinoic acid potentiates the chemotherapeutic effect of cisplatin by inducing differentiation of tumor initiating cells in liver cancer. J Hepatol. 2013;59:1255–1263. doi: 10.1016/j.jhep.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 33.Friedman MD, Jeevan DS, Tobias M, Murali R, Jhanwar-Uniyal M. Targeting cancer stem cells in glioblastoma multiforme using mTOR inhibitors and the differentiating agent all-trans retinoic acid. Oncol Rep. 2013;30:1645–1650. doi: 10.3892/or.2013.2625. [DOI] [PubMed] [Google Scholar]
- 34.Moro M, Bertolini G, Tortoreto M, et al. Patient-derived xenografts of non-small cell lung cancer: Resurgence of an old model for investigation of modern concepts of tailored therapy and cancer stem cells. J Biomed Biotechnol. 2012;2012:568567. doi: 10.1155/2012/568567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kondo J, Endo H, Okuyama H, et al. Retaining cell-cell contact enables preparation and culture of spheroids composed of pure primary cancer cells from colorectal cancer. Proc Natl Acad Sci USA. 2011;108:6235–6240. doi: 10.1073/pnas.1015938108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bertolini G, Moro M, Tortoreto M, et al. The subset of CD133+/CXCR4+/EpCAM(−) cancer initiating cells is responsible for lung tumor metastatic spreading. Eur. J Cancer. 2012;48:S90. Abstract No 372. [Google Scholar]
- 37.Kusumbe AP, Bapat SA. Cancer stem cells and aneuploid populations within developing tumors are the major determinants of tumor dormancy. Cancer Res. 2009;69:9245–9253. doi: 10.1158/0008-5472.CAN-09-2802. [DOI] [PubMed] [Google Scholar]
- 38.Basu D, Nguyen TT, Montone KT, et al. Evidence for mesenchymal-like sub-populations within squamous cell carcinomas possessing chemoresistance and phenotypic plasticity. Oncogene. 2010;29:4170–4182. doi: 10.1038/onc.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rothwell DG, Li Y, Ayub M, et al. Evaluation and validation of a robust single cell RNA-amplification protocol through transcriptional profiling of enriched lung cancer initiating cells. BMC Genomics. 2014;15:1129. doi: 10.1186/1471-2164-15-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pine SR, Ryan BM, Varticovski L, Robles AI, Harris CC. Microenvironmental modulation of asymmetric cell division in human lung cancer cells. Proc Natl Acad Sci USA. 2010;107:2195–2200. doi: 10.1073/pnas.0909390107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao MQ, Choi YP, Kang S, Youn JH, Cho NH. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene. 2010;29:2672–2680. doi: 10.1038/onc.2010.35. [DOI] [PubMed] [Google Scholar]
- 42.Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008;10:R25. doi: 10.1186/bcr1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dembinski JL, Krauss S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin Exp Metastasis. 2009;26:611–623. doi: 10.1007/s10585-009-9260-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Degos L, Wang ZY. All trans retinoic acid in acute promyelocytic leukemia. Oncogene. 2001;20:7140–7145. doi: 10.1038/sj.onc.1204763. [DOI] [PubMed] [Google Scholar]
- 45.McGowan S, Jackson SK, Jenkins-Moore M, et al. Mice bearing deletions of retinoic acid receptors demonstrate reduced lung elastin and alveolar numbers. Am J Respir Cell Mol Biol. 2000;23:162–167. doi: 10.1165/ajrcmb.23.2.3904. [DOI] [PubMed] [Google Scholar]
- 46.Mao JT, Tashkin DP, Belloni PN, et al. All-trans retinoic acid modulates the balance of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 in patients with emphysema. Chest. 2003;124:1724–1732. doi: 10.1378/chest.124.5.1724. [DOI] [PubMed] [Google Scholar]
- 47.Arrieta O, González-De la Rosa CH, Aréchaga-Ocampo E, et al. Randomized phase II trial of all-trans-retinoic acid with chemotherapy based on paclitaxel and cisplatin as first-line treatment in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2010;28:3463–3471. doi: 10.1200/JCO.2009.26.6452. [DOI] [PubMed] [Google Scholar]
- 48.Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 49.Silinsky J, Grimes C, Driscoll T, et al. CD 133+ and CXCR4+ colon cancer cells as a marker for lymph node metastasis. J Surg Res. 2013;185:113–118. doi: 10.1016/j.jss.2013.05.049. [DOI] [PubMed] [Google Scholar]
- 50.Domanska UM, Kruizinga RC, Nagengast WB, et al. A review on CXCR4/CXCL12 axis in oncology: No place to hide. Eur J Cancer. 2013;49:219–230. doi: 10.1016/j.ejca.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 51.Wang L, Wang Z, Liu X, Liu F. High-level C-X-C chemokine receptor type 4 expression correlates with brain-specific metastasis following complete resection of non-small cell lung cancer. Oncol Lett. 2014;7:1871–1876. doi: 10.3892/ol.2014.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xie S, Zeng W, Fan G, et al. Effect of CXCL12/CXCR4 on increasing the metastatic potential of non-small cell lung cancer in vitro is inhibited through the downregulation of CXCR4 chemokine receptor expression. Oncol Lett. 2014;7:941–947. doi: 10.3892/ol.2014.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]