

Abstract
The dopamine D3 receptor (D3R) is a target of pharmacotherapeutic interest in a variety of neurological disorders including schizophrenia, Parkinson's disease, restless leg syndrome, and drug addiction. A common molecular template used in the development of D3R-selective antagonists and partial agonists incorporates a butylamide linker between two pharmacophores, a phenylpiperazine moiety and an extended aryl ring system. The series of compounds described herein incorporates a change to that chemical template, replacing the amide functional group in the linker chain with a 1,2,3-triazole group. Although the amide linker in the 4-phenylpiperazine class of D3R ligands has been previously deemed critical for high D3R affinity and selectivity, the 1,2,3-triazole moiety serves as a suitable bioisosteric replacement and maintains desired D3R-binding functionality of the compounds. Additionally, using mouse liver microsomes to evaluate CYP450-mediated phase I metabolism, we determined that novel 1,2,3-triazole-containing compounds modestly improves metabolic stability compared to amide-containing analogues. The 1,2,3-triazole moiety allows for the modular attachment of chemical subunit libraries using copper-catalyzed azide-alkyne cycloaddition click chemistry, increasing the range of chemical entities that can be designed, synthesized, and developed toward D3R-selective therapeutic agents.
Keywords: Dopamine D3 receptor; Click chemistry; 1,2,3-Triazole; Bioisoteric replacement; Metabolic stability; Structure–activity relationships
1. Introduction
The neurotransmitter dopamine signals through a family of five (D1–D5) G protein-coupled receptors (GPCRs), subdivided into D1- like (D1R and D5R) and D2-like (D2R, D3R and D4R) sub-families on the basis of pharmacological and sequence similarity.1 D3R-selective ligands may be used to treat a variety of neuropsychiatric disorders associated with aberrant dopamine signaling, including schizophrenia,2 Parkinson's disease and associated dyskinesias,3,4 and drug addiction.5–7 Since it was cloned and characterized in 1990,8 the D3R, in particular, has been a target of pharmacotherapeutic interest in the treatment of drug addiction due to its relatively localized expression within mesolimbic neurocircuitry, including the nucleus accumbens, islands of Calleja and ventral striatum.9–11 Because these regions encode rewarding and motivational aspects of addictive drugs, pharmacological modification of D3R signaling is an appealing treatment option for psychostimulant addiction, for which there is currently no FDA-approved medication.
Many research groups have developed D3R-selective antagonists and partial agonists.12–14 One common template used to develop D3R-selective ligands incorporates two pharmacophores, a substituted 4-phenylpiperazine and an extended aryl ring system, connected via a butylamide linker chain.7 Previous structure–activity relationship (SAR) studies have determined that the butyl linker is an optimal length15,16 and that the amide function is necessary17 for maximal D3R over D2R selectivity (for review, see references5,7). The amide, however, is a potential site for metabolism: amides can be hydrolyzed by systemic esterases and hepatic amidases.18,19 As such, development of bioisosteric substitutes for the amide bond may be beneficial to the translational potential of substituted 4-phenylpiperazines. 1,2,3-Triazoles have been successfully used as bioisosteres for amides in HIV-1 and mammalian protease inhibitors20,21 and acetylcholinesterase inhibitors.22 Indeed, triazole ureas have been designed to inhibit hydrolases.23 It should be noted that others have recently investigated the 1,2,3-triazole template for novel D3R ligands,24,25 and the 1,2,4-triazole motif has also provided a rich set of D3R-selective antagonists,26,27 exemplified by the clinically investigated GSK598,809.9,28
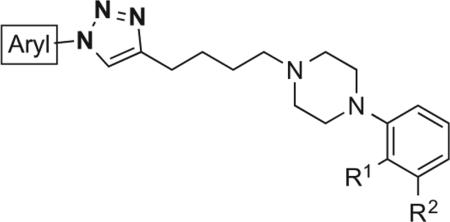
In this study, we evaluated the 1,2,3-triazole as a bioisosteric substitute for the amide functional group in a series of substituted 4-phenylpiperazines targeting the D3R, which were synthesized using a copper-catalyzed azide-alkyne cycloaddition (CuAAC).29–31 Herein, we report the design and synthesis of novel 1,2,3-triazole-containing compounds with high D3R affinity and subtype selectivity. This library was comprised of two series of compounds: those of template A and those of template B (Fig. 1). Analogues designed around template A contained an N1-aryl substituent and a C4-butyl linker. Inversely, those designed around template B possessed an N1-butyl linker and a C4-aryl substituent. Radioligand competition binding assays were performed to evaluate compound affinity for D2-like receptors. Furthermore, CYP450-mediated phase I metabolic stability in mouse microsomes was evaluated for selected amide and triazole analogues.
Figure 1.

Bioisosteric replacement of the amide with the 1,2,3-triazole in D3R-selective ligands, providing analogues of templates A and B.
2. Chemistry
Triazoles 4–5 (Scheme 1) were prepared starting from tosylate 1,32 which was displaced using commercially available 1-(2-methoxyphenyl)piperazine and 1-(2,3-dichlorophenyl)piperazine to give acetylene-containing piperazines 2 and 3, respectively. Using a CuAAC reaction,33 these acetylenes 2 and 3 were coupled to azides, formed in situ from the appropriate commercially available amine (or in a preceding step for compound 4d), which provided the desired template A triazoles 4–5.
Scheme 1.

Reagents and conditions: (a) K2CO3, NaI, appropriate phenylpiperazine, acetone, reflux, 12 h; (b) (i) t-BuONO, trimethylsilyl azide, appropriate amine, CH3CN, rt, 2 h; (ii) CuI, DIPEA, rt, 12 h; (c) appropriate amine, NaNO2, concd H2SO4, H2O, NaN3, 10 min. rt; (d) 5-azidoquinoline, CuI, DIPEA, appropriate azide, THF, rt, 12 h.
A library of template B triazoles was prepared in a similar manner, as shown in Scheme 2. Tosylate 634 was treated with the appropriate phenylpiperazine to provide the corresponding N-alkyl piperazines 7–8. CuAAC with aryl acetylenes using either a Cu(I)-catalyzed method or a Cu(II) and sodium ascorbate-catalyzed method resulted in triazoles 9–10.
Scheme 2.

Reagents and conditions: (a) K2CO3, NaI, appropriate phenylpiperazine, acetone, reflux, 12 h; (b) CuI, DIPEA, appropriate acetylene, THF, rt, 12 h; (c) sodium ascorbate, CuSO4·5H2O, appropriate acetylene, THF:H2O (1:1), 50 °C, 12 h.
Racemic 3-OH butyl-linked derivatives 12–13 (Scheme 3) were prepared from a one-pot epoxide-opening click chemistry reaction. 2-(2-Azidoethyl)oxirane (11)35 was treated with the appropriate phenylpiperazine hydrochloride and the prepared aryl acetylene in the presence of CuSO4·5H2O, sodium ascorbate, K2CO3, and β-cyclodextrin to facilely provide compounds 12–13.36,37
Scheme 3.

Reagents and Conditions: (a) Sodium ascorbate, CuSO4·5H2O, K2CO3, β-cyclodextrin, appropriate acetylene, appropriate phenylpiperazine hydrochloride, H2O, rt for 30 min then 80 °C for 2 h.
Racemic indoles 16 and 17a (Scheme 4), possessing the 3-OH butyl linker, were prepared, starting with the opening of epoxide 11 with the appropriate phenylpiperazine in refluxing acetonitrile to give 14 and 15. Next, CuAAC with 5-ethynyl-1H-indole38 gave compounds 16 and 17a. The structure of 17a was confirmed by X-ray crystallography (Fig. 2).
Scheme 4.

Reagents and conditions: (a) K2CO3, NaI, appropriate phenylpiperazine, acetonitrile, reflux, 16 h; (b) sodium ascorbate, CuSO4·5H2O, ethynyl-1H-indole, THF:H2O (3:1), rt, 16 h; (c) sodium ascorbate, CuSO4·5H2O, 3-ethynylquinoline, TBTA THF:H2O (3:1), rt, 16 h.
Figure 2.
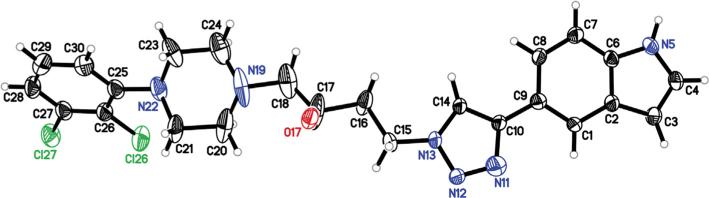
Structure of 2,3-diClPh-containing indolyltriazole 17a as determined by X-ray crystallography. Displacement ellipsoids are at the 50% level.
Lastly, the synthesis of racemic quinoline 17b (Scheme 4) was accomplished using a tris(benzyltriazolylmethyl)amine (TBTA)-catalyzed CuAAC method.39,40 The role of the polytriazole, TBTA, is to stabilize the copper intermediates formed throughout the catalytic cycle.41 Therefore, while the coupling of azide 15 with 3-ethynylquinoline in the absence of TBTA did not provide the triazole 17b, the addition of the polytriazole to the same reaction conditions cleanly yielded the desired 17b.42
3. Binding results and SAR discussion
To evaluate the D3R binding affinities and receptor subtype selectivity of this series of compounds, we used [3H]N-methylspiperone as a competitive radioligand for binding site competition in membranes prepared from HEK293 cells expressing either the human D2R, D3R or D4R. The results of these in vitro tests are listed in Tables 1 and 2 for template A and template B compounds, respectively.
Table 1.
In vitro dopamine receptor binding data for template A 1,2,3-triazoles
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | No. | Aryl | R1 | R2 | c Log P | D2R Ki ± SEM† (nM) | D3R Ki ± SEM† (nM) | D4R Ki ± SEM† (nM) | Receptor selectivity | |
| D2/D3 | D4/D3 | |||||||||
| BAK 04-70 | 4a |
|
OMe | H | 5.6 | 31.5 ± 2.06 | 2.93 ± 0.221 | 299 ± 28.6 | 11 | 102 |
| BAK 04-29 | 4b |
|
OMe | H | 6.3 | 57.2 ± 7.29 | 1.59 ± 0.439 | 329 ± 110 | 36 | 207 |
| BAK 04-71 | 4c |
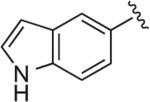
|
OMe | H | 4.6 | 51.8 ± 7.59 | 3.10 ± 0.802 | 353 ± 64.8 | 17 | 114 |
| BAK 04-74 | 4d |
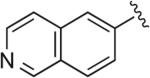
|
OMe | H | 4.8 | 83.4 ± 0.990 | 13.1 ± 2.72 | >1 μM (n = 1) | 6.4 | – |
| BAK 04-27 | 4e |
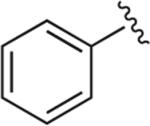
|
OMe | H | 4.4 | 53.6 ± 3.89 | 5.42 ± 0.447 | 486 ± 114 | 10 | 90 |
| BAK 04-89 | 5a |
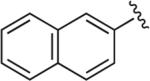
|
Cl | Cl | 7.1 | 80.8 ± 15.1 | 3.80 ± 0.760 | >2 μM (n = 1) | 21 | – |
| BAK 04-90 | 5b |
|
Cl | Cl | 7.8 | 424 ± 118 | 5.05 ± 0.141 | 4450 ± 1740 | 84 | 881 |
| BAK 04-92 | 5c |
|
Cl | Cl | 6.1 | 46.6 ± 16.1 | 1.00 ± 0.0960 | 510 ± 110 | 47 | 510 |
Ki values were determined via radioligand competition binding experiments using [3H]N-methylspiperone (0.4 nM) and membranes from HEK 293 cells stably expressing human D2R, D3R or D4R. Unless otherwise noted, data are presented as mean ± SEM and derived from 3 or more independent experiments.
Table 2.
