

Abstract
Valvular and vascular calcification are common causes of cardiovascular morbidity and mortality. Developing effective treatments requires understanding the molecular underpinnings of these processes. Shear stress is thought to play a role in inhibiting calcification. Furthermore, NOTCH1 regulates vascular and valvular endothelium, and human mutations in NOTCH1 can cause calcific aortic valve disease. Here, we determined the genome-wide impact of altering shear stress and NOTCH signaling on aortic valve endothelium. mRNA-sequencing of human aortic valve endothelial cells (HAVECs) with or without knockdown of NOTCH1, in the presence or absence of shear stress, revealed NOTCH1-dependency of the atherosclerosis-related gene connexin 40 (GJA5), and numerous repressors of endochondral ossification. Among these, Matrix GLA Protein (MGP) is highly expressed in aortic valve and vasculature, and inhibits soft tissue calcification by sequestering bone morphogenetic proteins (BMPs). Altering NOTCH1 levels affected MGP mRNA and protein in HAVECs. Furthermore, shear stress activated NOTCH signaling and MGP in a NOTCH1-dependent manner. NOTCH1 positively regulated endothelial MGP in vivo through specific binding motifs upstream of MGP. Our studies suggest that shear stress activates NOTCH1 in primary human aortic valve endothelial cells leading to downregulation of osteoblast-like gene networks that play a role in tissue calcification.
Keywords: Valve Calcification, Valve Endothelium, NOTCH1, NOTCH Signaling, Matrix GLA Protein
1. Introduction
Calcific aortic valve disease (CAVD) affects roughly 2% of the U.S. population and is the leading cause for valve replacement [1-3]. This incidence climbs in an age-dependent fashion, with 13% of individuals between 75 and 85 years of age suffering from CAVD [3-6]. In addition, 25% of patients born with congenitally bicuspid aortic valves, rather than the normal three-leaflet aortic valve, develop valve calcification as they age, suggesting a common genetic link between bicuspid aortic valves and a predisposition to calcification [7-10]. While valve replacement can successfully address the stenosis and regurgitation associated with valve disease, the procedure is invasive, expensive, and carries significant morbidity. In addition, 50% of individuals with CAVD also develop vascular stenosis and calcification, suggesting a mechanistic link [11]. The late onset and progressive nature of CAVD make it an ideal candidate for therapeutic intervention to slow progression of the calcification; however, there are currently no such therapies available. A prerequisite to developing effective therapies is a better understanding of the molecular events that lead to aortic valve calcification.
Aortic valve calcification is epidemiologically associated with multiple risk factors including age, gender, height, hypertension, smoking, and cholesterol levels in older patients [3,12]. The hallmarks of valve stenosis include inflammation, extracellular matrix remodeling including fibrosis, valve thickening, and angiogenesis. Ultimately, aberrant activation of osteoblast-like gene networks that lead to calcification are observed in the valve, which is very similar to the osteoblastic gene expression reported in vascular calcification [3,13]. In both settings, hemodynamic shear stress has been implicated in preventing the progression of calcification [10,14]. Laminar shear stress seems to be a protective anti-calcification force that is sensed by the endothelial lining of the arterial system, while regions of disturbed flow are more likely to calcify. Turbulent or oscillatory blood flow is typically observed at vascular bifurcations and on the aortic side of aortic valve leaflets, coinciding with sites of calcification [11,15].
While there are many factors associated with calcification, a hallmark of the disease process is the conversion of interstitial cells to osteoblast-like cells. The aortic valve consists of endothelial cells, mesenchymal interstitial cells and extracellular matrix components, including collagen, elastin and glycosaminoglycans, that provide strength and flexibility [16]. During valve calcification, interstitial cells of unknown origin begin to express α-smooth muscle actin and subsequently transform into osteoblast-like cells. Recent evidence suggests that these calcifying cells may originate from the valve endothelium [17]. These cells express the osteoblast-specifying transcription factor, RUNX2, and other calcification promoting proteins like ALP (alkaline phosphatase), IBSP (bone sialoprotein) and SPARC (osteonectin) [3,13]. While alterations of many cellular pathways, including TGFβ, NOTCH, and WNT signaling, result in the activation of RUNX2 in vitro, the molecular mediators of this activation are largely unknown [3].
We previously reported two families with aortic valve disease attributed to heterozygous non-sense mutations in NOTCH1 that suffered from severe, early calcification of the aortic valve and presence of a bicuspid aortic valve in many subjects [5]. These mutations result in truncated NOTCH1 mRNA transcripts that are predicted to undergo nonsense-mediated decay, suggesting haploinsufficiency of NOTCH1 [5]. In addition, multiple mouse models with decreased NOTCH signaling have shown increases in valve calcification [18,19]. NOTCH1 is a transmembrane protein that is cleaved upon binding to its ligands (Delta-like 1,3,4 and Jagged 1,2), which are expressed on neighboring cells [1,2]. Upon cleavage, the intracellular domain translocates to the nucleus and binds to the DNA-binding adaptor protein CSL [1]. Subsequently, co-activators MAML and histone acetyl transferases are recruited to the CSL complex, resulting in chromatin modification and target gene activation [1]. A candidate approach revealed that decreased in vitro expression of the NOTCH targets HEY1 and HEY2 in COS7 cells resulted in de-repression of RUNX2 [5]. However, as a transcription factor, NOTCH1 is likely to affect numerous target genes in vivo.
In order to understand the intersection between NOTCH signaling and shear stress in human valve disease, we used a multi-omics approach involving primary human endothelial cells. Here, we show that many genes within the endochondral ossification pathway are negatively regulated by NOTCH1 in a shear stress-dependent manner in primary human aortic valve endothelial cells, determined by RNA-sequencing (RNA-seq). Furthermore, ChIP-seq of the NOTCH1 intracellular domain (NICD) identified potential direct targets of NOTCH1 that are important for repression of calcification. In addition to negatively regulating osteoblastic gene networks in endothelial cells, we found NOTCH1 positively regulates the secreted Matrix Gla Protein (MGP), which is known to sequester BMP and calcium and inhibit soft tissue calcification in the vasculature and aortic valve [20]. These findings may provide novel targets for therapeutic intervention in CAVD.
2. Materials and Methods
2.1. Primary Cell Culture
Human Aortic Endothelial cells (HAECs)(Sciencell, San Diego, CA) and Human Aortic Valve Endothelial cells (HAVECs) were placed on plates coated with 10 μg/mL fibronectin from bovine plasma (Sigma, St Louis, MO) and grown in endothelial cell media (ECM)(Sciencell) at 5% CO2. Media was changed every other day and cells were passaged with 0.05% Trypsin EDTA when confluent. All experiments were performed with cells with fewer than 8 passages. Cells were cultured in unidirectional pulsatile shear stress conditions using μSlide I 0.8 Luer channel slides and flow kit (Ibidi, Verona, WI) and a L/S® Modular brushless digital dispensing drive peristaltic pump with 6-roller cartridge pump head (Cole Parmer, Vernon Hills, IL). Cells in shear stress conditions modeling laminar flow were exposed to media flow at 15 dynes/cm2 and cells in the static condition were grown in well plates. While only one flow rate was used, multiple flow rate conditions (i.e., low vs. high) may uncover even more gene expression differences.
2.2. Isolating Valve Cells
Aortic valves were obtained from patients undergoing heart-lung transplants for pulmonary hypertension according to the IRB at Stanford and UCSF. The valves were classified as “normal” by the transplant surgeon and neither patient had pre-existing pulmonary disease from childhood. Leaflets were removed from the discarded hearts and placed on ice in PBS. One leaflet was divided between Trizol reagent and RIPA buffer for later RNA and protein studies. One half of a leaflet was fixed in formalin for paraffin embedding and sectioning and the remaining leaflet pieces were processed for live cell isolation. The valves were rinsed five times in ice cold PBS and then digested in 2.5 mg/ml collagenase type IV (C5138, Sigma, St. Louis, MO) in M199 media (Lonza Biosciences, Walkersville, MD) at 37 °C for 30 min. The valve tissue was vortexed to remove valve endothelial cells, the supernatant was removed and spun at 500 × g for 2 min, and endothelial cells were then plated in ECM on fibronectin. This method results in an endothelial cell mixture that includes endothelial cells from both sides of the valve, which have been shown to have differential gene and protein expression levels [5,6]. The remaining valve tissue was digested further with 0.8 mg/ml collagenase in M199 for 3 hours at 37 °C with vortexing every 30 min. The tissue was vortexed and pipetted repeatedly to break up the tissue mass and then spun at 500 × g for 2 min. The supernatant containing freed cells was transferred to a new tube and spun down at 1100 × g for 8 min at 4 °C. The cells were resuspended in M199 containing 10% heat inactivated FBS and 1× pen/strep, and plated onto two t75s. After 3 to 4 days, endothelial cells were purified using anti-CD31 labeled magnetic beads (Miltenyi Biotec, Auburn, CA). To check cell type purity, cells were stained with mouse anti-human CD31-488 and anti-human CD144-PE conjugated antibodies (BD Biosciences) and checked by fluorescence activated cell analysis on an LSR II (BD Biosciences, San Jose, CA).
