

Abstract
Traumatic brain injury (TBI) is induced by mechanical forces which initiate a cascade of secondary injury processes, including inflammation. Therapies which resolve the inflammatory response may promote neural repair without exacerbating the primary injury. Specific derivatives of omega-3 fatty acids loosely grouped as specialized pro-resolving lipid mediators (SPMs) and termed resolvins promote the active resolution of inflammation. In the current study, we investigate the effect of two resolvin molecules, RvE1 and AT-RvD1, on post-traumatic sleep and functional outcome following diffuse TBI through modulation of the inflammatory response.
Adult, male C57BL/6 mice were injured using a midline fluid percussion injury (mFPI) model (6-10 min righting reflex time for brain-injured mice). Experimental groups included mFPI administered RvE1 (100ng daily), AT-RvD1 (100ng daily), or vehicle (sterile saline) and counterbalanced with uninjured sham mice. Resolvins or saline were administered daily for seven consecutive days beginning 3 days prior to TBI to evaluate proof-of-principle to improve outcome. Immediately following diffuse TBI, post-traumatic sleep was recorded for 24 hours post-injury. For days 1-7 post-injury, motor outcome was assessed by Rotarod. Cognitive function was measured at 6 days post-injury using Novel Object Recognition (NOR). At 7 days post-injury, microglial activation was quantified using immunohistochemistry for Iba-1.
In the diffuse brain-injured mouse, AT-RvD1 treatment, but not RvE1, mitigated motor and cognitive deficits. RvE1 treatment significantly increased post-traumatic sleep in brain-injured mice compared to all other groups. RvE1 treated mice displayed a higher proportion of ramified microglia and lower proportion of activated rod microglia in the cortex compared to saline or AT-RvD1 treated brain-injured mice. Thus, RvE1 treatment modulated post-traumatic sleep and the inflammatory response to TBI, albeit independently of improvement in motor and cognitive outcome as seen in AT-RvD1-treated mice. This suggests AT-RvD1 may impart functional benefit through mechanisms other than resolution of inflammation alone.
Keywords: TBI, inflammation, resolvins, protectins, sleep, mouse, behavior, aspirin-triggered resolvin
1. INTRODUCTION
Each year over 1.7 million people sustain traumatic brain injuries (TBI) in the United States alone1. The consequences of TBI for the individual can include diminished quality of life manifested through a range of symptoms including acute and chronic pain2, loss of neurological function3, and impaired cognitive or emotional ability3-5. Few therapies are available to treat these debilitating morbidities and many promising therapeutic agents have failed to achieve efficacy in clinical trials6. Animal models have been established to reproduce the complex pathophysiology of TBI and offer valuable means through which therapies can be tested on the resulting physiological, sensorimotor, and cognitive deficits. Pharmacological attempts to lessen the burden of TBI include a wide swath of treatment approaches, routinely using experimental compounds largely designed to target the cellular and molecular cascades which contribute to secondary injury in the hours to days following TBI6, 7.
TBI is induced by mechanical forces which initiate a cascade of secondary injury processes, including inflammation8. To complicate our understanding of underlying processes, cerebral inflammation has been shown to contribute to both beneficial and detrimental effects on outcome (for review, see Morganti-Kossmann et al. 20029). Following TBI, the brain is inundated with inflammation-mediating cytokines10-12 which prompt a spectrum of responses including cell differentiation, immune activation, and cell death13. While the role of early-onset inflammation in the pathophysiology of TBI is debatable, clinical and experimental data suggest that chronic over-production of inflammatory cytokines aggravate the primary injury14-16, and thereby impact outcome. Our lab has previously reported that the cortical primary somatosensory barrel fields (S1BF) are a primary site of neuropathology following experimental diffuse traumatic brain injury17, 18, with pronounced microglial activation and the induction of a previously understudied rod morphology of activated microglia 16, 19, 20. Rod microglia appear to constitute a phenotypically distinct class of activated microglia which present following acute neurological injury including diffuse TBI, infection and seizure 19, 21, 22. The relative distribution of microglial morphologies can thereby serve as an indicator neuroinflammation and underlying pathological and reparative processes. The current study further assesses neuroinflammation after diffuse TBI using a semi-quantification method of microglial activation to determine proportions of ramified (unactivated) microglia in relation to morphological phenotypes activated in response to injury— activated and rod microglia.
Cytokines are elevated and regulate inflammation following TBI12, 23. These cytokines, including pro-inflammatory interleukin-1 beta (IL-1), can also serve dual roles as sleep regulatory substances (SRSs) which influence sleep-wake behavior through action on the sleep circuits within the brain24-26. The elevation of IL-1 following midline fluid percussion brain injury in the mouse has previously been documented to correspond temporally with an acute increase in post-traumatic sleep during the first six hours post-injury27. These data suggest a relationship between inflammation and SRSs such that post-traumatic sleep may serve as an indicator of SRS action.
While inflammation is an essential component of the repair process, excessive or prolonged inflammation can aggravate injury-related damage28. Therapies which resolve the inflammatory response may promote neural recovery without exacerbating the primary injury. Certain derivatives of omega-3 fatty acids loosely grouped as specialized pro-resolving lipid mediators (SPMs) have been shown to promote the resolution of inflammation resulting from multiple induction pathways (for review, see Recchiuti 201229). These lipid mediators of inflammation are described as eicosanoids or docsanoids depending on their derivation from either eicosapentaenoic acid (EPA) or docosahexaenoic acid (DHA) respectively30 and encompass subclasses including resolvins, protectins, lipoxins, and maresins29. Dietary supplementation with precursors of SPMs, particularly DHA, has shown therapeutic potential by reducing lesion size and improving neurological function following a model of ischemic stroke31 and TBI27, 28 in rats by decreasing axonal injury and preserving cognitive function. Further, DHA supplementation after experimental stroke led to increased brain levels of another docosanoid, neuroprotectin D1 (NPD1)32, a lipid mediator of inflammation which was then shown to ameliorate the functional and histological consequences of experimental stroke on its own33. Therefore, pharmacotherapy targeting SPMs provides a pleiotropic approach to regulating inflammation resulting from TBI, rather than genetically or chemically targeting a single signaling pathway.
Considering the similar inflammatory pathophysiologies of stroke and TBI, these data suggest that SPMs may regulate inflammation resulting from TBI. In the current study, two lipid mediators of inflammation, resolvin E1 (RvE1) and aspirin-triggered resolvin D1 (AT-RvD1), are tested for their impact upon post-traumatic sleep, sensorimotor and cognitive outcome, and microglial activation after diffuse experimental brain injury in the mouse. These endogenous SPMs are proposed to bring the injury-induced inflammatory response to conclusion as evidenced by a reduction in post-traumatic sleep, shift in the microglial morphology profile, and thereby improvement in functional outcome. We hypothesize that RvE1 and AT-RvD1 will improve functional outcome from diffuse TBI through modulation of the inflammatory response.