In vitro dopamine receptor binding data for template B 1,2,3-triazoles
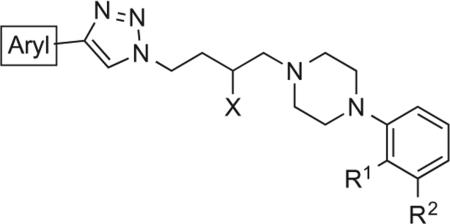
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | No. | Ar | R1 | R2 | X | c Log P | D2R Ki ± SEM† (nM) | D3R Ki ± SEM† (nM) | D4R Ki ± SEM† (nM) | Receptor selectivity | |
| D2/D3 | D4/D3 | ||||||||||
| BAK 05-21 | 9a |
|
OMe | H | H | 5.2 | 23.3 ± 1.38 | 11.1 ± 3.71 | 232 ± 92.3 | 2.1 | 21 |
| BAK 05-19 | 9b |
|
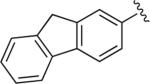
OMe | H | H | 5.9 | 50.1 ± 3.82 | 5.55 ± 1.38 | 588 ± 147 | 9.0 | 106 |
| BAK 05-16 | 9c |
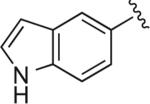
|
OMe | H | H | 4.0 | 43.7 ± 4.96 | 5.48 ± 0.430 | 222 ± 43.4 | 8.0 | 41 |
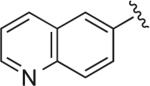
| BAK 05-27 | 9d |
|
OMe | H | H | 3.9 | 42.5 ± 5.45 | 6.73 ± 0.445 | 269 ± 66.6 | 6.3 | 40 |
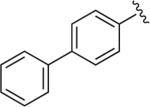
| BAK 05-18 | 9e |
|
OMe | H | H | 5.9 | 69.0 ± 14.0 | 5.88 ± 1.02 | 425 ± 87.8 | 12 | 72 |
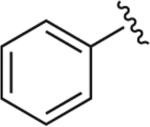
| BAK 04-81 | 9f |
|
OMe | H | H | 4.0 | 137 ± 6.86 | 22.9 ± 5.38 | 480 ± 58.8 | 6.0 | 21 |
| BAK 05-26 | 10a |
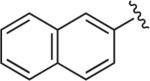
|
Cl | Cl | H | 6.7 | 103 ± 14.0 | 7.7 ± 1.87 | >1 μM (n = 1) | 13 | – |
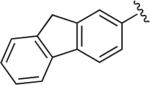
| BAK 05-24 | 10b |
|
Cl | Cl | H | 7.4 | 885 ± 298 | 14.0 ± 4.96 | >20 μM (n = 1) | 63 | – |
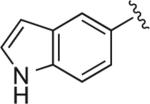
| BAK 05-25 | 10c |
|
Cl | Cl | H | 6.1 | 56.1 ± 7.20 | 1.47 ± 0.462 | >2 μM (n = 1) | 38 | – |
| BAK 05-28 | 10d |
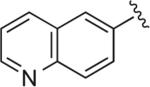
|
Cl | Cl | H | 5.4 | 53.9 ± 11.8 | 1.35 ± 0.184 | >1 μM (n = 1) | 40 | – |
| BAK 05-23 | 10e |
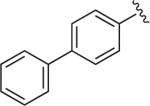
|
Cl | Cl | H | 7.4 | 2590 ± 920 | 8.32 ± 1.20 | >10 μM (n = 1) | 311 | – |
| BAK 05-52 | 12a |
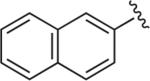
|
OMe | H | OH§ | 4.4 | 189 ± 23.6 | 40.7 ± 5.34 | >2 μM (n = 1) | 4.6 | – |
| BAK 05-47 | 12b |
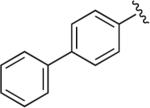
|
OMe | H | OH§ | 5.2 | 819 ± 87.1 | 30.5 ± 6.99 | >2 μM (n = 1) | 27 | – |
| BAK 05-51 | 12c |
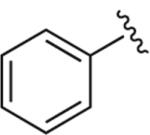
|
OMe | H | OH§ | 3.3 | 1210 ± 103 | 173 ± 9.60 | >3 μM (n = 1) | 7.0 | – |
| BAK 05-44 | 12d |
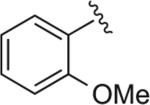
|
OMe | H | OH§ | 2.7 | 1020 ± 66.9 | 493 ± 24.1 | >3 μM (n = 1) | 2.1 | – |
| BAK 05-43 | 12e |
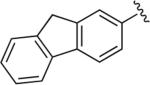
|
OMe | H | OH§ | 5.2 | 607 ± 54.9 | 18.5 ± 0.248 | >1 μM (n = 1) | 33 | – |
| BAK 05-56 | 12f |
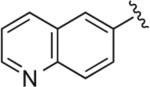
|
OMe | H | OH§ | 3.2 | 424 ± 25.9 | 68.2 ± 6.72 | >3 μM (n = 1) | 6.2 | – |
| BAK 05-53 | 13a |
|
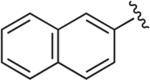
Cl | Cl | OH§ | 6.0 | 18,800 ± 8960 | 25.2 ± 2.17 | >20 μM (n = 1) | 745 | – |
| BAK 05-55 | 13b |
|
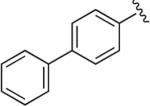
Cl | Cl | OH§ | 6.8 | 15,100 ± 2770 | 55.8 ± 12.9 | >7 μM (n = 1) | 270 | – |
| RDS 02-33 | 16 |
|
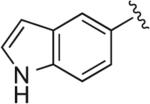
OMe | H | OH§ | 3.3 | 789 ± 79.4 | 33.6 ± 7.27 | NT | 23 | – |
| RDS 02-28 | 17a |
|
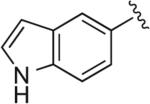
Cl | Cl | OH§ | 4.9 | 963 ± 168 | 5.85 ± 0.780 | NT | 165 | – |
| RDS 02-32 | 17b |
|
Cl | Cl | OH§ | 4.9 | 244 ± 22.8 | 18.0 ± 5.74 | NT | 14 | – |
| PG64844§ |
|
4.9 | 746 ± 123 | 1.88 ± 0.112 | 2600 ± 660 | 397 | 1380 | ||||
| OMO5-057§ |
|
4.9 | 321 ± 9.34 | 2.91 ± 0.499 | NT | 110 | – | ||||
| NGB290415,43 |
|
6.7 | 54.7 ± 5.20 | 0.233 ± 0.00893 | 3670 ± 626 | 235 | 15,800 | ||||
Compounds are racemic.
Ki values were determined via radioligand competition binding experiments using [3H]N-methylspiperone (0.4 nM) and membranes from HEK 293 cells stably expressing human D2R, D3R or D4R. Unless otherwise noted, data are presented as mean ± SEM and derived from 3 or more independent experiments.
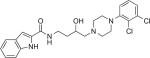
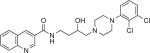
Binding studies showed that several of these butyltriazole-linked analogues had affinities in the low nanomolar range for D3R (Tables 1 and 2). These compare favorably with previous compounds we have synthesized and evaluated from the substituted 4-phenylpiperazine class using the butylamide linker, including NGB2904,15,43 PG648,44 and OMO5-057 [Manuscript, in preparation], presented in Table 2 for comparison. These results indicate that the 1,2,3-triazole substitution for the amide group is generally tolerated.
In general, template A triazoles were well-tolerated at D3R, with Ki values in the range of 1–13 nM and D3R over D2R selectivity between 10- and 84-fold; 5b had the best binding profile in this series, comparable to its amide analogue, NGB2904 (Table 2). Template B triazoles that were unsubstituted in the butyl linking chain showed a similar range of affinities, with 10c and 10d having the highest D3R affinities. However, it must be noted that the analogues with highest D3R selectivity (e.g., 5d, 10e, 13a, and 13b) were highly lipophilic with cLogP values of 6 or greater. As seen previously with the amide series,44,45 when a 3-OH group was added, both D3R and D2R affinities tended to decrease compared to the unsubstituted analogues. However, 17a retained high D3R affinity (Ki = 5.85 nM) and was 165-fold selective for D3R over D2R. Further, its cLogP value of 4.9 was equivalent to its amide analogue, PG648. The quinoline homologue (17b) of indole 17a, showed reduced D3R affinity and selectivity. This is consistent with observations we have made in a large series of quinoline amide analogues [Manuscript, in preparation] in which OMO5-57 had the most promising affinity and subtype selectivity profiles: Ki = 2.91 at D3R and >100-fold D3R versus D2R selectivity (Table 2).
Orientation of the 1,2,3-triazole appeared to have a small impact on the affinity at D2R or D3R. For example, template A compounds 4b and 5d, when compared to analogous template B compounds 9b and 10b, respectively, had slightly improved D3R affinity and subtype selectivity; the pattern holds for other comparisons as well [e.g., 4a and 5a (template A) compared to 9a and 10a (template B), respectively]. This suggests that, for the purposes of designing high-affinity D3R ligands with selectivity over D2R, the N1-aryl triazoles may be slightly preferred. We have reported previously that the D3R affinities of the 4-phenylpiperazines appear to be largely dictated by the primary pharmacophore (e.g., 2,3-dichlorophenylpiperazine vs 2-methoxyphenylpiperazine), whereas selectivity over D2R depends on the terminal arylamide.46 This is due to a larger secondary binding pocket in the D3R that features a critical Gly94 residue.16 Hence, in the present series of compounds both the nature and orientation of the terminal aryl moiety affect D3R-selectivity by decreasing affinity at D2R; the orientation afforded in template A may be optimal. However, it should be noted that the 3-OH analogues in the template A series could not be prepared, thus direct comparison to highly D3R-selective template B analogues (e.g., 13a, 13b, and 17a) was not possible and may be a limitation of this template. Modification of the butyl linking chain with the 3-OH generally improves D3R selectivity and reduces lipophilicity.
4. Microsomal metabolism results
The triazole moiety has convenient properties for modular synthesis of novel compounds and may offer additional protection against drug metabolism in comparison to the amide bond. Five compounds were tested for phase I metabolism in mouse liver microsomal incubations in the presence of NADPH, two featuring the amide linker (PG648 and OMO5-57), and three with the 1,2,3-triazole linker (16, 17a, and 17b). Compound stability over one hour incubation is presented in Figure 3.
Figure 3.

Phase I metabolic stability. Data is presented as percent original compound remaining (mean ± SEM) at 30 and 60 min. following incubation with mouse liver microsomes in presence of NADPH. Note, the three most stable compounds tested are from the 1,2,3-triazole set.
The 1,2,3-triazole analogues demonstrated modestly improved microsomal stability over the amide analogues. This is particularly apparent when comparing OMO5-057 and 17b, homologous structures that feature a quinoline at the extended aryl position and differ only in the presence of an amide or 1,2,3-triazole linker, respectively: after 60 min incubation in the presence of micro-somes, 26% of 17b remained, contrasted with only 4% remaining of OMO5-057. Similarly, 17a was more stable than PG648 (35% vs 11% remaining after 60 min incubation, respectively); however the orientation of the indole differs between PG648 and 17a, prohibiting a direct comparison. Finally, there was little stability difference between homologous structures differing only in substitutions at the 4-phenylpiperazine: 17a, with a 2,3-dichlorophenylpiperazine, and 16, with 2-methoxyphenylpiperazine, were present at similar amounts after 30 min of incubation (45% vs 44%, respectively) and after 60 min (35% vs 27%, respectively). Liver microsomes measure primarily CYP450-mediated metabolism and thus cannot directly test the hypothesis that our triazole-containing molecules are more resistant to amidase metabolism. The present results, however, demonstrate that the triazole substitution provides some resistance to CYP450-mediated phase I metabolism.
5. Conclusions
In summary, a series of 1,2,3-triazole-containing substituted 4-phenylpiperazines were synthesized to explore the triazole as a bioisosteric substitution for the amide linker commonly found in this chemical class of D3R-selective ligands. Several 1,2,3-triazole-containing compounds had high (Ki = 1–10 nM) affinity for D3R and >80-fold selectivity for D3R over D2R; however, several of these are likely too lipophilic to be reasonable drug candidates. An analysis of compound stability in mouse liver microsomes indicates that replacement of the amide linker with a 1,2,3-triazole may provide some protection against hepatic metabolism. In the present series, 2,3-dichlorophenyl-containing indolyltriazole 17a had the best overall profile in terms of D3R affinity, selectivity, lipophilicity and metabolic stability. Thus, using this molecular template for future D3R drug design is warranted.