2.3. siRNA Delivery
siRNAs were delivered into endothelial cells using RNAiMax (Life Technologies, Carlsbad, CA). The siRNAs used were NOTCH1 siRNA (cat# 4392422 s9633 and control scrambled siRNA (cat# 4390843) (Ambion/Life Technologies). Endothelial cell media was changed to optimem with no supplements (Gibco/Life Technologies), and siRNA/RNAiMax was added for 4 hours. Then, the media was changed to full ECM and the cells were harvested for analysis 48 hours later.
2.4. DAPT Treatment
For gene expression experiments, experimental cells were treated with 5 μM DAPT (Sigma) in DMSO and control cells were treated with an equivalent amount of DMSO. Cells were harvested in Trizol reagent 48 hours after addition of DAPT. For Western blots, endothelial cells were treated for 48 hours in DMSO or DAPT and then trypsinized, washed with PBS, and re-plated for 96 hours before harvesting in RIPA buffer (Sigma, St. Louis, MO) + 1× Complete Mini protease inhibitors (Roche, Madison, WI).
2.5. RNA-seq and Analysis Pipeline
RNA from HAVECs from each condition was purified using TRIzol extraction followed by RNeasy MinElute Cleanup Kit with column DNAse digestion (Qiagen, Valencia, CA). Then, RNA from the two biological replicates was pooled into one sample per condition prior to library generation. 50 ng of RNA was used with the Ovation® RNA-Seq System (7100-08)(NuGEN, San Carlos, CA) and manufacturer's protocol to synthesize and SPIA amplify cDNA, which was then sheared with a covaris S-series. After end repair, purification and P1 P2 adaptor ligation, templated beads were generated with the EZ beads system, and libraries were then paired end sequenced (50 bp forward, 35 bp reverse) on a SOLiD4 DNA sequencing system (Life Technologies). RNA-seq reads were aligned to the human hg19 genome using TopHat [21]. TopHat was supplied with GTF-format annotation of known exon positions. Novel exon discovery was disallowed—only exons and splice junctions that were already present in the annotation were included. Duplicate reads (reads mapping to the exact same start / stop position) were removed using the Picard tool MarkDuplicates (http://picard.sourceforqe.net/). Secondary mapping locations were discarded. For each read, only the top alignment was retained. For paired-end reads, any “singleton” reads (where both ends did not map successfully) were discarded. Sequencing depth for the four libraries ranged from 48.4 to 63.9 million unique aligned reads. Cufflinks was used to obtain a reads-per-gene total and provide normalized expression levels, reported as RPKM (reads mapped to a gene per thousand bases of the gene, per million mapped reads) [22]. Differentially expressed genes were identified using Cuffdiff, part of the Cufflinks suite. Only genes with RPKMs > 1 and CuffDiff output of “OK” were included in subsequent analysis. Relative expression levels for heatmaps were normalized to the control siRNA, and no flow condition and values were transformed to a log base 2 scale. UCSC Genome Browser tracks were generated using wiggle tracks and indicate the number of fragments that mapped to a particular locus. RNA-seq data are available from Gene Expression Omnibus (accession numbers GSE47157 & GSE47160).
2.6. ChIP-seq and ChIP-qRT-PCR
ChIP-seq was performed as described [3,8,12]. Briefly, 2.5 million HAVECs were infected with either Luciferase-myc tag or NICD1-myc tag lentivirus and fixed with formaldehyde after 48–72 hours. All myc tag ChIPs were done with a Goat polyclonal to Myc tag-ChIP grade antibody (ab9132, Abeam, Cambridge, MA). ChIPs were done in triplicate with Protein G magnetic beads and then pooled for subsequent ChIP-seq library preparation with the standard Illumina ChIP-seq library protocol. High-throughput sequencing was performed on an Illumina GAIIxs machine. Tags were mapped back to the genome using the bowtie aligner [10,21] and ChIP tag densities were calculated using bedops [11,23]. Peaks in ChIP signal were identified using SPP with an enrichment score cutoff of 10, based on known targets of NOTCH1 [3,12,24]. Genome feature enrichment analysis was implemented in perl and R, utilizing bedops in conjunction with Ensemble annotations [3,13,25]. Custom Taqman probes (Life Technologies) (Integrated DNA Technologies, Coralville, IA) were designed to verify selected NICD binding sites via qRT-PCR. Specific probe sequences are provided in supplemental data. ChIP-seq data are available from Gene Expression Omnibus (accession number GSE47152).
2.7. Gene Ontology and Pathway Analysis
Individual gene expression RPKMs were used to generate relative differential expression values. Only genes with at least 2 fold change in expression were considered for further analysis. Ensemble gene annotations were combined with ChIP-seq to identify the largest peak within 20 kb up or downstream of each gene transcriptional start site. We chose to search within 20 kb of the TSS since the majority of NOTCH 1 peaks occurred within this range. Patterns of differential gene expression combined with ChIP peak scores were then used to compile gene lists. GO Elite and GenMAPP-CS were used to predict GO terms and pathways overrepresented in the gene sets [7,9]. GO Elite was run with 2000 ORA permutations, Z-score cutoff of 1.96, permuted p-value cutoff of 0.05, and minimum of 3 genes changed per term.
2.8. Electrophoretic mobility shift assays (EMSAs)
DNA binding assays were carried out as previously described [16,26]. Oligos were annealed, labeled with 32P-dCTP, and gel purified on a 40% acrylamide gel. CSL protein was generated with a TNT T7/Sp6 Coupled Reticulocyte Lysate System (Promega, Madison, WI). Each DNA binding reaction was performed in 40 mM KCL, 15 mM HEPES pH7.9, 1 mM EDTA, 5 mM DTT and 5% Glycerol and then run on a 6% acrylamide gel before film exposure to visualize the 32P. Oligos used for the EMSA were as follows: HES1 Wt For, 5′ -GGATTACTATTTCCCACACATCTT-3′; HES1 Wt Rev, 5′ -GGTAAGATGTGTGGGAAATAGTAAT-3′; HES1 Mut For, 5′ -GGATTACTATTAACCACACATCTTA-3′; HES1 Mut Rev, 5′ -GGTAAGATGTGTGGTTAATAGTAAT-3′; MGP WT For, 5′ -GGTCCAAGGGCTTTTGGGAACAGATTTGTGAGAAAAGAGTGAAAAG-3′; MGP WT Rev, 5′ -GGCTTTTCACTCTTTTCTCACAAATCTGTTCCCAAAAGCCCTTGGA-3′; MGP 2× Mut For, 5′ -GGTCCAAGGGCTTTGTCGAACAGATTTGGTCGAAAAGAGTGAAAAG-3′; MGP 2× Mut Rev, 5′ - GGCTTTTCACTCTTTTCGACCAAATCTGTTCGACAAAGCCCTTGGA-3′. MGP 1× Mut For, 5′ - GGTCCAAGGGCTTTTGGGAACAGATTTGTGTCAAAAGAGTGAAAAG-3′. MGP 1× Mut Rev, 5′ - GGCTTTTCACTCTTTTGACACAAATCTGTTCCCAAAAGCCCTTGGA-3′.
2.9. qRT-PCR
RNA was purified from cells in Trizol reagent and cDNA was generated from 1 μg of RNA using Superscript III First Strand Synthesis Supermix (Life Technologies). Gene expression analysis was performed by quantitative reverse transcriptase-PCR (qRT-PCR) using standard Taq-Man primer sets and Taqman Gene Expression Mastermix on an ABI-7900 HT (Applied Biosystems).
2.10. Immunohistochemistry
Human aortic valves were fixed with 10% formalin overnight at 4 °C, paraffin embedded, and sectioned. Valve sections were subjected to antigen retrieval under heat and pressure for 2 min in antigen retrieval buffer (10 mM Tris, 1 mM EDTA, and 0.05% tween). The sections were permeabilized with PBS Triton (0.1%) and blocked with serum from the secondary antibody source. Primary antibodies were diluted 1:50 in PBS Triton (0.1%) and were incubated at 4 °C overnight. The sections were thoroughly washed with PBS and incubated with secondary antibody diluted in PBS triton (0.1%) 1:200 for 2 hours at room temperature. Sections were thoroughly washed in PBS, nuclei stained with DAPI or Hoescht and sections were then mounted under coverslips and imaged. Primary antibodies used were NOTCH1 intracellular domain (ab8925, Abeam), cMGP (clone G8A#1), ucMGP (clone B11#1, VitaK BV, Maastricht, The Netherlands), and phospho-SMAD 1/5/8 (#9511, Cell Signaling Technology, Danvers, MA). All tissue sections were imaged on a Leica DM4000B microscope using a Leica HC plan apo 20× / 0.7NA objective at room temperature. A Leica DFC310FX (PC) Color high sensitivity camera and Leica Application Suite (LAS) imaging software were used for image capture. The same software and camera were used on a Leica MZ16F dissecting microscope for mouse embryo imaging.
