

Abstract
The P2Y14 receptor (P2Y14R) is a Gi protein-coupled receptor that is activated by uracil nucleotides UDP and UDP-glucose. The P2Y14R structure has yet to be solved through X-ray crystallography, but the recent agonist-bound crystal structure of the P2Y12R provides a potentially suitable template for its homology modeling for rational structure-based design of selective and high-affinity ligands. In this study, we applied ligand docking and molecular dynamics refinement to a P2Y14R homology model to qualitatively explain structure-activity relationships of previously published synthetic nucleotide analogues and to probe the quality of P2Y14R homology modeling as a template for structure-based design. The P2Y14R model supports the hypothesis of a conserved binding mode of nucleotides in the three P2Y12-like receptors involving functionally conserved residues. We predict phosphate group interactions with R2536.55, K2777.35, Y2566.58 and Q2606.62 and nucleobase (anti-conformation) π-π stacking with Y1023.33 and the role of F1915.42 as a means for selectivity among P2Y12-like receptors. The glucose moiety of UDP-glucose docked in a secondary subpocket at the P2Y14R homology model. Thus, P2Y14R homology modeling may allow detailed prediction of interactions to facilitate the design of high affinity, selective agonists as pharmacological tools to study the P2Y14R.
Keywords: G protein-coupled receptor, homology modeling, docking, uracil nucleotides, P2Y14 receptor
Graphical abstract
INTRODUCTION
The P2Y receptors (P2YRs) are a family of membrane proteins that belong to the G protein-coupled receptor (GPCR) superfamily. Eight P2YR subtypes have been identified so far in human tissues and are further classified as P2Y1-like (P2Y1,2,4,6,11 subtypes) or P2Y12-like (P2Y12,13,14 subtypes) receptor subfamilies based on structural homology, sequence alignments, and the identification of conserved residues with key functions in each subtype [1,2]. The P2YRs are involved in specific signaling pathways; five P2Y1-like receptors couple to a Gq protein to modulate a phospholipase C signaling pathway, while three P2Y12-like receptors couple to Gi to inhibit adenylyl cyclase activity. Specifically, the P2Y14R is involved in the mast cell degranulation pathway, which suggests it may be a potential therapeutic target for asthma treatment [3,4]. The P2Y14R is also likely to be involved in the regulation of neuroimmune functions, but the lack of selective and stable agonists for this receptor have made it difficult to evaluate pharmacologically [5,6].
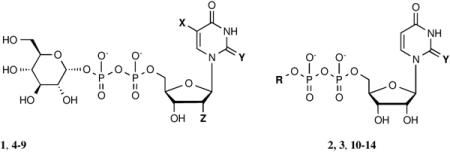
The P2Y12-like receptors, also including P2Y13R and P2Y14R, share significant sequence identity and structural homology. As a result, the P2Y12-like subtypes have been suggested to share a similar binding mode distinct from P2Y1-like receptors. Uridine 5′-diphosphoglucose (UDP-glucose or UDPG, 1, Table 1) and other UDP-sugars and uridine 5′-diphosphate (UDP, 2) have previously been revealed to be endogenous agonists for the P2Y14R. Yet, the fast degradation of nucleotides has made it difficult to study this receptor [7,8]. Previous structure-activity relationship (SAR) studies of synthetic nucleotide analogues have yielded highly potent agonist compounds by probing the base, ribose, phosphate, and terminal sugar moieties [9-11]. Such studies have revealed the functionally distinct regions of the ligand that contribute to receptor activation. Thio modification at the 2 position of the uracil base was found to significantly increase potency, the 2′ and 3′ hydroxyl groups of the ribose moiety were found to be necessary for receptor activation, and the hexose moiety was shown to be the most structurally permissive region. So far it has been difficult to interpret these SAR findings structurally due to the lack of a P2Y14R crystal structure. Homology modeling serves as a means for predicting ligand-receptor interactions and understanding ligand conformations in the binding pocket of a receptor for which a structure has not yet been solved. However, a limitation of this method is that the quality of a homology model is dependent on the templates that are available.
Table 1.