2. MATERIALS AND METHODS
2.1 Animals
Male C57BL/6 mice (Harlan Laboratories, Inc., Indianapolis, IN) were used for all experiments (n=73). Mice were housed in a 12 h light/12h dark cycle at a constant temperature (23°C ± 2° C) with food and water available ad libitum according to the Association for Assessment and Accreditation of Laboratory Animal Care International. All mice used in this study were singly housed. Mice were acclimated to their environment following shipment for at least three days prior to any experiments. After surgery, mice were evaluated daily during post-operative care by a physical examination and documentation of each animal’s condition. Animal care was approved by the Institutional Animal Care and Use Committees at St. Joseph’s Hospital and Medical Center (Phoenix, AZ).
2.2 Midline Fluid Percussion Injury (mFPI)
Mice (20-24g) were subjected to midline fluid percussion injury (mFPI) consistent with methods previously described27, 34-37. Group sizes are indicated in the results section and figure legends for individual studies. Mice were anesthetized using 5% isoflurane in 100% oxygen for five minutes and the head of the mouse was placed in a stereotaxic frame with continuously delivered isoflurane at 2.5% via nosecone. While anesthetized, body temperature was maintained using a Deltaphase® isothermal heating pad (Braintree Scientific Inc., Braintree, MA). A midline incision was made exposing bregma and lambda, and fascia was removed from the surface of the skull. A trephine (3 mm outer diameter) was used for the craniotomy, centered on the sagittal suture between bregma and lambda without disruption of the dura. An injury cap prepared from the female portion of a Luer-Loc needle hub was fixed over the craniotomy using cyanoacrylate gel and methyl-methacrylate (Hygenic Corp., Akron, OH). The incision was sutured at the anterior and posterior edges and topical Lidocaine ointment was applied. The injury hub was closed using a Luer-Loc cap and mice were placed in a heated recovery cage and monitored until ambulatory before being returned to their sleep cage.
For injury induction 24 hours post-surgery, mice were re-anesthetized with 5% isoflurane delivered for five minutes. The cap was removed from the injury-hub assembly and the dura was visually inspected through the hub to make sure it was intact with no debris. The hub was then filled with normal saline and attached to an extension tube connected to the male end of the fluid percussion device (Custom Design and Fabrication, Virginia Commonwealth University, Richmond, VA). An injury of moderate severity for our injury model (1.4 atm) was administered by releasing the pendulum onto the fluid-filled cylinder. Sham-injured mice underwent the same procedure except the pendulum was not released. Mice were monitored for the presence of a forearm fencing response and righting reflex times were recorded for the injured mice as indicators of injury severity 38. The righting reflex time is the total time from the initial impact until the mouse spontaneously rights itself from a supine position. The fencing response is a tonic posturing characterized by extension and flexion of opposite arms that has been validated as an overt indicator of injury severity 38. The injury hub was removed and the brain was inspected for uniform herniation and integrity of the dura. The dura was intact in all mice; none were excluded as technical failures. The incision was cleaned using saline and closed using sutures. Moderate brain-injured mice had righting reflex recovery times greater than six minutes and a positive fencing response. Sham injured mice recovered a righting reflex within 20 seconds. After spontaneously righting, mice were placed in a heated recovery cage and monitored until ambulatory (approximately 5 to 15 minutes) before being returned to their piezoelectric sleep cage. Adequate measures were taken to minimize pain or discomfort.
2.3 Pharmacological Treatment
Mice were treated with sterile saline (100μl, 0.9% NaCL), 100ng 17(R)-Resolvin D1 (AT-RvD1; Item # 13060, Cayman Chemical, Ann Arbor, MI), or 100ng Resolvin E1 (RvE1; Item # 10007848, Cayman Chemical, Ann Arbor, MI). Resolvins were administered intraperitoneally in 100μl of sterile saline for seven consecutive days beginning three days before mFPI (see Fig. 1). The seven day dosing schedule was selected to evaluate proof-of-principle for resolvin efficacy by covering both prophylactic and treatment approaches. Dosing was selected with regard to published studies using protectins in cerebral ischemia in mice33.
Figure 1. Study Design.
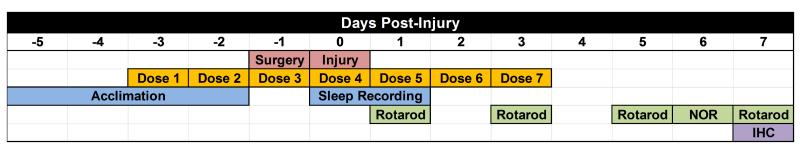
Mice were acclimated to their individual sleep cages before surgery and injury (blue). Mice were pre-treated with saline, AT-RvD1, or RvE1, for three days before surgery, and received post-injury injections for 4 consecutive days (orange). All mice received a midline craniectomy (red). One day post-surgery mice received either midline fluid percussion brain injury or sham injury (red). Immediately following injury, mice were placed back in their sleep cage and post-traumatic sleep was non-invasively recorded for 24 hours (blue). Following injury, mice were assessed for sensorimotor function using the rotarod task (1, 3, 5, 7 days post-injury). Cognitive performance was assessed using novel object recognition (NOR) at 6 days post-injury. Following behavior tasks, tissue was collected at 7 days post-injury and prepared for neuroinflammation analysis using immunohistochemistry (IHC; purple).
2.4 Sleep Recordings
The non-invasive sleep cage system (Signal Solutions, Lexington, KY) used in this study consisted of 16 separate units which simultaneously monitor sleep and wake states, as previously published39-41. Each cage unit housed a single mouse inside 18 × 18 centimeter walled compartments with attached food and water structures39. The cages had open bottoms resting on Polyvinylidine Difluoride (PVDF) sensors serving as the cage floor39. The non-invasive high-throughput PVDF sensors were coupled to an input differential amplifier and pressure signals were generated and classified by the classifier as motions consistent with either activity related to wake or inactivity and regular breathing movements associated with sleep39, 40. Briefly, sleep was characterized primarily by periodic (3 Hz) and regular amplitude signals recorded from the PVDF sensors, typical of respiration from a sleeping mouse. In contrast, signals characteristic of wake were both the absence of characteristic sleep signals and higher amplitude, irregular signals associated with volitional movements, even during quiet wake. The piezoelectric signals in two second epochs were classified by a linear discriminant classifier algorithm based on multiple signal variables to assign a binary label of “sleep” or “wake”39. Mice sleep in a polycyclic manner (often more than 40 sleep episodes per hour if short arousals are recorded)42 and therefore mouse sleep was quantified as the minutes spent sleeping per hour, presented as a percentage for each hour.
The current study was designed to avoid initial post-traumatic sleep recordings during a light transition period. As a result, sleep recordings began 6 hours into the light period, covered the light:dark transition, and continued for 6 hours into the dark period. Data collected from the cage system were binned over specified time periods (e.g. 1 hour) using the average of percent sleep, as well as binned by length of individual bouts of sleep and the median bout lengths were calculated. Where applicable, sleep metrics were compared between 6 hours of light and 6 hours of dark.
2.5 Behavioral Testing
2.5.1 Rotarod
Sensorimotor function was assessed using the Economex Rotarod system from Columbus Instruments (Columbus, OH). Mice were pre-trained for three consecutive days (60 sec at 4 RPM for 3 trials) prior to injury, following pre-injury drug treatment. For the test (1, 3, 5, 7 days post-injury), mice were placed on the rod with a starting speed of 4 RPM, and rod rotation speed was continuously increased over 5 minutes up to a max speed of 28 RPM, as previously published28, 35, 37. The trial ended when the mouse fell from the rod or 5 minutes elapsed. Two trials were performed at each time point. Data are presented as latency to fall in seconds (average of two trials).