2.11. Lentiviral Production
Expression of NICD was achieved by OmicsLink expression construct Lv140 (GeneCopoeia, Rockville, MD) with a CMV promoter driving NICD1 (UniProtKB/Swiss-Prot: P46531.4, aa1769- 2555 2364bp) with a myc tag and IRES mCherry to check the percent of cells infected. A control lentivirus contained a Luciferase gene in place of NICD in the same LV140 construct. Lentiviral particles were produced using the Lenti-Pac HIV expression packaging kit (Genecopoeia) and HEK-293t cells.
2.12. Western Blot
Western blots were performed using the Licor quantitative western blot protocol and visualized on an Odyssey Fc (Licor, Lincoln, NE). Antibodies used were anti-β-Actin clone AC-15 (A1978, Sigma), anti-GAPDH (ab8245, Abeam), anti-cMGP (clone G8A#1) and anti-ucMGP (clone B11#1, VitaK BV, Maastricht, The Netherlands).
2.13. Transgenic Animals
The wildtype (Wt) 821 bp MGP putative enhancer region was TOPO cloned into pCR2.1-TOPO vector (Life Technologies) using the following primers: MGP-Wt-800 bp For, 5′- AATGCCATAAGGGTCCTTCC-3′; MGP-Wt-800 bp Rev, 5′-TGAAGAAGCGAGCCACATC-3′. The pCR2.1-MGP-821 bp enhancer region was then cut with Nhe1 (New England Biolabs, Ipswich, MA), blunted with T4 DNA polymerase and then cut with Not1-HF (New England Biolabs). The MGP-821bp enhancer was then ligated into the plasmid pENTR1A (Life Technologies), which was cut with Xmn1 and Not1-HF. The pENTR1A-MGP-Wt-800 bp plasmid was cut with BglII and SanD1 (Thermo Scientific, Waltham, MA) and Cold Fusion Cloning Kit was used to introduce a double stranded oligo containing mutations in the CSL binding sites: MGP-3XMut-800 bp for 5′ -AAGTGGTCAGGTGTAGTACAGAGATCTGGGGCAATCATTGCATGTTGGAGTCGACGATTCCAAGGGCTTTGTCGAACAGATTTGGTCGAAAAGAGTGAAA-3′. The MGP Wt-and 3XMut-821 bp enhancers were then subcloned into an hsp68 lacZ-DV minimal promoter plasmid using LR clonase (Life Technologies). These final plasmids were cut with HindIII and Nael before pronuclear injection in FVB/N mice. LacZ transgenes were detected in pups by PCR from tail biopsies (Transnetyx, Cordova, TN). All experiments using animals were reviewed and approved by the UCSF Institutional Animal Care and Use Committee and complied with all institutional and federal guidelines.
2.14. Statistical Analysis
All experiments were performed at least three times unless otherwise stated. All statistical analysis was completed using GraphPad Prism software. Two-way analysis was completed with two-tailed unpaired t tests with a 95% confidence interval. Multiple comparison tests were performed by one-way ANOVA with Tukey's multiple comparison post-test (* P<0.05, ** P<0.01, *** P<0.001).
2.15 Supplemental Materials
ChIP-qPCR primers, cloning sequences, siRNA sequences and lentiviral NICD sequence are available in the supplemental section.
3. Results
3.1. Shear Stress and NOTCH1 Signaling in HAVECs
Normal human aortic valve sections were stained with an antibody specific to NOTCH1 intracellular domain (NICD) in order to identify the cellular compartment of active NOTCH1 signaling. We observed high expression of NICD in both the valve interstitial cells and endothelial cells lining the valve (Figure 1A). Since endothelial cells would sense shear stress and Notch signaling is known to be critical for endothelial cell development and homeostasis, we focused our attention on these cells. To test the interplay between NOTCH1 signaling and shear stress forces, primary human aortic valve endothelial cells (HAVECs) were isolated from explanted human valves and transfected with either control or NOTCH1 specific siRNAs to mimic the decreased NOTCH1 activity in previously reported patients [5,17]. Cells from these two conditions were then divided into static or approximately 15 dynes/cm2 media flow (shear stress) conditions for a total of four conditions (Figure 1B). A previous comparison of endothelial cells explanted from opposite sides of the valve and cultured under identical shear stress conditions revealed limited variation in gene expression between the two cell populations, in spite of dramatic differences, in vivo [3,13,27]. This highlights the importance of flow to replicate endogenous gene expression and was an important consideration in our experimental design. qRT-PCR confirmed that the NOTCH1 gene expression was indeed activated by shear stress and was effectively knocked down using NOTCH1 siRNA (Figure 1C). In addition, the canonical NOTCH1 targets, HEY2 and HES1, and a known shear responsive gene KLF2, were activated by shear stress (Figure 1C). Although HEY2 remained responsive to decreased NOTCH1 in the shear stress condition, HES1 and KLF2 expression was not affected by NOTCH1 knockdown in the presence of flow, illustrating the differential sensitivity of various Notch1 targets to both flow and variations in NOTCH1 expression.
Figure 1.
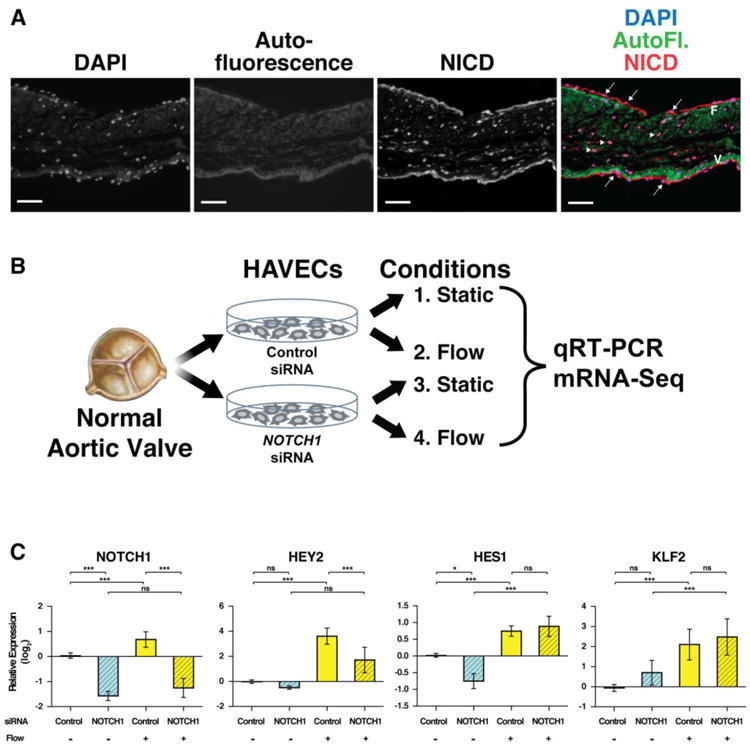
Activated NOTCH1 is detected in valve cells in a flow dependent manner. (A) Normal human aortic valve section stained with an antibody specific to the active form of NOTCH1 (NICD, red). NICD was found in both endothelial (arrows) and interstitial cells (arrowheads). Autofluorescence (AutoFI, green) of collagen and elastin highlight the fibrosa layer (F) and ventricularis layer (V), respectively. Nuclei, DAPI (blue). Scale bars indicate 100 μm. (B) Schematic of experimental procedure. Normal HAVECs were transfected with control or NOTCH1 siRNA, then cultured in static or fluid flow conditions. Gene expression was compared by qRT-PCR or mRNA-seq. (C) qRT-PCR analysis of HAVECs from four conditions. NOTCH1, two canonical direct targets, HES1 and HEY2 and a known flow responsive gene, KLF2, were analyzed. Graphs show mean gene expression relative to the static, control siRNA condition with error bars representing standard deviation. (n=3; *, p<0.05; ** p<0.01; *** p<0.001; NS, Not Significant).
RNA-seq was performed on RNA that was pooled from two biological replicates for each of the four conditions to understand how shear stress and NOTCH1 signaling may be controlling global gene transcription. GO-Elite and GenMAPP-CS were used to identify over-represented pathways among the dysregulated genes with a permuted P value of < 0.05.