6. Experimental methods
6.1. Synthesis
Reaction conditions and yields were not optimized. Anhydrous solvents were purchased from Aldrich and were used without further purification except for tetrahydrofuran, which was freshly distilled from sodium-benzophenone ketyl. All other chemicals and reagents were purchased from Sigma–Aldrich Co. LLC, Combi-Blocks, TCI America, OChem Incorporation, Acros Organics, May-bridge, and Alfa Aesar. Spectroscopic data and yields refer to the free base. Flash column chromatography was performed using silica gel (EMD Chemicals, Inc.; 230–400 mesh, 60 Å) or a Teledyne ISCO CombiFlash® Rf instrument. 1H and 13C NMR spectra were acquired using a Varian Mercury Plus 400 spectrometer at 400 and 100 MHz, respectively. Chemical shifts are reported in parts-per-million (ppm) and referenced according to a deuterated solvent standard [for 1H spectra (CDCl3, 7.26 or DMSO-d6, 2.50) and 13C spectra (CDCl3, 77.2 or DMSO-d6, 39.5)]. Gas chromatography-mass spectrometry (GC/MS) data were acquired (where obtainable) using an Agilent Technologies (Santa Clara, CA) 6890N GC equipped with an HP-5MS column (cross-linked 5% PH ME siloxane, 30 m × 0.25 mm i.d. × 0.25 μm film thickness) and a 5973 mass-selective ion detector in electron-impact mode. Ultra-pure grade helium was used as the carrier gas at a flow rate of 1.2 mL/min. The injection port and transfer line temperatures were 250 and 280 °C, respectively, and the oven temperature gradient used was as follows: the initial temperature (100 °C) was held for 3 min and then increased to 295 °C at 15 °C/min over 13 min, and finally maintained at 295 °C for 10 min. Combustion analysis was performed by Atlantic Microlab, Inc. (Norcross, GA) and the results agree within ±0.4% of calculated values. Melting point determination was conducted using a Thomas-Hoover melting point apparatus and are uncorrected. On the basis of NMR and combustion data, all final compounds are ≥95% pure.
6.1.1. 1-(Hex-5-yn-1-yl)-4-(2-methoxyphenyl)piperazine (2)
A catalytic amount of NaI (5.0 mg) was added to a solution of tosylate 1 (4.2 g, 17 mmol),32 commercially available 1-(2-methoxyphenyl)piperazine (3.3 g, 17 mmol), and K2CO3 (4.7 g, 34 mmol) in acetone (50 mL) at room temperature. This reaction mixture was stirred at reflux for 12 h. The resulting mixture was cooled to room temperature, filtered, and the K2CO3 residue was washed with additional acetone. The filtrate was concentrated and the crude product was purified by flash column chromatography (50% EtOAc/Hexanes) to give acetylene 2 in 97% yield (4.5 g, 16.5 mmol). 1H NMR (400 MHz, CDCl3) δ 7.01–6.88 (m, 3H), 6.85 (dd, J = 8.0, 1.6 Hz, 1H), 3.89 (s, 3H), 3.09 (br s, 4H), 2.65 (br s, 4H), 2.42 (t, J = 7.2 Hz, 2H), 2.25–2.21 (m, 2H), 1.95 (t, J = 2.8 Hz, 1H), 1.67–1.55 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 152.43, 141.55, 123.0, 121.13, 118.34, 111.31, 84.52, 58.31, 55.49, 53.63, 50.82, 26.66, 26.13, 18.54; GC–MS (EI) m/z 272 (M+).
6.1.2. 1-(2,3-Dichlorophenyl)-4-(hex-5-yn-1-yl)piperazine (3)
A catalytic amount of NaI (5.0 mg) was added to a solution of tosylate 1 (3.4 g, 13.5 mmol),32 commercially available 1-(2,3-dichlorophenyl) piperazine (3.12 g, 13.5 mmol), and K2CO3 (3.7 g, 27 mmol) in acetone (50 mL) at room temperature. This reaction mixture was stirred at reflux for 12 h. The resulting mixture was cooled to room temperature, filtered, and the K2CO3 residue was washed with additional acetone. The filtrate was concentrated and the crude product was purified by flash column chromatography (50% EtOAc/Hexanes) to give acetylene 3 in 98% yield (4.2 g, 13.2 mmol). 1H NMR (400 MHz, CDCl3) δ 7.16–7.13 (m, 2H), 6.83 (dd, J = 6.4, 3.2 Hz, 1H), 3.07 (br s, 4H), 2.64 (br s, 4H), 2.44 (t, J = 7.2 Hz, 2H), 2.26–2.22 (m, 2H), 1.95 (t, J = 2.8 Hz, 1H), 1.68– 1.56 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 151.43, 134.08, 127.58, 124.58, 118.67, 84.42, 68.57, 58.10, 53.40, 51.44, 26.54, 26.03, 18.46; GC–MS (EI) m/z 310 (M+).
6.1.3. General Method A
The appropriate arylamine (1.0 equiv) was dissolved in CH3CN (0.25 M) and cooled to 0 °C. To the solution was added t-BuONO (1.5 equiv) followed by the dropwise addition of azidotrimethylsi-lane (1.2 equiv). The reaction was stirred at room temperature for 2 h before the addition of the appropriate acetylene substrate (0.5 equiv), CuI (0.2 equiv), and DIPEA (5 equiv). The resulting mixture was stirred at room temperature for 12 h before it was concentrated in vacuo. The crude compound was subjected directly to flash column chromatography to provide the desired template A analogues.
6.1.4. 1-(2-Methoxyphenyl)-4-(4-(1-(naphthalen-2-yl)-1H-1,2,3-triazol-4-yl)butyl)piperazine (BAK 04-70; 4a)
Synthesized using General Method A in 80% yield (350 mg, 0.80 mmol) from commercially available naphthalen-1-amine (286 mg, 2 mmol) and acetylene 2 (272 mg, 1 mmol). Mp 167– 168 °C (oxalate salt); 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 2.0 Hz, 1H), 8.01–7.98 (m, 1H), 7.93–7.88 (m, 4H), 7.60–7.53 (m, 2H), 7.02–6.89 (m, 3H), 6.86 (dd, J = 7.6, 1.2 Hz, 1H), 3.86 (s, 3H), 3.11 (br s, 4H), 2.89 (t, J = 7.2 Hz, 2H), 2.49 (t, J = 7.6 Hz, 2H), 1.87–1.80 (m, 2H), 1.72–1.65 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.4, 149.0, 141.5, 134.8, 133.4, 132.9, 130.1, 128.3, 128.1, 127.5, 126.9, 123.0, 121.1, 119.2, 119.1, 118.3, 118.3, 111.3, 58.6, 55.5, 53.7, 50.7, 27.5, 26.6, 25.8; Anal. (C27H37N5O·C2H2O4·1/2H2O) C, H, N.
6.1.5. 1-(4-(1-(9H-Fluoren-2-yl)-1H-1,2,3-triazol-4-yl)butyl)-4-(2-methoxyphenyl)piperazine (BAK 04-29; 4b)
Synthesized using General Method A in 86% yield (335 mg, 0.86 mmol) from commercially available 9H-fluoren-2-amine (362 mg, 2 mmol) and acetylene 2 (272 mg, 1 mmol). Mp 207–209 °C (oxalate salt); 1H NMR (400 MHz, CDCl3) δ 7.93 (s, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.82–7.80 (m, 2H), 7.70 (dd, J = 8.0, 2.4 Hz, 1H), 7.57 (d, J = 7.2 Hz, 1H), 7.43–7.33 (m, 2H), 7.01–6.88 (m, 3H), 6.85 (dd, J = 8.0, 1.2 Hz, 1H), 3.98 (s, 2H), 3.86 (s, 3H), 3.13 (br s, 4H), 2.89 (t, J = 7.2 Hz, 2H), 2.69 (br s, 4H), 2.50 (t, J = 7.6 Hz, 2H), 1.87–1.79 (m, 2H), 1.77–1.66 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.4, 148.9, 144.8, 143.6, 142.2, 141.4, 140.5, 135.9, 127.5, 127.2, 125.3, 123.0, 121.1, 120.7, 120.3, 119.4, 119.4, 118.3, 117.6, 111.3, 58.8, 55.5, 53.9, 50.8, 37.2, 27.5, 26.6, 25.75; Anal. (C30H33N5O·C2H2O4) C, H, N.
6.1.6. 5-(4-(4-(4-(2-Methoxyphenyl)piperazin-1-yl)butyl)-1H-1,2,3-triazol-1-yl)-1H-indole (BAK 04-71; 4c)
Synthesized using General Method A in 68% yield (292 mg, 0.68 mmol) from commercially available 1H-indol-5-amine (264 mg, 2 mmol) and acetylene 2 (272 mg, 1 mmol). Mp 195–196 °C (oxalate salt); 1H NMR (400 MHz, CDCl3) δ 8.48 (br s, 1H), 7.91 (d, J = 2.0 Hz, 1H), 7.54 (dd, J = 7.2, 2.0 Hz, 1H), 7.50–7.47 (m, 1H), 7.33 (t, J = 3.2 Hz, 1H), 7.01–6.89 (m, 3H), 6.85 (dd, J = 7.6, 1,2 Hz, 1H), 6.65–6.63 (m, 1H)3.86 (s, 3H), 3.10 (br s, 4H), 2.86 (t, J = 7.6 Hz, 2H), 2.67 (br s, 4H), 2.48 (t, J = 7.6 Hz, 2H), 1.85–1.77 (m, 2H), 1.71–1.63 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.4, 148.5, 141.5, 135.6, 130.8, 128.2, 126.5, 123.0, 121.1, 120.0, 118.3, 115.9, 113.3, 112.0, 111.3, 103.3, 58.6, 55.5, 53.7, 50.8, 27.6, 26.7, 25.8; Anal. (C25H30N6O·C2H2O4·H2O) C, H, N.
6.1.7. 5-(4-(4-(4-(2-Methoxyphenyl)piperazin-1-yl)butyl)-1H-1,2,3-triazol-1-yl)quinoline (BAK 04-74; 4d)
To a solution of 5-azidoquinoline47 (170 mg, 1.0 mmol) and acetylene 2 (272 mg, 1.0 mmol) in THF (10 mL) were added DIPEA (0.87 mL, 5 mmol), CuI (38 mg, 0.2 mmol). The reaction was heated to 50 °C until TLC analysis indicated the full consumption of starting material. The resulting reaction mixture was concentrated and the crude compound was subjected directly to flash column chromatography (EtOAc followed by 90% CMA) to afford the desired product in 65% yield (287 mg, 0.65 mmol). Mp 150–152 °C (oxalate salt); 1H NMR (400 MHz, CDCl3) δ 9.01 (dd, J = 4.4, 1.6 Hz, 1H), 8.28 (dt, J = 8.8, 1.2 Hz, 1H), 8.15–8.12 (m, 1H), 7.84–7.80 (m, 1H), 7.69 (s, 1H), 7.64 (dd, J = 8.6, 1.2 Hz, 1H), 7.01–6.84 (m, 4H), 3.86 (s, 3H), 3.11 (br s, 4H), 2.92 (t, J = 7.6 Hz, 2H), 2.67 (br s, 4H), 2.50 (t, J = 7.6 Hz, 2H), 1.90–1.82 (m, 2H), 1.74–1.66 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.4, 151.6, 148.6, 148.5, 141.5, 133.8, 131.7, 131.6, 128.5, 124.1, 123.4, 123.3, 123.0, 122.6, 121.1, 118.3, 111.3, 58.6, 55.5, 53.7, 50.8, 27.5, 26.7, 25.7; Anal. (C26H30N6O·2C2H2O4·1/2H2O) C, H, N.
6.1.8. 1-(2-Methoxyphenyl)-4-(4-(1-phenyl-1H-1,2,3-triazol-4-yl)butyl)piperazine (BAK 04-27; 4e)
Synthesized using General Method A in 87% yield (340 mg, 0.87 mmol) from commercially available aniline (186 mg, 2 mmol) and acetylene 2 (272 mg, 1 mmol). Mp 172–173 °C (oxalate salt); 1H NMR (400 MHz, CDCl3) δ 7.74–7.71 (m, 3H), 7.53–7.49 (m, 2H), 7.44–7.40 (m, 1H), 7.01–6.89 (m, 2H), 6.85 (d, J = 8.0 Hz, 1H), 3.86 (s, 3H), 3.10 (br s, 4H), 2.85 (t, J = 7.2 Hz, 2H), 2.47 (t, J = 7.2 Hz, 3H), 1.84–1.76 (m, 2H), 1.72–1.64 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.4, 149.0, 141.5, 137.4, 129.8, 128.6, 123.0, 121.1, 120.6, 119.1, 118.3, 111.3, 58.6, 55.5, 53.7, 50.8, 27.5, 26.7, 25.8; Anal. (C23H29N5O·C2H2O4) C, H, N.
6.1.9. 1-(2,3-Dichlorophenyl)-4-(4-(1-(naphthalen-2-yl)-1H-1,2,3-triazol-4-yl)butyl)piperazine (BAK 04-89; 5a)
Synthesized using General Method A in 72% yield (346 mg, 0.72) from commercially available naphthalen-1-amine (286 mg, 2 mmol) and acetylene 3 (311 mg, 1 mmol). Mp 195–197 °C (oxa-late salt); 1H NMR (400 MHz, CDCl3) δ 8.16 (s, 1H), 8.0–7.88 (m, 5H), 7.60–7.53 (m, 2H), 7.15–7.11 (m, 2H), 6.96–6.93 (m, 2H), 3.07 (br s, 4H), 2.89 (t, J = 7.6 Hz, 2H), 2.66 (br s, 4H), 2.50 (t, J = 6.8 Hz, 2H), 1.87–1.80 (m, 2H), 1.72–1.65 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 151.39, 134.72, 134.10, 133.35, 132.86, 130.05, 128.32, 128.04, 127.55, 127.50, 126.95, 124,63, 119.19, 119.04, 118.69, 118.22, 58.46, 53.48, 51.41, 27.49, 26.63, 25.7; Anal. (C26H27Cl2N5·C2H2O4·1/2H2O) C, H, N.