2.5.2 Novel Object Recognition (NOR)
Cognitive impairment was tested using the NOR test as previously published35, 43, 44. The test consisted of three phases: habituation, training, and testing. On day 6 post-injury, mice were placed in an open field (42 cm, 21 cm, 21 cm) for one hour of habituation. Mice were removed and two identical objects were placed in opposing quadrants of the field for the training phase. Mice were placed in the center of the open field and given 5 minutes to explore the objects. Following training, mice were returned to their piezoelectric sleep cages. Testing began 4 hours after training. One familiar object was placed in an original location and one novel object was placed in the opposing quadrant of the open field. Mice were placed into the center and given 5 minutes to explore. For testing, the times spent actively investigating the novel and familiar object were quantified. Investigation of an object included the mice sniffing, touching, or climbing onto an object while facing the object. If an animal climbed onto an object and sniffed into the air, this time was not calculated into the exploration of the novel object. Testing data are displayed as the percentage of total investigation time spent with each object and as a discrimination index (DI) in which DI = (Tnovel − Tfamiliar)/(Tnovel + Tfamiliar).
2.6 Analysis of Microglial Activation
2.6.1 Tissue Preparation for Immunofluorescence
At 7 days post-injury or sham operation, mice were given an overdose of sodium pentobarbital (i.p.) and transcardially perfused with 4% paraformaldehyde after a phosphate buffered saline (PBS) flush. Brains were removed and placed in 4% paraformaldehyde overnight. Brains were immersed in serial dilutions (15% and 30%) of sucrose for 24 hours each. The brains were removed from the 30% sucrose and frozen at −20° C. After freezing, brains were cryosectioned in the coronal plane at 20μm, mounted onto glass slides, and stored at −80° C.
2.6.2 Iba-1 Immunofluorescence
Slides were removed from −80°C, placed in an oven at 60°C for approximately 4 hours and then rinsed three times for 5 minutes each in PBS. Next, the slides were incubated in 4% goat serum blocking solution for 1 hour. Slides were incubated with the primary antibody (rabbit anti-ionized calcium binding adaptor molecule 1, IBA-1; 1:1000, Item # 0199-19741, Wako Chemicals, Richmond, VA) and stored at 4°C overnight. Slides were rinsed three times in PBS and the secondary antibody (biotinylated horse anti-rabbit; 1:250, Vector Laboratories, Burlingame, CA) was added and slides were incubated on a rocker at room temperature for 1 hour. Slides were washed in PBS three times for 5 minutes each and tertiary stain was applied (streptavidin Alexa© Fluor 594; 1:1000, Jackson Immunoresearch, Westgrove, PA) and slides were incubated for 1 hour at room temperature. Lastly, slides were rinsed three times in PBS and coverslipped with anti-fade medium (Fluoromount G; Southern Biotech, Birmingham, AL). Sections were examined for microglial activation in response to brain-injury using a Zeiss LSM 710 Confocal Laser Scanning Microscope with attached digital camera.
2.6.3 Microglial Identification & Semi-Quantification
Stained sections (from 3-4 mice per group) were analyzed following Iba-1 staining to determine the proportion of microglial morphologies post-injury. The area of interest, the primary somatosensory barrel fields (S1BF), was chosen based on previous work demonstrating a multi-focal concentration of neuropathology and microglial activation in the S1BF following midline fluid percussion brain injury in the rat16, 17, 19. Sections were screened using a Zeiss (AXIO imager A2) microscope with attached digital camera (AxioCam MRc5). Images were captured with proprietary Zen software (Carl Zeiss, Germany) at 20X magnification. The area of interest was examined in both hemispheres and in two different coronal planes: an anterior section (approximately Bregma - 1.555mm) and a posterior section (approximately Bregma −2.255mm). A total of 4 photos per brain per area of interest were analyzed. Photomicrographs were analyzed in Image J software (National Institutes of Health, Bethesda, MD). On each photomicrograph, 250,000 pixel2 grid lines were placed and quantification was limited to 4 pre-defined boxes. Microglia within these boxes were classified as either having ramified (small soma, high defined processes), activated (hypertrophied soma, fewer processes), or rod (soma twice as long as width in one direction) morphologies. A minimum of 50 microglia were counted per section per region. Sham treatment groups were combined.
2.7 Statistical Analysis
Data are shown as mean ± SEM and analyzed using GraphPad Prism 6, with statistical significance assigned when p<0.05, unless otherwise indicated. For each test, shams receiving each treatment were statistically compared. No differences were detected between sham groups (treated and vehicle). To maintain comparable group sizes, a pooled sham group (n=12) was constructed using equal numbers of mice randomly selected from each treatment. Differences in righting reflex times were measured with a one-way analysis of variance (ANOVA). Differences in mortality were evaluated by chi-square analysis between both resolvin and saline-treated brain-injured groups. Percent sleep was analyzed using a repeated measure two-way ANOVA. Cumulative sleep measured in minutes and cumulative bout lengths of sleep were analyzed with a one-way ANOVA. All significant sleep data were further analyzed using Tukey’s multiple comparisons test. Differences in rotarod performance at 24 hours post-injury were determined with a one-way ANOVA followed by Dunnett’s multiple comparisons test. Differences in motor performance following TBI over the first week post-injury were determined with a repeated measure two-way ANOVA followed by Dunnett’s multiple comparisons test. Differences in time spent exploring the novel versus familiar object were determined for each treatment group by paired t-test, using a modified p value of 0.0125 to account for four separate tests. Proportional differences in rod microglial morphologies were subjected to chi-square analysis (Fisher’s exact test) in which significance was defined by a modified p value of 0.0083 to correct for the 6 tests made within each morphological comparison. Statistical values are included in the figure legends.
3. RESULTS
3.1 Resolvin treatment did not influence the initial induction of diffuse TBI
We have previously reported suppression of the righting reflex response in mice following mFPI35 as an injury-induced deficit and indicator of injury severity. Diffuse brain injury resulted in a significant suppression of the righting reflex in brain-injured mice, compared to anesthetized, uninjured shams, regardless of drug treatment (Fig 2). A one-way ANOVA comparing only brain-injured treatment groups showed no significant difference of righting reflex times, indicating that all brain-injured mice received injuries of similar severities. Of the 73 mice in this study, 48 were brain-injured. As a result of injury, 19 mice died (40% mortality). Of the 18 mortalities, 5 saline-treated mice died within 15 minutes of injury (33% mortality); 4 AT-RvD1-treated mice died within 15 minutes of injury (27% mortality); 9 RvE1-treated animals died within 15 minutes of injury (50% mortality); all presumably from respiratory arrest. Differences in acute post-injury mortality were compared with chi-square analysis, revealing no significant treatment effects on mortality.
Figure 2. TBI suppressed righting reflex regardless of drug treatment.

