

Abstract
Optimization of the sulfonamide-based kappa opioid receptor (KOR) antagonist probe molecule ML140 through constraint of the sulfonamide nitrogen within a tetrahydroisoquinoline moiety afforded a marked increase in potency. This strategy, when combined with additional structure-activity relationship exploration, has led to a compound only six-fold less potent than norBNI, a widely utilized KOR antagonist tool compound, but significantly more synthetically accessible. The new optimized probe is suitably potent for use as an in vivo tool to investigate the therapeutic potential of KOR antagonists.
Keywords: Kappa Opioid Receptor, Antagonist, Molecular constraint, Potency enhancement, Tetrahydroisoquinoline
1. Introduction
Activation of the kappa opioid receptor (KOR) by endogenous neuropeptides, primarily dynorphin, initiates complex signaling cascades. The downstream effects of KOR agonism vary greatly and include antinociception, dysphoria and anxiety, though the details of the pharmacological pathways are still being elucidated.12 In contrast, KOR antagonists have been investigated as potential therapeutic treatments for addiction, depression, post traumatic stress disorder, eating disorders and other conditions related to anxiety or aversion-reward responses.3, 4, 5 Many canonical KOR antagonists (Fig. 1) are derived from, or bear a structural element of morphinan opioids, such as the widely-utilized tool compounds norBNI,6 5′-GNTI7 and JDTic.8 Some KOR antagonists that are widely used, such as NorBNI, have a remarkable long duration of action in animal models (weeks), which could introduce difficulties in interpreting the effect of chronic antagonist exposure, or some other effects of the drug that are not fully understood.9, 10, 11, 12 Accordingly, one goal of our laboratories is to discover and evaluate structurally-distinct KOR antagonists with novel pharmacological properties, including a focus on KOR antagonists that are more rapidly cleared in vivo. We recently disclosed a new sulfonamide series of KOR antagonists, exemplified by the Molecular Libraries probe molecule ML140 (Figure 1).13
Figure 1.

Representative widely utilized KOR antagonist ligands and ML140.
Although ML140 only exhibited modest potency in the DiscoveRx βarrestin2 PathHunter™ assay (403 nM), it was selective against the μ and δ receptors (IC50 values of > 24 and >32 uM, respectively). In contrast to the better-established chemotypes noted, ML140 is highly modular, possesses no stereogenic centers and bears little structural similarity to known opioid ligands. Together, these properties inspired us to further investigate the structure–activity relationship (SAR) of this chemotype. In our original report,13 we evaluated the effect on biological potency through ca. forty structural changes focused primarily on the “eastern” portion of the molecule (Figure 1). We investigated both replacing the substituents of the basic nitrogen as well as introducing constraint into the eastern region of the molecule. Perhaps most interestingly, we found that replacing the N-isopropyl with N-tert-butyl can increase the potency by as much as 14-fold. Less successful were our attempts to enhance antagonist potency through constraint of the nitrogen within various ring systems as illustrated by the structures in Figure 2.
Figure 2.

Conformational constraints explored in ML140.
Each of these changes eradicated essentially all KOR activity, suggesting that the constraints examined placed this portion of the molecule into a conformation inconsistent with receptor activity, introduced unpalatable steric clashes, deprived the compound of an important hydrogen bond, or some combination of these effects. Herein we describe a more fruitful approach to conformational constraint by focusing on the modification of the sulfonamide portion of the molecule while retaining the promising substitution patterns around the basic nitrogen. This process afforded a series of antagonists with single-digit nanomolar potency in G protein coupling and consistent SAR trends across the series.
2. Results and discussion
In this investigation we measured the potency of all test compounds by three complementary methods for assessing KOR antagonist potency:14 [35S]GTPγS binding,15 ERK activation,16 and β-arrestin2 recruitment.17 Since our compounds, like norBNI, are found to be most potent in the [35S]GTPγS assay over the other two, we chose this assay for primary SAR determination. The analogues presented here were prepared in the manner of those previously reported,11 which is summarized in Scheme 1. Briefly, the tetrahydroisoquinoline core was reacted with the appropriate sulfonyl chloride and subsequently saponified to afford the western fragment bearing a carboxylic acid. Activation of the carboxylic acid to the acid chloride and coupling with the diamine fragments afforded the final analogues. All final compounds were purified by mass-directed, reverse-phase HPLC and submitted to biological assays at ≥94% purity.
Scheme 1.
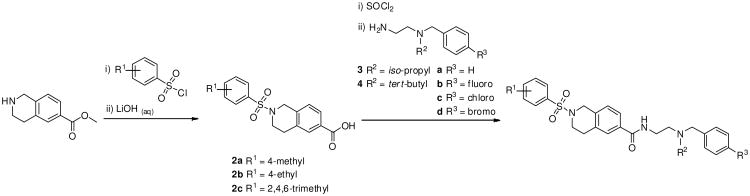
General scheme for the preparation of 2-(Arylsulfonyl)-1,2,3,4-tetrahydroisoquinoline-6-carboxamide derivatives.
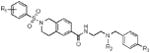
We began this investigation by preparing the analogue where both the sulfonamide and amide nitrogens of ML140 were methylated, which resulted in the loss of all KOR antagonist activity (Table 1, entry 1a) and suggesting that at least one of or both N-H groups were essential for activity. To determine if modification at the sulfonamide nitrogen was allowable, we first investigated the effect of tethering the sulfonamide nitrogen to the central benzene ring. We chose the tetrahydroisoquinoline carboxylate scaffold since it was both readily available and mapped directly onto the template established by ML140. Thus, we synthesized twelve tetrahydroisoquinoline analogues bearing modifications around the basic nitrogen fragment and substitution on the aryl sulfonamide group. The results of screening these analogues in the three KOR antagonist assays are presented in Table 1. To facilitate comparison to our previous SAR efforts and the broader KOR antagonist literature, norBNI, ML140, and the bromo analogue of ML140, 1b, were included in this study. Notably, the tetrahydroisoquinoline analogues were all significantly more potent than either ML140 or 1b. Directly comparing 1b to the tetrahydroisoquinolinone with the same substitution, 1f, shows an eight-fold improvement in potency. A further two-fold increase in potency was achieved through a bromo to chloro replacement (i.e., 1f to 1e). The effect of changing from isopropyl to tert-butyl was even more pronounced than was observed in the context of ML140, providing for the first time in this series compounds possessing single-digit nanomolar potency. Halogen incorporation further improved the potency, resulting in the nearly equipotent fluoro analogue 1j, chloro analogue 1l and the corresponding bromide 1m. To date, compound 1l is the most potent compound in the series, as measured by its activity in inhibiting G protein function. Throughout this study, 2,4,6-trimethyl aryl sulfonamide substitution led to notably less potent compounds when compared to either the benzene sulfonamide, 4-methyl- or 4-ethyl-substituted aryl sulfonamide analogues. However they are still more potent than ML140, nicely illustrating the positive influence of the tetrahydroisoquinoline moiety on the SAR of this chemotype.
Table 1.
KOR antagonist potency of ML140 analogues.
| entry/cmpd |
|
IC50 (nM) ± S.E.Ma | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| R1 | R2 | R3 | [35S]GTPγS | ERK | βarr2 | |
| norBNI | – | – | – | 0.28 ± 0.03 | 4.6 ± 0.7 | 2.5 ± 0.3 |
| ML140 | – | – | – | 138 ± 54 | 591 ± 87 | 403 ± 86b |
| 1a |
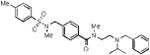
|
inactive | inactive | inactive | ||
| 1b |
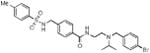
|
168 ± 48 | 750 ± 170 | 937 ± 190c | ||
| 1c | 2,4,6-Me | i-Pr | H | 113 ± 42 | 1,120 ± 110 | 930 ± 150 |
| 1d | 4-Me | i-Pr | F | 29.1 ± 7.9 | 406 ± 80 | 478 ± 110 |
| 1e | 4-Me | i-Pr | Cl | 10.0 ± 1.4 | 400 ± 87 | 392 ± 91 |
| 1f | 4-Me | i-Pr | Br | 19.7 ± 6.6 | 258 ± 93 | 388 ± 85 |
| 1g | 4-Et | i-Pr | Br | 33 ± 11 | 314 ± 56 | 350 ± 68 |
| 1h | 4-Me | tert-Bu | H | 5.3 ± 0.6 | 49.1 ± 9.7 | 104 ± 8 |
| 1i | 4-Et | tert-Bu | H | 3.4 ± 0.4 | 143 ± 23 | 186 ± 57 |
| 1j | 4-Me | tert-Bu | F | 3.7 ± 1.2 | 76 ± 24 | 104 ± 32 |
| 1k | 2,4,6-Me | tert-Bu | F | 85 ± 20 | 1,200 ± 300 | 880 ± 67 |
| 1l | 4-Me | tert-Bu | Cl | 1.6 ± 0.5 | 107 ± 17 | 84 ± 20 |
| 1m | 4-Me | tert-Bu | Br | 2.9 ± 1.2 | 87 ± 17 | 93 ± 30 |
| 1n | 2,4,6-Me | tert-Bu | Br | 106 ± 16 | 1,340 ± 410 | 710 ± 150 |
n≥3. All compounds fully blocked U69,593 (>86%). Test compounds were run against the approximate EC80 of U69,593.