We first considered genes that were either activated or repressed at least 2-fold in response to decreased NOTCH1 signaling in static conditions. We found that these included genes involved in transmembrane receptor activity, gated/substrate specific channel activity, chemokine activity, and extracellular matrix structural constituents (Table S1). Transmembrane receptors and chemokines are involved in recruitment of inflammatory cells and ECM proteins like collagens and fibulins. These processes have been shown to play a role in the progression of valve calcification or atherosclerosis. In addition, genes involved in the response to steroid hormone stimulus, the acute inflammatory response, and integrin mediated cell adhesion were dysregulated.
We next considered genes that were down regulated by at least 2-fold in response to decreased NOTCH1 signaling and found that they included genes in the NOTCH signaling pathway, blood vessel morphogenesis, and regulation of BMP signaling, the most potent bone-inducing morphogens (Table S2). Also downregulated were gene ontologies involved with inflammation and immune activity, including monocyte chemotaxis, leukocyte tethering and rolling, and chemokine activity, all of which play a critical role in the onset and progression of atherosclerosis. Finally, genes involved in vasoconstriction, many of which play a role in coronary artery disease, were down regulated. Conversely, genes activated in the NOTCH1 knock-down static condition included voltage gated sodium channels, fibroblast growth factors, and myosin complex genes (Table S3).
Because regions of the arterial system with laminar blood flow seem relatively resistant to calcification, similar to the ventricular side of the aortic valve, we investigated genes that were activated or repressed by at least 2-fold when exposed to flow (Table S4). Many genes implicated in activin, retinoid, and SMAD receptor binding were represented, each involving a signaling pathway that can stimulate calcification. Metallopeptidase activity, calcium ion binding, as well as extracellular matrix protein binding (glycosaminoglycan, collagen, and fibronectin proteins) were also affected. These processes are vital to extracellular matrix remodeling and when perturbed can lead to increased ECM deposition, an early step in the progression towards valve stenosis and eventual calcification. Regulation of cell-cell adhesion, VEGF receptor signaling, leukocyte proliferation, and wound healing were also positively affected by shear stress. Other pathways affected by shear stress included GPCR signaling, endochondral ossification, heart development, and complement and coagulation cascades.
Finally, we investigated genes that were activated by flow and were affected by decreased NOTCH1 in HAVECs, potentially indicating direct NOTCH1 targets as NOTCH1 is canonically viewed as a transcriptional activator (Table S5). Gene ontological networks included receptor binding activity such as the pro-inflammatory interleukin-1 receptors, as well as thrombin receptors, which are involved in platelet activation and inflammatory cell recruitment after formation of vascular lesions, eventually leading to atherosclerosis. In addition, pathways including NOTCH signaling and VEGF signaling were affected (Table S3). Developmentally important genes involved in vascular development and skeletal system development were also affected by decreased NOTCH1 signaling. Immune function related gene sets were represented by leukocyte chemotaxis and negative regulation of immune response. All of these genes may play a role in the protective effect of shear stress that is perturbed in the case of defective NOTCH1 signaling.
Major pathways activated by shear stress and affected by decreased NOTCH1 signaling included heart development, complement activation and endochondral ossification (Table 1). The genes involved in heart development included important valve development regulators such as TWIST1, HEY1, HEY2, and FOXC1 [2,3]. Complementation Activation gene members were C9, CD55, and MASP1. Most importantly from the standpoint of calcification, the endochondral ossification pathway members (Figure S1) included many genes important for maintaining non-calcified regions of growing bones, known as epiphyseal plates, including SOX6, PLAT (Tissue Plasminogen Activator), PTHrP (Parathyroid hormone-related protein), FGF18, and MGP (Matrix Gla Protein) (Figure 2, S2).
Table 1. Pathways Activated by Shear Stress in NOTCH1 Dependent Manner.
| MAPP Name | # Changed | # Measured | # on MAPP | Z Score | P Value | Gene symbols |
|---|---|---|---|---|---|---|
| Heart Development: WP1591 | 8 | 30 | 44 | 7.03 | 0.000 | BHLHE40, BMP2, FOXC1, GATA6, HEY1, HEY2, SHH, VEGFA |
| Complement Activation, Classical Pathway: WP545 | 3 | 5 | 17 | 6.95 | 0.001 | C9, CD55, MASP1 |
| Endochondral Ossification: WP474 | 9 | 41 | 64 | 6.56 | 0.000 | ALPL, FGF18, FGFR3, MGP, PLAT, PTHLH, SOX6, TGFB1, VEGFA |
| Complement and Coagulation Cascades: WP558 | 5 | 19 | 62 | 5.49 | 0.001 | C9, F2R, MASP1, PLAT, THBD |
| GPCRs, Class A Rhodopsin-like: WP455 | 7 | 59 | 261 | 3.60 | 0.003 | F2R, F2RL2, F2RL3, GPR173, LPAR5, OR10A4, P2RY6 |
| Adipogenesis: WP236 | 7 | 84 | 131 | 2.51 | 0.037 | BMP2, EGR2, GATA3, LIF, PTGIS, TGFB1,TWIST1 |
Genes activated by at least 2 fold in shear stress conditions and down regulated by at least 2 fold upon NOTCH1 knockdown in shear stress conditions were analyzed by GO Elite to identify potential, shear stress activated pathways that depend on NOTCH1. Significantly enriched pathways were determined by a Z score >1.96 and P vale >0.05.
Figure 2.
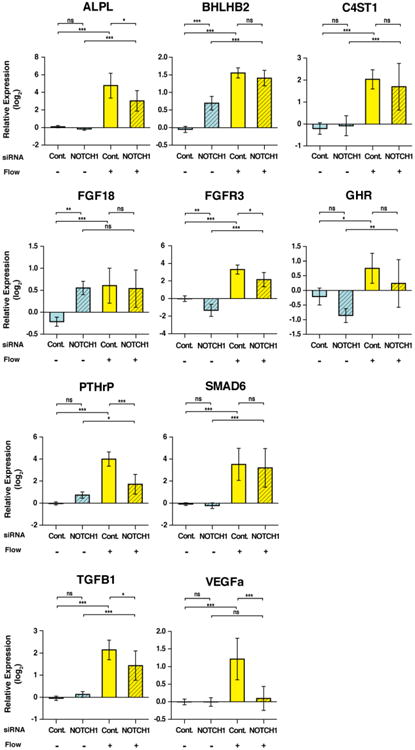
Expression of endochondral ossification genes is affected by shear stress and NOTCH1 signaling. Graphs show mean gene expression relative to the static, control siRNA condition with error bars representing standard deviation. All numbers are shown in log2 scale. (n=3; *, p< 0.05; **, p< 0.01; ***, p< 0.001, NS, Not Significant).
3.2. Genome-wide NOTCH1 DNA-Binding in HAVECs
To determine the direct transcriptional targets of NOTCH1, we performed ChIP-seq to identify genome-wide DNA occupancy of NOTCH1 in primary HAVECs isolated from fresh human aortic valve tissue. To overcome potential limitations of existing ChIP-grade NICD antibodies and low levels of NICD protein expression, we infected HAVECs with a lentivirus expressing a myc-tagged NOTCH1 intracellular domain (NICD) and used anti-myc antibodies to precipitate DNA regions bound by NOTCH1, followed by DNA sequencing. Roughly 8500 NICD peaks were identified and approximately 6400 genes had a significant NOTCH1 peak within 20 kb upstream or downstream of the transcriptional start site (Table S6). Gene Ontology analysis was performed on these 6400 potential direct gene targets of NOTCH1 to identify gene networks that may be controlled by NOTCH1. Molecular functions potentially targeted by NOTCH1 included transmembrane receptor phosphorylation, T cell receptor binding, SMAD and beta-catenin binding, and extracellular matrix binding such as integrin, glycoprotein, collagen and cadherin binding (Table S7). Enriched biological processes included wound healing, vasculogenesis, response to fluid shear stress, regulation of TGFβ 2 production, ossification/chondroblast differentiation, and endothelial cell proliferation/migration.
Data from the NOTCH1 ChIP-seq and RNA-seq of control and NOTCH1-deficient cells were integrated to identify potential direct targets of NOTCH1 in HAVECs. Based on our observation that NOTCH1 is activated by flow in HAVECS (1C), we focused on genes that were simultaneously NOTCH1-dependent and flow responsive. Pattern 1 contained genes that were repressed upon Notch1 depletion that were also activated by shear stress in a NOTCH1-dependent manner and Pattern 2 contained genes induced by NOTCH1 depletion and repressed by shear stress in a NOTCH1-dependent manner (Figure 3A and Table S8). Of the genes displaying either pattern, we focused specifically on those for which our ChIP-seq identified NOTCH1 occupancy within 20 kb of the TSS, which might indicate direct transcriptional control by NOTCH1 (Figure 3B). Potential targets activated both by NOTCH1 and by shear stress in a NOTCH1-dependent manner (Figure S6) were validated by ChIP qPCR (Figure S7) and included genes important for maintaining non-calcified bone growth plates, repressing osteogenesis, and preventing atherosclerosis, vascular calcification, and angiogenesis (Figure S2-6 and Table S9). Among them, we identified MGP as an interesting candidate whose role in human valve calcification has not been explored. Targeted deletion of this secreted factor in mice causes the most severe ectopic calcification phenotype reported in any mouse model, including calcification of epiphyseal growth plates, the arterial system, and aortic valve within weeks of birth [5,20]. For this reason, we investigated the regulation of MGP by NOTCH1 in greater detail.