Diffuse TBI resulted in a significant suppression of the righting reflex (mean ±SEM; F(3,34)=15.09, p<0.0001) compared to uninjured shams. Further, by excluding the uninjured shams, the one-way ANOVA revealed no significant differences between brain-injured mice, regardless of treatment (F(2,23)=0.06; p=0.9). (sham n=12, saline-treated injury n=9, AT-RvD1-treated injury n=9, RvE1-treated injury n=8; #, p<0.05 compared to sham).
3.2 RvE1 increased TBI-induced post-traumatic sleep
As expected, we found that uninjured shams, regardless of drug treatment, slept similarly (data not shown). During the first 12 hours following injury, a repeated-measure two-way ANOVA revealed a significant main effect of both time and treatment group on percent sleep. Further, Tukey’s multiple comparisons test revealed that RvE1-treated mice slept significantly more than all other groups during the first 12 hours following injury (Fig 3A). A more detailed analysis was performed by calculating the number of cumulative minutes spent sleeping during the first light and dark periods after brain injury. RvE1-treated brain-injured mice slept significantly more during the light period immediately following injury compared to saline-treated brain-injured mice (Fig 3B). During the following dark period, RvE1-treated brain-injured mice continued to sleep significantly more compared to uninjured sham and both other brain-injured groups (Fig 3C). Overall, brain-injured mice treated with RvE1 slept significantly more than other brain-injured mice, since uninjured mice treated with RvE1 slept similar to the remaining uninjured mice, indicating a brain injury rather than a treatment effect.
Figure 3. RvE1 increased post-traumatic sleep.

During the first 12 hours following injury (A), a two-way ANOVA revealed a significant main effect of treatment group on sleep (F(3,31)=7.6, p<0.0006). Specifically, Tukey’s multiple comparison test indicated that RvE1-treated mice slept significantly more than all other groups. RvE1-treated brain-injured mice slept more during the first light (B) and dark (C) period following injury. RvE1-treated brain-injured mice slept significantly more compared to saline-treated brain-injured mice in the light period (F(2,20)=5.0, p=0.02) and dark period (F(3,31)=9.1, p=0.0002) immediately following injury. Post-traumatic sleep showed a significant injury effect on number of sleep bouts during the first light period (D) (F(3,31)=7.0, p=0.001) and dark period (E) (F(3,31)=16.3, p<0.0001). (saline-treated injury n=7, AT-RvD1-treated injury n=9, RvE1-treated injury n=7, sham n=12) (*, p<0.05 compared to sham; #, p<0.05 compared to vehicle; +, p<0.05 compared to AT-RvD1)
Further, post-traumatic sleep showed a significant injury effect on number of sleep bouts during the first light period post-injury (Fig 3D). Post-hoc analysis indicated that regardless of treatment, all brain-injured groups slept significantly more sleep bouts compared to uninjured shams (Fig 3D). Overall, there was a group effect on the number of bouts in the dark period (Fig 3E). Tukey’s multiple comparison test revealed RvE1-treated brain-injured mice had significantly more bouts during the dark period compared to uninjured shams, saline-treated brain-injured mice, and AT-RvD1-treated brain-injured mice. These data show that RvE1 increased sleep through an increase in the number of sleep bouts, but not necessarily the length of the sleep bout.
3.3 AT-RvD1 mitigated TBI-induced sensorimotor deficits on the rotarod task
To assess motor function, the rotarod task was used as previously published28, 35, 37. Motor function was tested as the latency to stay on the rotarod over 7 days post-injury, with significant effects on both time post-injury and between groups. Tukey’s post-hoc analysis indicated that compared to uninjured shams, brain-injured mice treated with saline or RvE1 demonstrated significantly shorter latencies to fall from the rotarod. However, AT-RvD1-treated brain-injured mice did not show a significant deficit compared to uninjured shams and displayed significantly longer latencies than RvE1-treated mice. Overall, diffuse brain injury reduced motor function measured on the rotarod task. AT-RvD1-treated brain-injured mice did not show injury-induced impairments measured by latency (Fig 4). While AT-RvD1-treated mice did not perform significantly better than saline-treated mice, they did not demonstrate a deficit in comparison to sham. Together, these data suggest that AT-RvD1 improved or accelerated recovery from TBI, but perhaps not to an uninjured level.
Figure 4. AT-RvD1 prevented TBI-induced motor deficits measured by the rotarod task.

There was a significant effect of time after injury on motor performance F(3,99)=32.0, p<0.0001). There was also a significant group effect on latency to stay on the rotarod (F(3,33)=4.6, p=0.008). Tukey’s post-hoc analysis indicated that compared to uninjured shams, brain-injured mice treated with saline or RvE1 demonstrated significantly shorter latencies to fall from the rotarod. However, AT-RvD1-treated brain-injured mice performed comparably to uninjured shams and displayed significantly longer latencies than RvE1-treated mice. saline-treated injury n=11, AT-RvD1-treated injury n=6, RvE1-treated n=8, sham n=12). (*, p<0.05 compared to sham; +, p<0.05 compared to AT-RvD1)
3.4 TBI-induced cognitive impairment was improved with AT-RvD1 treatment
Cognitive function was measured using the novel object recognition task45. Paired t-tests indicated significant differences between time spent exploring the novel and familiar objects for uninjured shams and AT-RvD1-treated brain-injured mice, indicating recall of the familiar object (Fig 5A). However, saline-treated and RvE1-treated brain-injured mice spent equivalent times investigating both objects, indicating no recall of the familiar object (Fig 5A). Statistical comparisons between investigation times of the groups were not performed. A significant difference in investigation of novel and familiar was treated as the criterion for ‘recognition;’ only sham and AT-RvD1-treated groups reached recognition criterion. To further analyze cognitive performance on the NOR task, the discrimination index was calculated as the time spent with the novel object minus the time spent with the familiar object divided by the total time spent exploring for each mouse. There was no significant difference in the discrimination index between groups (Fig 5B).
Figure 5. AT-RvD1 prevented TBI-induced cognitive impairment measured by novel object recognition.

Differences in time spent exploring the novel versus familiar object were determined for each treatment group by paired t-test, using a modified p value of 0.0125 to account for four separate tests. These tests revealed a significant differences between investigation time of novel and familiar objects among sham (t(11)=4.6, p=0.0008) and AT-RvD1 treated brain-injured mice (t(8)=3.8, p=0.006) (A), indicating recall of the familiar object. However, saline-treated (t(7)=1.1, p=0.3) and RvE1-treated brain-injured mice (t(7)=1.7, p=0.1) spent similar times investigating both objects indicating no recall of the familiar object.The discrimination index (B) was calculated as the time spent with the novel object minus the time spent with the familiar object divided by the total time spent exploring. While sham and AT-RvD1 trended toward object recognition, there was no significant difference in discrimination indices between groups (F(3,33)=0.9, p=0.4). (saline-treated injury n=8, AT-RvD1-treated injury n=9, RvE1-treated injury n=8, shams n=12) (*, p<0.0125 between novel and familiar)
3.5 RvE1 increased the proportion of ramified microglia and decreased the proportion of rod microglia in the sensory cortex
Resolvin treatment did not alter the proportions of microglia phenotypes among saline and treated sham animals in somatosensory cortex (chi-square, p>0.05; data not shown). All brain-injured groups demonstrated a lower proportion of ramified microglia than uninjured sham mice at 7 days post-injury (chi-square, p<0.0001). RvE1-treated brain-injured mice had a significantly higher proportion of ramified microglia compared to saline-treated (p=0.0001) and AT-RvD1 mice (p<0.0001). All brain-injured groups demonstrated a greater proportion of activated microglia than uninjured sham mice (chi-square, p<0.0001). RvE1-treated brain-injured mice had a significantly lower proportion of activated microglia than AT-RvD1 treated mice (p=0.005). As expected, sham mice demonstrated no rod microglia. RvE1-treated brain-injured mice had a significantly lower proportion of rod microglia than saline-treated brain-injured mice (p=0.0002). These data show RvE1 significantly altered the inflammatory profile of microglia in somatosensory cortex, whereas microglial activation after AT-RvD1 treatment remained unchanged from the untreated condition.