6.1.10. 1-(4-(1-(9H-Fluoren-2-yl)-1H-1,2,3-triazol-4-yl)butyl)-4-(2,3-dichlorophenyl)piperazine (BAK 04-90; 5b)
Synthesized using General Method A in 82% yield (450 mg, 0.82 mmol) from commercially available 9H-fluoren-2-amine (362 mg, 2 mmol) and acetylene 3 (311 mg, 1 mmol). Mp 209– 210 °C (oxalate salt); 1H NMR (400 MHz, CDCl3) δ 7.93(s, 1H), 7.87 (d, J = 7.6 Hz, 1H), 7.82 (d, J = 7.2 Hz, 2H), 7.79 (s, 1H), 7.70 (dd, J = 8.0, 1.6 Hz, 1H), 7.58 (d, J = 7.6 Hz, 1H), 7.44–7.34 (m, 2H), 7.16–7.11 (m, 2H), 6.95 (dd, J = 6.8, 2.8 Hz, 1H), 3.99 (s, 2H), 3.07 (br s, 4H), 2.87 (t, J = 7.2 Hz, 2H), 2.65 (br s, 4H), 2.49 (t, J = 7.6 Hz, 2H), 1.86–1.78 (m, 2H), 1.70–1.63 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 151.41, 148.83, 144.87, 143.56, 142.21, 140.55, 135.97, 134.10, 127.59, 127.56, 127.51, 127.21, 125.30, 124.64, 120.74, 120.34, 119.41, 119.22, 118.70, 117.60, 58.49, 53.51, 51.44, 37.17, 27.53, 26.65, 25.76; Anal. (C29H29Cl2N5·C2H2O4·1/4H2O) C, H, N.
6.1.11. 5-(4-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butyl)-1H-1,2,3-triazol-1-yl)-1H-indole (BAK 04-92; 5c)
Synthesized using General Method A in 82% yield (450 mg, 0.83 mmol) from commercially available 1H-indol-5-amine (264 mg, 2 mmol) and acetylene 3 (311 mg, 1 mmol). Mp 183– 185 °C (oxalate salt); 1H NMR (400 MHz, DMSO) d 11.37 (br s, 1H), 8.46 (s, 1H), 7.96 (s, 1H), 7.53 (s, 2H), 7.47 (t, J = 2.8 Hz, 1H), 7.28–7.25 (m, 2H), 7.11 (dd, J = 5.6, 4.0 Hz, 1H), 6.53 (t, J = 2.4 Hz, 1H), 2.96 (br s, 4H), 2.71 (t, J = 7.2 Hz, 2H), 2.51 (br s, 4H), 2.37 (t, J = 6.8 Hz, 2H), 1.73–1.66 (m, 2H), 1.57–1.52 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 151.40, 135.59, 134.11, 130.80, 128.15, 127.58, 126.47, 124.65, 119.98, 118.72, 115.89, 113.28, 112.02, 103.42, 58.49, 53.49, 51.41, 27.59, 26.63, 25.78; GC–MS (EI) m/z 170 (M+); Anal. (C24H26Cl2N6·9/8C2H2O4·1/2H2O) C, H, N.
6.1.12. 1-(4-Azidobutyl)-4-(2-methoxyphenyl)piperazine (7)
A catalytic amount of NaI (5.0 mg) was added to a solution of tosylate 634 (3.23 g, 12 mmol), commercially available 1-(2-methoxyphenyl)piperazine (2.31 g, 12 mmol), and K2CO3 (3.32 g, 24 mmol) in acetone (35 mL) at room temperature. This reaction mixture was stirred at reflux for 12 h. The resulting mixture was cooled to room temperature, filtered, and the K2CO3 residue was washed with additional acetone. The filtrate was concentrated and the crude product was purified by flash column chromatography (50% EtOAc/hexanes) to give the pure product in 98% yield (3.4 g, 11.8 mmol). 1H NMR (400 MHz, CDCl3) δ 7.02–6.89 (m, 3H), 6.86 (dd, J = 8.0, 1.6 Hz, 1H), 3.86 (s, 3H), 3.31 (t, J = 6.8 Hz, 2H), 3.1 (br s, 4H), 2.65 (br s, 4H), 2.43 (t, J = 7.2 Hz, 2H), 1.68– 1.60 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 152.31, 141.39, 122.94, 121.02, 128.23, 111.17, 58.09, 55.39, 53.51, 51.45, 50.72, 27.0, 24.14; GC–MS (EI) m/z 289 (M+).
6.1.13. 1-(4-Azidobutyl)-4-(2,3-dichlorophenyl)piperazine (8)
A catalytic amount of NaI (5.0 mg) was added to a solution of tosylate 634 (3.23 g, 12 mmol), commercially available 1-(2,3-dichlorophenyl)piperazine (2.77 g, 12 mmol), and K2CO3 (3.32 g, 24 mmol) in acetone (35 mL) at room temperature. This reaction mixture was stirred at reflux for 12 h. The resulting mixture was cooled to room temperature, filtered, and the K2CO3 residue was washed with additional acetone. The filtrate was concentrated and the crude product was purified by flash column chromatography (50% EtOAc/hexanes) to give the pure product in 97% yield (3.8 g, 11.6 mmol). 1H NMR (400 MHz, CDCl3) δ 7.16–7.11 (m, 2H), 6.96 (dd, J = 6.4, 3.2 Hz, 1H), 3.31 (t, J = 6.8 Hz, 2H), 3.06 (br s, 4H), 2.63 (br s, 4H), 2.44 (t, J = 7.2 Hz, 2H), 1.66–1.60 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 151.36, 134.06, 127.54, 127.53, 124.60, 118.66, 57.97, 53.35, 51.45, 51.40, 26.96, 24.13; GC–MS (EI) m/z 299 (M+ −N2).
6.1.14. General Method B
To a suspension of CuI (0.2 equiv) in THF (10 mL) was added the appropriate azidobutylphenylpiperazine (1.0 equiv) and ethynylaryl substrate (1.0 equiv). The reaction was stirred at room temperature for 12 h before the resulting mixture was concentrated in vacuo and subjected directly to flash column chromatography to afford the pure product.
6.1.15. 1-(2-Methoxyphenyl)-4-(4-(4-(naphthalen-2-yl)-1H-1,2,3-triazol-1-yl)butyl)piperazine (BAK 05-21; 9a)
Synthesized using General Method B in 85% yield (338 mg, 0.77 mmol) from azidobutylphenylpiperazine 7 (260 mg, 0.9 mmol) and 2-ethynylnaphthalene (137 mg, 0.9 mmol).48 Mp 194–195 °C (oxalate salt). 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 7.95–7.83 (m, 5H), 7.52–7.46 (m, 2H), 7.01–6.84 (m, 4H), 4.49 (t, J = 6.8 Hz, 2H), 3.85 (s, 3H), 3.08 (br s, 4H), 2.63 (br s, 4H), 2.47 (t, J = 7.6 Hz, 2H), 2.09–2.01(m, 2H), 1.66–1.59 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.30, 147.86, 141.32, 133.62, 133.19, 128.65, 128.25, 128.10, 127.84, 126.52, 126.19, 124.40, 123.94, 122.99, 121.03, 119.84, 118.23, 111.19, 57.82, 55.40, 53.52, 50.70, 50.42, 28.43, 23.89; Anal. (C27H31N5O·C2H2O4·1/5H2O) C, H, N.
6.1.16. 1-(4-(4-(9H-Fluoren-2-yl)-1H-1,2,3-triazol-1-yl)butyl)-4-(2-methoxyphenyl)piperazine (BAK 05-19; 9b)
Synthesized using General Method B in 91% yield (393 mg, 0.82 mmol) from azidobutylphenylpiperazine 7 (260 mg, 0.9 mmol) and 2-ethynyl-9H-fluorene (171 mg, 0.9 mmol).49 Mp 211–212 °C (oxalate salt). 1H NMR (400 MHz, CDCl3) δ 8.07 (s, 1H), 7.83–7.94 (m, 4H), 7.56 (d, J = 7.2 Hz, 1H), 7.39 (t, J = 7.2 Hz, 1H), 7.32 (dt, J = 7.6, 1.2 Hz, 1H), 7.02–6.84 (m, 2H), 4.47 (t, J = 7.2 Hz, 2H), 3.96 (s, 2H), 3.85 (s, 3H), 3.09 (br s, 4H), 2.63 (br s, 4H), 2.47 (t, J = 7.2 Hz, 2H), 2.08–2.01(m, 2H), 1.66–1.58 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.28, 148.17, 143.95, 143.53, 141.71, 141.37, 141.31, 129.19, 126.91, 126.89, 125.14, 124.53, 122.98, 122.35, 121.01, 120.24, 120.02, 119.42, 118.21, 111.17, 57.80, 55.39, 53.50, 50.68, 50.34, 36.98, 28.40, 23.86; Anal. (C30H33N5O·C2H2O4) C, H, N.
6.1.17. 5-(1-(4-(4-(2-Methoxyphenyl)piperazin-1-yl)butyl)-1H-1,2,3-triazol-4-yl)-1H-indole (BAK 05-16; 9c)
Synthesized using General Method B in 72% yield (263 mg, 0.65 mmol) from azidobutylphenylpiperazine 7 (260 mg, 0.9 mmol) and 5-ethynyl-1H-indole (127 mg, 0.9 mmol).50 Mp 214–215 °C (oxalate salt). 1H NMR (400 MHz, CDCl3) δ 8.35 (br s, 1H), 8.10 (m, 1H), 7.74 (s, 1H), 7.70 (dd, J = 8.4, 1.6 Hz, 1H), 7.45– 7.42 (m, 1H), 7.23 (t, J = 2.8 Hz, 1H), 7.01–6.84 (m, 4H), 6.60–6.58 (m, 1H), 4.45 (t, J = 7.2 Hz, 2H), 3.85 (s, 3H), 3.08 (br s, 4H), 2.63 (br s, 4H), 2.46 (t, J = 8.0 Hz, 2H), 2.06–1.99 (m, 2H), 1.65–1.58 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.35, 149.24, 141.36, 135.90, 128.30, 125.14, 123.04, 122.66, 121.07, 120.43, 118.86, 118.31, 118.12, 111.58, 111.22, 103.0, 57.89, 55.43, 53.54, 50.73, 50.33, 28.48, 23.92; Anal. (C25H30N6O·C2H2O4·1/2H2O) C, H, N.
6.1.18. 5-(4-(4-(4-(2-Methoxyphenyl)piperazin-1-yl)butyl)-1H-1,2,3-triazol-1-yl)quinoline (BAK 05-27; 9d)
Sodium ascorbate (27 mg, 15 mol %) and CuSO4·5H2O (11 mg, 5 mol %) were added to a solution of azidobutylphenylpiperazine 7 (260 mg, 0.9 mmol) and 6-ethynylquinoline (138 mg, 0.9 mmol)51 in THF/H2O (1:1, 6 mL). The reaction was stirred at 50 °C for 12 h. The resulting solution was cooled to room temperature, diluted with H2O (5 mL) and extracted with CHCl3 (3 × 10 mL). The combined organic layer was dried over MgSO4, filtered, and concentrated. The crude compound was purified by flash column chromatography to give the pure compound in 79% yield (314 mg, 0.71 mmol). Mp 186–188 °C (oxalate salt). 1H NMR (400 MHz, CDCl3) δ 8.91 (dd, J = 4.4, 2.0 Hz, 1H), 8.38 (d, J = 1.2 Hz, 1H), 8.21 (dd, J = 8.0, 1.6 Hz, 1H), 8.17–8.10 (m, 2H), 7.92 (s, 1H), 7.43 (dd, J = 8.4, 4.4 Hz, 1H), 7.02–6.84 (m, 4H), 4.50 (t, J = 7.2 Hz, 2H), 3.85 (s, 3H), 3.09 (br s, 4H), 2.64 (br s, 4H), 2.48 (t, J = 7.6 Hz, 2H), 2.10–2.03 (m, 2H), 1.67–1.60 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.30, 150.60, 148.11, 147.15, 141.28, 136.35, 130.16, 128.85, 128.59, 127.54, 124.13, 123.03, 121.75, 121.04, 120.15, 118.23, 111.20, 57.79, 55.41, 53.52, 50.68, 50.50, 28.42, 23.87; Anal. (C26H30N6O·2C2H2O4·H2O) C, H, N.