Previous testing of this compound at Sanford-Burnham Medical Research Institute measured a βarrestin2 PathHunter™ assay potency of 860 nM; here we report the contemporary and optimized value of 403 nM.13
Previous testing of this compound at Sanford-Burnham Medical Research Institute measured a DiscoveRx βarrestin2 PathHunter™ assay potency of 660 nM; here we report the contemporary and optimized value of 937 nM.13
Another interesting feature is the differential potencies noted for many of the analogs against G protein function, as revealed in the [35S]GTPγS assay, relative to βarrestin2 recruitment or ERK phosphorylation. For example, simple comparison of the relevant IC50 values obtained in each assay suggests that the compounds may be more potent in inhibiting G protein function as opposed to βarrestin2 recruitment, although they are less potent than norBNI in both assays. Whether this suggests a bias in the affinity to imparting differential active states for KOR remains to be determined as such mathematical modeling parameters have yet to be defined for comparing compounds that do not reveal stimulatory efficacy. Differences also appear in relative potencies for inhibiting ERK phosphorylation, but may vary because the ERK is downstream in both the G protein and βarrestin2 pathways. Although possibly intriguing in light of current interest in functional selectivity in GPCR agonism, the functional relevance of any such differences in the present series of antagonists is currently unknown.
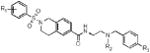
We had previously profiled the lead compound for this study, ML140, in radioligand binding assays and observed selectivity for the KOR compared to the δ opioid receptor (DOR) and modest selectivity compared to the μ opioid receptor (MOR).13 To determine if ML140 analogues had off target effects, 1f, 1h and 1l were also profiled through the Psychoactive Drug Screening Program at the University of North Carolina, Chapel Hill for KOR, DOR and MOR binding affinity (Table 2). Gratifyingly, the more potent antagonists were found to be more selective in radioligand binding assays than ML140. For example, 1l possessed a binding affinity (Ki) at KOR of 53 nM and binding affinity (Ki) of >10,000 nM at both the DOR and MOR.
Table 2.
KOR, DOR and MOR binding affinity for ML140 and select analogues.
| entry/cmpd |
|
receptor binding affinity, Ki values (nM)a | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| R1 | R2 | R3 | KOR | DOR | MOR | |
| ML140 | – | – | – | 50 | 1,088 | 306 |
| 1f | 4-Me | i-Pr | Br | 73 | 5,190 | 8,170 |
| 1h | 4-Me | tert-Bu | H | 37 | 2,328 | 1,749 |
| 1l | 4-Me | tert-Bu | Cl | 53 | >10,000 | >10,000 |
Ki determinations calculated from a 12-point concentration response curve (n=1) conducted by the Psychoactive Drug Screening Program using the rat KOR expressed in HEK-293 cells. In those studies, the KD of U69,593 was determined to be 1 nM.
3. Conclusion
We have improved the antagonist potency of the sulfonamide-based KOR antagonist probe molecule ML140 from 138 nM to 1.6 nM as measured by [35S]GTPγS binding to afford a new molecular tool appropriate to investigate the physiological role of the KOR and the therapeutic potential of KOR antagonists. The constrained sulfonamide analogue 1l is almost 100-fold more potent than ML140 and permits for the first time the potential use of this chemotype in behavioral or in vivo efficacy models. These experiments are currently planned as well as further pharmacological characterization and the synthesis of additional analogues. The results of these studies will be disclosed as appropriate.
4. Experimental
4.1 General methods
All reagents and materials were purchased from commercial vendors (Sigma, Alfa Aesar, Oakwood or ASW Medchem) and used as received. Ethyl ether, toluene, THF, MeCN and CH2Cl2 were degassed with nitrogen and passed through two columns of basic alumina on an Innovative Technology solvent purification system. 1H and 13C NMR spectra were recorded on a Bruker AM 400 spectrometer (operating at 400 and 100 MHz respectively) in CDCl3 with 0.03% TMS as an internal standard, unless otherwise specified. Chemical shifts are reported in parts per million (ppm) downfield from TMS. 13C multiplicities were determined with the aid of an APT pulse sequence, differentiating the signals for methyl and methane carbons as “d” from methylene and quarternary carbons as “u”. The infrared (IR) spectra were acquired as thin films using a universal ATR sampling accessory on a PerkinElmer Spectrum 100 FT-IR spectrometer and the absorption frequencies are reported in cm-1. Melting points were determined on a Stanford Research Systems Optimelt automated melting point system interfaced through a PC and are uncorrected. ML140 and 1b were synthesized as previously described.13
HPLC/MS analysis was carried out with gradient elution (5% CH3CN to 100% CH3CN) on an Agilent 1200 RRLC with a photodiode array UV detector and an Agilent 6224 TOF mass spectrometer (also used to produce high resolution mass spectra). HPLC purification was carried out by mass-directed fractionation (MDF) with gradient elution (a narrow CH3CN gradient was chosen based on the retention time of the target from LCMS analysis of the crude sample) on an Agilent 1200 instrument with photodiode array detector, an Agilent 6120 quadrupole mass spectrometer, and a HTPAL LEAP autosampler. Fraction collection was triggered using an MS and UV threshold determined by HPLC/MS analysis of the crude sample. One of two column/mobile phase conditions was chosen for both analysis and purification to promote the target compound's neutral state (0.02% formic acid with Waters Atlantis T3 5um, 19 × 150 mm; or pH 9.8 NH4OH with Waters XBridge C18 5 um, 19 × 150 mm).
4.2 Synthesis of carboxylic acid fragments 2a-2c
4.2.1.1. Methyl 2-tosyl-1,2,3,4-tetrahydroisoquinoline-6-carboxylate
To a solution of methyl 1,2,3,4-tetrahydroisoquinoline-6-carboxylate (171 mg, 0.894 mmol) and triethylamine (271 mg, 2.68 mmol, 3.0 equiv) in CH2Cl2 (15 mL) was added p-toluenesulfonyl chloride (256 mg, 1.34 mmol, 1.5 equiv). The reaction was stirred at rt for 15 h, diluted with 1 N HCl and extracted with CH2Cl2. The combined organics were dried (Na2SO4), concentrated and purified by silica gel chromatography to afford the sulfonamide product as a white solid (231 mg, 0.669 mmol, 75% yield). Mp = 143–145 °C; Rf = 0.34 (25% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 2.41 (s, 3H), 2.96 (t, J = 6.0 Hz, 2H), 3.36 (t, J = 6.0 Hz, 2H), 3.88 (s, 3H),4.28 (s, 2H), 7.10 (d, J = 8.0 Hz, 1H), 7.33 (d, J = 8.0 Hz, 2H), 7.72 (d, J = 8.0 Hz, 2H), 7.77 (s, 1H), 7.79 (d, J = 8.0 Hz, 1H); 13C NMR (101 MHz, CDCl3, APT pulse sequence) δ d 21.5, 52.1, 126.6, 127.4, 127.7, 129.8, 130.2; u 28.8, 43.6, 47.7, 128.7, 133.2, 133.4, 136.9, 143.9, 166.7; IR 1718 cm-1; HRMS (ESI) m/z calcd for C18H20NO4S ([M+H]+) 346.1108, found 346.1116.