In vitro pharmacological data for UDPG (1), UDP (2) and their respective analogues in the stimulation of phospholipase Cβ by activation of the recombinant hP2Y14R expressed in COS-7 cells and transiently transfected with Gαqi. X = H, Y = O and Z = O, unless noted.
| |||
|---|---|---|---|
| Compound | Structure | Substitutiona | EC50 at hP2Y14R (nM)b |
| 1 | UDP-[1]glucose | - | 400±90 [21] |
| 2 | UDP | R= H | 160±40 [21] |
| 3 | 2-thio-UDP | R= H; Y= S | 1.92±0.69 [21] |
| 4 | 2-Thio-UDP-[1″]glucose | Y= S | 11±5 [21] |
| 5 | 5-Iodo-UDP-[1″]glucose | X= I | NE [19] |
| 6 | 5-Azido-UDP-[1″]glucose | X= N3 | NE [19] |
| 7 | 5-Amino-UDP-[1″]glucose | X= NH2 | NE [19] |
| 8 | 2′-deoxy-2′-azido-UDP-[1″]glucose | X= H; Z= N3 | NE [19] |
| 9 | 2′-deoxy-2′-amino-UDP-[1″]glucose | X= H; Z=NH2 | NE [19] |
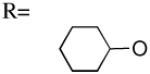
| 10 | UDP-0-cyclohexyl |
|
5160±1830 [21] |
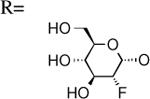
| 11 | UDP-2″-F[1″]2″-deoxyglucose |
|
2500±900 [20] |
| 12 | UDP-3″-F[1″]3″-deoxyglucose |
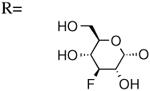
|
361±94 [20] |
| 13 | UDP-4″-F[1″]4″-deoxyglucose |
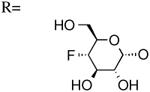
|
567±156 [20] |
| 14 | UDP-6″-F[1″]6″-deoxyglucose |
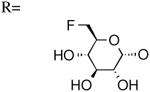
|
905±429 [20] |
| 15 | (S)mc-UDP-[1″]glucose | NE [21] | |
| 16 | (N)mc-UDP-[1″]glucose | NE [21] | |
See Figure S2 (Supporting information) for structures of methanocarba analogues 15 and 16.
NE, no effect at 10 μM.
Structures of the P2Y12R have recently been determined using X-ray crystallography, and they may serve as the best templates yet for modeling other P2Y12-like receptors [12,13]. P2YRs have been previously modeled using class A GPCRs as templates: Rhodopsin and β2-adrenergic and CXCR4 chemokine receptors [14-24]. However, the P2YRs belong to a different branch (δ) of class A GPCRs than the aforementioned receptor templates. PAR1 is the only other δ-branch GPCR determined structurally, but even this receptor has some unique distinguishing features, such as the placement of proline residues [25].
Therefore, a homology model based on an agonist-bound structure of P2Y12R (complex with 2-methylthioadenosine 5′-diphosphate, 2MeSADP, 17) may serve as an appropriate template to test the hypothesis that another member of the P2Y12-like subfamily shares a conserved binding mode. We recently constructed a P2Y14R homology model based on the agonist-bound P2Y12R structure to explore nonnucleotide antagonist binding, but this was not applied to the analysis of nucleotide binding [26]. Molecular modeling studies may help to explain the patterns observed in previous SAR studies of P2Y14R agonists while providing additional insight into the binding pocket regions that can be exploited for the design of selective agonist ligands for the P2Y14R [27].
In this study we have constructed several human (h) P2Y14R homology models based on the agonist-bound P2Y12R structure, and we have docked previously published synthetic nucleotide agonists to structurally rationalize SAR findings, to understand the nature of the P2Y14R binding pocket and to probe ligand-receptor interactions. In fact, a GPCR homology model is typically validated first using known ligand SAR prior to its application to the discovery of novel ligands [9-11].
Yet, a consequence of ligand docking to a homology model is the limitation of a perceived static model, which does not represent the dynamic nature of protein structures. To overcome this limitation of ligand docking, molecular dynamics is traditionally applied as a way to refine GPCR homology models through simulation in a solvated phospholipid bilayer [28-32]. However, molecular dynamics is at present limited to short timescales due to the significant amount of computing power that is required to solve Newton’s second law for each atom in an explicit solvent protein-membrane system. Regardless, molecular dynamics permits the exploration of a rugged energy landscape at even short timescales, which can extract useful information when applied to the refinement of ligand poses already docked in an orthosteric binding pocket. Thus, we applied molecular dynamics to the refinement of 2 docked in the orthosteric binding pocket of the P2Y14R.
Our models have suggested a similar nucleotide binding mode in the P2Y14R as in the P2Y12R, thus supporting a conserved binding mode hypothesis for P2Y12-like receptors involving functionally conserved residues in the binding pocket. Furthermore, an analysis of the binding pocket topology within the P2Y14R model provides a qualitative explanation for previously observed SAR patterns. As a result, the P2Y14R model may assist in the structure-based design of selective agonist compounds for studying the pharmacology of the P2Y14R.