6.1.19. 1-(4-(4-([1,1′-Biphenyl]-4-yl)-1H-1,2,3-triazol-1-yl)butyl)-4-(2-methoxyphenyl)piperazine (BAK 05-18; 9e)
Synthesized using General Method B in 82% yield (345 mg, 0.74 mmol) from azidobutylphenylpiperazine 7 (260 mg, 0.9 mmol) and 4-ethynyl-1,1′-biphenyl (160 mg, 0.9 mmol).52 Mp 181–182 °C (oxalate salt). 1H NMR (400 MHz, CDCl3) δ 7.92–7.90 (m, 2H), 7.80 (s, 1H), 7.68–7.63 (m, 2H), 7.45 (t, J = 7.6 Hz, 2H), 7.38–7.34 (m, 1H), 7.01–6.84 (m, 4H), 4.47 (t, J = 7.2 Hz, 2H), 3.86 (s, 3H), 3.08 (br s, 4H), 2.63 (br s, 4H), 2.46 (t, J = 7.2 Hz, 2H), 2.07–2.0 (m, 2H), 1.65–1.57 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.22, 147.37, 141.25, 140.72, 140.49, 129.67, 128.83, 127.47, 127.43, 126.93, 126.05, 122.90, 120.95, 119.55, 118.14, 111.12, 57.72, 55.33, 53.43, 50.61, 50.27, 28.32, 23.79; Anal. (C29H33N5O·C2H2O4·1/2H2O) C, H, N.
6.1.20. 1-(2-Methoxyphenyl)-4-(4-(4-phenyl-1H-1,2,3-triazol-1-yl)butyl)piperazine (BAK 04-81; 9f)
Synthesized using General Method B in 89% yield (209 mg, 0.53 mmol) from azidobutylphenylpiperazine 7 (173 mg, 0.6 mmol) and commercially available ethynylbenzene (61 mg, 0.6 mmol). Mp 176–177 °C (oxalate salt). 1H NMR (400 MHz, CDCl3) δ 7.84 (dd, J = 8.8, 1.2 Hz, 2H), 7.77 (s, 1H), 7.43 (t, J = 7.6 Hz, 2H), 7.34 (tt, J = 7.6, 1.6 Hz, 1H), 7.02–6.97 (m, 1H), 6.94–6.91 (m, 2H), 6.86 (dd, J = 8.0, 1.2 Hz, 1H), 4.45 (t, J = 6.8 Hz, 2H), 3.86 (s, 3H), 3.08 (br s, 4H), 2.63 (br s, 4H), 2.07–1.99 (m, 2H), 1.64–1.57 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.39, 147.93, 141.42, 130.83, 128.96, 128.22, 125.83, 121.11, 119.53, 118.31, 111.30, 57.87, 55.47, 53.59, 50.75, 50.43, 28.48, 23.93; Anal. (C23H29N5O·C2H2O4) C, H, N.
6.1.21. 1-(2,3-Dichlorophenyl)-4-(4-(4-(naphthalen-2-yl)-1H-1,2,3-triazol-1-yl)butyl)piperazine (BAK 05-26; 10a)
Synthesized using General Method B in 88% yield (380 mg, 0.79 mmol) from azidobutylphenylpiperazine 8 (295 mg, 0.9 mmol) and commercially available 2-ethynylnaphthalene (137 mg, 0.9 mmol). Mp 190–192 °C (oxalate salt). 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 7.95–7.83 (m, 5H), 7.52–7.48 (m, 2H), 7.16–7.09 (m, 2H), 6.94–6.91 (m, 1H), 4.48 (t, J = 6.8 Hz, 2H), 3.04 (br s, 4H), 2.63 (br s, 4H), 2.47 (t, J = 6.8 Hz, 2H), 2.09–2.01 (m, 2H), 1.66–1.59 (m,2H); 13C NMR (100 MHz, CDCl3) δ 151.22, 147.80, 133.98, 133.57, 133.15, 128.63, 128.21, 128.06, 127.82, 127.51, 127.44, 126.52, 126.18, 124.58, 124.35, 123.89, 119.86, 118.63, 57.61, 53.29, 51.28, 50.33, 28.33, 23.79; Anal. (C26H27Cl2N5·C2H2O4·1/10H2O) C, H, N.
6.1.22. 1-(4-(4-(9H-Fluoren-2-yl)-1H-1,2,3-triazol-1-yl)butyl)-4-(2,3-dichlorophenyl)piperazine (BAK 05-24; 10b)
Synthesized using General Method B in 87% yield (406 mg, 0.78 mmol) from azidobutylphenylpiperazine 8 (295 mg, 0.9 mmol) and 2-ethynyl-9H-fluorene (171 mg, 0.9 mmol). Mp 228–230 °C (oxalate salt). 1H NMR (400 MHz, CDCl3) δ 8.07 (s, 1H), 7.83–7.93 (m, 4H), 7.56 (d, J = 7.2 Hz, 1H), 7.39 (t, J = 7.2 Hz, 1H), 7.32 (m, 1H), 7.16–7.10 (m, 2H), 6.93 (dd, J = 7.6, 2.4 Hz, 1H), 4.47 (t, J = 7.2 Hz, 2H), 3.96 (s, 2H), 3.05 (br s, 4H), 2.62 (br s, 4H), 2.48 (t, J = 7.2 Hz, 2H), 2.08–2.01 (m, 2H), 1.65–1.58 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 151.31, 148.26, 144.01, 143.57, 141.79, 141.40, 134.08, 129.22, 127.55, 126.98, 126.95, 125.19, 124.65, 124.57, 122.41, 120.30, 120.06, 119.43, 118.67, 57.70, 53.37, 51.37, 50.37, 37.03, 28.42, 23.89; Anal. (C29H29Cl2N5·C2H2O4·1/4H2O) C, H, N.
6.1.23. 5-(1-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butyl)-1H-1,2,3-triazol-4-yl)-1H-indole (BAK 05-25; 10c)
Synthesized using General Method B in 72% yield (263 mg, 0.65 mmol) from azidobutylphenylpiperazine 8 (295 mg, 0.9 mmol) and 5-ethynyl-1H-indole (127 mg, 0.9 mmol). Mp 214– 215 °C (oxalate salt). 1H NMR (400 MHz, CDCl3) δ 8.32 (br s, 1H), 8.11 (m, 1H), 7.75 (s, 1H), 7.70 (dd, J = 8.4, 1.6 Hz, 1H), 7.46–7.43 (m, 1H), 7.24 (t, J = 2.8 Hz, 1H), 7.16–7.12 (m, 2H),6.92 (dd, J = 7.6, 2.4 Hz, 1H), 6.60–6.58 (m, 1H), 4.45 (t, J = 7.2 Hz, 2H), 3.04 (br s, 4H), 2.61 (br s, 4H), 2.46 (t, J = 7.6 Hz, 2H), 2.07–1.99 (m, 2H), 1.66–1.57 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 151.34, 149.26, 135.90, 134.09, 128.31, 127.57, 125.13, 124.66, 122.72, 120.47, 118.84, 118.71, 118.15, 111.58, 103.08, 57.76, 53.39, 51.40, 50.33, 28.46, 23.94; Anal. (C24H26Cl2N6·3/4C2H2O4) C, H, N.
6.1.24. 6-(1-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butyl)-1H-1,2,3-triazol-4-yl)quinoline (BAK 05-28; 10d)
Sodium ascorbate (27 mg, 15 mol %) and CuSO4·5H2O (11 mg, 5 mol %) were added to a solution of azidobutylphenylpiperazine 8 (295 mg, 0.9 mmol) and 6-ethynylquinoline (138 mg, 0.9 mmol) in THF/H2O (1:1, 6 mL). The reaction was stirred at 50 °C for 12 h. The resulting solution was cooled to room temperature, diluted with H2O (5 mL) and extracted with CHCl3 (3 × 10 mL). The combined organic layer was dried over MgSO4, filtered, and concentrated. The crude compound was purified by flash column chromatography to give the pure compound in 67% yield (290 mg, 0.60 mmol). Mp 165–167 °C (oxalate salt). 1H NMR (400 MHz, CDCl3) δ 8.90 (m, 1H), 8.38 (s, 1H), 8.21–8.10 (m, 3H), 7.91 (s, 1H), 7.42 (dd, J = 8.4, 4.0 Hz, 1H), 7.15–7.10 (m, 2H), 6.92 (d, J = 7.6 Hz, 1H), 4.50 (t, J = 7.2 Hz, 2H), 3.04 (br s, 4H), 2.61 (br s, 4H), 2.47 (t, J = 7.2 Hz, 2H), 2.10–2.02 (m, 2H), 1.66–1.58 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 151.24, 150.61, 148.10, 147.14, 136.31, 134.03, 130.15, 128.82, 128.57, 127.53, 127.49, 124.62, 124.11, 121.75, 120.14, 118.63, 57.64, 53.33, 51.33, 50.46, 28.38, 23.86; Anal. (C25H26Cl2N6·2C2H2O4) C, H, N.
6.1.25. 1-(4-(4-([1,1′-Biphenyl]-4-yl)-1H-1,2,3-triazol-1-yl)-butyl)-4-(2,3-dichlorophenyl)piperazine (BAK 05-23; 10e)
Synthesized using General Method B in 78% yield (355 mg, 0.70 mmol) from azidobutylphenylpiperazine 8 (295 mg, 0.9 mmol) and 4-ethynyl-1,1′-biphenyl (160 mg, 0.9 mmol). Mp 196–197 °C (oxalate salt). 1H NMR (400 MHz, DMSO) d 8.67 (s, 1H), 7.95 (d, J = 8.4 Hz, 2H), 7.78–7.71 (m, 4H), 7.48 (t, J = 8.0 Hz, 2H), 7.40–7.36 (m, 1H), 7.30–7.28 (m, 2H), 7.13–7.11 (m, 1H), 4.45 (t, J = 7.2 Hz, 2H), 2.97 (br s, 4H), 2.51 (br s, 4H), 2.38 (t, J = 6.8 Hz, 2H), 1.95–1.91 (m, 2H), 1.52–1.44 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 151.27, 147.54, 140.89, 140.60, 134.06, 129.73, 128.92, 127.59, 127.55, 127.04, 126.14, 124.65, 119.58, 118.67, 57.68, 53.35, 51.33, 50.37, 28.39, 23.85; Anal. (C28H29Cl2N5·C2H2O4) C, H, N.
6.1.26. General Method C
Sodium ascorbate (15 mol %) and CuSO4·5H2O (5 mol %) were added to a solution of 2-(2-azidoethyl)oxirane (11, 1.0 equiv), the appropriate phenylpiperazine hydrochloride (1.0 equiv), the appropriate arylacetylene (1.0 equiv), β-cyclodextrin (3 mol %), and K2CO3 (1.0 equiv) in H2O (0.25 M). The reaction mixture was stirred at room temperature for 30 min and heated to 80 °C for 2 h. The resulting mixture was diluted with water (5 mL), extracted with CH2Cl2 (3 × 10 mL). The combined organic layer was dried over MgSO4, filtered, and concentrated under vacuum. The crude solid was purified by flash column chromatography.
6.1.27. 1-(4-(2-Methoxyphenyl)piperazin-1-yl)-4-(4-(naphthalen-2-yl)-1H-1,2,3-triazol-1-yl)butan-2-ol (BAK 05-52; 12a)
Synthesized using General Method C in 61% yield (280 mg, 0.61 mmol) from commercially available 1-(2-methoxyphenyl)piperazine hydrochloride (220 mg, 1.0 mmol) and commercially available 2-ethynylnaphthalene (152 mg, 1 mmol). Mp 155–156 °C (free base); 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 7.97–7.83 (m, 5H), 7.52–7.45 (m, 2H), 7.02–6.98 (m, 1H), 6.93– 6.86 (m, 2H), 6.85 (d, J = 8.0 Hz, 1H), 4.68–4.64 (m, 2H), 3.85 (s, 3H), 3.71 (m, 2H), 3.08 (br s, 4H), 2.84 (m, 2H), 2.58 (m, 2H), 2.46–2.36 (m, 2H), 2.22–2.14 (m, 2H), 1.97–1.89 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.21, 147.57, 141.03, 133.55, 133.12, 128.60, 128.19, 128.03, 127.79, 126.48, 126.14, 124.34, 123.89, 123.08, 120.89, 120.70, 118.18, 111.20, 63.82, 63.16, 55.36, 53.42, 50.67, 47.17, 35.23; Anal. (C27H31N5O2) C, H, N.
