

Abstract
Unresolved inflammation is a major contributor to the development of heart failure following myocardial infarction (MI). Pro-resolving lipid mediators, such as resolvins (e.g. RvD1), are biosynthesized endogenously. The role of RvD1 in resolving post-MI inflammation has not been elucidated due to its unstable nature. Here, we have tested the role for two forms of RvD1, after incorporation into liposomes (Lipo-RvD1) and its free acid form (RvD1) in left ventricle (LV) and splenic remodeling post-MI. 8 to 12-week old male, C57BL/6J-mice were subjected to coronary artery ligation and Lipo-RvD1 or RvD1 (3µg/kg/day) was injected 3 hr post-MI for day (d)1 or until d5. No-MI mice and saline-injected MI mice served as controls. RvD1 injected groups showed improved fractional shortening post-MI; preserving transient changes in the splenic reservoir compared to MI-saline. RvD1-groups showed an early exit of neutrophils from LV and spleen at d5 post-MI with an increased expression of lipoxin A4 receptor; (ALX; synonym formyl peptide receptor; FPR2) compared to MI-saline group. The levels of pro-resolving mediators RvD1, RvD2, Maresin 1 (MaR1) and Lipoxin A4 (LXA4) were increased in spleens from RvD1 injected mice at d5 post-MI. RvD1 administration reduced macrophage density, ccr5 and cxcl5 levels at d5 post-MI compared to saline injected mice (both, p<0.05). Increased transcripts of mrc-1, arg-1 and Ym-1; (all, p<0.05) suggest macrophage-mediated clearance of necrotic cells in RvD1-groups. RvD1 reduced the pro-fibrotic genes (colla1, coll2a1 and tnc (all; p<0.05) and decreased collagen deposition, thereby reducing post-MI fibrosis and thus stabilizing the extracellular matrix. In summary, RvD1 and Lipo-RvD1 promote the resolution of acute inflammation initiated by MI, thereby delaying the onset of heart failure.
Keywords: Resolvin D1, resolution of inflammation, myocardial infarction, neutrophils, splenic remodeling, liposomes, metabololipidomics
Graphical abstract
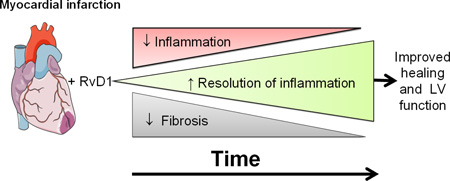
1. Introduction
Delayed or diminished resolution of inflammation is the key process that advances the pathophysiology of many diseases, including congestive heart failure, atherosclerosis and peripheral artery disease [1–3]. Persistent, unresolved inflammation is the major unifying component in the progression of heart failure following myocardial infarction (MI). Post-MI cytokine inhibitory anti-inflammatory treatments have been unsuccessful to treat defective resolution of inflammation leading to congestive heart failure. With the failure of cyclooxygenase (COX)-2 inhibitors, rofecoxib and celecoxib, [4, 5] and TNF-α inhibitor trials [6] in clinical settings, novel directions to resolve inflammation are necessary. Post-MI resolution of inflammation is an active, dynamic process following an acute inflammatory response, demonstrated by the synchronic temporal and spatial activities of neutrophils and macrophages. The overactive neutrophils are associated with major secretions of pro-inflammatory mediators and directly promote cardiomyocyte death [7]. In cardiac healing, it is necessary to discontinue the overactive priming of neutrophils in order to resolve inflammation. Thus, as a part of the post-MI resolution process, several specialized pro-resolving lipid mediators (SPM) are produced during the neutrophils-monocyte trafficking and cell-cell interaction. The interactions of neutrophils, platelets and macrophages promotes the generation of specialized pro-resolving lipids, including lipoxins, resolvins, protectins, and maresins, with their functions being to limit neutrophil recruitment and promote the resolution of acute inflammation [3].
Excessive intake of n-6 polyunsaturated fatty acids promotes non-resolving post-MI inflammation, leading to early left ventricle (LV) dysfunction [2, 8]. However, the role of novel pro-resolving lipid mediators, generated endogenously from omega-3 essential fatty acids, is unaddressed in post-MI remodeling. Our previous study suggests that docosahexaenoic acid (DHA) is more potent than eicosapentaenoic acid (EPA) in controlling chronic inflammation in lupus-prone mice [9]. Analysis of the DHA bioactive metabolome revealed novel bioactive molecules, named as Resolvins (RvD1-RvD6), which control neutrophil priming and re-program inflammatory macrophages towards the healing response. Resolvins are formed by enzymatic conversion of DHA (D-series) and EPA (E-series) [10]. Another pathway to form the D-series resolvins is triggered by aspirin, which acetylates COX-2 that converts DHA into 17R-HDHA. 17R-HDHA is then converted by 5-LOX to create AT-RvD1 [3]. The pro-resolving role of RvD1 has been studied [11] in a number of systems including metabolic disease and infections [12, 13]. The rapid conversion of RvD1 by eicosanoid oxidoreductase to 8-oxo- and 17-oxo-RvD1 dramatically lowers its bioactivity [14]. In the present investigation, we determined the role of liposomal RvD1 (Lipo-RvD1) and RvD1 in post-MI resolution of inflammation and LV remodeling. RvD1 was formulated into multilamellar liposomes to increase the stability of RvD1. Furthermore, we determined the impact of RvD1 on splenic remodeling post-MI. Our study delineates that both Lipo-RvD1 and RvD1 activate the post-MI resolving phase by limiting neutrophil-activation at the LV and spleen, furthermore clearing macrophages from the injured LV to improve LV function. RvD1 discontinued the priming of neutrophils and promoted the biosynthesis of pro-resolving bioactive lipid mediators, including RvD2, MaR1, and LXA4, in the spleen. Thus, our results demonstrate the host protective actions of RvD1, as well as a novel route of administration via incorporation into multilamellar liposomes. In summary, RvD1 altered neutrophils kinetics at the splenic reservoir and LV, leading to improved resolution of inflammation and attenuated cardiac remodeling post-MI.
2. Methods
2.1 Animal care and compliance
All animal surgery procedures and treatments were conducted according to the “Guide for the Care and Use of Laboratory Animals” (8th Edition. 2011), and AVMA Guidelines for the Euthanasia of Animals: (2013 Edition) and were approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham, USA.
2.2 Materials
Egg phosphatidylcholine (EPC-35), DSPE-PEG (2000) (1,2-distearoyl-sn-glycero-3-phosphoethanolamine (polyethylene glycol)-2000] were obtained from Avanti polar lipids (Alabaster, AL, USA). Cholesterol and RvD1 were obtained from Sigma, (St. Louis, MO, USA) and Cayman chemicals, (Ann Arbor, MI, USA) respectively.
2.3 Preparation and characterization of RvD1-liposomes
PEGylated liposomes containing RvD1 were prepared from a mixture of phospholipids by utilizing the film extrusion method. [15] The liposomes were characterized for size and polydispersity index using dynamic light scattering for colloidal size measurements.
2.4 RvD1-liposomes size measurements
The average hydrodynamic diameter and the polydispersity index of the liposome dispersions were determined by dynamic light scattering using a Malvern zetasizer nano (Malvern Instruments Ltd.; Worcestershire, United Kingdom) at 25°C using an argon-ion laser (488 nm) operating at 10.4 mW (Uniphase). The polydispersity index was calculated according to the method of Zhao et al [16]. The size of the PEGylated liposomes was found to be in between 100 and 150 nm with a polydispersity index below 0.20, indicating a relatively homogenous size distribution.
2.5 Zeta potential measurements
Electrophoretic mobility measurements (Zetasizer Nano-Z, Malvern instruments, UK) were performed after dilution of the liposomes in HEPES buffer pH 7.5. The instruments were calibrated using polystyrene latex beads of defined zeta potential. The mean zeta potential of PEGylated liposomes was found to be −25 mV.