4. DISCUSSION
The current study was designed with the proof-of-principle goals to: 1) test the administration of SPMs AT-RvD1 and RvE1 for therapeutic effect on translational outcomes of motor and cognitive function; 2) investigate a potential mechanism for SPM interactions in the injured brain by evaluating their effects upon microglial activation; 3) further probe the relationship between sleep and inflammation following diffuse brain injury in the mouse. One SPM, AT-RvD1, showed significant efficacy in mitigating the motor and cognitive deficits resulting from diffuse TBI. The other, RvE1, elicited a dramatic increase in post-traumatic sleep and tempered microglia activation without significantly influencing behavioral outcome compared to saline treatment. The divergence in histological (microglial activation) and physiological (sleep activity) outcomes from functional (motor and cognitive performance) outcomes suggests that microglial reactivity may contribute to post-traumatic sleep and that AT-RvD1 may achieve therapeutic efficacy through means other than inflammation resolution alone. Overall, SPMs demonstrate proof-of-principle as a therapeutic approach for diffuse brain injury, where additional studies are warranted, as described below.
Diffuse brain injury in humans results in long-term disabilities, including motor and cognitive impairments, which appreciably diminish quality of life3, 5. These injury-induced impairments can be modeled in rodents. The rotarod test has been used in the evaluation of motor function in rats46 and mice37, 40, 47, demonstrating impairments to remain on the accelerating rod following diffuse TBI. In the current study, saline-treated brain-injured mice exhibited significant rotarod impairment compared to sham-injured mice. This impairment was also evident in RvE1-treated mice, but not in AT-RvD1-treated mice. Similar results were found in cognitive performance using the novel object recognition (NOR) test, where sham-injured and brain-injured AT-RvD1-treated mice investigated novel objects significantly more than familiar objects—indicating short term episodic recognition48—but saline-treated and RvE1-treated brain-injured mice failed to recognize the familiar object. Although no literature is available on the effects of SPMs on functional outcome following TBI, there are several studies which show that DHA and fish oil (containing both EPA and DHA) supplementation attenuate TBI-induced motor49, 50 and cognitive51, 52 deficits. The current set of studies were undertaken, because the neuroprotective effects of DHA and fish oil may be in part due to actions of their SPM derivatives, including the resolvins, lipoxins, and protectins. We selected two resolvins to evaluate in diffuse brain injury. To this effect, one study tested the therapeutic efficacy of aspirin-triggered neuroprotectin D1 (AT-NPD1), an SPM derived from DHA, in a rat model of experimental stroke. One i.v. dose of AT-NPD1 at three hours after onset of experimental stroke was sufficient to improve performance on a battery of tests of neurological function compared to rats treated with saline33. These data, in light of the functional improvements elicited by AT-RvD1 in the present manuscript, suggest that SPM derivatives may mediate the protective effects of omega-3 fatty acids.
Clinical studies have provided evidence that TBI contributes to disorders in sleep regulation, including chronic sleep disturbance as well as excessive daytime sleepiness53-56. Chronic sleep issues in the rodent are unclear, with evidence both for57, 58 and against36 sustained sleep issues. We have previously demonstrated that sleep is increased in the first 3-6 hours following diffuse TBI in the mouse (post-traumatic sleep) compared to sham, when injury was induced 3 hours after light onset 27. In the present study, diffuse brain injury was delayed until 6 hours after the light onset and did not result in increased post-traumatic sleep among vehicle-treated mice. Therefore, the time of injury relative to the light:dark transition may influence post-traumatic sleep more than the time of day. Similarly, another study subjected mice to CCI and found that TBI resulted in acute decreases in wakefulness59. It is unclear the cellular benefit or detriment of this acute post-traumatic sleep on recovery following TBI, however, we have previously shown immediate disruption of post-traumatic sleep does not worsen injury-induced motor or cognitive deficits35 in the mouse. While the role of post-traumatic sleep in the pathophysiology of TBI remains unclear, data support a mechanistic link between inflammation and the induction of sleep24-27. SPMs including RvE1 and AT-RvD1 may resolve inflammation resulting from TBI, as incorporated into the extended drug delivery design, and subsequently attenuate post-traumatic sleep disorders.
Presently, RvE1 treatment of TBI significantly increased post-traumatic sleep, without functional improvement, compared to saline-treated and AT-RvD1-treated brain-injured mice. Further, microglial activation showed a lower proportion of rod microglia, with a higher proportion of ramified microglia, at 7 days post-injury when treated with RvE. The increase in sleep behavior and attenuation of the post-acute inflammatory response appear contradictory. However, inflammation and, more recently, microglial activation have been implicated in sleep regulation (for review, see Ingiosi60). It has been proposed that some drugs, such as methamphetamine, exert sleep regulatory effects through actions on microglia61. For example, genetic depletion of microglia attenuated the wake-promoting effect of methamphetamine in mice, suggesting that the presence of microglia contributes to sleep regulation following cellular stress61. Clinically, intracellular monocyte pro-inflammatory cytokine production was correlated with sleep modulation62. In the present study, post-traumatic sleep could increase in RvE1-treated mice with reduced post-acute microglial activation because of the delay between measurements, non-inflammatory sleep pressures, or unknown physiological mechanisms that also contributed to the higher mortality in RvE1 mice. Further, analysis of microglia morphology is only one indirect index of inflammation; direct measurement of inflammation-mediating cytokines would improve our understanding of the cellular processes involved. It remains likely that increased post-traumatic sleep of RvE1-treated mice, possibly a protective response, may promote resolution of inflammation as indicated by fewer activated rod microglia, albeit without functional benefit. Since sleep disruption results in microglial activation63 without worsening functional outcome after diffuse TBI in mice35, the effect of disrupting post-traumatic sleep in RvE1 treated mice would be intriguing.
While electroencephalographic (EEG) recording offers insight into sleep quality and stage, the piezoelectric sleep monitoring system offers a non-invasive approach to only quantify wakefulness and sleep durations, concordant with EEG analysis41. It is unclear how TBI or resolvin treatment may independently alter respiratory rate and confound the algorithmic detection of sleep, but we have previously demonstrated that the piezoelectric sleep system detects discrete bouts of 3 Hz breathing classified as sleep following mFPI. Further, post-traumatic sleep was not continuous and was interleaved with periods classified as wake and movement27. Sham-injured mice treated with either RvE1 or AT-RvD1 did not show changes in sleep pattern compared to vehicle treated mice.