6.1.28. 4-(4-([1,1′-Biphenyl]-4-yl)-1H-1,2,3-triazol-1-yl)-1-(4-(2-methoxyphenyl)piperazin-1-yl)butan-2-ol (BAK 05-47; 12b)
Synthesized using General Method C in 65% yield (312 mg, 0.65 mmol) from commercially available 1-(2-methoxyphenyl)piperazine hydrochloride (220 mg, 1.0 mmol) and 4-ethynyl-1,1′-biphenyl (178 mg, 1.0 mmol). Mp 159–160 °C (oxalate salt); 1H NMR (400 MHz, CDCl3) δ 7.94–7.91 (m, 2H), 7.89 (s, 1H), 7.69–7.62 (m, 4H), 7.48–7.44 (m, 2H), 7.38–7.34 (m, 1H), 7.03– 6.98 (m, 1H), 6.94–6.91 (m, 2H), 6.86 (d, J = 7.6 Hz, 1H), 4.66–4.63 (m, 2H), 3.85 (s, 3H), 3.68 (m, 2H), 3.08 (br s, 4H), 2.83 (m, 2H), 2.58 (m, 2H), 2.46–2.35 (m, 2H), 2.21–2.12 (m, 2H), 1.96–1.87 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.33, 147.35, 141.16, 140.91, 140.69, 129.79, 128.94, 127.62, 127.54, 127.09, 126.19, 123.18, 121.07, 120.45, 118.27, 111.28, 63.87, 63.12, 55.46, 53.51, 50.82, 47.21, 35.30; Anal. (C29H33N5O2·C2H2O4·1/4H2O) C, H, N.
6.1.29. 1-(4-(2-Methoxyphenyl)piperazin-1-yl)-4-(4-phenyl-1H-1,2,3-triazol-1-yl)butan-2-ol (BAK 05-51; 12c)
Synthesized using General Method C in 73% yield (300 mg, 0.73 mmol) from commercially available 1-(2-methoxyphenyl)piperazine hydrochloride (220 mg, 1.0 mmol) and commercially available ethynylbenzene (102 mg, 1 mmol). Mp 129– 130 °C (free base); 1H NMR (400 MHz, CDCl3) δ 7.85–7.83 (m, 3H), 7.43 (t, J = 8.0 Hz, 2H), 7.35–7.31 (m, 1H), 7.03–6.84 (m, 4H), 4.63 (t, J = 7.2 Hz, 2H), 3.85 (s, 3H), 3.68 (m, 2H), 3.07 (br s, 4H), 2.83 (m, 2H), 2.57 (m, 2H), 2.43–2.34 (m, 2H), 2.18–2.11 (m, 2H), 1.95–1.87 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.24, 147.56, 141.08, 130.74, 128.87, 128.11, 125.72, 123.09, 121.0, 120.37, 118.19, 111.22, 63.83, 63.11, 55.38, 53.43, 50.72, 47.11, 35.23; Anal. (C23H29N5O2) C, H, N.
6.1.30. 4-(4-(2-Methoxyphenyl)-1H-1,2,3-triazol-1-yl)-1-(4-(2-methoxyphenyl)piperazin-1-yl)butan-2-ol (BAK 05-44; 12d)
Synthesized using General Method C in 65% yield (284 mg, 0.65 mmol) from commercially available 1-(2-methoxyphenyl)piperazine hydrochloride (220 mg, 1.0 mmol) and 1-ethynyl-2-methoxybenzene (132 mg, 1.0 mmol).53 Mp 158–160 °C (oxalate salt); 1H NMR (400 MHz, CDCl3) δ 8.35 (dd, J = 7.2, 1.2 Hz, 1H), 8.10 (s, 1H), 7.33–7.29 (m, 1H), 7.08 (t, J = 7.6 Hz, 1H), 7.10–6.84 (m, 6H), 4.62 (m, 2H), 3.94 (s, 3H), 3.85 (s, 3H), 3.71 (m, 2H), 3.07 (br s, 4H), 2.83 (m, 2H), 2.57 (m, 2H), 2.44– 2.34 (m, 2H), 2.20–2.11 (m, 2H), 1.98–1.88 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 155.68, 152.28, 143.0, 141.14, 128.89, 127.69, 123.73, 123.13, 121.10, 121.02, 119.58, 118.23, 111.25, 110.83, 63.84, 63.26, 55.43, 53.46, 50.78, 46.98, 35.38; Anal. (C24H31N5O3·C2H2O4·1/10H2O) C, H, N.
6.1.31. 4-(4-(9H-Fluoren-2-yl)-1H-1,2,3-triazol-1-yl)-1-(4-(2-methoxyphenyl)piperazin-1-yl)butan-2-ol (BAK 05-43; 12e)
Synthesized using General Method C in 65% yield (321 mg, 0.65 mmol) from commercially available 1-(2-methoxyphenyl)piperazine hydrochloride (220 mg, 1.0 mmol) and 2-ethynyl-9H-fluorene (190 mg, 1.0 mmol). Mp 153–155 °C (oxalate salt); 1H NMR (400 MHz, CDCl3) δ 8.08 (m, 1H), 7.89 (m, 1H), 7.83 (m, 2H), 7.80 (d, J = 7.2 Hz, 1H), 7.56 (d, J = 7.2 Hz, 1H), 7.39 (t, J = 7.2 Hz, 1H), 7.34–7.30 (m, 1H), 7.03–6.98 (m, 2H), 6.92 (m, 2H), 6.86 (d, J = 8.0 Hz, 1H), 4.64 (t, J = 7.2 Hz, 2H), 3.96 (s, 2H), 3.85 (s, 3H), 3.67 (m, 2H), 3.08 (br s, 4H), 2.84 (m, 2H), 2.45–2.35 (m, 2H), 2.21–2.13 (m, 2H), 1.96–1.88 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.29, 148.01, 143.98, 143.55, 141.74, 141.39, 141.13, 129.23, 126.94, 125.16, 124.57, 123.14, 122.39, 121.04, 120.27, 120.28, 120.04, 118.23, 111.25, 63.85, 63.14, 55.43, 53.51, 50.78, 47.18, 37.01, 35.29; Anal. (C30H33N5O2·C2H2O4·1/2H2O) C, H, N.
6.1.32. 1-(4-(2-Methoxyphenyl)piperazin-1-yl)-4-(4-(quinolin-6-yl)-1H-1,2,3-triazol-1-yl)butan-2-ol (BAK 05-56; 12f)
Synthesized using General Method C in 87% yield (400 mg, 0.87 mmol) from commercially available 1-(2-methoxyphenyl)piperazine hydrochloride (220 mg, 1.0 mmol) and 6-ethynylquinoline (153 mg, 1.0 mmol). Mp 172–174 °C (free base); 1H NMR (400 MHz, CDCl3) δ 8.91 (dd, J = 4.4, 1.6 Hz, 1H), 8.37 (d, J = 1.2 Hz, 1H), 8.21 (dd, J = 8.0, 1.6 Hz, 1H), 8.17–8.12 (m, 2H), 7.43 (dd, J = 8.0, 4.0 Hz), 7.02–6.97 (m, 2H), 6.93–6.91 (m, 2H), 6.85 (d, J = 7.6 Hz, 1H), 4.69–4.66 (m, 2H), 3.86 (s, 3H), 3.71 (m, 2H), 3.08 (br s, 4H), 2.84 (m, 2H), 2.58 (m, 2H), 2.43–2.36 (m, 2H), 2.23–2.15 (m, 2H), 1.98–1.89 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 152.09, 150.35, 147.82, 146.67, 140.93, 136.17, 129.87, 128.75, 128.39, 127.40, 123.90, 122.94, 121.57, 120.93, 120.86, 118.03, 111.09, 63.81, 63.13, 55.25, 53.36, 50.56, 47.17, 35.13; Anal. (C26H30N6O·1/4H2O) C, H, N.
6.1.33. 1-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-4-(4-(naphthalen-2-yl)-1H-1,2,3-triazol-1-yl)butan-2-ol (BAK 05-53; 13a)
Synthesized using General Method C in 65% yield (320 mg, 0.65 mmol) from commercially available 1-(2,3-dichlorophenyl)piperazine hydrochloride (268 mg, 1.0 mmol) and commercially available 2-ethynylnaphthalene (152 mg, 1.0 mmol). Mp 165–166 °C (free base); 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 7.97–7.83 (m, 5H), 7.52–7.45 (m, 2H), 7.18–7.11 (m, 2H), 6.93 (dd, J = 7.6, 2.4 Hz, 1H), 4.68–4.64 (m, 2H), 3.71 (m, 2H), 3.05 (br s, 4H), 2.83 (m, 2H), 2.58 (m, 2H), 2.48–2.37 (m, 2H), 2.23–2.14 (m, 2H), 1.98–1.89 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 151.10, 147.80, 134.22, 133.69, 133.26, 128.72, 128.32, 128.16, 127.92, 127.67, 127.59, 126.60, 126.27, 124.89, 124.48, 124.01, 120.72, 118.70, 63.81, 63.18, 53.39, 51.45, 47.25, 35.30; Anal. (C26H27Cl2N5O) C, H, N.
6.1.34. 4-(4-([1,1’-Biphenyl]-4-yl)-1H-1,2,3-triazol-1-yl)-1-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-2-ol (BAK 05-55; 13b)
Synthesized using General Method C in 60% yield (314 mg, 0.60 mmol) from commercially available 1-(2,3-dichlorophenyl)piperazine hydrochloride (268 mg, 1.0 mmol) and 4-ethynyl-1,1′-biphenyl (178 mg, 1.0 mmol). Mp 212–213 °C (free base); 1H NMR (400 MHz, CDCl3) δ 7.94–7.91 (m, 2H), 7.88 (s, 1H), 7.69–7.62 (m, 4H), 7.48–7.44 (m, 2H), 7.38–7.34 (m, 1H), 7.17–7.12 (m, 2H), 6.94 (dd, J = 7.6, 2.4 Hz, 1H), 4.67–4.63 (m, 2H), 3.68 (m, 2H), 3.05 (br s, 4H), 2.83 (m, 2H), 2.57 (m, 2H), 2.47–2.36 (m, 2H), 2.21–2.12 (m, 2H), 1.97–1.88 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 151.12, 147.41, 140.99, 140.71, 134.22, 129.79, 128.96, 127.66, 127.60, 127.11, 126.21, 124.88, 120.88, 120.45, 118.70, 63.81, 63.17, 53.41, 51.49, 47.22, 35.28; Anal. (C28H29Cl2N5O·1/2H2O) C, H, N.
6.1.35. 4-Azido-1-(4-(2-methoxyphenyl)piperazin-1-yl)butan-2-ol (14)
To a 15 mL round bottom flask was added 1-(2-methoxyphenyl)piperazine (400.0 mg, 2.08 mmol) dissolved in CH3CN (4.4 mL). Sodium iodide (6 mg, 0.042 mmol) and K2CO3 (632.4 mg, 4.58 mmol) were added and allowed to stir for 10 min at room temperature. A solution of azide 11 (470.6 mg, 4.16 mmol) in CH3CN (0.6 mL) was added and the mixture was heated to reflux and stirred for 16 h. The reaction was cooled to room temperature and filtered through a pad of celite, which was washed thoroughly with CH3CN. The filtrate was collected, concentrated in vacuo, redissolved in 9% MeOH/1% NH4OH/90% CH2Cl2 and filtered through a 200 silica plug. The filtrate was collected and concentrated under reduced pressure to yield 14, which was used directly in the next step.
6.1.36. 4-Azido-1-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-2-ol (15)
To a 15 mL round bottom flask was added 1-(2,3-dichlorophenyl)piperazine (400.0 mg, 1.74 mmol) dissolved in CH3CN (4.4 mL). Sodium iodide (5 mg, 0.035 mmol) and K2CO3 (529.1 mg, 3.83 mmol) were added and allowed to stir for 10 min at room temperature. A solution of azide 11 (393.2 mg, 3.48 mmol) in CH3CN (0.6 mL) was added and the mixture was heated to reflux and stirred for 16 h. The reaction was cooled to room temperature and filtered through a pad of celite, which was washed thoroughly with CH3CN. The filtrate was collected, concentrated in vacuo, redissolved in 9% MeOH/1% NH4OH/90% CH2Cl2 and filtered through a 2′′ silica plug. The filtrate was collected and concentrated under reduced pressure to yield 15, which was used directly in the next step.