2.6 Coronary artery ligation surgery in mice and RvD1 treatment plan
C57BL/6J mice of 8–12 weeks old were obtained from Jackson Laboratory (Bar Harbor, Maine, USA) and were maintained under constant temperature (19.8–22.2°C). The mice were given free access to water and standard chow diet. The mice were divided into 4 groups- (1) Group-1 as a control group with no surgery (day 0: no-MI control), (2) Group-2 as MI-saline group having MI surgery with vehicle treatment, (3) Group-3 administered liposomal-RvD1, 3 hr post-MI (Lipo-RvD1), (4) Group-4 treated RvD1, 3 hr post-MI (RvD1). To induce MI, mice were subjected to the surgical ligation of the left anterior descending coronary artery, as described previously [17]. In brief, the mice were anesthetized with 2% isoflurane and the left anterior descending coronary artery was permanently ligated using nylon 8-0 sutures (ARO Surgical Instruments Corporation, CA, USA) in a minimally invasive surgery. Prior to MI surgery, carprofen (5 mg/kg; subcutaneous (SQ) and buprenorphine (0.1 mg/kg, SQ) were administered to reduce pain. The mice were injected with either Lipo-RvD1 (3 µg/kg/day; SQ) or RvD1 (3 µg/kg/day; SQ) 3 hours post-MI and monitored for day (d)1 or d5 necropsy samples.
2.7 Echocardiography
For the echocardiography analysis, mice were anesthetized using 1.5–2.0% isoflurane in a 100% oxygen mix. Electrocardiograms and heart rates were monitored using a surface electrocardiogram. Images were acquired using the Vevo 770 in vivo imaging system (Visual Sonics, Canada) equipped with probes up to 40 MHz and a resolution of 30 µm. Short and long axis images were acquired at heart rates >400 beats/min to achieve physiologically relevant measurements. Measurements were taken from the two dimensional parasternal long-axis (B-mode) and short-axis (M-mode) recordings from the mid-papillary region. Echocardiographic studies were performed before necropsy for d0 control mice and for d1 and d5 post-MI mice. For each variable, three images from consecutive cardiac cycles were measured and averaged by operator blinded to genotype [17].
2.8 Necropsy and infarct area analysis
No-MI control day (d0), d1 or d5 post-MI RvD1 treated, and saline injected mice were anesthetized under 2% isoflurane anesthesia in 100% oxygen mix. To collect plasma, heparin (4 IU/g; I.P.) injection was used. The blood was collected from the carotid artery after 5 minutes post heparin administration, and centrifuged for 5 min to isolate plasma. The lungs and left and right ventricles were collected, weighed and processed as previously described [8]. The spleen was dissected by making incision in left of the peritoneal wall. The spleen was weighed and photographed using canon DSLR camera. The spleen was divided into two halves and the broad and concave portion was fixed in 10% zinc formalin for IHC and the rest of the spleen was snap-frozen for biochemical and molecular analysis.
2.9 LV and spleen histology and immunohistochemistry
For histological measurements, LV transverse and spleen sections were embedded in paraffin and sectioned. For assessment of neutrophils and macrophages, paraffin-embedded sections were deparaffinized in citrisolv and rehydrated through graded ethanol. Heat-mediated antigen retrieval was performed to expose antigen epitopes (Target Retrieval Solution, Dako S1699) using a pressure cooker (BioSB Tinto Retriever). Sections, blocked with normal rabbit or goat serum as per antibody, were incubated with rat anti-mouse neutrophils (CL 8993AP, clone 7 1:50; Cedarlane) and rat anti-Mac-3 monoclonal antibody (CL 8943AP, clone M3/84; 1:100 dilution; Cedarlane). Neutrophils and macrophages staining were followed with the Vectastain Elite ABC kit (Vector). The slides were mounted using permount and allowed to dry for use in image analysis [17].
2.10 Metabololipidomics using liquid chromatography-tandem mass spectrometry (LC-MS/MS)
Briefly, spleens were suspended in 1.0 ml cold methanol and gently homogenized, afterwards they were kept on ice for 1h to allow for protein precipitation. Lipid mediators (LM) were profiled as described in Colas et al [18]. LM were extracted using SPE on C18 columns and methyl formate fractions were taken for LC-MS/MS-based lipidomics. The spleen samples from no-MI control, MI-saline and RvD1-treated group were analyzed on a LC -MS/MS system, QTRAP 6500 (ABSciex). Calibration curves were obtained using synthetic and authentic LM mixtures. Linear calibration curves for each were obtained with r2 values in the range 0.98–0.99. Quantification was carried out based on peak area of the MRM transition and the linear calibration curve for each compound and biosynthetic pathway marker as in Colas et al [18].
2.11 Picrosirius red staining
For PSR staining, paraffin-embedded unstained sections of LV tissue were deparaffinized in citrisolv (Fisher Scientific, USA) and rehydrated through subsequent washes of ethanol. After a wash with water, phosphomolybdic acid (0.2% in water) was placed on the section. A subsequent wash with water was followed by addition of sirius red, 0.1 % in saturated picric acid (26357-02) and then application of 0.01N hydrochloric acid. Then, slides were dehydrated and mounted using permount. The slides were allowed to dry for image acquisition and analysis. [19].
2.12 Image analysis for IHC and PSR staining
For each slide per mouse, a total of 4–6 images were acquired from spleen and LV infarct area including border zone using a microscope (BX43) with an attached camera (Olympus DP73). The images were captured using the cellSens Dimension program (Olympus version 1.9) and then analyzed for percent area stained using Image-Pro Premier 64-bit analyzer software (Image Pro Premier, Cybernatics). The percent stained area was determined by the image analysis software and averaged to determine the percent area stained for neutrophils (spleen and LV), macrophages and collagen (PSR) density in the infarcted area.
2.13 RT2profiler inflammatory and extracellular matrix (ECM) PCR array
Gene expression profiling was done using inflammatory and ECM PCR arrays in frozen infarcted (LVI) samples for no-MI control, MI saline, Lipo-RvD1 and RvD1-injected mice as previously described [8]. Briefly, RNA was isolated and cDNA synthesis was performed. The samples were loaded on RT2-PCR plate (Inflammatory Cytokine and Receptor by Qiagen PAMM-011E and Mouse ECM & adhesion molecules PAMM-013E) and ran on an ABI 7900HT. Gene levels were normalized to hypoxanthine phosphoribosyltransferase (Hprt-1) as the housekeeping gene. The results were reported as 2−ΔCt (ΔΔCt) values.
2.14 Quantitative real-time PCR for measurements of gene transcripts
For qPCR, reverse transcription was performed with 2.0 µg of total RNA using SuperScript® VILO cDNA Synthesis kit (Invitrogen, CA, USA). Quantitative PCR for ALX/FPR2, ptgs-1, ptgs-2, hmox-1, Alox-12, Alox-5, tnf-α, IL-6, ccl2, IL-1β, CD163, arg-1, mrc-1 and Ym-1 genes was performed using taqman probes (Applied Biosystems, CA, USA) on master cycler ABI, 7900HT. Gene levels were normalized to Hprt-1 as the housekeeping control gene. The results were reported as 2−ΔCt (ΔΔCt) values. All the experiments were performed in duplicates with n=3–4/group.
2.15 LV protein extraction for immunoblotting
The LVI tissues were processed for protein extraction as previously described [8].