Of the SPMs investigated in the current study, RvE1 solely tempered microglial reactivity while AT-RvD1 solely reduced the functional consequences of diffuse brain injury. Because selective results were obtained, the mechanism through which SPMs imparts functional benefit may not be through resolution of inflammation alone. In various experimental paradigms, resolvins and protectins have been implicated in reducing the deleterious effects of oxidative stress64, a recognized component of TBI pathophysiology. Further research is warranted to determine the mechanism(s) through which AT-RvD1 improved functional outcome from TBI. Regardless, the omega-3 fatty acids and likely their derivatives can impart functional recovery from TBI65, indicating that each derivative may target a specific pathophysiological process in a combination therapy manner.
A systematic isobolographic approach could combine the two (or more) treatments in varying concentrations for different groups of animals to better study synergistic drug effects66. This method has been suggested as a valid method of detecting beneficial drug-drug interactions in the development of therapies for TBI67. An isobolographic analysis of RvE1 and AT-RvD1 could potentially shed light on the interactions among post-traumatic sleep, inflammation, and functional outcome. Yet, this study may serve to caution against dissecting a pleotropic drug at the expense of separating specific effects and mechanisms.
As the first study to specifically assess the effects of resolvins in experimental TBI, several factors should be taken into consideration in future studies. The current proof of principle design could be refined to optimize the resolvin dosing with respect to the time of injury. Additionally, no literature is available on the most effective or biologically applicable dosing of synthetic resolvins with respect to acute neurological injury. One study administered AT-RvD1 (i.v. 300ng/mouse) in a mouse model of fibromyalgia68 while another study administered the comparable docosanoid neuroprotectin D1 (i.v. at 333 μg/kg) in a rat model of cerebral ischemia33. The current study falls between these wide margins of 12.5 μg/kg and 333 μg/kg respectively with a cumulative intraperitoneal dose of ~30μg/kg (700 ng/mouse). Further studies are warranted on the most appropriate administration and dosing schedules of lipid mediators.
5. CONCLUSION
In the diffuse brain-injured mouse, AT-RvD1 treatment, but not RvE1, mitigated motor and cognitive deficits. RvE1 treatment significantly increased post-traumatic sleep in comparison to all other groups. RvE1 treated mice displayed a higher proportion of ramified microglia and lower proportion of rod microglia in the cortex compared to saline or AT-RvD1 treated brain-injured mice. Increased post-traumatic sleep in the RvE1-treated brain-injured mice may be associated with active resolution of acute inflammation. We demonstrate that modulation of post-traumatic sleep and the inflammatory response to TBI seen in RvE1-treated mice were accomplished independently of improvement in motor and cognitive outcome as seen in AT-RvD1-treated mice. This suggests AT-RvD1 may impart neuroprotection through mechanisms other than inflammation resolution alone.
Diffuse TBI resulted in motor and cognitive impairments in the mouse.
AT-RvD1, but not RvE1, facilitated functional recovery.
RvE1-treated brain-injured mice slept significantly more immediately after TBI.
RvE1, but not AT-RvD1, decreased the injury-induced activation of microglia to TBI.
AT-RvD1 & RvE1 independently altered sleep, inflammation, and outcome after TBI.
Figure 6. RvE1 increased the proportion of ramified microglia and decreased the proportion of rod microglia.

Representative microscopic fields abundant with (A) ramified microglia (carets), (B) activated microglia (arrowheads), and (C) rod microglia (arrows) in the primary somatosensory barrel fields at 7 days post-injury. (D) Sham mice demonstrated a higher proportion of ramified microglia than all brain-injured groups (p<0.0001). RvE1-treated brain-injured mice had a significantly higher proportion of ramified microglia compared to saline-treated (p=0.0001) and AT-RvD1 mice (p<0.0001). (E) Sham mice demonstrated a lower proportion of activated microglia than all brain-injured groups (p<0.0001). RvE1-treated brain-injured mice had a significantly lower proportion of activated microglia than AT-RvD1 treated mice (p=0.005). (F) Sham mice demonstrated a lower proportion of rod microglia than all brain-injured groups (p<0.0001). RvE1-treated brain-injured mice had a significantly lower proportion of rod microglia than saline-treated brain-injured mice (p=0.0002). (G) A combined representation of observed microglial morphological distributions. (*, p<0.05 compared to sham; #, p<0.05 compared to vehicle; +, p<0.05 compared to AT-RvD1)
ACKNOWLEDGMENTS
Research reported in this manuscript was supported, in part, by National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award numbers R21 NS072611 and RO1 NS-065052, PCH Mission Support Funds, and Kentucky Spinal Cord & Head Injury Research Trust award 10-5A. The generosity of the Diane and Bruce Halle Foundation supported JLH during the conduct of these studies. RKR was supported by a Science Foundation Arizona Bisgrove Scholarship during the conduct of these studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Faul M, Xu L, Wald MM, Coronado VG. In: Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002 – 2006. Centers for Disease Control and Prevention NCfIPaC, editor. Atlanta, GA: 2010. [Google Scholar]
- 2.Tham SW, Palermo TM, Wang J, et al. Persistent pain in adolescents following traumatic brain injury. The journal of pain: official journal of the American Pain Society. 2013;14:1242–9. doi: 10.1016/j.jpain.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arciniegas DB, Topkoff J, Silver JM. Neuropsychiatric Aspects of Traumatic Brain Injury. Current treatment options in neurology. 2000;2:169–86. doi: 10.1007/s11940-000-0017-y. [DOI] [PubMed] [Google Scholar]
- 4.Albensi BC, Janigro D. Traumatic brain injury and its effects on synaptic plasticity. Brain injury: [BI] 2003;17:653–63. doi: 10.1080/0269905031000107142. [DOI] [PubMed] [Google Scholar]
- 5.Masel BE, DeWitt DS. Traumatic brain injury: a disease process, not an event. Journal of neurotrauma. 2010;27:1529–40. doi: 10.1089/neu.2010.1358. [DOI] [PubMed] [Google Scholar]
- 6.Margulies S, Hicks R. Combination therapies for traumatic brain injury: prospective considerations. Journal of neurotrauma. 2009;26:925–39. doi: 10.1089/neu.2008.0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McIntosh TK, Juhler M, Wieloch T. Novel pharmacologic strategies in the treatment of experimental traumatic brain injury: 1998. Journal of neurotrauma. 1998;15:731–69. doi: 10.1089/neu.1998.15.731. [DOI] [PubMed] [Google Scholar]
- 8.Werner C, Engelhard K. Pathophysiology of traumatic brain injury. British journal of anaesthesia. 2007;99:4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- 9.Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Inflammatory response in acute traumatic brain injury: a double-edged sword. Current opinion in critical care. 2002;8:101–5. doi: 10.1097/00075198-200204000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Morganti-Kossmann MC, Rancan M, Otto VI, Stahel PF, Kossmann T. Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock. 2001;16:165–77. doi: 10.1097/00024382-200116030-00001. [DOI] [PubMed] [Google Scholar]