6.1.37. 4-(4-(1H-Indol-5-yl)-1H-1,2,3-triazol-1-yl)-1-(4-(2-methoxyphenyl)piperazin-1-yl)butan-2-ol (RDS 02-33; 16)
To a 10 mL round bottom flask, charged freshly prepared solutions of 1 M sodium ascorbate (0.26 mL, 0.051 mmol) and CuSO4·5H2O (0.15 mL, 0.031 mmol) in THF/H2O (3:1, 4 mL) was added 5-ethynyl-1H-indole (77.7 mg, 0.51 mmol) and azide 14 (155 mg, 0.51 mmol). The reaction mixture was stirred for 16 h at room temperature before it was diluted with H2O (2 mL) and extracted with EtOAc (3 × 3 mL). The combined organic extracts were washed with brine and concentrated under reduced pressure. The resulting crude oil was purified by flash column chromatography (9% MeOH/1% NH4OH/90% CH2Cl2) to afford 1,2,3-triazole 16 in 78% yield (177 mg, 0.40 mmol). Mp 82–83 °C (free base) 1H NMR (400 MHz, CDCl3) δ 9.21 (s, 1H), 8.19–8.02 (m, 1H), 7.73 (s, 1H), 7.63 (dd, J = 8.4, 1.7 Hz, 1H), 7.37 (d, J = 8.4 Hz, 1H), 7.18 (t, J = 2.8 Hz, 1H), 6.99 (ddd, J = 8.4, 6.2, 2.9 Hz, 1H), 6.95–6.73 (m, 3H), 6.53 (t, J = 2.6 Hz, 1H), 4.67–4.38 (m, 2H), 3.81 (s, 3H), 3.69 (q, J = 6.2 Hz, 1H), 3.04 (s, 4H), 2.77 (dt, J = 10.3, 4.6 Hz, 2H), 2.62–2.46 (m, 2H), 2.35 (d, J = 6.6 Hz, 2H), 2.07 (ddt, J = 16.4, 8.2, 5.2 Hz, 1H), 1.87 (ddt, J = 14.4, 9.2, 5.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 152.20, 148.87, 141.05, 135.71, 128.16, 124.90, 123.04, 122.69, 120.94, 120.40, 119.56, 118.16, 118.03, 111.37, 111.14, 103.03, 77.33, 77.21, 77.01, 76.69, 63.75, 63.01, 55.34, 50.71, 46.97, 35.23; Anal. (C25H30N6O2·1/2CH2Cl2) C, H, N.
6.1.38. 4-(4-(1H-Indol-5-yl)-1H-1,2,3-triazol-1-yl)-1-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-2-ol (RDS 02-28; 17a)
To a 10 mL round bottom flask, charged freshly prepared solutions of 1 M sodium ascorbate (0.26 mL, 0.051 mmol) and CuSO4·5H2O (0.15 mL, 0.031 mmol) in THF/H2O (3:1, 4 mL) was added 5-ethynyl-1H-indole (77.7 mg, 0.51 mmol) and azide 15 (175.0 mg, 0.51 mmol). The reaction mixture was stirred for 16 h at room temperature before it was diluted with H2O (2 mL) and extracted with EtOAc (3 × 3 mL). The combined organic extracts were washed with brine and concentrated under reduced pressure. The resulting crude oil was purified by flash column chromatography (9% MeOH/1% NH4OH/90% CH2Cl2) to afford 1,2,3-triazole 17a in 74% yield (185.0 mg, 0.38 mmol). Mp 157–159 °C (free base); 1H NMR (400 MHz, CDCl3) δ 8.22 (s, 1H), 8.12 (dd, J = 1.6, 0.8 Hz, 1H), 7.82 (d, J = 0.7 Hz, 1H), 7.72 (dd, J = 8.5, 1.7 Hz, 1H), 7.52–7.38 (m, 1H), 7.25–7.22 (m, 2H), 7.19–7.05 (m, 2H), 6.93 (dd, J = 7.1, 2.6 Hz, 1H), 6.60 (td, J = 2.2, 1.1 Hz, 1H), 4.63 (t, J = 7.0 Hz, 2H), 3.70 (t, J = 9.7 Hz, 1H), 3.05 (s, 4H), 2.93–2.72 (m, 2H), 2.57 (s, 2H), 2.50–2.28 (m, 2H), 2.25–1.81 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 154.83, 153.19, 140.08, 137.77, 132.09, 131.42, 129.39, 128.57, 125.16, 123.93, 123.47, 122.51, 121.63, 115.56, 105.76, 68.01, 57.40, 55.01, 51.82, 50.98, 39.36; Anal. (C24H26Cl2N6O·1/2H2O) C, H, N.
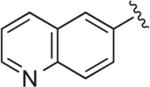
6.1.39. 1-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-4-(4-(quinolin-3-yl)-1H-1,2,3-triazol-1-yl)butan-2-ol (RDS 02-32; 17b)
To a 10 mL round bottom flask, charged freshly prepared solutions of 1 M sodium ascorbate (0.26 mL, 0.051 mmol) and CuSO4·5H2O (0.15 mL, 0.031 mmol), TBTA (5.41 mg, 0.01 mmol) in THF/H2O (3:1, 4 mL) was added 3-ethynylquinoline (78.1 mg, 0.51 mmol)42 and azide 15 (175.0 mg, 0.51 mmol). The reaction mixture was stirred for 16 h at room temperature before it was diluted with H2O (2 mL) and extracted with EtOAc (3 × 3 mL). The combined organic extracts were washed with brine and concentrated under reduced pressure. The resulting crude oil was purified by flash column chromatography (9% MeOH/1% NH4OH/90% CH2Cl2) to afford 1,2,3-triazole 17b in 89% yield (226 mg, 0.45 mmol). Mp 147–149 °C (free base); 1H NMR (400 MHz, CDCl3) δ 9.28 (d, J = 2.1 Hz, 1H), 8.57 (d, J = 2.1 Hz, 1H), 8.05 (d, J = 10.3 Hz, 2H), 7.79 (d, J = 8.1 Hz, 1H), 7.64 (t, J = 7.7 Hz, 1H), 7.49 (t, J = 7.5 Hz, 1H), 7.19–6.94 (m, 2H), 6.82 (dd, J = 7.5, 2.1 Hz, 1H), 4.63 (dd, J = 8.0, 5.9 Hz, 2H), 3.84–3.59 (m, 1H), 2.95 (t, J = 4.7 Hz, 4H), 2.74 (dd, J = 10.7, 5.1 Hz, 2H), 2.51 (dd, J = 10.6, 4.8 Hz, 2H), 2.43–2.30 (m, 2H), 2.13 (dtd, J = 11.3, 8.2, 2.6 Hz, 1H), 2.03–1.82 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 150.91, 148.39, 147.76, 144.76, 134.07, 131.83, 129.60, 129.33, 128.09, 127.96, 127.50, 127.46, 127.20, 124.76, 123.89, 120.84, 118.55, 100.36, 77.34, 77.22, 77.02, 76.70, 63.68, 63.03, 51.30, 47.29, 35.10.; Anal. (C25H26Cl2N6O·1/2H2O) C, H, N.
6.2. X-ray crystal structure of RDS 02-28 (17a)
Single-crystal X-ray diffraction data on RDS 02-28 (17a) was collected at 150 K using MoKα radiation (α = 0.71073 Å) and a Bruker APEX 2 CCD area detector. The sample was prepared for data collection by coating with high viscosity microscope oil (Par-atone-N, Hampton Research). The oil-coated crystal was mounted on a MicroMesh mount (MiTeGen, Inc.) and transferred immediately to the diffractometer. The 0.301 × 0.144 × 0.089 mm3 crystal was monoclinic in space group P21/c with unit cell dimensions a = 13.5627(10) Å, b = 12.8948(7) Å, c = 12. 9598(9) Å, and b = 93.733(4)°. Data were 99.8% complete to 26.404°q. Corrections were applied for Lorentz, polarization, and absorption effects. The structure was solved by direct methods and refined by full-matrix least squares on F2 values using the programs found in the SHELXTL suite (Bruker, SHELXTL v6.10, 2000, Bruker AXS Inc., Madison, WI). The asymmetric unit contains a single molecule. Parameters refined included atomic coordinates and anisotropic thermal parameters for all non-hydrogen atoms. Hydrogen atoms on carbons were included using a riding model [coordinate shifts of C applied to H atoms] with C–H distance set at 0.96 Å.
Atomic coordinates for this compound have been deposited with the Cambridge Crystallographic Data Centre (deposition numbers 1026851). Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK [fax: +44(0) 1223 336033 or deposit@ccdc.cam.ac.uk].
6.3. Radioligand competition binding
Binding at dopamine D2-like receptors was determined using previously described methods.54,55 Membranes were prepared from HEK293 cells expressing human D2R, D3R or D4R, grown in a 50:50 mix of DMEM and Ham's F12 culture media, supplemented with 20 mM HEPES, 2 mM l-glutamine, 0.1 mM non-essential amino acids, 1X antibiotic/antimycotic, 10% heat-inactivated fetal bovine serum, and 200 μg/mL hygromycin (Life Technologies, Grand Island, NY) and kept in an incubator at 37°C and 5% CO2. Upon reaching 80–90% confluence, cells were harvested using pre-mixed Earle's Balanced Salt Solution (EBSS) with 5 μM EDTA (Life Technologies) and centrifuged at 3000 rpm for 10 min at 21 °C. The supernatant was removed and the pellet was resuspended in 10 mL hypotonic lysis buffer (5 mM MgCl2·6H2O, 5 mM Tris, pH 7.4 at 4 °C) and centrifuged at 20,000 rpm for 30 min at 4 °C. The pellet was then resuspended in fresh EBSS buffer made from 8.7 g/L Earle's Balanced Salts without phenol red (US Biological, Salem, MA), 2.2 g/L sodium bicarbonate, pH to 7.4. A Bradford protein assay (Bio-Rad, Hercules, CA) was used to determine the protein concentration and membranes were diluted to 500 μg/mL and stored in a −80 °C freezer for later use.
Radioligand competition binding experiments were conducted using thawed membranes. Test compounds were freshly dissolved in 30% DMSO and 70% H2O to a stock concentration of 100 μM. To assist the solubilization of free-base compounds, 10 μl of glacial acetic acid was added along with the DMSO. Each test compound was then diluted into 13 half-log serial dilutions using 30% DMSO vehicle; final test concentrations ranged from 10 μM to 10 pM. Previously-frozen membranes were diluted in fresh EBSS to a 100 μg/ mL (for hD2R or hD3R) or 200 μg/mL (hD4R) stock for binding. Radioligand competition experiments were conducted in glass tubes containing 300 μl fresh EBSS buffer with 0.2 mM sodium metabisulfite, 50 μl of diluted test compound, 100 μl of membranes (10 μg total protein for hD2R or hD3R, 20 μg total protein for hD4R), and 50 μl of [3H]N-methylspiperone (0.4 nM final concentration; Perkin Elmer). Nonspecific binding was determined using 10 μM butaclamol (Sigma–Aldrich, St. Louis, MO) and total binding was determined with 30% DMSO vehicle. All compound dilutions were tested in triplicate and the reaction incubated for one hour at room temperature. The reaction was terminated by filtration through Whatman GF/B filters, presoaked for one hour in 0.5% polyethylenimine, using a Brandel R48 filtering manifold (Brandel Instruments, Gaithersburg, MD). The filters were washed 3 times with 3 mL of ice cold EBSS buffer and transferred to scintillation vials. 3 mL CytoScint liquid scintillation cocktail (MP Biomedicals, Solon, OH) was added and vials were counted using a Perkin Elmer Tri-Carb 2910 TR liquid scintillation counter (Waltham, MA). IC50 values for each compound were determined from dose–response curves and Ki values were calculated using the Cheng–Prusoff equation;56 these analyses were performed using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA). Reported Ki values were determined from least three independent experiments.
6.4. Mouse microsomal stability assay
Phase I metabolic stability assays were conducted in mouse liver microsomes. The reaction was carried out with 100 mM potassium phosphate buffer, pH 7.4, in the presence of NADPH regenerating system (1.3 mM NADPH, 3.3 mM glucose 6-phosphate, 3.3 mM MgCl2, 0.4 U/mL glucose-6-phosphate dehydrogenase, 50 μM sodium citrate). Reactions in triplicate were initiated by addition of the liver microsomes to the incubation mixture (compound final concentration was 1 μM; 0.2 mg/mL micro-somes). Negative controls in the absence NADPH were carried out to determine the non-CYP mediated metabolism. Positive controls for phase I metabolism (testosterone) were also evaluated. At predetermined times (0, 30 and 60 min), aliquots of the mixture were removed and the reaction quenched by addition of two times the volume of ice cold acetonitrile spiked with the internal standard. Compound disappearance was monitored over time using a liquid chromatography and tandem mass spectrometry (LC/MS/ MS) method. All reactions were performed in triplicate.