2.16 LV protein immunoblotting
Electrophoresis of 10 µg of LVI protein were performed using criterion XT bis-tris 4–12 % gel (Bio-Rad Inc.) in MOPs Buffer (Bio-Rad) and transferred on nitrocellulose membrane (BioRad Inc.). The total protein stain was acquired using pierce reversible protein stain, nitrocellulose membranes kit (Thermo Scientific Inc.). The membrane was blocked for 1 hr at room temperature, using 5% non-fat milk powder (Bio-Rad) dissolved in TPBS and probed with primary antibody [COX-2 1:1000, COX-1 1:1000, 5-LOX 1:200 (abcam) and ALX/FPR2 1:500 (Santa cruz)] overnight at 4°C followed by secondary antibody (Biorad). The proteins were detected using the femto chemiluminescence detection system (Pierce Chemical, Rockford, IL, USA). Densitometry was performed using Image J software (NIH, USA).
2.17 Venn diagram, hierarchical clustering of inflammatory and ECM array
Venn diagram, hierarchical cluster, and heat maps were generated for RT2 profiler inflammatory and ECM PCR array data using cluster 3.0 and java tree view software. The values of genes were normalized by taking geometric mean of the genes for statistical significance. For Venn diagrams the values were normalized to no-MI d0 control at the respective time points and evaluated for statistical significance.
2.18 Statistical analysis
Data are expressed as mean and SEM. Statistical analyses were performed using Graphpad prism 5. Analysis of variance (ANOVA) followed by Newman–Keuls post-hoc test was used for multiple comparisons of post-MI d1, d5 groups (RvD1 and saline injected compared to no-MI control. All immunoblotting densitometry data was normalized to total protein/lane. For 2 groups comparison, the student-t test (unpaired) was applied, p<0.05 was considered as statistically significant.
3. Results
3.1 RvD1 reduced LV hypertrophy and preserved transient depletion of splenic reservoir post-MI
To determine the post-MI gravimetric changes after RvD1 treatment, LV, spleen and lung masses were measured. Both Lipo-RvD1 and RvD1-injected groups reduced the LV mass to body weight ratio compared to the MI-saline group at d5-post-MI, indicating a reduced post-MI LV remodeling response (Table 1A). Post-MI spleen mass was decreased within 24 hr (post-MI d1) in MI-saline group, suggesting a depletion feature of the splenic reservoir in post-MI remodeling. We observed a significant decrease in spleen mass at d1 and increase at d5 post-MI in MI-saline compared to no-MI control group. Interestingly, Lipo-RvD1 and RvD1 treated mice did not display significant differences in spleen mass at d1 compared to no-MI control and decrease by d5 post-MI compared to saline-injected MI mice (Table 1A). Thus, gravimetric analyses indicated that RvD1 administration decreased LV hypertrophy and reduced transient changes in the splenic reservoir post-MI.
Table 1.
Necropsy and echocardiography parameters in controls and post-MI RvD1 injected mice.
| Table 1A | d0 Control | day 1 MI | day 5 MI | ||||
|---|---|---|---|---|---|---|---|
| Necropsy parameters |
No-MI Control | MI-Saline | Lipo-RvD1 | RvD1 | MI-Saline | Lipo-Rvd1 | RvD1 |
| n | 6 | 7 | 8 | 8 | 6 | 3 | 8 |
| Body weight (g) | 23±2 | 20±1 | 19±0.5 | 20±1 | 21±1 | 20±1 | 20±0.5 |
| Left ventricle (mg) | 74±5 | 79±4 * | 76±4 | 72±5 | 108±5* | 77±7$ | 78±4$ |
| LV/BW (mg/g) | 3.2 ± 0.04 | 4.0 ± 0.24* | 3.9±0.2* | 3.8 ± 0.2* | 5.3 ± 0.31* | 3.8±0.3*$ | 3.6±0 .2*$ |
| Spleen weight (mg) | 71 ± 3 | 39 ± 2* | 62±2*$ | 52 ± 4*$ | 82 ± 5* | 66±6$ | 66 ± 6 $ |
| Right ventricle (mg) | 17±2 | 19±1 | 17±1 | 20±1 | 19±2 | 19±2 | 17±1 |
| Lung weight (mg) | 127±8 | 184±22* | 146±16* | 176±17* | 211±12* | 119±8$ | 136±10$ |
| Lung wt (dry; mg) | 31±1 | 34±2 | 30±1 | 32±2 | 42±2* | 28±1$ | 27±1$ |
| Tibia (mm) | 16±0.7 | 16±0.3 | 15±0.4 | 16±0.3 | 17±0.2 | 16±0.5 | 16±0.1 |
| Ratio-LV/BW:LV Tibia | 0.71± 0.03 | 0.81±0.04* | 0.80±0.03* | 0.78 ±0.01* | 0.81±0.03* | 0.79±0.02* | 0.80 ±0.02* |
| RV mass/ BW | 0.74± 0.03 | 0.95±0.06* | 0.87±0.03* | 0.96 ±0.04* | 0.93±0.11* | 0.91±0.09* | 0.83±0.03* |
| Lung mass mg/ W body weight (g) |
5±0.2 | 9±0.1* | 8±0.9* | 8±0.9* | 18±0.7* | 6±0.4$ | 7±0.5$ |
| % infarct area | ND | 47 ± 1 | 44 ± 2 | 53 ± 4 | 49 ± 1 | 55 ± 4 | 46 ± 2 |
| Echocardiography parameters | |||||||
| Table 1B | d0 Control | day 1 MI | day 5 MI | ||||
| Echocardiography parameters |
No-MI Control | MI-Saline | Lipo-RvD1 | RvD1 | MI-Saline | Lipo-Rvd1 | RvD1 |
| Heart Rate (bpm) | 454±16 | 473±18 | 490±18 | 453±10 | 491±16 | 479±15 | 466±18 |
| Fractional shortening (%) |
37 ± 1.9 | 7 ± 0.8* | 10± 0.2*$ | 11 ±1.0*$ | 5 ± 0.4* | 19± 0.5*$ | 15±2*$ |
| EDD (mm) | 3.7±0.12 | 4.5±0.12 * | 4.3 ± 0.1*$ | 4.3±0.03*$ | 6.5±0.02* | 3.8±0.12 *$ | 4.1±0.1*$ |
| ESD (mm) | 2.3±0.14 | 4.2 ± 0.13* | 3.8± 0.1* | 3.7±0.07*$ | 6.1 ± 0.18* | 3.6 ±0.08*$ | 3.5±0.2*$ |
| PWTs (mm) | 1.05± 0.02 | 0.56 ± 0.06* | 0.53± 0.06* | 0.40±0.06* | 0.47 ± 0.05* | 0.55± 0.05*$ | 0.54±0.05*$ |
Values are mean± SEM; n indicates sample size. LV, left ventricle; BW, body weight; bpm, beats per minute; EDD, end-diastolic dimension; ESD, end-systolic dimension; FS, fractional shortening; PWT, Infarcted wall thickness, S,systole; mm, millimeter
p<0.05 vs day 0 control;
p<0.05 vs MI-saline at respective time point
3.2 RvD1 improved LV function and pulmonary edema post-MI
To assess the actions of RvD1 on LV function post-MI, echocardiography measurements were performed. Echocardiography data reflected a reduced fractional shortening at d1 and d5 in MI-saline group compared with no-MI control, indicating post-MI LV dysfunction. Both Lipo-RvD1 and RvD1 injected groups improved their fractional shortening compared to MI- saline group (Table 1B). Both Lipo-RvD1 and RvD1 groups showed lower dilation in LV geometry as evidenced by their decreased end systolic dimension (ESD) and end diastolic dimension (EDD) at d5 post-MI. Lipo-RvD1 and RvD1 treated mice showed a significant decrease in lung mass to body weight ratios compared to MI-saline injected mice at d5 post-MI (Table 1A). The echocardiography and gravimetric measurement outcomes suggested that RvD1 reduced both LV dysfunction and pulmonary edema post-MI.