4.2.1.2. 2-Tosyl-1,2,3,4-tetrahydroisoquinoline-6-carboxylicacid (2a)
To a solution of the above methyl ester (298 mg, 0.765 mmol) in THF:MeOH:water (3:1:1, 10 mL) was added lithium hydroxide monohydrate (160 mg, 3.83 mmol, 5 equiv) and the reaction stirred at rt for 15 h. The THF and MeOH were removed in vacuo and the reaction concentrate was acidified with 2 N HCl, precipitating the carboxylic acid product as a sparingly soluble white solid (213 mg, 0.643 mmol, 84% yield), which was filtered, washed with water, dried under vacuum and used without further purification. Mp = 234–240 °C; Rf = 0.58 (10% MeOH and 2% AcOH in CH2Cl2); 1H NMR (400 MHz, DMSO-d6) δ 2.39 (s, 3H), 2.91 (t, J = 6.0 Hz, 2H), 3.30 (t, J = 6.0 Hz, 2H), 4.25 (s, 2H), 7.28 (d, J = 8.0 Hz, 1H), 7.44 (d, J = 8.0 Hz, 2H), 7.70–7.74 (complex, 4H), 12.89 (br s, 1H); 13C NMR (101 MHz, DMSO-d6, APT pulse sequence) δ d 21.0, 126.7, 126.9, 127.4, 129.7, 129.9; u 27.9, 43.3, 47.3, 129.1, 133.0, 133.4, 136.7, 143.7, 167.0; IR 1678 cm-1; HRMS (ESI) m/z calcd for C17H16NO4S ([M-H]-) 330.0806, found 330.0807.
4.2.2.1. Methyl 2-((4-ethylphenyl)sulfonyl)-1,2,3,4-tetrahydroisoquinoline-6-carboxylate
Methyl 1,2,3,4-tetrahydroisoquinoline-6-carboxylate (182 mg, 0.952 mmol) and 4-ethyl-benzenesulfonyl chloride (292 mg, 1.43 mmol, 1.5 equiv) were reacted according to the protocol in section 4.2.1.1. to afford the sulfonamide product as a white solid (285 mg, 0.793 mmol, 83% yield). Mp =128–130 °C; Rf = 0.40 (25% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 1.23 (t, J = 7.6 Hz, 3H), 2.69 (q, J = 7.6 Hz, 2H), 2.95 (t, J = 6.0 Hz, 2H), 3.36 (t, J = 6.0 Hz, 2H), 3.87 (s, 3H), 4.28 (s, 2H), 7.09 (d, J = 8.4 Hz, 1H), 7.35 (d, J = 8.4 Hz, 2H), 7.74–7.79 (complex, 4H); 13C NMR (101 MHz, CDCl3, APT pulse sequence) δ d 14.9, 51.9, 126.4, 127.2, 127.7, 128.5, 130.0; u 28.62, 28.65, 43.5, 47.6, 128.6, 133.2, 133.3, 136.8, 149.8, 166.5; IR 1717 cm-1; HRMS (ESI) m/z calcd for C19H22NO4S ([M+H]+) 360.1264, found 360.1274.
4.2.2.2. 2-((4-Ethylphenyl)sulfonyl)-1,2,3,4-tetrahydroisoquinoline-6-carboxylic acid (2b)
The above methyl ester (252 mg, 0.701 mmol) was reacted according the protocol in section 4.2.1.2. to afford the carboxylic acid product as a sparingly soluble white solid (216 mg, 0.625 mmol, 89% yield), which was filtered, washed with water, dried under vacuum and used without further purification. Mp = 208– 213 °C; Rf = 0.58 (10% MeOH and 2% AcOH in CH2Cl2); 1H NMR (400 MHz, DMSO-d6) δ 1.18 (t, J = 7.6 Hz, 3 H), 2.68 (q, J = 7.6 Hz, 2H), 2.91 (t, J = 6.0 Hz, 2H), 3.31 (t, J = 6.0 Hz, 2H), 4.26 (s, 2H), 7.28 (d, J = 8.0 Hz, 1H), 7.46 (d, J = 8.0 Hz, 2H), 7.69–7.76 (complex, 4H), 12.91 (br s, 1H); 13C NMR (101 MHz, DMSO-d6, APT pulse sequence) δ d 15.0, 126.7, 126.8, 127.5, 128.7, 129.7; u 28.0, 43.3, 47.3, 129.1, 133.2, 133.4, 136.6, 149.6, 167.0; IR 1683 cm-1; HRMS (ESI) m/z calcd for C18H20NO4S ([M+H]+) 346.1113, found 346.1114.
4.2.3.1. Methyl 2-((2,4,6-trimethylphenyl)sulfonyl)-1,2,3,4-tetrahydroisoquinoline-6-carboxylate
Methyl 1,2,3,4-tetrahydroisoquinoline-6-carboxylate (191 mg, 0.999 mmol) and 2,4,6-trimethyl-benzenesulfonyl chloride (328 mg, 1.50 mmol, 1.5 equiv) were reacted according to the protocol in section 4.2.1.1. to afford the sulfonamide product as a white solid (188 mg, 0.503 mmol, 50% yield). Mp = 142–143 °C; Rf = 0.50 (25% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 2.29 (s, 3H), 2.63 (s, 6H), 2.91 (t, J = 6.0 Hz, 2H), 3.47 (t, J = 6.0 Hz, 2H), 3.89 (s, 3H), 4.40 (s, 2H), 6.96 (s, 2H), 7.12 (d, J = 8.0 Hz, 1H), 7.81 (m, 2H); 13C NMR (101 MHz, CDCl3, APT pulse sequence) δ d 21.0, 22.8, 52.1, 126.6, 127.4, 130.3, 132.0; u 28.6, 41.9, 45.9, 128.7, 131.7, 133.8, 137.3, 140.5, 142.9, 166.7; IR 1718 cm-1; HRMS (ESI) m/z calcd for C20H24NO4S ([M+H]+) 374.1426, found 374.1424.
4.2.3.2. 2-((2,4,6-Trimethylphenyl)sulfonyl)-1,2,3,4-tetrahydroisoquinoline-6-carboxylic acid (2c)
The above methyl ester (146 mg, 0.391 mmol) was reacted according to the protocol in section 4.2.1.2. to afford the carboxylic acid product as a sparingly soluble white solid (137 mg, 0.381 mmol, 97% yield), which was filtered, washed with water, dried under vacuum and used without further purification. Mp = 222–233 °C; Rf = 0.55 (10% MeOH and 2% AcOH in CH2Cl2); 1H NMR (400 MHz, DMSO-d6) δ 2.28 (s, 3H), 2.55 (s, 6H), 2.87 (t, J = 5.6 Hz, 2H), 3.43 (t, J = 5.6 Hz, 2H), 4.36 (s, 2H), 7.08 (s, 2H), 7.31 (d, J = 7.6 Hz, 1H), 7.71–7.73 (m, 2H); 13C NMR (101 MHz, DMSO-d6, APT pulse sequence) δ d 20.5, 22.3, 126.8, 126.9, 129.9, 131.9; u 27.8, 41.6, 45.5, 129.2, 133.8, 137.2, 139.6, 142.6, 167.1; IR 1683 cm-1; HRMS (ESI) m/z calcd for C19H22NO4S ([M+H]+) 360.1264, found 360.1270.
4.3. Conversion of carboxylic acids 2a-2c to the acid chlorides
4.3.1. 2-Tosyl-1,2,3,4-tetrahydroisoquinoline-6-carbonylchloride
The carboxylic acid 2a (145 mg, 0.438 mmol) was dissolved in thionyl chloride (0.95 mL, 13.13 mmol, 30 equiv) and heated at 65 °C for 4 h. Excess thionyl chloride was removed in vacuo and the residue azeotropically dried with toluene (3 × 5 mL) to afford the acid chloride as a white solid (147 mg, 0.420 mmol, 96% yield), which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 2.36 (s, 3H), 2.94 (t, J = 5.9 Hz, 2H), 3.32 (t, J = 5.9 Hz, 2H), 4.25 (s, 2H), 7.11 (d, J = 8.2 Hz, 1H), 7.25–7.30 (m, 2H), 7.63–7.69 (m, 2H), 7.77–7.85 (m, 2H).
4.3.2. 2-((4-Ethylphenyl)sulfonyl)-1,2,3,4-tetrahydroisoquinoline-6-carbonyl chloride
The carboxylic acid 2b (303 mg, 0.878 mmol) was reacted according to the protocol in section 4.3.1. to afford the acid chloride as a white solid (322 mg, 0.860 mmol, 98% yield), which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 1.19 (t, J = 7.6 Hz, 3H), 2.65 (q, J = 7.6 Hz, 2H), 2.95 (t, J = 5.9 Hz, 2H), 3.33 (t, J = 5.9 Hz, 2H), 4.25 (s, 2H), 7.11 (d, J = 8.2 Hz, 1H), 7.27–7.32 (m, 2H), 7.65–7.71 (m, 2H), 7.77- 7.85 (m, 2H).