Figure 3.
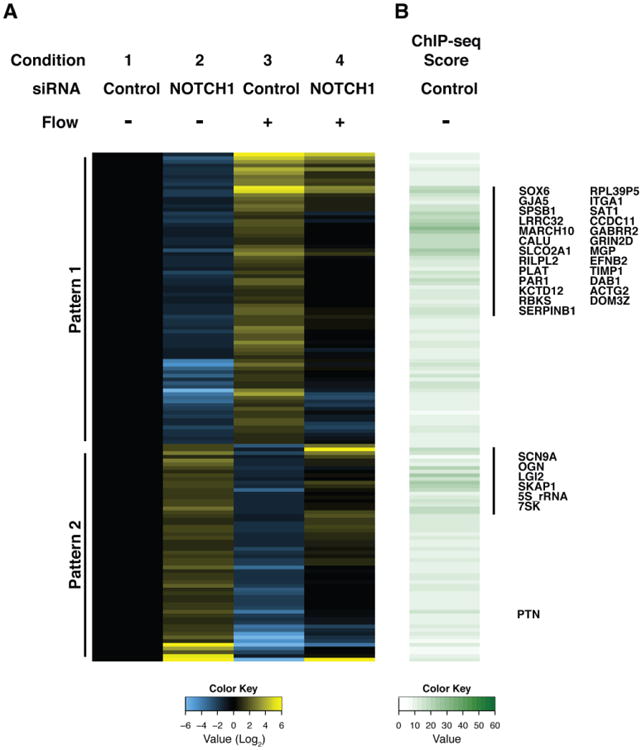
Heatmap and clustering analysis of RNA-seq and ChIP-seq from HAVECs reveal likely NOTCH1 direct targets. (A) The expression of each gene was normalized to the static, control siRNA condition and then Log2 transformed. Displayed genes were selected as matching one of two patterns reflecting coordinate regulation by both NOTCH1 and shear stress: Pattern 1) at least 2-fold down upon NOTCH1 siRNA knockdown in both static and flow conditions, and at least 2-fold up in flow control siRNA condition compared to static, or Pattern 2) at least 2 fold up in NOTCH1 siRNA knockdown in both static and flow conditions, and at least 2 fold down in flow control siRNA condition compared to static. (B) ChIP-seq score shows the value for the highest ChIP peak +/− 20 kb from the transcriptional start site (TSS) of each gene. Annotated genes represent potential direct targets of NOTCH1 based on ChIP-seq score.
3.3. NOTCH1 Directly Regulates MGP
Localization of MGP protein expression in human aortic valves has not been reported to date. MGP exists in an uncarboxylated (inactive) form that can be enzymatically activated by a Vitamin K-dependent gamma carboxylase to yield λ-carboxylated MGP (cMGP). Expression levels of both were assayed via immunohistochemistry using conformation specific antibodies. Both forms of MGP were expressed in the valve endothelial and interstitial cells, though the active form seemed higher in the endothelium and the inactive form more prevalent in the interstitial cells (Figure 4A). Secreted MGP sequesters bone morphogenetic protein 2 (BMP2), a protein that activates SMAD signaling leading to phosphorylation of SMADs 1, 5 and 8 (pSMAD1/5/8). Active BMP signaling as observed by pSMAD1/5/8 staining was present in both endothelial and interstitial cells, with highest expression on the aortic side of the valve similar to results reported in Ankeny et al. (Figure 4A) [5,28]. qRT-PCR validation in HAVECs showed that MGP expression levels were activated by shear stress in a NOTCH1-dependent manner (Figure 4B). Lentiviral over-expression of NICD1 induced higher levels of cMGP in endothelial cells but not in interstitial cells, suggesting primary regulation of cMGP by NOTCH1 in endothelial cells (Figure 4C-E). In contrast, chemical inhibition of NOTCH1 with DAPT in static conditions decreased the levels of active cMGP (Figure 4F, G). HAECs were utilized for these analyses due to the large number of cells required for Western blotting, which could not be obtained from cultured HAVECs. Likewise, we were unable to analyze cMGP levels in cells treated with NOTCH1 siRNA and shear stress because the flow chambers used in this study have a very limited capacity, precluding us from applying flow-mediated shear stress to enough cells for subsequent Western analysis. Nevertheless, these data illustrate that human endothelial cells, but not interstitial cells, modulate cMGP levels in response to NOTCH signaling.
Figure 4.
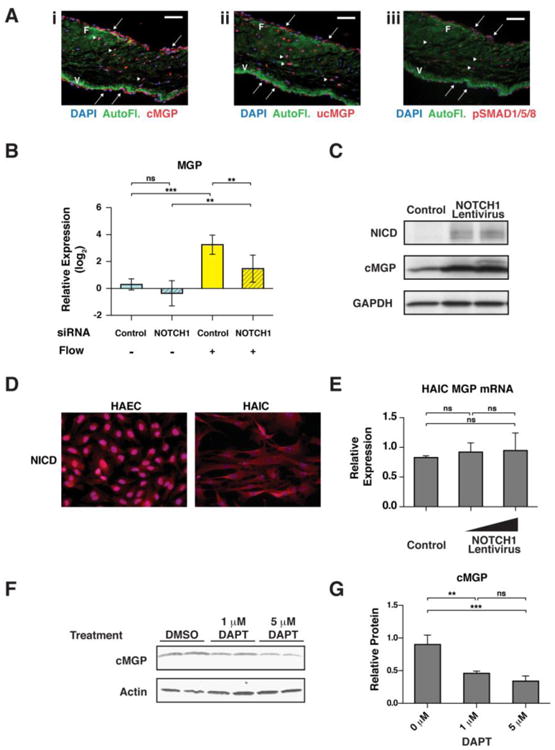
MGP expression is NOTCH1 dependent. (A) Normal human aortic valve sections stained with antibodies specific to: i. cMGP, the active, carboxylated form of MGP; ii. ucMGP, the inactive, uncarboxylated form of MGP; and iii. phosphoSMAD 1/5/8 (pSMAD1/5/8) cMGP, ucMGP and pSMAD1/5/8 (red) were found in both endothelial (arrows) and interstitial (arrowheads) cells. Autofluorescence (AutoFI, green) of collagen and elastin highlight the fibrosa layer (F) and ventricularis layer (V), respectively. Nuclei, DAPI (blue). Scale bars indicate 100 μm. (B) qRT-PCR analysis of MGP mRNA expression in HAVECs under four conditions. Graph shows mean gene expression relative to the no flow, no siRNA condition with error bars representing the standard deviation. (n=3; *, p< 0.05). (C) Western blot detecting NICD, active MGP (cMGP) and GAPDH from HAECs infected in vitro with increasing amounts of myc-tagged NOTCH1 intracellular domain (NICD). (D) Immunostaining for NICD (red) in cultured Human Aortic Endothelial Cells (HAEC) or Human Aortic Interstitial Cells (HAIC). (E) qRT-PCR to measure MGP mRNA in control or NOTCHI-overexpressing HAICs. (F) Western blot detecting active MGP in control or γ-secretase inhibitor (DAPT) treated HAECs. (G) Quantification of cMGP protein in (F) normalized to Actin. Error bars show standard deviation (n=3; **, p< 0.01; ***, p< 0.001; NS, Not Significant).
Gene and protein expression level changes and ChIP-seq data suggested that NOTCH1 might be directly activating MGP. One significant NOTCH1 ChIP-seq peak identified was 20 kb upstream of MGP, between MGP and its closest gene, ERP27 (Figure S8). Data from the ENCODE project involving Human Umbilical Vein Endothelial Cells (HUVECs) indicated that this NOTCH1 ChIP-seq peak was within a region of the genome that displays many markers of an active regulatory region and is predicted as a “strong enhancer” [1,29]. These include markers of accessible open chromatin (DNAse1 hypersensitivity, FAIRE) and H3K27ac/H3K4me1 histone marks, indicating a region bound by transcription factors that displace nucleosomes [2,30]. In addition, this region is conserved throughout vertebrate evolution from human to zebrafish and Genomic Evolutionary Rate Profiling (GERP) shows evolutionary constraint for a substitution deficit. Finally, the NICD DNA binding partner, CSL, is predicted to bind to a pair of strong CSL motifs 6 bp apart and to a third weaker site 15 bp upstream in the putative enhancer (Figure S9). Together, these observations led us to test this region as a NOTCH-responsive MGP enhancer.