3.3 RvD1 reduced neutrophil recruitment in LV
To investigate the impact of RvD1 on neutrophil infiltration post-MI, neutrophils density was measured. In response to MI-mediated acute inflammation, neutrophil infiltration is an immediate onset event during wound healing to resolve inflammation [20]. Post-MI, there was a robust neutrophil priming effect in all the groups which peaked at d1 in MI-saline group. Lipo-RvD1 and RvD1 injected groups showed unaltered and immediate neutrophil priming infiltration at d1 compared to MI-saline group. However, RvD1 group exhibited higher neutrophils infiltration at d1 post-MI when compared with Lipo-RvD1 and MI-saline groups. Presumably, this may be due to the immediate unavailability of RvD1 from liposomal formulation and owing to RvD1 encapsulation in Lipo-RvD1 compared to RvD1 at d1 post-MI. As expected, no-MI naïve control did not show neutrophil infiltration in LV (Figure 1A and C). The chemical structure of RvD1 is provided in Figure 1B. Of note, neutrophils priming was reduced to the infarct area in the Lipo-RvD1 and RvD1 injected group, while neutrophils recruitment continued in the MI-saline group at d5 post-MI. This was complemented by decreased expression of ccr5, ccl7 and cxcl5 gene expression in the infarcted area (Supplementary table 1). RvD1 is known to interact with ALX/FPR2 [21, 22] therefore ALX/FPR2 mRNA and protein expression was measured. At d1 to d5 post-MI, there was a delay to increase ALX/FPR2 expression in MI-saline group, accompanied with higher neutrophil density. Both Lipo-and RvD1 treated groups showed a more rapid increase in ALX/FPR2expression from d1 to d5 post-MI compared with MI-saline group; supporting the discontinuation of neutrophil priming for these groups (Figure 1D and supplementary Figure 2) as a result of the pro-resolving action of RvD1. Thus, reduced density of neutrophils at d5 clearly indicated that RvD1 administration post-MI reduced neutrophil recruitment and attenuated inflammation, leading to the early initiation of resolution.
Figure 1. RvD1 reduced LV neutrophil density and activated ALX/FPR2 expression at d5 post-MI.

(A) Representative neutrophil IHC images of LV transverse section for MI-saline, Lipo-RvD1 and RvD1 injected group at d1 and d5 post-MI (Magnification 40X, 4–6 images/slide were quantified/mouse, 3–5 mice/group, LV middle section at 1.25x, scale bar =50 µm). The arrow indicate the neutrophils at d5 post-MI in MI-saline, Lipo-RvD1 and RvD1 injected group (B) Chemical structure of RvD1, (C) Quantification of percentage stained area of neutrophils in the LV IHC at d1 and d5 post-MI in MI-saline, Lipo-RvD1 and RvD1 injected groups, the absence of quantifiable neutrophils in the LV are presented as “undetermined”. (D) mRNA expression of ALX/FPR2 in infarcted area of MI-saline, Lipo-RvD1 and RvD1 injected groups. *p<0.05 vs d0 no-MI control, $p<0.05 vs MI-saline at respective day, n=3–5 mice/group/day.
3.4 RvD1 reduced neutrophils density in spleen that coordinated with LV
Splenic metabolic activation is well documented in the pathogenesis of cardiac remodeling and subsequent chronic heart failure [23–25]. Neutrophils are known to be present around B lymphocytes in the spleen without inflammation [26] and their number increases in response to inflammation. Thus, we evaluated the density of splenic neutrophils post-MI. We observed a continuous increase in neutrophil population in the spleen at d1 post-MI (2.4%±0.6%) to d5 post-MI (4.1%±0.1%) in MI-saline group compared to no-MI control. However, consistent with the neutrophil density in LV, both Lipo-RvD1 and RvD1 injected mice showed a robust increase in neutrophils, 3.4±0.5% and 4.1±0.4% respectively, compared to the MI-saline group at d1 post-MI. By d5, both Lipo-RvD1 and RvD1 group showed rapid clearance of neutrophils, 2.1±0.3% and 2.4±0.2% respectively, compared to MI-saline group (Figure 2A and B). We next investigated whether ALX/FPR2 may be involved in mediating the actions of RvD1 in these spleens, perhaps in a similar manner as that observed in the LV infarcted area. RvD1 stimulated relatively higher expression of ALX/FPR2 in the spleen compared with infarcted area at d5 post-MI (Figure 2C). The data indicated that RvD1 expanded neutrophils activation at d1 post-MI for priming the immune cells. However, at d5 post-MI, RvD1 reduced neutrophil recruitment by activating ALX/FPR2 to pave the way for other immune cells and to expedite effective resolution thereby, limiting ventricular remodeling.
Figure 2. RvD1 reduced spleen neutrophils density and activated FPR2/ALX expression at d5 post-MI.

(A) H& E images of the spleen of d0 controls and at d1 and d5 post-MI in MI-saline, Lipo-RvD1 and RvD1 injected group; scale bar= 50 µm, magnification=40X, 4–6 images/slide were quantified/mouse, 3–5 mice/group. (B) Quantification of the percentage stained area of neutrophils in the spleen IHC at d1 and d5 post-MI in MI-saline, Lipo-RvD1 and RvD1 injected groups. (C) mRNA expression of ALX/FPR2 in spleen in MI-saline, Lipo-RvD1 and RvD1 treated groups in control and at d1 and d5 post-MI. *p<0.05 vs d0 no-MI control, $p<0.05 vs MI-saline at respective day; n=3-5/group/day.
3.5 RvD1 promoted the production of SPM in spleen to resolve post-MI inflammation
Our results showed that RvD1 acted on the spleen and LV to resolve post-MI inflammation. We then questioned whether RvD1 regulated lipid mediators in the spleen post-MI. Using LC-MS/MS-based lipid mediator metabololipidomics, we identified SPMs in the spleen at d5 post-MI in both MI-saline and RvD1 treated mice and compared their levels to no-MI control mice. In accordance to previous findings [27] with spleens from no-MI mice, we identified mediators from all three major bioactive lipid mediator metabolomes (Figure 3 and Supplementary Table 2), which includes the DHA derived D-series resolvins, protectins and maresins, the EPA derived E-series resolvins and the arachidonic acid derived lipoxins. These were all identified in accordance with published criteria that include matching retention times and MS-MS fragmentation (Figure 3A and B) [18]. The lipid mediator profiles of spleens from mice at d5 post-MI showed elevated levels of a number of pro-inflammatory mediators, including the arachidonic acid derived leukotriene B4 and prostaglandin D2. Spleens from RvD1 treated mice at d5 post-MI showed increase in a number of pro-resolving mediators, including RvD2, 17R–RvD1, MaR1, LXA4 and LXB4 (Supplementary Table 2). These results suggested that administration of RvD1 post-MI promotes the formation of SPM in the spleen that may contribute to organ protection and improved resolution of inflammation.
Figure 3. Mass spectrometry profiling of SPM (specialized pro-resolving lipid mediators) from DHA, EPA and arachidonic acid bioactive metabolomes in spleen at d5 post-MI.

(A) Representative Multiple Reaction Monitoring (MRM) traces for the identified lipid mediators accompanying MS/MS spectra used for identification of LXA4 and MaR1. (B) Accompanying MS/MS spectra used for identification of LXA4 and MaR1.