4.3.3. 2-(Mesitylsulfonyl)-1,2,3,4-tetrahydroisoquinoline-6-carbonyl chloride
The carboxylic acid 2c (38 mg, 0.106 mmol) was reacted according to the protocol in section 4.3.1. to afford the acid chloride as a white solid (39 mg, 0.103 mmol, 98% yield), which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 2.31 (s,, 3H), 2.63 (s, 6H), 2.95 (t, J = 6.0 Hz, 2H), 3.493 (t, J = 6.0 Hz, 2H), 4.43 (s, 2H), 6.97 (s, 2H), 7.21 (d, J = 8.0 Hz, 1H), 7.88 (s, 1H), 7.90 (d, J = 8.4 Hz, 1H).
4.4. Synthesis of diamine fragments 4a-4d
4.4.1.1. 2-(Benzyl(tert-butyl)amino)acetonitrile
To a solution of N-benzyl-tert-butylamine (930 mg, 5.70 mmol) in MeCN (15 mL) was added K2CO3 (1,575 mg, 11.40 mmol, 2 equiv), potassium iodide (946 mg, 5.70 mmol, 1 equiv) and chloroacetonitrile (1,720 mg, 22.79 mmol, 4 equiv). The reaction was heated at 75 °C for 16 h, cooled to rt and partitioned between water (150 mL) and ethyl ether (3 × 75 mL). The combined organic layers were washed with brine (50 mL), dried (Na2SO4), adsorbed onto celite and purified by silica gel chromatography to afford the nitrile product as a colorless oil (846 mg, 4.18 mmol, 73% yield). Rf = 0.49 (10% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 1.29 (s, 9H), 3.43 (s, 2H), 3.82 (s, 2H), 7.23–7.28 (m, 1H), 7.29–7.36 (complex, 4H); 13C NMR (101 MHz, CDCl3, APT pulse sequence) δ d 27.4, 127.5, 128.5, 128.6; u 35.7, 51.4, 55.2, 118.0, 138.9; IR 2975, 1454, 1366, 1202 cm-1; HRMS (ESI) m/z calcd for C13H19N2 ([M+H]+) 203.1543, found 203.1536.
4.4.1.2. N1-benzyl-N1-(tert-butyl)ethane-1,2-diamine (4a)
To a solution of the nitrile from 4.4.1.1. (255 mg, 1.26 mmol) in THF (25 mL) was added LiAlH4 solution (1 M in THF, 5.0 mL, 5.04 mmol, 4 equiv) at rt. The reaction was stirred at rt for 4 h and quenched by the careful addition of Na2SO4•10H2O (20 g). The reaction was filtered and the solids washed with ethyl ether (2 × 10 mL). The combined organic layers were washed with brine (50 mL), dried (Na2SO4) and purified by silica gel chromatography to afford the diamine fragment 4a as a colorless oil (64 mg, 0.31 mmol, 25% yield). Rf = 0.49 (10% MeOH + 2% NH4OH in CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 1.12 (s, 9H), 2.43 (t, J = 6.4 Hz, 2H), 2.62 (t, J = 6.4 Hz, 2H), 3.67 (s, 2H), 7.18 (t, J = 7.2 Hz, 1H), 7.27 (t, J = 7.2 Hz, 2H), 7.35 (d, J = 6.8 Hz, 2H); 13C NMR (101 MHz, CDCl3, APT pulse sequence) δ d 27.5, 126.4, 127.7, 128.1; u 42.2, 54.4, 55.1, 55.3, 143.6; IR 2967, 2869, 1576, 1469, 1452, 1361 cm-1; HRMS (ESI) m/z calcd for C13H23N2 ([M+H]+) 207.1861, found 207.1848.
4.4.2.1. 2-((4-Fluorobenzyl)(tert-butyl)amino)acetonitrile
N-(4-Fluoromobenzyl)-tert-butylamine (900 mg, 4.97 mmol) was reacted according to the protocol in section 4.4.1.1. to afford the nitrile product as a colorless oil (1,038 mg, 4.71 mmol, 95% yield). Rf = 0.49 (10% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 1.29 (s, 9H), 3.42 (s, 2H), 3.80 (s, 2H), 7.00 (t, J = 8.8 Hz, 2H), 7.31 (dd, J = 5.6, 8.8 Hz, 2H); 13C NMR (101 MHz, CDCl3, APT pulse sequence) δ d 27.4, 115.5 (d, J = 21.5 Hz), 130.1 (d, J = 8.0 Hz); u 35.6, 50.7, 55.3, 117.9, 134.5, 162.3 (d, J = 246.3 Hz); IR 2975, 1604, 1508, 1367, 1219 cm-1; HRMS (ESI) m/z calcd for C13H18FN2 ([M+H]+) 221.1449, found 221.1438.
4.4.2.2. N1-(tert-Butyl)-N1-(4-fluorobenzyl)ethane-1,2-diamine (4b)
The nitrile from 4.4.2.1. (582 mg, 2.64 mmol) was reacted according to the protocol in section 4.4.1.2. to afford the diamine fragment 4b as a colorless oil (422 mg, 1.88 mmol, 71% yield). Rf = 0.46 (10% MeOH + 2% NH4OH in CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 1.11 (s, 9H), 2.42 (t, J = 6.4 Hz, 2H), 2.61 (t, J = 6.4 Hz, 2H), 3.63 (s, 2H), 6.96 (t, J = 8.8 Hz, 2H), 7.30 (dd, J = 6.4, 8.8 Hz, 2H); 13C NMR (101 MHz, CDCl3, APT pulse sequence) δ d 27.4, 114.8 (d, J = 21.2 Hz), 129.0 (d, J = 7.8 Hz); u 42.1, 54.3, 54.5, 55.1, 139.1, 161.6 (d, J = 244.6 Hz); IR 2970, 2870, 1603, 1506, 1362 cm-1; HRMS (ESI) m/z calcd for C13H22FN2 ([M+H]+) 225.1767, found 225.1756.
4.4.3.1. 2-((4-Chlorobenzyl)(tert-butyl)amino)acetonitrile
N-(4-Chlorobenzyl)-tert-butylamine (700 mg, 3.54 mmol) was reacted according to the protocol in section 4.4.1.1. to afford the nitrile product as a white solid (685 mg, 2.89 mmol, 82% yield). Mp = 65–67 °C; Rf = 0.49 (10% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 1.27 (s, 9H), 3.41 (s, 2H), 3.78 (s, 2H), 7.27 (s, 4H); 13C NMR (101 MHz, CDCl3, APT pulse sequence) δ d 27.2, 128.6, 129.8; u 35.6, 50.7, 55.2, 117.7, 133.0, 137.4; IR 2975, 1597, 1490, 1368, 1201 cm-1; HRMS (ESI) m/z calcd for C13H18ClN2 ([M+H]+) 237.1153, found 237.1141.
4.4.3.2. N1-(tert-Butyl)-N1-(4-chlorobenzyl)ethane-1,2-diamine (4c)
The nitrile from 4.4.3.1. (384 mg, 1.62 mmol) was reacted according to the protocol in section 4.4.1.2. to afford the diamine fragment 4c as a colorless oil (248 mg, 1.03 mmol, 64% yield). Rf = 0.51 (10% MeOH + 2% NH4OH in CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 1.10 (s, 9H), 2.44 (t, J = 6.4 Hz, 2H), 2.61 (t, J = 6.4 Hz, 2H), 3.63 (s, 2H), 7.22 – 7.30 (complex, 4H); 13C NMR (101 MHz, CDCl3, APT pulse sequence) δ d 27.4, 128.2, 128.9; u 42.2, 54.5, 54.6, 55.2, 131.9, 142.2; IR 2969, 2869, 1488, 1363 cm-1; HRMS (ESI) m/z calcd for C13H22ClN2 ([M+H]+) 241.1472, found 241.1462.
4.4.4.1. 2-((4-Bromobenzyl)(tert-butyl)amino)acetonitrile A
N-(4-Bromobenzyl)-tert-butylamine (920 mg, 3.80 mmol) was reacted according to the protocol in section 4.4.1.1. to afford the nitrile product as a white solid (906 mg, 3.22 mmol, 85% yield). Mp = 64–66 °C; Rf = 0.49 (10% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 1.26 (s, 9H), 3.40 (s, 2H), 3.76 (s, 2H), 7.21 (d, J = 8.4 Hz, 2H), 7.42 (d, J = 8.0 Hz, 2H); 13C NMR (101 MHz, CDCl3, APT pulse sequence) δ d 27.2, 130.1, 131.5; u 35.6, 50.7, 55.1, 117.6, 121.1, 137.9; IR 2976, 1592, 1485, 1366, 1203 cm-1;HRMS (ESI) m/z calcd for C13H18BrN2 ([M+H]+) 281.0648, found 281.0644.