- 11.Frugier T, Morganti-Kossmann MC, O’Reilly D, McLean CA. In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. Journal of neurotrauma. 2010;27:497–507. doi: 10.1089/neu.2009.1120. [DOI] [PubMed] [Google Scholar]
- 12.Semple BD, Bye N, Rancan M, Ziebell JM, Morganti-Kossmann MC. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2−/− mice. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2010;30:769–82. doi: 10.1038/jcbfm.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nature reviews. Neuroscience. 2001;2:734–44. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt OI, Heyde CE, Ertel W, Stahel PF. Closed head injury--an inflammatory disease? Brain research. Brain research reviews. 2005;48:388–99. doi: 10.1016/j.brainresrev.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 15.Lloyd E, Somera-Molina K, Van Eldik LJ, Watterson DM, Wainwright MS. Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small molecule improves long-term neurologic outcome in a mouse model of traumatic brain injury. Journal of neuroinflammation. 2008;5:28. doi: 10.1186/1742-2094-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao T, Thomas TC, Ziebell JM, Pauly JR, Lifshitz J. Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience. 2012;225:65–75. doi: 10.1016/j.neuroscience.2012.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lifshitz J, Lisembee AM. Neurodegeneration in the somatosensory cortex after experimental diffuse brain injury. Brain structure & function. 2012;217:49–61. doi: 10.1007/s00429-011-0323-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hall KD, Lifshitz J. Diffuse traumatic brain injury initially attenuates and later expands activation of the rat somatosensory whisker circuit concomitant with neuroplastic responses. Brain research. 2010;1323:161–73. doi: 10.1016/j.brainres.2010.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ziebell JM, Taylor SE, Cao T, Harrison JL, Lifshitz J. Rod microglia: elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. Journal of neuroinflammation. 2012;9:247. doi: 10.1186/1742-2094-9-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor SE, Morganti-Kossmann C, Lifshitz J, Ziebell JM. Rod microglia: a morphological definition. PloS one. 2014;9:e97096. doi: 10.1371/journal.pone.0097096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wirenfeldt M, Clare R, Tung S, Bottini A, Mathern GW, Vinters HV. Increased activation of Iba1+ microglia in pediatric epilepsy patients with Rasmussen’s encephalitis compared with cortical dysplasia and tuberous sclerosis complex. Neurobiology of disease. 2009;34:432–40. doi: 10.1016/j.nbd.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mukherjee S, Zeitouni S, Cavarsan CF, Shapiro LA. Increased seizure susceptibility in mice 30 days after fluid percussion injury. Frontiers in neurology. 2013;4:28. doi: 10.3389/fneur.2013.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics. 2010;7:22–30. doi: 10.1016/j.nurt.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krueger JM, Takahashi S, Kapas L, et al. Cytokines in sleep regulation. Advances in neuroimmunology. 1995;5:171–88. doi: 10.1016/0960-5428(95)00007-o. [DOI] [PubMed] [Google Scholar]
- 25.Krueger JM, Rector DM, Churchill L. Sleep and Cytokines. Sleep Med Clin. 2007;2:161–9. doi: 10.1016/j.jsmc.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krueger JM, Obal FJ, Fang J, Kubota T, Taishi P. The role of cytokines in physiological sleep regulation. Annals of the New York Academy of Sciences. 2001;933:211–21. doi: 10.1111/j.1749-6632.2001.tb05826.x. [DOI] [PubMed] [Google Scholar]
- 27.Rowe RK, Striz M, Bachstetter AD, et al. Diffuse brain injury induces acute post-traumatic sleep. PloS one. 2014;9:e82507. doi: 10.1371/journal.pone.0082507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bachstetter AD, Rowe RK, Kaneko M, Goulding D, Lifshitz J, Van Eldik LJ. The p38alpha MAPK Regulates Microglial Responsiveness to Diffuse Traumatic Brain Injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:6143–53. doi: 10.1523/JNEUROSCI.5399-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Recchiuti A, Serhan CN. Pro-Resolving Lipid Mediators (SPMs) and Their Actions in Regulating miRNA in Novel Resolution Circuits in Inflammation. Frontiers in immunology. 2012;3:298. doi: 10.3389/fimmu.2012.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serhan CN, Hong S, Gronert K, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. The Journal of experimental medicine. 2002;196:1025–37. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belayev L, Khoutorova L, Atkins KD, Bazan NG. Robust docosahexaenoic acid-mediated neuroprotection in a rat model of transient, focal cerebral ischemia. Stroke; a journal of cerebral circulation. 2009;40:3121–6. doi: 10.1161/STROKEAHA.109.555979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belayev L, Khoutorova L, Atkins KD, et al. Docosahexaenoic Acid Therapy of Experimental Ischemic Stroke. Translational stroke research. 2011;2:33–41. doi: 10.1007/s12975-010-0046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bazan NG, Eady TN, Khoutorova L, et al. Novel aspirin-triggered neuroprotectin D1 attenuates cerebral ischemic injury after experimental stroke. Experimental neurology. 2012;236:122–30. doi: 10.1016/j.expneurol.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lifshitz J. Fluid Percussion Injury. In: Chen J, X-MXaJZ ZX, editors. Animal Models of Acute Neurological Injuries. The Humana Press Inc.; Totowa, NJ: 2008. [Google Scholar]
- 35.Rowe RK, Harrison JL, O’Hara BF, Lifshitz J. Recovery of neurological function despite immediate sleep disruption following diffuse brain injury in the mouse: clinical relevance to medically untreated concussion. Sleep. 2014;37:743–52. doi: 10.5665/sleep.3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rowe RK, Harrison JL, O’Hara BF, Lifshitz J. Diffuse brain injury does not affect chronic sleep patterns in the mouse. Brain injury: [BI] 2014;28:504–10. doi: 10.3109/02699052.2014.888768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrison JL, Rowe RK, O’Hara BF, Adelson PD, Lifshitz J. Acute over-the-counter pharmacological intervention does not adversely affect behavioral outcome following diffuse traumatic brain injury in the mouse. Experimental brain research. 2014 doi: 10.1007/s00221-014-3948-3. [DOI] [PubMed] [Google Scholar]