3.6 RvD1 promoted macrophages clearance by d5 post-MI
Macrophages are versatile, heterogeneous, big eaters whose role is to engulf necrotic cardiomyocytes and apoptotic neutrophils. Macrophages clear tissue debris and initiate ECM deposition in the infarcted area post-MI [20]. To understand the impact of RvD1 on macrophage kinetics post-MI, we further assessed the macrophage density and phenotypes post-MI. LV IHC results showed that RvD1 injected mice decreased macrophage density (mac-3) compared to MI-saline group at d5 post MI (Figure 4A–B). The decreased expression of chemokine receptors ccl4 (mip-1β), ccr2 and cx3cr1 (all p<0.05) indicated the rapid clearance of macrophages with a simultaneous decrease in the macrophage-derived chemokine (MDC/ccl22, p<0.05) (Figure 4; Supplementary table 1) in RvD1-injected groups. Thus, RvD1 stimulated macrophages emigration and thus prevented accumulation in the infarcted area at d5 post-MI by regulating both monocyte trafficking and their clearance from LV infarct area.
Figure 4. RvD1 reduced macrophage density at d5 post-MI.

(A) Representative Mac-3 IHC images of LV transverse section at d1 and d5 post-MI in MI-saline, Lipo-RvD1 and RvD1 respectively. (Magnification 40X, 4–6 images/slide were quantified/mouse, 3–5 mice/group, LV middle section at 1.25x, scale =50 µm), *p<0.05 vs MI-saline at d5 post-MI for Lipo-RvD1 and RvD1 treated groups (n=3–4 mice/group/day); (B) Macrophages (mac-3) stained area in the LV IHC at d5 post-MI in MI-saline, Lipo-RvD1 and RvD1 treated groups. (C) Venn diagram representing post-MI inflammatory gene expression in LVI regions of MI-saline, Lipo-RvD1 and RvD1 mice at d1 post-MI. (D) Venn diagram representing post-MI inflammatory gene expression in infarcted area of MI-saline, Lipo-RvD1 and RvD1 mice at d1 and d5 post-MI. Red indicates ted genes expression and green indicates led genes. Black indicates no change in genes. All the genes are normalized to no-MI control.
3.7 RvD1 contained inflammation and promoted inflammation resolution post-MI
To evaluate the impact of RvD1 on acute inflammatory response superimposed on the resolving response in post-MI settings, we measured 84 inflammation-associated genes. Array data showed that 20 genes were upregulated and 11 genes downregulated for the MI-saline group in MI settings. However, the Lipo-RvD1 treated group showed 8 upregulated and 18 down-regulated genes, while in the RvD1 group only 1 gene was upregulated while 16 were down-regulated. Interestingly, at d5 post-MI both Lipo-RvD1 and RvD1 injected mice exhibited down regulation of pro-inflammatory genes (Figure 4C–D). The decreased cxcl5 and ccl2 (both p<0.05) expression in both RvD1 groups confirmed the reduction of neutrophils and early clearance of macrophages at d5 post-MI. The Venn diagram (Figure 4C–D) reflects the down regulation of pro-inflammatory genes in RvD1-injected groups compared to MI-saline, which delineated the faster resolution of inflammation.
3.8 RvD1 promoted alternative macrophage polarization at d5 post-MI
Macrophages are heterotypic populations in the post-MI setting depending upon milieu, whether to promote or limit LV remodeling [23]. We explored the action of RvD1 on macrophage polarization at d5 post-MI. Tnf-α, IL-1β, ccl2 and IL-6 were classified as classical macrophage markers (M1; Figure 5A) while mrc-1, arg-1 and Ym-1 were classified as alternative macrophage (M2; Figure 5B). Results from M1 and M2 gene expression measurements revealed that RvD1 played a dual role in macrophage activation and polarization at specific time points. RvD1 rapidly increased the initial neutrophil infiltration, displayed by an increase in expression of tnf-α and ccl2 at d1 post-MI while at d5 post-MI it reduced neutrophil recruitment and expedited the clearance of neutrophils by macrophages (Figure 5A). This was supported by the increase in expression of resolving M2 macrophages markers mrc-1, arg-1 and Ym-1 (Figure 5B) in Lipo-RVD1 and RvD1-treated groups, suggesting the clearance of apoptotic neutrophils, fibroblast debris and dead myocytes. Since spleens of RvD1-treated groups generated SPMs at d5 post-MI, we asked the question of whether there is a change in macrophages polarization in the spleen as well. Gene expression data suggests that both Lipo-and RvD1 treated groups increased the expression of tnf-α at d1, which decreased at d5 post-MI with a simultaneous increase in the expression of arg-1 and mrc-1 (Supplementary Figure 3). These results indicated that RvD1 initiated resolution by expediting M2 gene expression and SPMs without altering the acute proinflammatory phase post-MI.
Figure 5. RvD1 promoted alternative macrophages polarization without altering pro-inflammatory response.

Gene expression of (A) Classical M1 markers, (B) Alternative M2 markers in MI-saline, Lipo-RvD1 and RvD1 treated groups. *p<0.05 vs d0 control, $p<0.05 vs MI-saline at respective day; n=4 mice/group/day, Lowest gene expression values of IL-6, Arg-1 and Ym-1 at d0 control are presented as “undetermined”.
3.9 RvD1 stimulated 5-LOX and ALX/FPR2 leading to attenuated cardiac remodeling post-MI
5-LOX has a supporting role in wound healing post-MI [28]. Since, 5-LOX is one of the enzyme involved in RvD1 biosynthesis, we measured the action of RvD1 on 5-LOX. As expected, 5-LOX was upregulated in the MI-saline group from d1 to d5 post-MI. Both RvD1-injected groups showed higher expressions of 5-LOX at the mRNA and protein levels compared to MI-saline group post-MI (Figure 6A–B) along with increases in ALX/FPR2 protein expression (Figure 6C–D). These results suggest that RvD1 mediated resolution by stimulating 5-LOX and ALX/FPR2 in order to facilitate the resolution of inflammation as part of the post-MI repair mechanism.
Figure 6. RvD1 activated 5-LOX, ALX/FPR2 and modulated COX-1 and COX-2.

(A) Immunoblot representing 5-LOX expression (B) Densitometric analysis and mRNA expression of 5-LOX (C) Immunoblot representing ALX/FPR2 expression (D) Densitometric analysis and mRNA expression of ALX/FPR2 (E) Immunoblot representing COX-1 expression (F) Densitometric analysis and mRNA expression of COX-1 (G) Immunoblot representing COX-2 expression. (H) Densitometric analysis and mRNA expression of COX-2. The protein and mRNA expression is measured in LV infarct of MI-saline, Lipo-RvD1 and RvD1 injected mice at d1 and d5 post-MI. *p<0.05 vs d0 no-MI control, $p<0.05 vs MI-saline at respective day; n=3-4/group/day.
3.10 RvD1 reduced COX-1 and COX-2 expression in resolving phase
COX-1 and COX-2 resolves inflammation post-MI by generating pro-inflammatory and pro-resolving mediators in order to clear the post-MI debris of dead cells [29]. RvD1 did not alter COX-1 and −2 levels at d1 post-MI, however, it did decrease the expression of both COX-1 and −2 at d5 post-MI (Figure 6E–H). Thus, the limited expression of COX-1 and-2 in both Lipo-RvD1 and RvD1 treated groups indicated reduced inflammation at d5 post-MI without alteration of the acute inflammatory phase.
3.11 RvD1 reduced collagen deposition post-MI
Collagen deposition is a subsequent event occurring after the acute inflammatory response which leads to myocardium stiffness and ultimately LV dysfunction post-MI. In order to evaluate whether RvD1 could limit myocardial ECM deposition post-MI, we measured PSR stained area in the infarct zone at d0, d1 and d5 post-MI in MI-saline and RvD1-treated groups. At d5, MI-saline group showed 23±4% increase in collagen levels compared to d1 or d0 controls. However, both Lipo- and RvD1 treated groups reduced collagen deposition to 13±1% and 14±1% respectively at d5 post-MI (Figure 7A–B). Thus, RvD1 treatment initiated the early reparative phase to limit collagen deposition and improve the LV healing process.
Figure 7. RvD1 reduced post-MI LV collagen content and extracellular matrix transcripts.