4.4.4.2. N1-(tert-Butyl)-N1-(4-bromobenzyl)ethane-1,2-diamine (4d)
To a solution of the nitrile from 4.4.4.1. (195 mg, 0.69 mmol) in THF (20 mL) was added LiAlH4 solution (1 M in THF, 0.7 mL, 0.69 mmol, 1 equiv) at 0 °C. The reaction was stirred at 0 °C for 4 h and quenched by the careful addition of Na2SO4•10H2O (20 g). The reaction was filtered and the solids washed with ethyl ether (2 × 10 mL). The combined organic layers were washed with brine (50 mL), dried (Na2SO4) and purified by silica gel chromatography to afford the diamine fragment 4d as a colorless oil (106 mg, 0.37 mmol, 54% yield). Rf = 0.46 (10% MeOH + 2% NH4OH in CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 1.10 (s, 9H), 2.44 (t, J = 6.4 Hz, 2H), 2.61 (t, J = 6.0 Hz, 2H), 3.61 (s, 2H), 7.23 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 8.4 Hz, 2H); 13C NMR (101 MHz, CDCl3, APT pulse sequence) δ d 27.4, 129.4, 131.4; u 42.2, 54.5, 54.7, 55.3, 120.1, 142.6; IR 2968, 2868, 1596, 1473,1451, 1360 cm-1; HRMS (ESI) m/z calcd for C13H22BrN2([M+H]+) 285.0966, found 285.0960.
4.5. Synthesis of compounds 1a, 1c-1m
4.5.1. N-(2-(Benzyl(isopropyl)amino)ethyl)-N-methyl-4-((N,4-dimethylphenylsulfonamido)methyl)benzamide (1a)
To a solution of ML140 (41 mg, 0.085 mmol) in DMF (1 mL) was added sodium hydride, 60% dispersion in mineral oil (10 mg, .256 mmol, 3 equiv). The reaction was stirred for 10 min at rt and methyl iodide (30 mg, 0.215 mmol, 2.5 equiv) was added. The reaction was stirred at rt for 17 h and partitioned between aqueous NaHCO3 and CH2Cl2. The organics were separated and the aqueous layer extracted with CH2Cl2 (3 × 5 mL) and the combined organic layers were dried (Na2SO4), concentrated and purified by silica gel chromatography to afford 1a as a light yellow oil (36 mg, 0.071 mmol, 83% yield). Rf = 0.49 (75% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) (ca. 1:1 mixture of rotomers) δ 0.92 (d, J = 6.4 Hz, 3H), 1.06 (d, J = 6.4 Hz, 3H), 2.46 (s, 3H), 2.50 (t, J = 7.2 Hz, 1 H), 2.54 (s, 1.5H), 2.59 (s, 1.5H), 2.73 (m, 1.5H), 2.85 (s, 1.5H), 2.93 (s, 1.5H), 3.04 (m, 0.5H), 4.14 (m, 1 H), 3.41 (s, 1 H), 3.49 (t, J = 6.0 Hz, 1 H), 3.63 (s, 1 H), 4.13 (d, J = 3.6 Hz, 2 H), 7.19–7.37 (complex, 11H), 7.73 (d, J = 8.0 Hz, 2 H); 13C NMR (101 MHz, CDCl3, APT pulse sequence) (mixture of rotomers) δ d 17.9, 21.5, 33.6, 34.3, 34.5, 38.6, 49.8, 50.5, 126.7, 126.9, 127.0, 127.3, 127.5, 128.2, 128.4, 128.7, 129.8; u 46.9, 47.1, 47.9, 51.0, 53.9, 54.6, 134.4, 136.4, 136.5, 136.9, 137.0, 140.4, 140.9, 143.6, 170.7, 171.5; IR 2964, 2928, 1629 cm-1; HRMS (ESI) m/z calcd for C29H38N3O3S([M+H]+) 508.2628, found 508.2629; HPLC purity = 97.4%.
4.5.2. N-(2-(Benzyl(isopropyl)amino)ethyl)-2-(mesitylsulfonyl)-1,2,3,4-tetrahydroisoquinoline-6-carboxamide (1c)
To a solution of 2-(mesitylsulfonyl)-1,2,3,4- tetrahydroisoquinoline-6-carbonyl chloride (20 mg, 0.054 mmol) in CH2Cl2 (3 mL) was added diamine fragment 3a (10 mg, 0.054 mmol) and triethylamine (0.019 mL, 0.14 mmol, 2.6 equiv). The reaction was stirred for 48 h at rt, aqueous, saturated sodium bicarbonate solution (2 mL) was added and all solvents removed in vacuo. The residue was extracted with a solution of CH2Cl2:MeOH (5:1, 6 mL). The filtrate was dried (Na2SO4), evaporated and purified by mass-directed, reverse phase preparative HPLC to afford 1c (9 mg, 0.017 mmol, 31% yield). 1H NMR (400 MHz, DMSO-d6) δ 1.00 (d, J = 6.6 Hz, 6H), 2.24 (s, 3H), 2.57 (s, 6H), 2.61 (t, J = 5.8 Hz, 2H), 2.83 (t, J = 5.9 Hz, 2H), 2.94 (sep, J = 6.6 Hz, 1H), 3.29 (q, J = 5.0 Hz, 2H), 3.41 (t, J = 5.9 Hz, 2H), 3.49 (s, 2H), 4.33 (s, 2H), 6.47 (br s, 1H), 6.90 (s, 2H), 7.03 (d, J = 8.0 Hz, 1H), 7.15 – 7.30 (m, 6H), 7.39 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 18.0, 21.0, 22.9, 28.7, 37.5, 41.9, 45.7, 47.8, 49.7, 53.7, 124.4, 126.6, 127.0, 127.9, 128.5, 128.6, 131.6, 132.0, 133.3, 133.8, 135.4, 140.6, 140.8,142.9, 166.7; IR 2966, 1648, 1544, 1494 cm-1; HRMS (ESI) m/z calcd for C31H39N3O3S ([M+H]+), 534.2790, found 534.2804;HPLC purity = 98.7%.
4.5.3. N-(2-((4-Fluorobenzyl)(isopropyl)amino)ethyl)-2-tosyl-1,2,3,4-tetrahydroisoquinoline-6-carboxamide (1d)
2-Tosyl-1,2,3,4-tetrahydroisoquinoline-6-carbonyl chloride (20 mg, 0.054 mmol) and diamine fragment 3b (11 mg, 0.054 mmol) were reacted according the protocol in section 4.5.2. to afford 1d (15 mg, 0.028 mmol, 52% yield). 1H NMR (400 MHz, DMSO-d6) δ 0.96 (d, J = 6.4 Hz, 6H), 2.39 (s, 3H), 2.51 (m, 2H), 2.82 – 2.90 (m, 3H), 3.22 (m, 2H), 3.29 (t, J = 6.4 Hz, 2H), 3.54 (s, 2H), 4.21 (s, 2H), 7.06 (t, J = 8.8 Hz, 2H), 7.23 (d, J = 8.4 Hz, 1H), 7.36 (dd, J = 6.4, 8.8 Hz, 2H), 7.43 (d, J = 8.0 Hz, 2H), 7.56 (m, 2H), 7.72 (d, J = 8.4 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 17.9, 21.0, 28.0, 40.4, 43.4, 47.2, 48.4, 49.3, 53.0, 114.7 (d, J = 21.1 Hz), 124.7, 126.4, 127.45, 127.49, 129.9 130.0, 132.90, 132.96, 132.99, 134.6, 137.0 (d, J = 2.8 Hz), 143.7, 161.0 (d, J = 242.3 Hz), 165.6; IR 2966, 1649, 1508 cm-1; HRMS (ESI) m/z calcd for C29H35FN3O3S ([M+H]+), 524.2383, found 524.2401; HPLC purity = 97.0%.