- 38.Hosseini AH, Lifshitz J. Brain injury forces of moderate magnitude elicit the fencing response. Med Sci Sports Exerc. 2009;41:1687–97. doi: 10.1249/MSS.0b013e31819fcd1b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Donohue KD, Medonza DC, Crane ER, O’Hara BF. Assessment of a non-invasive high-throughput classifier for behaviours associated with sleep and wake in mice. Biomedical engineering online. 2008;7:14. doi: 10.1186/1475-925X-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rowe RK, Harrison JL, O’Hara BF, Lifshitz J. Recovery of neurological function despite immediate sleep disruption following diffuse brain injury in the mouse: clinical relevance to medically untreated concussion. Sleep. 2013 doi: 10.5665/sleep.3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mang GM, Nicod J, Emmenegger Y, Donohue KD, O’Hara BF, Franken P. Evaluation of a piezoelectric system as an alternative to electroencephalogram/ electromyogram recordings in mouse sleep studies. Sleep. 2014;37:1383–92. doi: 10.5665/sleep.3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McShane BB, Galante RJ, Jensen ST, Naidoo N, Pack AI, Wyner A. Characterization of the bout durations of sleep and wakefulness. J Neurosci Methods. 2010;193:321–33. doi: 10.1016/j.jneumeth.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han X, Tong J, Zhang J, et al. Imipramine treatment improves cognitive outcome associated with enhanced hippocampal neurogenesis after traumatic brain injury in mice. Journal of neurotrauma. 2011;28:995–1007. doi: 10.1089/neu.2010.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ennaceur A, Aggleton JP. The effects of neurotoxic lesions of the perirhinal cortex combined to fornix transection on object recognition memory in the rat. Behav Brain Res. 1997;88:181–93. doi: 10.1016/s0166-4328(97)02297-3. [DOI] [PubMed] [Google Scholar]
- 45.Antunes M, Biala G. The novel object recognition memory: neurobiology, test procedure, and its modifications. Cognitive processing. 2012;13:93–110. doi: 10.1007/s10339-011-0430-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hamm RJ. Neurobehavioral assessment of outcome following traumatic brain injury in rats: an evaluation of selected measures. Journal of neurotrauma. 2001;18:1207–16. doi: 10.1089/089771501317095241. [DOI] [PubMed] [Google Scholar]
- 47.Fenn AM, Gensel JC, Huang Y, Popovich PG, Lifshitz J, Godbout JP. Immune Activation Promotes Depression 1 Month After Diffuse Brain Injury: A Role for Primed Microglia. Biological psychiatry. 2013 doi: 10.1016/j.biopsych.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav Brain Res. 1988;31:47–59. doi: 10.1016/0166-4328(88)90157-x. [DOI] [PubMed] [Google Scholar]
- 49.Russell KL, Berman NE, Gregg PR, Levant B. Fish oil improves motor function, limits blood-brain barrier disruption, and reduces Mmp9 gene expression in a rat model of juvenile traumatic brain injury. Prostaglandins, leukotrienes, and essential fatty acids. 2014;90:5–11. doi: 10.1016/j.plefa.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Desai A, Kevala K, Kim HY. Depletion of brain docosahexaenoic acid impairs recovery from traumatic brain injury. PloS one. 2014;9:e86472. doi: 10.1371/journal.pone.0086472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu A, Ying Z, Gomez-Pinilla F. The salutary effects of DHA dietary supplementation on cognition, neuroplasticity, and membrane homeostasis after brain trauma. Journal of neurotrauma. 2011;28:2113–22. doi: 10.1089/neu.2011.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu A, Ying Z, Gomez-Pinilla F. Exercise facilitates the action of dietary DHA on functional recovery after brain trauma. Neuroscience. 2013;248:655–63. doi: 10.1016/j.neuroscience.2013.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Castriotta RJ, Wilde MC, Lai JM, Atanasov S, Masel BE, Kuna ST. Prevalence and consequences of sleep disorders in traumatic brain injury. Journal of clinical sleep medicine: JCSM: official publication of the American Academy of Sleep Medicine. 2007;3:349–56. [PMC free article] [PubMed] [Google Scholar]
- 54.Baumann CR, Werth E, Stocker R, Ludwig S, Bassetti CL. Sleep-wake disturbances 6 months after traumatic brain injury: a prospective study. Brain: a journal of neurology. 2007;130:1873–83. doi: 10.1093/brain/awm109. [DOI] [PubMed] [Google Scholar]
- 55.Kempf J, Werth E, Kaiser PR, Bassetti CL, Baumann CR. Sleep-wake disturbances 3 years after traumatic brain injury. Journal of neurology, neurosurgery, and psychiatry. 2010;81:1402–5. doi: 10.1136/jnnp.2009.201913. [DOI] [PubMed] [Google Scholar]
- 56.Baumann CR. Traumatic Brain Injury and Disturbed Sleep and Wakefulness. Neuromolecular Med. 2012 doi: 10.1007/s12017-012-8178-x. [DOI] [PubMed] [Google Scholar]
- 57.Hazra A, Macolino C, Elliott MB, Chin J. Delayed thalamic astrocytosis and disrupted sleep-wake patterns in a preclinical model of traumatic brain injury. Journal of neuroscience research. 2014 doi: 10.1002/jnr.23430. [DOI] [PubMed] [Google Scholar]
- 58.Lim MM, Elkind J, Xiong G, et al. Dietary therapy mitigates persistent wake deficits caused by mild traumatic brain injury. Science translational medicine. 2013;5:215ra173. doi: 10.1126/scitranslmed.3007092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Willie JT, Lim MM, Bennett RE, Azarion AA, Schwetye KE, Brody DL. Controlled cortical impact traumatic brain injury acutely disrupts wakefulness and extracellular orexin dynamics as determined by intracerebral microdialysis in mice. Journal of neurotrauma. 2012;29:1908–21. doi: 10.1089/neu.2012.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ingiosi AM, Opp MR, Krueger JM. Sleep and immune function: glial contributions and consequences of aging. Current opinion in neurobiology. 2013;23:806–11. doi: 10.1016/j.conb.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wisor JP, Schmidt MA, Clegern WC. Cerebral microglia mediate sleep/wake and neuroinflammatory effects of methamphetamine. Brain, behavior, and immunity. 2011;25:767–76. doi: 10.1016/j.bbi.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 62.Thomas KS, Motivala S, Olmstead R, Irwin MR. Sleep depth and fatigue: role of cellular inflammatory activation. Brain, behavior, and immunity. 2011;25:53–8. doi: 10.1016/j.bbi.2010.07.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hsu JC, Lee YS, Chang CN, Chuang HL, Ling EA, Lan CT. Sleep deprivation inhibits expression of NADPH-d and NOS while activating microglia and astroglia in the rat hippocampus. Cells, tissues, organs. 2003;173:242–54. doi: 10.1159/000070380. [DOI] [PubMed] [Google Scholar]
- 64.Wang L, Yuan R, Yao C, et al. Effects of resolvin D1 on inflammatory responses and oxidative stress of lipopolysaccharide-induced acute lung injury in mice. Chin Med J (Engl) 2014;127:803–9. [PubMed] [Google Scholar]
- 65.Hasadsri L, Wang BH, Lee JV, et al. Omega-3 fatty acids as a putative treatment for traumatic brain injury. Journal of neurotrauma. 2013;30:897–906. doi: 10.1089/neu.2012.2672. [DOI] [PubMed] [Google Scholar]
- 66.Tallarida RJ. Revisiting the isobole and related quantitative methods for assessing drug synergism. The Journal of pharmacology and experimental therapeutics. 2012;342:2–8. doi: 10.1124/jpet.112.193474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shear DA, Tortella FC. A military-centered approach to neuroprotection for traumatic brain injury. Frontiers in neurology. 2013;4:73. doi: 10.3389/fneur.2013.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klein CP, Sperotto ND, Maciel IS, Leite CE, Souza AH, Campos MM. Effects Of D-Series Resolvins On Behavioral And Neurochemical Changes In A Fibromyalgia-Like Model In Mice. Neuropharmacology. 2014 doi: 10.1016/j.neuropharm.2014.05.043. [DOI] [PubMed] [Google Scholar]