(A) Representative images of PSR staining in LV infarct of MI-saline, Lipo-RvD1 and RvD1 injected mice at d1 and d5 post-MI (Magnification 40X, 4–6 images/slide were quantified/mouse, 3–5 mice/group, LV middle section at 1.25x, scale =50 µm). (B) Collagen percentage stained area in the LV at d5 post-MI in MI-saline, Lipo-RvD1 and RvD1 injected groups. *p<0.05 vs d5 MI-saline; n=3-4/group/day (C) Venn diagram representing post-MI ECM remodeling genes in LV infarct of MI-saline, Lipo-RvD1 and RvD1 injected mice at d1 post-MI. (D) Venn diagram representing post-MI ECM remodeling genes in LV infarct of MI-Saline, Lipo-RvD1 and RvD1 injected mice at d5 post-MI. Post-MI gene expression are normalized to respective d0 control. Red indicates ↑ed and green indicates ↓ed gene expression. Black indicates no change in genes.
3.12 RvD1 reduced ECM gene transcripts post-MI
To identify the manner in which RvD1 influences the ECM remodeling post-MI, we evaluated the RvD1 action on ECM remodeling. We used a gene array for 84 ECM and adhesion molecules in post-MI settings in the LV infarct area (Figure 7C–D). At d1 post-MI, 9 genes in Lipo-RvD1 and 1 gene in RvD1 group was upregulated compared to the MI-saline group where 21 genes were upregulated. Similarly, Lipo-RvD1 and RvD1 showed an increase in 6 genes and 1 gene respectively at d5 post-MI. The decreased expression of fn-1, Icam, CD44, coll1a1, tnc and mmp-9 (Supplementary table 3) and the increase in Timp-1 (all, p<0.05) at d5 post-MI in RvD1-injetced groups indicated a reduced ECM deposition in the LV. Thus, RvD1 activated early resolution of inflammation and modulated ECM regulatory genes to reduce post-MI fibrosis, thereby improved LV function.
4. Discussion
The post-MI resolution of inflammation is an active, dynamic process that dampens pro-inflammatory signals without altering the host defense, however, uncontrolled inflammation often leads to congestive heart failure [1–3]. Thus, in the present investigation, we have assessed the role of RvD1 and Lipo-RvD1 formulation in the post-MI setting. RvD1 treatment; 1) prevented early changes in the splenic reservoir and improved LV function; 2) reduced neutrophil density in the spleen and discontinued neutrophil recruitment in LV; 3) increased pro-resolving lipid mediators level in the spleen and stimulated macrophages clearance to promote early resolution in the LV; 4) initiated 5-LOX and the ALX/FPR2 as well as modulated COX-1 and COX-2 expression and 5) reduced ECM gene expression and collagen deposition, thereby limiting the progression towards LV dysfunction. Thus, the present study suggests that the DHA-derived bioactive lipid mediator, RvD1, exerts potent pro-resolving actions in MI-mediated injury.
Lipid mediators, like RvD1, are subject to rapid inactivation in vivo [30]. The stability of RvD1 and its delivery are the key challenges in formulation and use as therapeutic tool for long-term treatment. Owing to these local inactivation pathways, a delivery approach that reduces local metabolism, increasing the half-life and potency of this potent pro-resolving mediator, would be desirable. Herein, we encapsulated RvD1 in a liposomal formulation to offer stability and future translational use in the chronic inflammatory heart failure setting which demands a long term treatment. RvD1 is endogenously produced as a consequence of injury or infection at the intended site of action to act as an autacoid [3]. In this study we demonstrated that liposome incorporated RvD1 retained its potent pro-resolving actions, suggesting this methodology may also be employed in the delivery of other SPMs, including E-series resolvins, lipoxins, protectins or maresins.
The first action of RvD1 is a preserved splenic depletion, revealed by attenuated transient post-MI mass loss. Recent studies suggest that post-MI spleen metabolic activity and splenic monocytes are contributors in the development of chronic heart failure [31, 32]. In the current post-MI study, the RvD1 mechanism of action resolves the inflammation by remitting neutrophil infiltration in both the spleen and LV by conserving transient changes in splenic weight. We observed a major decrease in spleen weight at d1 post-MI, suggesting a quick depletion of immune cells from the splenic reservoir. Interestingly, mice treated with RvD1 retained their splenic mass consistently from post-MI d1 to d5, suggesting RvD1 serves to retain the splenic reservoir. This accounts for the notion that the spleen serves as a constant reservoir of immune cells. Presented immune cell kinetics align with the findings by Leuschner et al, that the spleen acts as an immediate reservoir of monocyte/ macrophages which patrol in the early phase of myocardium injury [33].
The second action of RvD1 is evidenced by the reduced neutrophils density, increased pro-resolving mediators and relatively higher expression of ALX/FPR2 in the spleen rather than the LV. In the spleen, neutrophils are located around B lymphocytes in order to help their activation and to offer a first rapid response to MI injury or infection-mediated inflammation. Puga et al have shown that neutrophils in the spleen inherit the ability to interact with B cells or B lymphocytes in order to produce antibodies, which blood lymphocytes cannot do [26]. Thus, splenic neutrophils immediately get activated in response to post-MI inflammation and their cell-cell interaction activates other cell types to resolve inflammation. RvD1 regulates B cell activation, thereby stimulating a rapid defense mechanism [34]. This correlates with the simultaneous rush of neutrophils at d1 post-MI in RvD1 group, both in the LV and spleens, indicating mononuclear cells coordinate between the LV and spleen, so as to expedite the resolution of inflammation. Reduced neutrophils priming was further supported by the activation of ALX/FPR2 in RvD1 injected mice. The activation of ALX/FPR2 at d5 post-MI is complemented with previous findings where neutrophils were decreased [35, 36], suggesting an increase in mononuclear cells phagocytosis. RvD1 stimulated the ALX/FPR2 not only at the site of wound healing, i.e LV, but it also had a robust action in the spleen, suggesting a dynamic role of the spleen in resolving post-MI inflammation by limiting pro-inflammation. Thus, an unaltered neutrophil response at d1 post-MI by RvD1 plays a pivotal role in resolving inflammation. This could be an effective pharmacological approach, as RvD1 increases the levels of bioactive lipid mediators in the spleen to expedite the resolution of inflammation. Here, we report for the first time that these SPMs are elevated in the spleen post-MI and are even further elevated by RvD1 administration, suggesting the spleen as one of the major organ that coordinates with the LV post-MI injury. Thus, post-MI administration of RvD1 shortens the span of the pro-inflammatory window due to an increase of pro-resolving lipid mediators in the spleen. Mass spectrometry results showed increased levels of RvD1, RvD2, MaR1 and LXA4; this indicates that RvD1 has multiple actions through increasing levels of different SPMs in order to activate the resolution phase. Our findings are in parallel with the enhanced diabetic wound healing capacity of RvD1 in diabetic (db/db) mice. This provides evidence on the utilization of bioactive lipid molecules for the effective resolution of inflammation in several cardiovascular complications and other unresolved inflammation-mediated disease [35].