Chromatographic analysis was performed using an Accela™ ultra high-performance system consisting of an analytical pump, and an autosampler coupled with TSQ Vantage mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA). Separation of the analyte from potentially interfering material was achieved at ambient temperature using Agilent Eclipse Plus column (100 × 2.1 mm i.d.) packed with a 1.8 μm C18 stationary phase. The mobile phase used was composed of 0.1% formic acid in acetonitrile and 0.1% formic acid in H2O with gradient elution, starting with 10% (organic) linearly increasing to 99% up to 2.5 min, maintaining at 99% (2.5–3.5 min) and reequlibrating to 10% by 4.5 min. The total run time for each analyte was 4.5 min. The mass transitions used for each compound are as follows; PG648 461.148 > 213.106, 143.989; OMO05-057 473.110 > 155.996; 17a 485.064 > 301.126, 129.998; 17b 496.850 > 301.126, 129.998; 16 447.130 > 263.198, 129.999
Supplementary Material
Acknowledgments
This research was funded by the NIDA Intramural Research Program. T.M.K., A.K.B., R.D.S., A.B. and O.M.O.-B. were supported by NIH Postdoctoral Intramural Research Training Award (IRTA) Fellowships. C.B. was supported by an NIH Post-baccalaureate IRTA Fellowship. Crystallographic studies were supported by NIDA contract Y1-DA1101. HEK 293 cells were generously provided by Dr. David Sibley of NINDS. Thanks to Ben Free and Theresa Kopajtic for help in developing the radioligand binding assay. Thanks to Catherine Schweppe and Michael Ellenberger for technical assistance with the radioligand binding studies. Thanks to Elie Pommier for technical assistance with the metabolic studies.
Abbreviations
- CuAAC
copper-catalyzed azide-alkyne cycloaddition
- DIPEA
diisopropylethylamine
- D2R
dopamine D2 receptor
- D3R
dopamine D3 receptor
- GPCR
G protein-coupled receptors
- SAR
structure–activity relationship
- TBTA
tris(benzyltriazolylmethyl)amine
Footnotes
Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2015.01.017.
References and notes
- 1.Beaulieu JM, Gainetdinov RR. Pharmacol. Rev. 2011;63:182. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- 2.Gross G, Drescher K. Handb. Exp. Pharmacol. 2012:167. doi: 10.1007/978-3-642-25758-2_7. [DOI] [PubMed] [Google Scholar]
- 3.Joyce JN, Millan MJ. Curr. Opin. Pharmacol. 2007;7:100. doi: 10.1016/j.coph.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 4.Bezard E, Ferry S, Mach U, Stark H, Leriche L, Boraud T, Gross C, Sokoloff P. Nat. Med. 2003;9:762. doi: 10.1038/nm875. [DOI] [PubMed] [Google Scholar]
- 5.Newman AH, Blaylock BL, Nader MA, Bergman J, Sibley DR, Skolnick P. Biochem. Pharmacol. 2012;84:882. doi: 10.1016/j.bcp.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newman AH, Grundt P, Nader MA. J. Med. Chem. 2005;48:3663. doi: 10.1021/jm040190e. [DOI] [PubMed] [Google Scholar]
- 7.Heidbreder CA, Newman AH. Ann. N. Y. Acad. Sci. 2010;1187:4. doi: 10.1111/j.1749-6632.2009.05149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sokoloff P, Giros B, Martres MP, Bouthenet ML, Schwartz JC. Nature. 1990;347:146. doi: 10.1038/347146a0. [DOI] [PubMed] [Google Scholar]
- 9.Searle G, Beaver JD, Comley RA, Bani M, Tziortzi A, Slifstein M, Mugnaini M, Griffante C, Wilson AA, Merlo-Pich E, Houle S, Gunn R, Rabiner EA, Laruelle M. Biol. Psychiatry. 2010;68:392. doi: 10.1016/j.biopsych.2010.04.038. [DOI] [PubMed] [Google Scholar]
- 10.Cho DI, Zheng M, Kim KM. Arch. Pharm. Res. 2010;33:1521. doi: 10.1007/s12272-010-1005-8. [DOI] [PubMed] [Google Scholar]
- 11.Gurevich EV, Joyce JN. Neuropsychopharmacology. 1999;20:60. doi: 10.1016/S0893-133X(98)00066-9. [DOI] [PubMed] [Google Scholar]
- 12.Keck TM, Burzynski C, Shi L, Newman AH. Adv. Pharmacol. 2014;69:267. doi: 10.1016/B978-0-12-420118-7.00007-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boeckler F, Gmeiner P. Pharmacol. Ther. 2006;112:281. doi: 10.1016/j.pharmthera.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 14.Micheli F, Heidbreder C. Expert Opin. Ther. Pat. 2013;23:363. doi: 10.1517/13543776.2013.757593. [DOI] [PubMed] [Google Scholar]
- 15.Robarge MJ, Husbands SM, Kieltyka A, Brodbeck R, Thurkauf A, Newman AH. J. Med. Chem. 2001;44:3175. doi: 10.1021/jm010146o. [DOI] [PubMed] [Google Scholar]
- 16.Michino M, Donthamsetti P, Beuming T, Banala A, Duan L, Roux T, Han Y, Trinquet E, Newman AH, Javitch JA, Shi L. Mol. Pharmacol. 2013;84:854. doi: 10.1124/mol.113.087833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banala AK, Levy BA, Khatri SS, Furman CA, Roof RA, Mishra Y, Griffin SA, Sibley DR, Luedtke RR, Newman AH. J. Med. Chem. 2011;54:3581. doi: 10.1021/jm200288r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Testa B, Mayer JM. The Hydrolysis of Amides In Hydrolysis in Drug and Prodrug Metabolism. Verlag Helvetica Chimica Acta; 2006. p. 81. [Google Scholar]
- 19.Fukami T, Yokoi T. Drug Metab. Pharmacokinet. 2012;27:466. doi: 10.2133/dmpk.dmpk-12-rv-042. [DOI] [PubMed] [Google Scholar]
- 20.Brik A, Alexandratos J, Lin YC, Elder JH, Olson AJ, Wlodawer A, Goodsell DS, Wong CH. ChemBioChem. 2005;6:1167. doi: 10.1002/cbic.200500101. [DOI] [PubMed] [Google Scholar]
- 21.Monceaux CJ, Hirata-Fukae C, Lam PC, Totrov MM, Matsuoka Y, Carlier PR. Bioorg. Med. Chem. Lett. 2011;21:3992. doi: 10.1016/j.bmcl.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 22.Bourne Y, Kolb HC, Radic Z, Sharpless KB, Taylor P, Marchot P. Proc. Natl. Acad. Sci. U. S. A. 2004;101:1449. doi: 10.1073/pnas.0308206100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adibekian A, Martin BR, Wang C, Hsu K-L, Bachovchin DA, Niessen S, Hoover H, Cravatt BF. Nat. Chem. Biol. 2011;7:469. doi: 10.1038/nchembio.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwak JM, Moon JS, Choi JI, Murugan RN, Park WK, Gong JY, Lee HY, Koh HY. Bull. Korean Chem. Soc. 2013;34:3467. [Google Scholar]
- 25.Insua I, Alvarado M, Masaguer CF, Iglesias A, Brea J, Loza MI, Carro L. Bioorg. Med. Chem. Lett. 2013;23:5586. doi: 10.1016/j.bmcl.2013.08.047. [DOI] [PubMed] [Google Scholar]
- 26.Micheli F. ChemMedChem. 2011;6:1152. doi: 10.1002/cmdc.201000538. [DOI] [PubMed] [Google Scholar]
- 27.Micheli F, Arista L, Bertani B, Braggio S, Capelli AM, Cremonesi S, Di-Fabio R, Gelardi G, Gentile G, Marchioro C, Pasquarello A, Provera S, Tedesco G, Tarsi L, Terreni S, Worby A, Heidbreder C. J. Med. Chem. 2010;53:7129. doi: 10.1021/jm100832d. [DOI] [PubMed] [Google Scholar]
- 28.Elrashidi MY, Ebbert JO. Expert Opin. Emerg. Drugs. 2014;19:243. doi: 10.1517/14728214.2014.899580. [DOI] [PubMed] [Google Scholar]
- 29.Tron GC, Pirali T, Billington RA, Canonico PL, Sorba G, Genazzani AA. Med. Res. Rev. 2008;28:278. doi: 10.1002/med.20107. [DOI] [PubMed] [Google Scholar]
- 30.Kolb HC, Sharpless KB. Drug Discov. Today. 2003;8:1128. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 31.Lutz JF, Zarafshani Z. Adv. Drug Deliv. Rev. 2008;60:958. doi: 10.1016/j.addr.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 32.Gryder BE, Akbashev MJ, Rood MK, Raftery ED, Meyers WM, Dillard P, Khan S, Oyelere AK. ACS Chem. Biol. 2013;8:2550. doi: 10.1021/cb400542w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem., Int. Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 34.Li H, Zhu Z, Fahrenbach AC, Savoie BM, Ke C, Barnes JC, Lei J, Zhao YL, Lilley LM, Marks TJ, Ratner MA, Stoddart JF. J. Am. Chem. Soc. 2013;135:456. doi: 10.1021/ja310060n. [DOI] [PubMed] [Google Scholar]
- 35.Carboni B, Vaultier M, Carrié R. Tetrahedron. 1987;43:1799. [Google Scholar]
- 36.Shin JA, Lim YG, Lee KH. J. Org. Chem. 2012;77:4117. doi: 10.1021/jo3000095. [DOI] [PubMed] [Google Scholar]
- 37.Azizi N, Saidi MR. Org. Lett. 2005;7:3649. doi: 10.1021/ol051220q. [DOI] [PubMed] [Google Scholar]
- 38.Li Y, Shen M, Zhang Z, Luo J, Pan X, Lu X, Long H, Wen D, Zhang F, Leng F, Li Y, Tu Z, Ren X, Ding K. J. Med. Chem. 2012;55:10033. doi: 10.1021/jm301188x. [DOI] [PubMed] [Google Scholar]
- 39.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Org. Lett. 2004;6:2853. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 40.Hein JE, Fokin VV. Chem. Soc. Rev. 2010;39:1302. doi: 10.1039/b904091a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Himo F, Lovell T, Hilgraf R, Rostovtsev VV, Noodleman L, Sharpless KB, Fokin VV. J. Am. Chem. Soc. 2005;127:210. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]
- 42.Rodríguez JG, de los Rios C, Lafuente A. Tetrahedron. 2005;61:9042. [Google Scholar]
- 43.Yuan J, Chen X, Brodbeck R, Primus R, Braun J, Wasley JW, Thurkauf A. Bioorg. Med. Chem. Lett. 1998;8:2715. doi: 10.1016/s0960-894x(98)00469-7. [DOI] [PubMed] [Google Scholar]
- 44.Newman AH, Grundt P, Cyriac G, Deschamps JR, Taylor M, Kumar R, Ho D, Luedtke RR. J. Med. Chem. 2009;52:2559. doi: 10.1021/jm900095y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grundt P, Prevatt KM, Cao J, Taylor M, Floresca CZ, Choi JK, Jenkins BG, Luedtke RR, Newman AH. J. Med. Chem. 2007;50:4135. doi: 10.1021/jm0704200. [DOI] [PubMed] [Google Scholar]
- 46.Newman AH, Beuming T, Banala AK, Donthamsetti P, Pongetti K, LaBounty A, Levy B, Cao J, Michino M, Luedtke RR, Javitch JA, Shi L. J. Med. Chem. 2012;55:6689. doi: 10.1021/jm300482h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pokhodylo NT, Matiichuk VS, Obushak ND. Russ. J. Org. Chem. 2010;46:894. [Google Scholar]
- 48.Hachiya S, Asai K, Konishi G-I. Tetrahedron Lett. 1839;2013:54. [Google Scholar]
- 49.Görl C, Alt HG. J. Organomet. Chem. 2007;692:4580. [Google Scholar]
- 50.Sirivolu VR, Vernekar SK, Ilina T, Myshakina NS, Parniak MA, Wang Z. J. Med. Chem. 2013;56:8765. doi: 10.1021/jm401232v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gulykina NS, Dolgina TM, Bondarenko GN, Beletskaya IP. Russ. J. Org. Chem. 2003;39:797. [Google Scholar]
- 52.Feng YS, Xie CQ, Qiao WL, Xu HJ. Org. Lett. 2013;15:936. doi: 10.1021/ol400099h. [DOI] [PubMed] [Google Scholar]
- 53.Nishi M, Kuninobu Y, Takai K. Org. Lett. 2012;14:6116. doi: 10.1021/ol302810u. [DOI] [PubMed] [Google Scholar]
- 54.Zou MF, Cao J, Kopajtic T, Desai RI, Katz JL, Newman AH. J. Med. Chem. 2006;49:6391. doi: 10.1021/jm060762q. [DOI] [PubMed] [Google Scholar]
- 55.Chen J, Levant B, Jiang C, Keck TM, Newman AH, Wang S. J. Med. Chem. 2014;57:4962. doi: 10.1021/jm401798r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng Y, Prusoff WH. Biochem. Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.