4.5.4. N-(2-((4-Chlorobenzyl)(isopropyl)amino)ethyl)-2-tosyl-1,2,3,4-tetrahydroisoquinoline-6-carboxamide (1e)
2-Tosyl-1,2,3,4-tetrahydroisoquinoline-6-carbonyl chloride (20 mg, 0.054 mmol) and diamine fragment 3c (12 mg, 0.054 mmol) were reacted according the protocol in section 4.5.2. to afford 1e (13 mg, 0.024 mmol, 45% yield). 1H NMR (400 MHz, DMSO-d6) δ 0.95 (d, J = 6.4 Hz, 6H), 2.31 (s, 3H), 2.55 (m, 2H), 2.85 (m, 2H), 3.24 – 3.28 (m, 4H), 3.41 (s, 2H), 4.17 (s, 2H), 6.97 (d, J = 8.0 Hz, 1H), 7.07 – 7.12 (complex, 4H), 7.15 (s, 1H), 7.19 (m, 1H), 7.22 (d, J = 8.0 Hz, 2H), 7.31 (br s, 1H), 7.62 (d, J = 8.4 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 17.9, 21.0, 28.0, 40.4, 43.4, 47.2, 48.6, 49.5, 53.0, 124.7, 126.4, 127.4, 127.5, 127.9, 129.86, 129.95, 130.2, 130.9, 132.9, 133.0, 134.6, 140.1 143.6, 165.6; IR 2966, 1647, 1543, 1491 cm-1; HRMS (ESI) m/z calcd for C29H35ClN3O3S ([M+H]+), 540.2088, found 540.2104; HPLC purity = 100.0%.
4.5.5. N-(2-((4-Bromobenzyl)(isopropyl)amino)ethyl)-2-tosyl-1,2,3,4-tetrahydroisoquinoline-6-carboxamide (1f)
2-Tosyl-1,2,3,4-tetrahydroisoquinoline-6-carbonyl chloride (20 mg, 0.054 mmol) and diamine fragment 3d (15 mg, 0.054 mmol) were reacted according the protocol in section 4.5.2. to afford 1f (18 mg, 0.30 mmol, 56% yield). 1H NMR (400 MHz, CDCl3) δ 0.98 (d, J = 6.4 Hz, 6H), 2.35 (s, 3H), 2.58 (t, J = 5.6 Hz, 2H), 2.88 – 2.94 (m, 3H), 3.26 – 3.31 (m, 4H), 3.42 (s, 2H), 4.21 (s, 2H), 6.39 (br s, 1 H), 7.01 (d, J = 8.0 Hz, 1H), 7.08 (d, J = 8.4 Hz, 1H), 7.22 – 7.28 (complex, 5H), 7.34 (s, 1H), 7.66 (d, J = 8.4 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 18.1, 21.6, 29.0, 37.8, 43.7, 47.6, 48.2, 49.9, 53.2, 120.7, 124.4, 126.6, 127.75, 127.82, 129.9, 130.4, 131.6, 133.2, 133.3, 133.7, 135.1, 139.9 144.0, 166.9; IR 2965, 1646, 1541, 1486 cm-1; HRMS (ESI) m/z calcdl for C29H34BrN3O3S ([M+H]+), 586.1562, found 586.1585; HPLC purity = 94.6%.
4.5.6. N-(2-((4-Bromobenzyl)(isopropyl)amino)ethyl)-2-((4-ethylphenyl)sulfonyl)-1,2,3,4-tetrahydroisoquinoline-6-carboxamide (1g)
2-((4-Ethylphenyl)sulfonyl)-1,2,3,4-tetrahydroisoquinoline-6-carbonyl chloride (160 mg, 0.44 mmol) and diamine fragment 3d (119 mg, 0.44 mmol) were reacted according the protocol in section 4.5.2. to afford 1g (122 mg, 0.21 mmol, 47% yield). 1H NMR (400 MHz, DMSO-d6) δ 0.95 (d, J = 6.8 Hz, 6H), 1.15 (t, J = 7.6 Hz, 3H), 2.55 (t, J = 6.0 Hz, 2H), 2.61 (q, J = 7.6 Hz, 2H), 2.83 – 2.90 (m, 3H), 3.22 – 3.28 (m, 4H) 3.40 (s, 2H), 4.18 (s, 2H), 6.52 (br s, 1H), 6.99 (d, j = 8.0 Hz, 1H), 7.07 (d, J = 8.4 Hz, 1H) 7.21 – 7.27 (complex, 5H), 7.33 (s, 1H), 7.65 (d, J = 8.4 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 15.0, 17.8, 27.98, 28.03, 40.4, 43.4, 47.2, 48.6, 49.5, 53.1, 119.3, 124.7, 126.3, 127.47,127.55, 128.7, 130.3, 130.8, 132.92, 132.93, 133.1, 134.6, 140.5149.5, 165.6; IR 2965, 1646, 1543, 1486 cm-1; HRMS (ESI) m/z calcd for C30H36BrN3O3S ([M+H]+), 600.1719, found 600.1740;HPLC purity = 98.1%.
4.5.7. N-(2-(Benzyl(tert-butyl)amino)ethyl)-2-tosyl-1,2,3,4-tetrahydroisoquinoline-6-carboxamide (1h)
2-Tosyl-1,2,3,4-tetrahydroisoquinoline-6-carbonyl chloride (20 mg, 0.054 mmol) and diamine fragment 4a (11 mg, 0.054 mmol) were reacted according the protocol in section 4.5.2. to afford 1h (26 mg, 0.049 mmol, 91% yield). 1H NMR (400 MHz, CDCl3) δ 1.10 (s, 9H), 2.36 (s, 3H), 2.76 (t, J = 6.0 Hz, 2H), 2.89 (t, J = 6.0 Hz, 2H), 3.12 (q, J = 5.6 Hz, 2H), 3.30 (t, J = 6.0 Hz, 2H), 3.64 (s, 2H), 4.21 (s, 2H), 6.22 (br s, 1H), 6.98 (d, J = 8.0 Hz, 1H), 7.09 (m, 1H), 7.16 – 7.22 (m, 3H), 7.27 – 7.29 (complex, 5H), 7.66 (d, J = 8.4 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 21.6, 27.5, 28.9, 39.9, 43.7, 47.6, 50.1, 55.2, 55.6, 124.5, 126.5, 126.8, 127.7. 127.8, 127.9, 128.5, 129.9, 133.2, 133.36, 133.42, 134.9,142.9, 143.9, 166.7; IR 2989, 1628, 1538, 1497, 1460 cm-1;HRMS (ESI) m/z calcd for C30H37N3O3S ([M+H]+), 520.2634,found 520.2650; HPLC purity = 97.5%.
4.5.8. N-(2-(Benzyl(tert-butyl)amino)ethyl) -((4-ethylphenyl)sulfonyl) -1,2,3,4-tetrahydroisoquinoline-6-carboxamide (1i)
2-((4-Ethylphenyl)sulfonyl)-1,2,3,4-tetrahydroisoquinoline-6-carbonyl chloride (160 mg, 0.44 mmol) and diamine fragment 4a (91 mg, 0.44 mmol) were reacted according the protocol in section 4.5.2. to afford 1i (66 mg, 0.12 mmol, 28% yield). 1H NMR (400 MHz, CDCl3) δ 1.11 (s, 9H), 1.20 (t, J = 7.6 Hz, 3H), 2.67 (q, J = 7.6 Hz, 2H), 2.77 (t, J = 6.1 Hz, 2H), 2.90 (t, J = 5.9 Hz, 2H), 3.12 (q, J = 5.9 Hz, 2H), 3.31 (t, J = 5.9 Hz, 2H), 3.65 (s, 2H), 4.21 (s, 2H), 6.37 (br s, 1H), 7.00 (d, J = 8.0 Hz, 1H), 7.07 – 7.11 (m, 1H), 7.16 – 7.21 (m, 2H), 7.24 – 7.27 (m, 1H), 7.29 – 7.33 (m, 5H), 7.67 – 7.71 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 15.0, 27.3, 28.7, 28.8, 39.9, 40.9, 43.6, 47.5, 50.0, 55.1, 124.5, 126.4, 126.6, 127.6, 127.76, 127.81, 128.3, 128.6, 133.1, 133.2, 134.7, 142.8, 149.9, 166.6; IR 2968, 1645, 1540, 1494 cm-1; HRMS (ESI) m/z calcd for C30H39N3O3S ([M+H]+), 534.2790, found 534.2801; HPLC purity = 94.7%.