The third main action of RvD1 identified in the present study is that it shortens the acute pro-inflammatory window. RvD1 did not alter the acute inflammatory response, marked by an increase in LV expression of cxcl9, ccl1, ccr10 and few M1 markers at d1 post-MI. Further, the RvD1 action decreased LV expression of chemokine receptors ccr2, ccr5, cxcr2 and cxcr5 at d5, indicating SPMs promote resolution by shortening the stay of neutrophils at the site of injury post-MI [37]. Administration of RvD1 reduced neutrophil recruitment at d5 post-MI in the spleen and LV, coinciding with the down regulation of pro-inflammatory cytokines tnf-α, IL-1β, ccl2 and IL-6 in the resolving phase without altering the acute inflammatory response at d1. Of note, at d1 tnf-a levels were increased in RvD1 injected mice, indicating an unaltered host-defense in response to MI. Both IL-1β and tnf-α prolongs the neutrophil life-span, which prolongs the proinflammatory environment to extend granulocyte survival. Further, at d5-MI granulocytes undergo apoptosis, a process which is capable of attenuating inflammation, indicating the resolving phase of inflammation [38]. Post-MI wound healing coordinates to increase spleen and LV neutrophils with IL-1β and tnf-α via RvD1 at d1 post-MI. Additional, down regulation of IL-1-β and tnf-α at d5 post-MI suggested that RvD1 shortens the prolonged inflammatory phase by decreasing the transmigration of neutrophils to the site of injury.
The fourth main action of RvD1 post-MI is the clearance of macrophages and expression of the resolving macrophage phenotype. The directed migration of neutrophils is referred to as chemotaxis, a process which regulates intracellular signaling, allowing neutrophils to detect a gradient of attractant, polarize and migrate rapidly towards the chemoattractant in order to resolve post-MI inflammation [39]. RvD1 promoted CD44 , which is important for the containment of chronic inflammation and enrichment of fibroblasts post-MI [40]. CD44 is associated with enhanced, prolonged neutrophil and macrophage infiltration and also an increase in expression of proinflammatory cytokines in the infarcted myocardium [40]. Activation of CD44 expression in MI-saline group was associated with overactive neutrophils at d5 post-MI. RvD1 administration post-MI gave rapid exodus of the macrophages from the LV along with a decreased cxcl5 and ccl2 expression. These results highlight the importance of limiting neutrophil infiltration in the local chemotaxis altered macrophage population and macrophage plasticity. Increased macrophage phagocytosis in RvD1 treated mice initiated apoptosis of neutrophils which expedites the resolution of inflammation. This was further supported by enhanced expression of resolving macrophage markers mrc-1, arg-1 and Ym-1, thereby improving resolution of inflammation.
The fifth action of RvD1 is a consequence of activated inflammation-resolution axis leading to subsequent dynamic alterations in the ECM. ECM deposition is the progression following the inflammatory response post-MI, coordinated by the up-regulation of ECM transcripts. Resolution of inflammation overlaps with reorganization of ECM deposition following the healing response post-MI. The poor regenerative capacity of the myocardium leads to the formation of an irreversible scar, ultimately leading to the adverse remodeling of the myocardium [41]. RvD1 administration in mice not only resolves the inflammatory response, but also reduced the expression of fibrotic genes, such as colla1, coll2a1 and tnc. These observations suggest that RvD1 stabilizes the ECM by resolving inflammation. LOX and COX are the key enzymes for generating potent lipid mediators, which primarily coordinates the resolution of inflammation in pathophysiological settings [42]. Fredman et al. reported that RvD1 favors synthesis of the pro-resolving lipid mediator, LXA4, via limiting nuclear translocation of 5-LOX in inflammation [43]. RvD1 critically balanced LOX enzymes by stimulating 5-LOX expression, which is essential for wound healing post-MI [28]. Chronic, dissipating, and unresolved inflammatory response leads to ventricular dysfunction post-MI. Thus, RvD1 has the potential to delay heart failure but still requires long-term studies in order to prove its utilization in chronic heart failure management.
In conclusion, we demonstrated that RvD1 mediates the pro-resolving actions, remitting ungoverned inflammation associated with post-MI. RvD1 limited neutrophil recruitment in the spleen and LV, increased resolving lipid mediators and altered the macrophages phenotype post-MI. RvD1 promotes the resolution of inflammation and stabilizes the ECM through regulating LOX and COX enzymes. Together, these results indicated that RvD1 has a high potential to modulate the resolution pathophysiology in order to limit cardiac remodeling and thereby delay the progression of heart failure following MI.
Supplementary Material
Highlights.
We provide evidence on Resolvin D1 (RvD1) action in healing heart following myocardial infarction (MI) where;
-
1)
RvD1 acted on ALX/FPR2 receptor and discontinued neutrophil priming in spleen and LV post-MI.
-
2)
RvD1 stimulated macrophages clearance from infarcted area to promote early resolution of inflammation post-MI.
-
3)
RvD1 reduced ECM gene expression and collagen deposition thereby limited left ventricle dysfunction post-MI.
Acknowledgement
This work was supported by grants from National Institutes of Health (NIH)-NCCAM R00AT006704, UAB start-up fund to G.V.H. and NIH-P01GM095467 to C.N.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement
C.N.S. is an inventor on patents [resolvins] assigned to BWH and licensed to Resolvyx Pharmaceuticals; scientific founder of Resolvyx Pharmaceuticals with equity ownership in the company; and has interests reviewed and managed by the Brigham and Women's Hospital and Partners Health Care in accordance with their conflict of interest policies.
References
- 1.Reina-Couto M, Carvalho J, Valente MJ, Vale L, Afonso J, Carvalho F, et al. Impaired resolution of inflammation in human chronic heart failure. Eur J Clin Invest. 2014;44:527–538. doi: 10.1111/eci.12265. [DOI] [PubMed] [Google Scholar]
- 2.Kain V, Prabhu SD, Halade GV. Inflammation revisited: inflammation versus resolution of inflammation following myocardial infarction. Basic Res Cardiol. 2014;109:444. doi: 10.1007/s00395-014-0444-7. [DOI] [PubMed] [Google Scholar]
- 3.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saito T, Rodger IW, Hu F, Robinson R, Huynh T, Giaid A. Inhibition of COX pathway in experimental myocardial infarction. J Mol Cell Cardiol. 2004;37:71–77. doi: 10.1016/j.yjmcc.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Kimmel SE, Berlin JA, Reilly M, Jaskowiak J, Kishel L, Chittams J, et al. Patients exposed to rofecoxib and celecoxib have different odds of nonfatal myocardial infarction. Ann Intern Med. 2005;142:157–164. doi: 10.7326/0003-4819-142-3-200502010-00005. [DOI] [PubMed] [Google Scholar]
- 6.Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL) Circulation. 2004;109:1594–1602. doi: 10.1161/01.CIR.0000124490.27666.B2. [DOI] [PubMed] [Google Scholar]
- 7.Zhang S, Dehn S, DeBerge M, Rhee KJ, Hudson B, Thorp EB. Phagocyte-myocyte interactions and consequences during hypoxic wound healing. Cell Immunol. 2014 doi: 10.1016/j.cellimm.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lopez EF, Kabarowski J, Ingle KA, Kain V, Barnes S, Crossman DK, et al. Obesity superimposed on aging magnifies inflammation and delays the resolving response following myocardial infarction. Am J Physiol Heart Circ Physiol. 2014 doi: 10.1152/ajpheart.00604.2014. ajpheart 00604 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halade GV, Rahman MM, Bhattacharya A, Barnes JL, Chandrasekar B, Fernandes G. Docosahexaenoic acid-enriched fish oil attenuates kidney disease and prolongs median and maximal life span of autoimmune lupus-prone mice. Journal of immunology (Baltimore, Md : 1950) 2010;184:5280–5286. doi: 10.4049/jimmunol.0903282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang MJ, Spite M. Resolvins: anti-inflammatory and proresolving mediators derived from omega-3 polyunsaturated fatty acids. Annu Rev Nutr. 2012;32:203–227. doi: 10.1146/annurev-nutr-071811-150726. [DOI] [PubMed] [Google Scholar]