ChIP followed by qPCR confirmed that the region containing CSL sites upstream of MGP was enriched for NICD protein to a similar degree as a canonical, validated HES1-NICD/CSL binding site (Figure 5A). In addition, electrophoretic mobility shift assays showed that in vitro transcribed and translated CSL protein was capable of binding a labeled DNA probe corresponding to the potential MGP enhancer (5B). This binding could be competed by addition of excess unlabeled oligonucleotides corresponding to either the validated HES1-NICD/CSL binding site or the potential MGP enhancer. Binding was not effectively competed by oligonucleotides in which the CSL binding sites were mutated, demonstrating the specificity of CSL for these sites.
Figure 5.
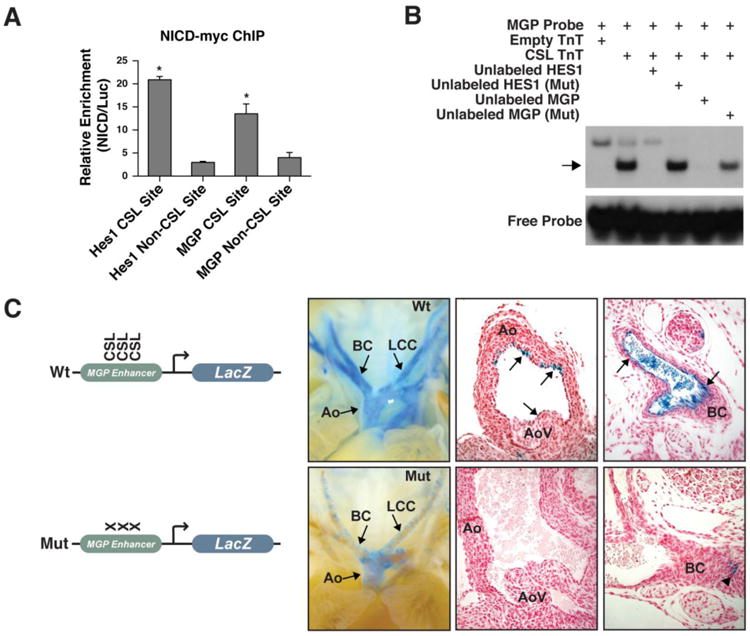
NOTCH directly regulates MGP through an endothelial enhancer. (A) NICD1-myc ChIP-qPCR. Relative enrichment is shown for four sites: validated HES1 NOTCH1/CSL binding site, validated HES1 non-CSL binding site, predicted CSL binding sites within the 821 bp putative MGP enhancer and an MGP non-CSL site. The HES1 and MGP non-CSL binding sites are located approximately 2 kb from the CSL binding sites. Error bars indicate standard deviation. (n=4; *, p< 0.05). (B): EMSA snowing CSL binding to putative MGP enhancer. A 46 bp oligo corresponding to the NICD1-myc ChIP-seq peak was labeled with 32P-dCTP (MGP Probe). Incubation with in vitro transcribed and translated CSL (CSL TnT) shifted the probe (arrow) This interaction could be competed by addition of unlabeled MGP probe (Unlabeled MGP) or oligo corresponding to a validated HES1 enhancer containing CSL sites (Unlabeled HES1), but not with unlabeled oligos in which the CSL sites were mutated (Mut), demonstrating specificity. Reticulocyte lysate (Empty TnT) caused a higher, non-specific shift. (C) Images of whole mount or histological sections from E16.0 transgenic embryos containing the 821 bp MGP enhancer upstream of the Hsp68 minimal promoter driving LacZ with or without mutation of the three predicted CSL binding sites. Mice containing the wildtype (Wt) enhancer had strong endothelial cell expression of LacZ (blue) in the large vessels of the arterial system (8/13) and some weak expression in the aortic valve (arrows) (4/13 embryos). Mutation of CSL sites (Mut) abolished endothelial expression (8/8); the mutant enhancer had weak expression in the smooth muscle layer (arrowhead) of 3/8 embryos. Sections were counterstained with Eosin (red). Aorta, Ao; branchiocephalic artery, BC; left common carotid, LCC; aortic valve, AoV.
In order to test the activity of the putative enhancer in vivo, we generated transgenic mice containing an 821 base-pair region encompassing the CSL sites (Wt enhancer) or with mutations of the 3 CSL sites (Mut enhancer) combined with a minimal promoter driving LacZ (Figure 5C). MGP is expressed in fibroblasts, smooth muscle, arterial endothelium and chondrocytes [20]. The Wt enhancer LacZ transgenics also showed expression within many cell types. Expression of the Wt enhancer LacZ reporter was consistent in the large vessels of the arterial system (8 out of 13 mice), including the aorta, branching arteries, pulmonary artery, and aortic valve, suggesting that this region could direct MGP expression in the arterial vasculature. Mutation of the 3 CSL binding sites abolished endothelial expression throughout the arterial system, microvessels, and valves in all Mut enhancer transgenic mice (n=8) (Figure 5C). Smooth muscle expression in the aorta was observed occasionally in both Wt enhancer mice (2/13) and Mut enhancer mice (3/8). Thus, in endothelial cells, NOTCH directly and positively regulates MGP, a secreted factor that is known to be essential for repressing the osteoblastic gene program in soft tissues, possibly contributing to the role of NOTCH1 in preventing calcification [1,20].
4. Discussion
This study provides genome-wide investigation of NOTCH1 and shear stress in primary human aortic valve endothelial cells and reveals NOTCH1-dependency of multiple ossification-repressing gene networks. Included in these networks was Matrix GLA Protein (MGP), a secreted factor that is highly expressed throughout the aortic valve and vasculature and inhibits soft tissue calcification by sequestering bone morphogenetic proteins (BMPs). We found that NOTCH1 positively regulated endothelial expression of MGP both in vitro and in vivo through a cluster of CSL binding sites upstream of the MGP locus. Our results implicate MGP as an effector of anti-calcific instruction by Notch1 and provide the impetus for further research into this Notch-MGP axis as a regulator of valve calcification in vivo.
Whole transcriptome profiling coupled with ChIP-seq provided evidence that gene networks similar to those responsible for maintaining a non-calcified region of proliferating chondrocytes in long bones, known as the epiphyseal or growth plate, were responsive to shear stress and affected by NOTCH1 dosage in primary human aortic valve endothelial cells (Figure 6). NOTCH1 also regulated multiple pathways previously implicated in the pathology of aortic valve calcification, including chondrocyte development, immune cell interactions, and BMP signaling. For example, our data show that GJA5 (Connexin 40) is a direct target of NOTCH1. Interestingly, GJA5 modulates arterial endothelial identity, is shear responsive, and is activated by NOTCH over-expression [31,32]. Furthermore, endothelial cell-specific knockout of GJA5 results in increased levels of atherosclerosis in mice [33]. It is intriguing to consider that the valve endothelium may act in a similar manner to the perichondral cell layer of the growth plate, secreting cytokines to control the proliferation and differentiation of a neighboring cell type. While disruption of the NOTCH ligand, JAGGED1, in the aortic valve endothelium has already been shown to cause calcific disease [19], our ongoing mouse studies will interrogate the impact of altering other endothelial NOTCH1 signaling components on aortic valve calcification.
Figure 6.
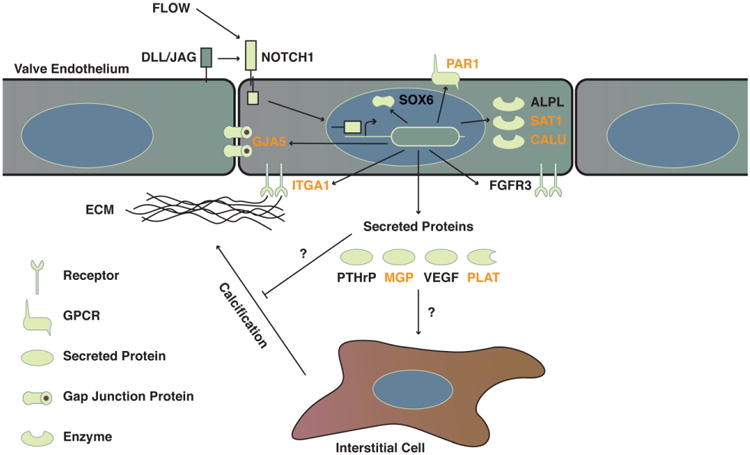
Model of NOTCH1 regulation of human endothelial cell calcification. Yellow shapes indicate different classes of molecules and their sub-cellular localization. Genes in black are regulated by shear stress and NOTCH1 signaling, while those in orange also have a significant NOTCH1 ChIP-seq peak within 20 kb of the transcriptional start site, suggesting potential direct transcriptional regulation by NOTCH1.