4.5.9. N-(2-((4-Fluorobenzyl)(tert-butyl)amino)ethyl)-2-tosyl-1,2,3,4-tetrahydroisoquinoline-6-carboxamide (1j)
2-Tosyl-1,2,3,4-tetrahydroisoquinoline-6-carbonyl chloride (20 mg, 0.054 mmol) and diamine fragment 4b (12 mg, 0.054 mmol) were reacted according the protocol in section 4.5.2. to afford 1j (28 mg, 0.052 mmol, 96% yield). 1H NMR (400 MHz, CDCl3) δ 1.08 (s, 9H), 2.35 (s, 3H), 2.73 (t, J = 6.1 Hz, 2H), 2.87 (t, J = 5.9Hz, 2H), 3.10 (q, J = 5.6 Hz, 2H), 3.29 (t, J = 5.9 Hz, 2H), 3.58 (s, 2H), 4.20 (s, 2H), 6.10 (br s, 1H), 6.77 – 6.84 (m, 2H), 6.97 (d, J = 8.0 Hz, 1H), 7.18 – 7.28 (m, 6H), 7.64 – 7.67 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 21.5, 27.4, 28.8, 40.1, 43.6, 47.5,50.0, 54.5, 55.5, 115.2 (d, J = 21.2 Hz), 124.3, 126.4, 127.6,127.7, 129.3 (d, J = 8.0 Hz), 129.8, 133.2, 133.3, 133.5, 135.0,138.3 (d, J = 4.0 Hz), 143.9, 161.6 (d, J = 245.4 Hz), 166.7; IR 2970, 1643, 1541, 1506 cm-1; HRMS (ESI) m/z calcd for C30H36FN3O3S ([M+H]+), 538.2540, found 538.2554; HPLC purity = 96.3%.
4.5.10. N-(2-((4-Fluorobenzyl)(tert-butyl)amino)ethyl)-2-(mesitylsulfonyl)-1,2,3,4-tetrahydroisoquinoline-6-carboxamide (1k)
2-(mesitylsulfonyl)-1,2,3,4-tetrahydroisoquinoline-6-carbonyl chloride (20 mg, 0.054 mmol) and diamine fragment 4b (12 mg, 0.054 mmol) were reacted according the protocol in section 4.5.2. to afford 1k (14 mg, 0.024 mmol, 45% yield). 1H NMR (400 MHz, CDCl3) δ 1.08 (s, 9H), 2.35 (s, 3H), 2.54 (s, 6H), 2.73 (t, J = 6.1 Hz, 2H), 2.87 (t, J = 5.9 Hz, 2H), 3.10 (q, J = 5.6 Hz, 2H), 3.29 (t, J = 5.9 Hz, 2H), 3.58 (s, 2H), 4.20 (s, 2H), 6.10 (br s, 1H), 6.77 – 6.84 (m, 2H), 6.97 (d, J = 8.0 Hz, 1H), 7.18 – 7.28 (m, 6H), 7.64 – 7.67 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 21.5, 27.4, 28.8, 40.1, 43.6, 47.5, 50.0, 54.5, 55.5, 115.2 (d, J = 21.2 Hz), 124.3, 126.4, 127.6, 127.7, 129.3 (d, J = 8.0 Hz), 129.8, 133.2, 133.3, 133.5, 135.0, 138.3 (d, J = 4.0 Hz), 143.9, 161.6 (d, J = 245.4 Hz), 166.7; IR 2971, 1644, 1604, 1543, 1507 cm-1; HRMS (ESI) m/z calcd for C32H41FN3O3S ([M+H]+), 566.2853, found 566.2871; HPLC purity = 99.1%.
4.5.11. N-(2-((4-Chlorobenzyl)(tert-butyl)amino)ethyl)-2-tosyl-1,2,3,4-tetrahydroisoquinoline-6-carboxamide (1l)
2-Tosyl-1,2,3,4-tetrahydroisoquinoline-6-carbonyl chloride (20 mg, 0.054 mmol) and diamine fragment 4c (13 mg, 0.054 mmol) were reacted according the protocol in section 4.5.2. to afford 1l (25 mg, 0.046 mmol, 85% yield). 1H NMR (400 MHz, CDCl3) δ 1.09 (s, 9H), 2.39 (s, 3H), 2.64 (m, 2H), 2.87 (t, J = 6.0 Hz, 2H), 3.04 (m, 2H), 3.28 (t, J = 6.0 Hz, 2H), 3.69 (s, 2H), 4.20 (s, 2H), 7.20 (d, J = 8.0 Hz, 1H), 7.31 (m, 2H), 7.39 – 7.45 (m, 4H), 7.53 (m, 2H), 7.71 (d, J = 8.4 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 21.0, 27.1, 28.0, 40.4, 43.4, 47.2, 49.9, 53.4, 54.8, 124.7, 126.3, 127.42, 127.45, 127.8, 129.3, 129.9, 130.5, 132.77, 132.84, 132.9, 134.6, 142.1, 143.6, 165.5; IR 2969, 1644, 1541, 1489 cm-1; HRMS (ESI) m/z calcd for C30H37ClN3O3S ([M+H]+), 554.2244, found 554.2261; HPLC purity = 99.2%.
4.5.12. N -(2-((4-Bromobenzyl)(tert-butyl)amino)ethyl)-2-tosyl-1,2,3,4-tetrahydroisoquinoline-6-carboxamide (1m)
2-Tosyl-1,2,3,4-tetrahydroisoquinoline-6-carbonyl chloride (20 mg, 0.054 mmol) and diamine fragment 4d (15 mg, 0.054 mmol) were reacted according the protocol in section 4.5.2. to afford 1m (7.8 mg, 0.013 mmol, 24% yield). 1H NMR (400 MHz, CDCl3) δ 1.08 (s, 9H), 2.35 (s, 3H), 2.74 (t, J = 6.0 Hz, 2H), 2.89 (t, J = 5.9 Hz, 2H), 3.13 (q, J = 5.8 Hz, 2H), 3.27 – 3.32 (m, 2H), 3.56 (s, 2H), 4.21 (s, 2H), 6.07 (br s, 1H), 7.00 (d, J = 8.0 Hz, 1H), 7.11 – 7.20 (m, 3H), 7.23 – 7.30 (m, 5H), 7.64 – 7.68 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 21.6, 27.3, 28.9, 40.1, 43.6, 47.5, 50.2, 54.6, 55.6, 120.3, 124.3, 126.5, 127.6, 127.7, 129.5, 129.8, 131.5, 133.21, 133.23, 133.6, 135.0, 141.9, 143.8, 166.7; IR 2980, 1652, 1521, 1489 cm-1; HRMS (ESI) m/z calcd for C30H36BrN3O3S ([M+H]+), 600.1719, found 600.1740; HPLC purity = 94.1%.
4.5.13. N-(2-((4-Bromobenzyl)(tert-butyl)amino)ethyl)-2-(mesitylsulfonyl)-1,2,3,4-tetrahydroisoquinoline-6-carboxamide (1n)
2-(mesitylsulfonyl)-1,2,3,4-tetrahydroisoquinoline-6-carbonyl chloride (20 mg, 0.054 mmol) and diamine fragment 4d (15 mg, 0.054 mmol) were reacted according the protocol in section 4.5.2. to afford 1n (10 mg, 0.016 mmol, 29% yield). 1H NMR (400 MHz, CDCl3) δ 1.17 (s, 9H), 2.33 (s, 3H), 2.66 (s, 6H), 2.83 (m, 2H), 2.92 (t, J = 6.0 Hz, 2H), 3.33 (q, J = 6.0 Hz, 2H), 3.50 (t, J = 6.0 Hz, 2H), 3.66 (s, 2H), 4.42 (s, 2H), 6.18 (br s, 1H), 6.99 (s, 2H), 7.12 (d, J = 8.0 Hz, 1H), 7.12 (d, J = 8.4 Hz, 2H), 7.28 (m, 1H), 7.34 (d, J = 8.4 Hz, 2H), 7.42 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 21.1, 23.0, 27.5, 28.8, 40.2, 42.0, 45.8, 50.3, 54.8, 55.7, 120.4, 124.3, 126.8, 127.9, 129.6, 131.6, 131.7, 132.1, 133.4, 134.0. 135.6, 140.7, 142.0, 143.0, 166.9; IR 2972, 1647, 1533,1486 cm-1; HRMS (ESI) m/z calcd for C32H40BrN3O3S ([M+H]+),628.2032, found 628.2049; HPLC purity = 98.6%.