- 11.Tang H, Liu Y, Yan C, Petasis NA, Serhan CN, Gao H. Protective Actions of Aspirin-Triggered (17R) Resolvin D1 and Its Analogue, 17R-Hydroxy-19-Para-Fluorophenoxy-Resolvin D1 Methyl Ester, in C5a-Dependent IgG Immune Complex-Induced Inflammation and Lung Injury. Journal of immunology (Baltimore, Md: 1950) 2014;193:3769–3778. doi: 10.4049/jimmunol.1400942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiang N, Fredman G, Backhed F, Oh SF, Vickery T, Schmidt BA, et al. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature. 2012;484:524–528. doi: 10.1038/nature11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rius B, Titos E, Moran-Salvador E, Lopez-Vicario C, Garcia-Alonso V, Gonzalez-Periz A, et al. Resolvin D1 primes the resolution process initiated by calorie restriction in obesity-induced steatohepatitis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2014;28:836–848. doi: 10.1096/fj.13-235614. [DOI] [PubMed] [Google Scholar]
- 14.Sun YP, Oh SF, Uddin J, Yang R, Gotlinger K, Campbell E, et al. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. The Journal of biological chemistry. 2007;282:9323–9334. doi: 10.1074/jbc.M609212200. [DOI] [PubMed] [Google Scholar]
- 15.Koning GA, Morselt HWM, Velinova MJ, Donga J, Gorter A, Allen TM, et al. Selective transfer of a lipophilic prodrug of 5-fluorodeoxyuridine from immunoliposomes to colon cancer cells. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1999;1420:153–167. doi: 10.1016/s0005-2736(99)00091-7. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y, Chen W, Hair D, Xu J, Wu C, Han CC. Light scattering studies of stereocomplex formation of stereoregular poly(methyl methacrylate) in solutions. European Polymer Journal. 2005;41:447–452. [Google Scholar]
- 17.Halade GV, Ma Y, Ramirez TA, Zhang J, Dai Q, Hensler JG, et al. Reduced BDNF attenuates inflammation and angiogenesis to improve survival and cardiac function following myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2013;305:H1830–H1842. doi: 10.1152/ajpheart.00224.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colas RA, Shinohara M, Dalli J, Chiang N, Serhan CN. Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. American journal of physiology Cell physiology. 2014;307:C39–C54. doi: 10.1152/ajpcell.00024.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zamilpa R, Ibarra J, de Castro Bras LE, Ramirez TA, Nguyen N, Halade GV, et al. Transgenic overexpression of matrix metalloproteinase-9 in macrophages attenuates the inflammatory response and improves left ventricular function post-myocardial infarction. J Mol Cell Cardiol. 2012;53:599–608. doi: 10.1016/j.yjmcc.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–173. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Norling LV, Dalli J, Flower RJ, Serhan CN, Perretti M. Resolvin D1 limits polymorphonuclear leukocyte recruitment to inflammatory loci: receptor-dependent actions. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:1970–1978. doi: 10.1161/ATVBAHA.112.249508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krishnamoorthy S, Recchiuti A, Chiang N, Yacoubian S, Lee CH, Yang R, et al. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1660–1665. doi: 10.1073/pnas.0907342107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ Res. 2014;114:266–282. doi: 10.1161/CIRCRESAHA.113.301720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emami H, Singh P, MacNabb M, Vucic E, Lavender Z, Rudd JH, et al. Splenic Metabolic Activity Predicts Risk of Future Cardiovascular Events: Demonstration of a Cardiosplenic Axis in Humans. JACC Cardiovascular imaging. 2014 doi: 10.1016/j.jcmg.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim EJ, Kim S, Kang DO, Seo HS. Metabolic activity of the spleen and bone marrow in patients with acute myocardial infarction evaluated by 18f-fluorodeoxyglucose positron emission tomograpic imaging. Circulation Cardiovascular imaging. 2014;7:454–460. doi: 10.1161/CIRCIMAGING.113.001093. [DOI] [PubMed] [Google Scholar]
- 26.Puga I, Cols M, Barra CM, He B, Cassis L, Gentile M, et al. B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat Immunol. 2012;13:170–180. doi: 10.1038/ni.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCauley LK, Dalli J, Koh AJ, Chiang N, Serhan CN. Cutting edge: Parathyroid hormone facilitates macrophage efferocytosis in bone marrow via proresolving mediators resolvin D1 and resolvin D2. Journal of immunology (Baltimore, Md : 1950) 2014;193:26–29. doi: 10.4049/jimmunol.1301945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blomer N, Pachel C, Hofmann U, Nordbeck P, Bauer W, Mathes D, et al. 5-Lipoxygenase facilitates healing after myocardial infarction. Basic Res Cardiol. 2013;108:367. doi: 10.1007/s00395-013-0367-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maskrey BH, Megson IL, Whitfield PD, Rossi AG. Mechanisms of resolution of inflammation: a focus on cardiovascular disease. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:1001–1006. doi: 10.1161/ATVBAHA.110.213850. [DOI] [PubMed] [Google Scholar]
- 30.Ji R-R, Xu Z-Z, Strichartz G, Serhan CN. Emerging roles of resolvins in the resolution of inflammation and pain. Trends in Neurosciences. 2011;34:599–609. doi: 10.1016/j.tins.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487:325–329. doi: 10.1038/nature11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ismahil MA, Prabhu SD. Cardiac immune cell remodeling after myocardial infarction. J Mol Cell Cardiol. 2013;62:142–143. doi: 10.1016/j.yjmcc.2013.05.017. [DOI] [PubMed] [Google Scholar]
- 33.Leuschner F, Rauch PJ, Ueno T, Gorbatov R, Marinelli B, Lee WW, et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med. 2012;209:123–137. doi: 10.1084/jem.20111009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramon S, Gao F, Serhan CN, Phipps RP. Specialized proresolving mediators enhance human B cell differentiation to antibody-secreting cells. Journal of immunology (Baltimore, Md : 1950) 2012;189:1036–1042. doi: 10.4049/jimmunol.1103483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang Y, Zhang MJ, Hellmann J, Kosuri M, Bhatnagar A, Spite M. Proresolution therapy for the treatment of delayed healing of diabetic wounds. Diabetes. 2013;62:618–627. doi: 10.2337/db12-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dalli J, Consalvo AP, Ray V, Di Filippo C, D’Amico M, Mehta N, et al. Proresolving and tissue-protective actions of annexin A1-based cleavage-resistant peptides are mediated by formyl peptide receptor 2/lipoxin A4 receptor. Journal of immunology (Baltimore, Md : 1950) 2013;190:6478–6487. doi: 10.4049/jimmunol.1203000. [DOI] [PubMed] [Google Scholar]
- 37.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 38.Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood. 1992;80:2012–2020. [PubMed] [Google Scholar]
- 39.Wang F. The signaling mechanisms underlying cell polarity and chemotaxis. Cold Spring Harb Perspect Biol. 2009;1:a002980. doi: 10.1101/cshperspect.a002980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huebener P, Abou-Khamis T, Zymek P, Bujak M, Ying X, Chatila K, et al. CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. Journal of immunology (Baltimore, Md : 1950) 2008;180:2625–2633. doi: 10.4049/jimmunol.180.4.2625. [DOI] [PubMed] [Google Scholar]
- 41.Yabluchanskiy A, Chilton RJ, Lindsey ML. Left ventricular remodeling: one small step for the extracellular matrix will translate to a giant leap for the myocardium. Congest Heart Fail. 2013;19:E5–E8. doi: 10.1111/chf.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng Y, Austin SC, Rocca B, Koller BH, Coffman TM, Grosser T, et al. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science. 2002;296:539–541. doi: 10.1126/science.1068711. [DOI] [PubMed] [Google Scholar]
- 43.Fredman G, Ozcan L, Spolitu S, Hellmann J, Spite M, Backs J, et al. Resolvin D1 limits 5-lipoxygenase nuclear localization and leukotriene B4 synthesis by inhibiting a calcium-activated kinase pathway. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:14530–14535. doi: 10.1073/pnas.1410851111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.