In addition to the endothelial contribution, it is thought that interstitial cells in calcifying valves become activated myofibroblasts and then trans-differentiate into RUNX2 positive osteoblast-like cells [3,13]. Bone Morphogenic Protein (BMP) signaling via BMP2 through the SMAD 1/5/8 pathway is thought to activate ectopic gene expression of the bone master regulator, RUNX2 in human valve interstitial cells [34]. NOTCH signaling also converges on Runx2 activity through the previously described Runx2 interacting partner, Hey 1/2 [5]. Future in vivo studies will address the interplay of NOTCH1 and BMP signaling, including how altered endothelial NOTCH signaling impacts valve calcification via the BMP pathway. Intriguingly, recent evidence suggests that enhanced BMP signaling can induce the endothelium to transdifferentiate into calcifying cells involved in vascular calcification [17,35]. It remains to be seen if a similar mechanism holds true in human aortic valve calcification and if NOTCH1 and/or BMP signaling play a role in the transdifferentiation of endothelial cells into osteoblast-like cells in the aortic valve.
It has also been reported that the endothelium can affect the osteogenesis of vascular smooth muscle cells undergoing calcification [8,36] and can signal to valve interstitial cells to maintain a quiescent phenotype characterized by lower expression levels of smooth muscle actin [37]. Our finding that NOTCH1 directly regulates MGP may provide further evidence for a non-cell autonomous function of NOTCH1 in repressing the osteogenic fate through modulation of BMP signaling. Ongoing in vivo studies will address the role of endothelial expressed MGP in vascular calcification. Mouse knockout models suggest MGP is the most potent inhibitor of calcification in the arterial system and reestablishment of smooth muscle MGP expression rescues the arterial calcification phenotype of MGP−/− mice [20,38]. Rescuing endothelial expression of MGP in the MGP knockout model may provide insight into the cell autonomous and non-autonomous roles of endothelial MGP.
It is worth noting that our previously reported NOTCH1+/− patients had no signs of vascular calcification. While we have shown that altering NOTCH1 impacts the level of endothelial MGP protein expression in endothelial cells, we have also observed that altering NOTCH signaling does not affect MGP expression in vascular smooth muscle cells (Figure 4D, E and data not shown). It is possible that due to this discrepancy, the smooth muscle compartment of the arterial system may be able to compensate for the loss of NOTCH1. In addition, other NOTCH receptors expressed in the vascular endothelium may compensate for decreased NOTCH1 signaling. This highlights the importance of future co-culture experiments to be conducted with multiple cell types from the valve and vasculature, as well as animal models to test the significance of these initial findings, in vivo. These limitations of our current study are being addressed in ongoing work using mouse models to evaluate the role of endothelial expressed MGP in vascular calcification.
We identified an enhancer that is dependent on a cluster of CSL-binding sites for MGP expression in the endothelium and showed that shear stress normally upregulates MGP. MGP must be activated by a Vitamin K dependent carboxylase [39]. Interestingly, use of the widely prescribed blood thinner, Warfarin, which inhibits Vitamin K-dependent enzymes, was recently associated with an increased incidence of aortic valve calcification [40]. Additionally, a large epidemiological trial showed an inverse relationship between high levels of dietary Vitamin K2 and all cause mortality, coronary heart disease, and atherosclerosis [41]. Although the western diet is deficient in Vitamin K2, there are no interventional studies addressing whether Vitamin K2 could prevent onset or progression of calcification diseases [42]. However, a recent study showed that an increase in the decarboxylated, inactive, form of MGP was associated with an increase in vascular calcification [43]. Based on the results of this study placed in the context of current knowledge, future animal studies investigating this potential relationship may be warranted.
Supplementary Material
Highlights.
Shear stress affects NOTCH signaling in human aortic valve endothelial cells (HAVECs).
RNA-seq on primary HAVECs in static, flow, and NOTCH1-depleted states reveals dysregulated networks.
Potential direct NOTCH1 targets were revealed by integrating RNA- and ChIP-seq data.
MGP, a secreted BMP regulator, is directly controlled by NOTCH in HAVECs.
Activated by shear stress, NOTCH1 inhibits osteoblast-like gene networks in HAVECs.
Acknowledgments
We are grateful to the Srivastava lab and Gladstone community for useful discussions. We thank A. Holloway, A. Williams, and S. Thomas from the Gladstone Bioinformatics core, S. Elmes from UCSF Laboratory for Cell Analysis, and the Gladstone Genomics and Stem Cell Cores for technical expertise. Finally, we are grateful for editorial help from G. Howard and manuscript preparation by B. Taylor. D.S. was supported by grants from NHLBI/NIH (U01 HL100405, U01 HL098179), L.K. Whittier Foundation, William Younger Family Foundation, Eugene Roddenberry Foundation, the California Institute for Regenerative Medicine (CIRM), and by NIH/NCRR grant (C06RR018928) to the Gladstone Institutes. X.M. was supported by grants from NHLBI/NIH and NIA/NIH grants (R01 HL106582, R01 AG039545). K.N.I, was supported by an American Heart Association (AHA) Scientist Development Grant. M.P.W. was supported by a Predoctoral fellowship from the AHA and a Winslow Postdoctoral Fellowship. C.T. was supported by an AHA Predoctoral Fellowship, UCSF Medical Scientist Training Program (NIH T32GM007618), and the UCSF Developmental and Stem Cell Biology Graduate Program (NIH T32HD007470).
Abbreviations
- CAVD
calcific aortic valve disease
- ALP
alkaline phosphatase
- IBSP
bone sialoprotein
- SPARC
osteonectin
- TGFβ
transforming growth factor beta
- WNT
wingless-type MMTV integration site family
- CSL
suppressor of hairless
- MAML
mastermind-like protein 1
- HEY1
hairy/enhancer-of-split related with YRPW motif protein 1
- HEY2
hairy/enhancer-of-split related with YRPW motif protein 2
- RNA-seq
RNA sequencing
- ChIP-seq
chromatin immuno precipitation sequencing
- NICD
NOTCH1 intracellular domain
- MGP
matrix gla protein
- BMP
bone morphogenic protein
- HAECs
human aortic endothelial cells
- HAVECs
human aortic valve endothelial cells
- ECM
endothelial cell media
- EMSA
electrophoretic mobility shift assays
- PBS
phosphate buffered saline
- cMGP
λ-carboxylated matrix gla protein
- ucMGP
uncarboxylated matrix gla protein
- DAPI
4′,6-diamidino-2-phenylindole
- SMAD
similar to mothers against decapentaplegic
- Wt
wildtype
- Mut
mutant
- KLF2
krüppel-like Factor 2
- PLAT
tissue plasminogen activator
- PTHrP
parathyroid hormone-related protein
- GJA5
gap junction alpha-5 protein or connexin 40
- SAT1
spermine N1-acetytransferase
- BMP2
bone morphogenetic protein 2
- HUVEC
human umbilical vein endothelial cells
- GERP
genomic evolutionary rate profiling
- LacZ
bacterial beta-galactosidase gene
- FGF18
fibroblast growth factor 18
- FGFR1
fibroblast growth factor receptor 1
- FGFR2
fibroblast growth factor receptor 2
- OGN
osteoglycin
- PTN
pleiotrophin
- BMP6
bone morphogenic protein 6
- BMP2
bone morphogenic protein 2
- BMPER
BMP binding endothelial regulator
Footnotes
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–89. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 2.La Pompa de JL. Notch signaling in cardiac development and disease. Pediatr Cardiol. 2009;30:643–50. doi: 10.1007/s00246-008-9368-z. [DOI] [PubMed] [Google Scholar]
- 3.Akat K, Borggrefe M, Kaden JJ. Aortic valve calcification: basic science to clinical practice. Heart. 2008;95:616–23. doi: 10.1136/hrt.2007.134783. [DOI] [PubMed] [Google Scholar]
- 4.Ge G. Bone Morphogenetic Protein-1/Tolloid-related Metalloproteinases Process Osteoglycin and Enhance Its Ability to Regulate Collagen Fibrillogenesis. Journal of Biological Chemistry. 2004;279:41626–33. doi: 10.1074/jbc.M406630200. [DOI] [PubMed] [Google Scholar]