4.6. In vitro assay methods
4.6.1. Compounds and reagents
(+)-(5α,7α,8β)-N-Methyl-N-(7-(1-pyrrolidinyl)-1-oxaspiro(4.5)dec-8-yl)-benzeneacetamide (U69,593) and nor-binaltorphimine dihydrochloride (norBNI) were purchased from Sigma Aldrich. U69,593 was prepared in ethanol as a 10 mM stock, norBNI was prepared in water as a 10 mM stock, and test compounds were prepared as 10 mM stocks in DMSO (Fisher). All compounds were then diluted to working concentrations in vehicle for each assay without exceeding 1% DMSO or ethanol concentrations. [35S]GTPγS was purchased from PerkinElmer Life Sciences. Phospho-ERK1/2 and total ERK1/2 antibodies were purchased from Cell Signaling (Beverly, MA) and Li-Cor secondary antibodies (anti-rabbit IRDye800CW and anti-mouse IRDye680LT) were purchased from Li-Cor Biosciences.
4.6.2. Cell lines and cell culture
Chinese hamster ovary (CHO) cells were virally transfected to express HA-tagged recombinant human kappa opioid receptors (CHO-hKOR cell line) and maintained in DMEM/F-12 media (Invitrogen) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 500 μg/ml geneticin as previously described.14 A DiscoveRx PathHunter™ U2OS cell line expressing βarrestin2 and hKOR (U2OS-hKOR-βarrestin2-DX) was purchased from DiscoveRx Corporation (Fremont, CA) and maintained MEM with 10% fetal bovine serum, 1% penicillin/streptomycin, 500 μg/ml geneticin and 250 μg/ml hygromycin B. All cells were grown at 37 °C (5% CO2 and 95% relative humidity).
4.6.3. G Protein coupling assay
[35S]GTPγS binding assay was performed following a previously published protocol.14,18 Briefly, cells were serum-starved for 1 hour and membranes were prepared. Each reaction was performed at room temperature and contained 15 μg of membrane protein, 40 μM GDP, ∼0.1 nM [35S]GTPγS and increasing concentrations of compounds in assay buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA). Directly after the addition of test compounds, 100 nM U69,593 was added to yield a total volume of 200 μL. After 1 hour, reactions were quenched by rapid filtration through GF/B filters and radioactivity was counted with a TopCount NXT high throughput screening microplate scintillation and luminescence counter (PerkinElmer Life Sciences). In some cases, the assays were also carried out as above except that 1 μM U69,593 was used; analogous results were obtained.
4.6.4. βArrestin2 recruitment (DiscoveRx PathHunter™)assay
The PathHunter™ assay was performed according to the manufacturer's protocol with slight modification and following previously published protocols.14,18 Briefly, 5,000 cells/well were plated overnight in Opti-MEM containing 1% fetal bovine serum, 1% penicillin/streptomycin. Cells were pretreated with antagonist for 15 min at 37 °C followed by the addition of 1 μM U69,593 and a 90 minute incubation at 37 °C. PathHunter™ detection reagent was added and cells incubated at room temperature for 60 minutes. Chemiluminescence was detected using a SpectraMax® M5e Multimode Plate Reader (Molecular Devices).
4.6.5. In-cell western ERK1/2 phosphorylation
Antagonist inhibition of U69,593-induced ERK phosphorylation was determined by in-cell westerns as previously described.14,18 Briefly, hKOR-CHO cells were plated in 384-well plate at 15,000 cells per well and incubated at 37 °C overnight. After an hour serum-starve, cells were treated with compound followed by the addition of 100 nM U69,593 and a 10 minute incubation at 37 °C. Cells were fixed, permeabilized, blocked, and stained with primary antibodies for phosphorylated ERK1/2 and total-ERK1/2 (1:300 and 1:400, respectively) at 4 °C overnight. Cells were then incubated with Li-Cor secondary antibodies (anti-rabbit IRDye800CW, 1:500; anti-mouse IRDye680LT, 1:1500) and imaged with the Odyssey Infrared Imager (Li-Cor Biosciences, Lincoln, NE) at 700 and 800 nm.
4.6.6. Data analysis and statistics
GraphPad Prism 6.01 software (GraphPad) was used to generate sigmoidal concentration-response curves using a three-parameter, non-linear regression analysis. All compounds were run in parallel assays in 2 – 4 replicates per individual experiment. All studies were performed n ≥ 3 independent experiments in multiple replicates. For determination of antagonist inhibition, each individual experiment was normalized to the percentage of maximal U69,593 stimulation. The efficacy and potency values were obtained from the averages of the nonlinear regression analysis performed on each individual curve and are reported as the mean ± S.E.M.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from the National Institute on Drug Abuse (grant R01 DA031927 to LMB and JA). We thank Ben Neuenswander for performing HPLC compound purification and high resolution mass determinations. Ki determinations were generously provided by the National Institute of Mental Health's Psychoactive Drug Screening Program, Contract # HHSN-271-2013-00017-C (NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and project officer Jamie Driscoll at NIMH, Bethesda MD, USA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ream AH, Bruchas MR. Anesthesiology. 2011;115:1363–1381. doi: 10.1097/ALN.0b013e318238bba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lamberts JT, Traynor JR. Curr Pharm Design. 2013;19:7333–7347. doi: 10.2174/138161281942140105160625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Metcalf MD, Coop A. AAPS J. 2005;7:E704–E722. doi: 10.1208/aapsj070371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carlezon WA, Béguin C, Knoll AT, Cohen BM. Pharmacol Ther. 2009;123:334–343. doi: 10.1016/j.pharmthera.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carroll FI, Carlezon WA. J Med Chem. 2013;56:2178–2195. doi: 10.1021/jm301783x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Portoghese PS, Lipkowski AW, Takemori AE. J Med Chem. 1987;30:238–239. doi: 10.1021/jm00385a002. [DOI] [PubMed] [Google Scholar]
- 7.Jones RM, Hjorth SA, Schwartz TW, Portoghese PS. J Med Chem. 1998;41:4911–4914. doi: 10.1021/jm9805182. [DOI] [PubMed] [Google Scholar]
- 8.Thomas JB, Atkinson RN, Rothman RB, Fix SE, Mascarella SW, Vinson NA, Xu H, Dersch CM, Lu Y, Cantrell BE, Zimmerman DM, Carroll FI. J Med Chem. 2001;44:2687–2690. doi: 10.1021/jm015521r. [DOI] [PubMed] [Google Scholar]
- 9.Bruchas MR, Yang T, Schreiber S, Defino M, Kwan SC, Li S, Chavkin C. J Biol Chem. 2007;282:29803–29811. doi: 10.1074/jbc.M705540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melief EJ, Miyatake M, Carroll FI, Béguin C, Carlezon WA, Cohen BM, Grimwood S, Mitch CH, Rorick-Kehn L, Chavkin C. Mol Pharmacol. 2011;80:920–929. doi: 10.1124/mol.111.074195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munro TA, Berry LM, Vant Veer A, Béguin C, Carroll FI, Zhao Z, Carlezon WA, Cohen BM. JMC Pharmacol. 2012;12:5. doi: 10.1186/1471-2210-12-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patkar KA, Wu J, Ganno ML, Singh HD, Ross NC, Rasakham K, Toll L, McLaughlin JP. J Pharmacol Exp Ther. 2013;346:545–554. doi: 10.1124/jpet.113.206086. [DOI] [PubMed] [Google Scholar]
- 13.Frankowski KJ, et al. ACS Chem Neurosci. 2012;3:231–236. doi: 10.1021/cn200128x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmid CL, Streicher JM, Groer CE, Munro TA, Zhou L, Bohn LM. J Biol Chem. 2013;288:22387–22398. doi: 10.1074/jbc.M113.476234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strange PG. Br J Pharmacol. 2010;161:1238–1249. doi: 10.1111/j.1476-5381.2010.00963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osmond RIW, Sheehan A, Borowicz R, Barnett E, Harvey G, Turner C, Brown A, Crouch MF, Dyer AR. J Biomol Screen. 2005;10:730–737. doi: 10.1177/1087057105277968. [DOI] [PubMed] [Google Scholar]
- 17.Zhao X, Jones A, Olson KR, Peng K, Wehrman T, Park A, Mallari R, Nebalasca D, Young SW, Xiao SH. J Biomol Screen. 2008;13:737–747. doi: 10.1177/1087057108321531. [DOI] [PubMed] [Google Scholar]
- 18.Zhou L, Lovell KM, Frankowski KJ, Slauson SR, Phillips AM, Streicher JM, Stahl E, Schmid CL, Hodder P, Madoux F, Cameron MD, Prisinzano TE, Aubé J, Bohn LM. J Biol Chem. 2013;288:36703–36716. doi: 10.1074/jbc.M113.504381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.