- 5.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–4. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 6.Simmons CA. Spatial Heterogeneity of Endothelial Phenotypes Correlates With Side-Specific Vulnerability to Calcification in Normal Porcine Aortic Valves. Circ Res. 2005;96:792–9. doi: 10.1161/01.RES.0000161998.92009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zambon AC, Gaj S, Ho I, Hanspers K, Vranizan K, Evelo CT, et al. GO-Elite: a flexible solution for pathway and ontology over-representation. Bioinformatics. 2012;28:2209–10. doi: 10.1093/bioinformatics/bts366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–20. doi: 10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salomonis N, Hanspers K, Zambon AC, Vranizan K, Lawlor SC, Dahlquist KD, et al. GenMAPP 2: new features and resources for pathway analysis. BMC Bioinformatics. 2007;8:217. doi: 10.1186/1471-2105-8-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberts WC. The congenitally bicuspid aortic valve. A study of 85 autopsy cases. The American Journal of Cardiology. 1970;26:72–83. doi: 10.1016/0002-9149(70)90761-7. [DOI] [PubMed] [Google Scholar]
- 11.Otto CM, Lind BK, Kitzman DW, Gersh BJ, Siscovick DS. Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. New England Journal of Medicine. 1999;341:142–7. doi: 10.1056/NEJM199907153410302. [DOI] [PubMed] [Google Scholar]
- 12.Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, et al. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. Journal of the American College of Cardiology. 1997;29:630–4. doi: 10.1016/s0735-1097(96)00563-3. [DOI] [PubMed] [Google Scholar]
- 13.Bossé Y, Mathieu P, Pibarot P. Genomics: the next step to elucidate the etiology of calcific aortic valve stenosis. Journal of the American College of Cardiology. 2008;51:1327–36. doi: 10.1016/j.jacc.2007.12.031. [DOI] [PubMed] [Google Scholar]
- 14.Butcher JT, Nerem RM. Valvular endothelial cells and the mechanoregulation of valvular pathology. Philosophical Transactions of the Royal Society of London Series B, Biological Sciences. 2007;362:1445–57. doi: 10.1098/rstb.2007.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weinberg EJ, Mack PJ, Schoen FJ, García-Cardeña G, Kaazempur Mofrad MR. Hemodynamic Environments from Opposing Sides of Human Aortic Valve Leaflets Evoke Distinct Endothelial Phenotypes In Vitro. Cardiovascular Engineering. 2010;10:5–11. doi: 10.1007/s10558-009-9089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schoen FJ. Evolving Concepts of Cardiac Valve Dynamics: The Continuum of Development, Functional Structure, Pathobiology, and Tissue Engineering. Circulation. 2008;118:1864–80. doi: 10.1161/CIRCULATIONAHA.108.805911. [DOI] [PubMed] [Google Scholar]
- 17.Yao Y, Jumabay M, Ly A, Radparvar M, Cubberly MR, Bostrom KI. A Role for the Endothelium in Vascular Calcification. Circ Res. 2013;113:495–504. doi: 10.1161/CIRCRESAHA.113.301792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nus M, Macgrogan D, Martínez-Poveda B, Benito Y, Casanova JC, Fernández-Avilés F, et al. Diet-Induced Aortic Valve Disease in Mice Haploinsufficient for the Notch Pathway Effector RBPJK/CSL. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011 doi: 10.1161/ATVBAHA.111.227561. [DOI] [PubMed] [Google Scholar]
- 19.Hofmann JJ, Briot A, Enciso J, Zovein AC, Ren S, Zhang ZW, et al. Endothelial deletion of murine Jag1 leads to valve calcification and congenital heart defects associated with Alagille syndrome. Development (Cambridge, England) 2012 doi: 10.1242/dev.084871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78–81. doi: 10.1038/386078a0. [DOI] [PubMed] [Google Scholar]
- 21.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011;27:2325–9. doi: 10.1093/bioinformatics/btr355. [DOI] [PubMed] [Google Scholar]
- 23.Neph S, Kuehn MS, Reynolds AP, Haugen E, Thurman RE, Johnson AK, et al. BEDOPS: high-performance genomic feature operations. Bioinformatics. 2012;28:1919–20. doi: 10.1093/bioinformatics/bts277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nature Biotechnology. 2008;26:1351–9. doi: 10.1038/nbt.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, et al. The Ensembl genome database project. Nucleic Acids Research. 2002;30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dodou E, Xu SM, Black BL. mef2c is activated directly by myogenic basic helix-loop-helix proteins during skeletal muscle development in vivo. Mechanisms of Development. 2003;120:1021–32. doi: 10.1016/s0925-4773(03)00178-3. [DOI] [PubMed] [Google Scholar]
- 27.Holliday CJ, Ankeny RF, Jo H, Nerem RM. Discovery of shear- and side-specific mRNAs and miRNAs in human aortic valvular endothelial cells. AJP: Heart and Circulatory Physiology. 2011;301:H856–67. doi: 10.1152/ajpheart.00117.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ankeny RF, Thourani VH, Weiss D, Vega JD, Taylor WR, Nerem RM, et al. Preferential Activation of SMAD1/5/8 on the Fibrosa Endothelium in Calcified Human Aortic Valves - Association with Low BMP Antagonists and SMAD6. PLoS ONE. 2011;6:e20969. doi: 10.1371/journal.pone.0020969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–9. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brütsch R, Liebler SS, Wüstehube J, Bartol A, Herberich SE, Adam MG, et al. Integrin cytoplasmic domain-associated protein-1 attenuates sprouting angiogenesis. Circ Res. 2010;107:592–601. doi: 10.1161/CIRCRESAHA.110.217257. [DOI] [PubMed] [Google Scholar]
- 32.Buschmann I, Pries A, Styp-Rekowska B, Hillmeister P, Loufrani L, Henrion D, et al. Pulsatile shear and Gja5 modulate arterial identity and remodeling events during flow-driven arteriogenesis. Development (Cambridge, England) 2010;137:2187–96. doi: 10.1242/dev.045351. [DOI] [PubMed] [Google Scholar]
- 33.Chadjichristos CE, Scheckenbach KEL, van Veen TAB, Richani Sarieddine MZ, de Wit C, Yang Z, et al. Endothelial-Specific Deletion of Connexin40 Promotes Atherosclerosis by Increasing CD73-Dependent Leukocyte Adhesion. Circulation. 2010;121:123–31. doi: 10.1161/CIRCULATIONAHA.109.867176. [DOI] [PubMed] [Google Scholar]
- 34.Yang X, Meng X, Su X, Mauchley DC, Ao L, Cleveland JC, et al. Bone morphogenic protein 2 induces Runx2 and osteopontin expression in human aortic valve interstitial cells: role of Smad1 and extracellular signal-regulated kinase 1/2. The Journal of Thoracic and Cardiovascular Surgery. 2009;138:1008–15. doi: 10.1016/j.jtcvs.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 35.Tang Z, Wang A, Yuan F, Yan Z, Liu B, Chu JS, et al. Differentiation of multipotent vascular stem cells contributes to vascular diseases. Nature Communications. 2012;3:1–13. doi: 10.1038/ncomms1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shin V, Zebboudj AF, Boström K. Endothelial Cells Modulate Osteogenesis in Calcifying Vascular Cells. Journal of Vascular Research. 2004;41:193–201. doi: 10.1159/000077394. [DOI] [PubMed] [Google Scholar]
- 37.Butcher JT, Nerem RM. Valvular endothelial cells regulate the phenotype of interstitial cells in co-culture: effects of steady shear stress. Tissue Engineering. 2006;12:905–15. doi: 10.1089/ten.2006.12.905. [DOI] [PubMed] [Google Scholar]
- 38.Murshed M, Schinke T, McKee MD, Karsenty G. Extracellular matrix mineralization is regulated locally; different roles of two gla-containing proteins. The Journal of Cell Biology. 2004;165:625–30. doi: 10.1083/jcb.200402046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Danziger J. Vitamin K-dependent proteins, warfarin, and vascular calcification. Clinical Journal of the American Society of Nephrology: CJASN. 2008;3:1504–10. doi: 10.2215/CJN.00770208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holden RM, Sanfilippo AS, Hopman WM, Zimmerman D, Garland JS, Morton AR. Warfarin and aortic valve calcification in hemodialysis patients. Journal of Nephrology. 2007;20:417–22. [PubMed] [Google Scholar]
- 41.Geleijnse JM, Vermeer C, Grobbee DE, Schurgers LJ, Knapen MHJ, van der Meer IM, et al. Dietary intake of menaquinone is associated with a reduced risk of coronary heart disease: the Rotterdam Study. The Journal of Nutrition. 2004;134:3100–5. doi: 10.1093/jn/134.11.3100. [DOI] [PubMed] [Google Scholar]
- 42.Shea MK, Holden RM. Vitamin K Status and Vascular Calcification: Evidence from Observational and Clinical Studies. Advances in Nutrition: an International Review Journal. 2012;3:158–65. doi: 10.3945/an.111.001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delanaye P, Krzesinski JM, Warling X, Moonen M, Smelten N, Médart L, et al. Dephosphorylated-uncarboxylated Matrix Gla protein concentration is predictive of vitamin K status and is correlated with vascular calcification in a cohort of hemodialysis patients. BMC Nephrol. 2014;15:145. doi: 10.1186/1471-2369-15-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.