RESULTS
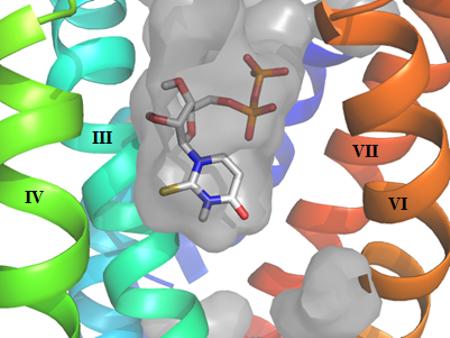
We built a homology model of the P2Y14R based on the 2MeSADP-bound P2Y12R crystal structure using the alignment shown in Figure S1 (Supporting information) showing an overall sequence identity of 45%. The Cα root-mean-square deviation (RMSD) between the P2Y14R model and the P2Y12R crystal structure was 0.8998 Å. A binding site comparison between the P2Y14R homology model and the P2Y12R template structure is shown in Figure 1. As in the P2Y12R, the binding pocket was observed to have a bifurcated shape; with a main pocket delimited by TMs 3, 4, 5, 6 and 7 (pocket 1) and a smaller pocket delimited by TMs 1, 2, 3 and 7 (pocket 2). To refine the model and especially the orientation of the binding site side chains we performed induced fit docking of 1 and 2 to the original P2Y14R model. The docking pose of 2 in the P2Y14R model was similar to the conformation of 2MeSADP in the P2Y12R and occupies the pocket delimited by TMs 3, 4, 5, 6 and 7 (Figure 2). The binding pocket where the nucleobase binds (left-half of each binding pocket in Figures 1 and 2) is noticeably larger in the P2Y12R compared to the P2Y14R. This observation was consistent with the preference of P2Y14Rs for binding uracil over adenine nucleotides. The 2MeSADP binding mode at the P2Y12R structure is stabilized by interactions with key conserved residues in the P2Y12-like receptors. 2MeSADP is stabilized in the P2Y12R crystal structure by π-π stacking with Y1053.33 (numbers in superscript refer to the Ballesteros-Weinstein notation [33]); hydrophobic interactions with Y1063.34, L1554.56, S1564.57, and V1905.39; bidentate interaction with N1915.40; H-bonds between the ribose 2′- and 3′-hydroxyl groups and K179EL2, H1875.36 and the backbone carbonyl of C973.25; and polar contacts between the phosphate groups and R2566.55, K2807.35, Y2596.58 and Q2636.62. Conserved residues between P2Y12R and P2Y14R were found to be involved in stabilizing the docking poses of 1 and 2 in the P2Y14R model (Table S1, Supporting information). In fact, the uridine moiety of 1 occupied the same region as in the docking pose of 2, while the glucose moiety was accommodated in the second half of the pocket between TMs 1, 2, 3 and 7. A 2D representation of the interactions formed by 1 at the P2Y14R is provided in Figure 3A. For comparison, the SAR of nucleotides at the P2Y14R is summarized (Figure 3B). Key interactions predicted from the docking pose of 1 docked in the orthosteric binding pocket are shown in Figure 4. The models obtained after induced fit docking of 1 and 2 were then used to perform docking of various nucleotide analogues previously tested in functional assays at the P2Y14R to structurally explain the main SAR findings. Structures and functional assay data of the nucleotide analogues analyzed in this study are listed in Table 1. In the following subsections, the analysis of the binding of each ligand moiety and the effect of substitutions are discussed.
Figure 1.
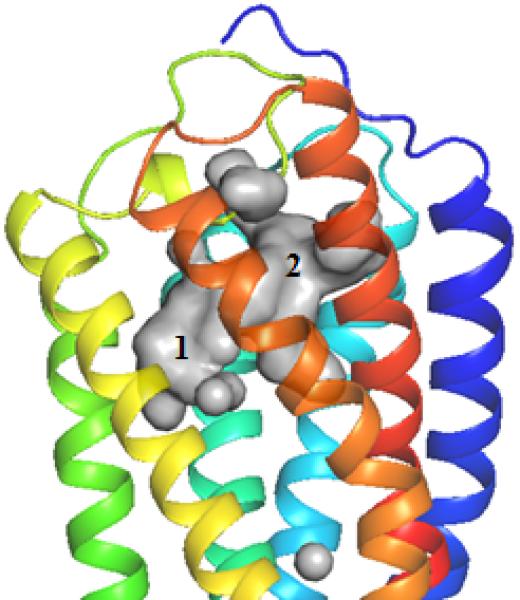

A. P2Y14R model binding site surface comparison with the B. Agonist-bound P2Y12R crystal structure (right) binding site surface. The gray areas represent the surface area in the receptor pocket predicted to be available for ligand binding. As expected, the nucleobase binding pocket delimited by TM3, TM4 and TM5 is larger in P2Y12R compared to P2Y14R (TM1=Blue, TM2=Cyan, TM3= Cyan/Green, TM4=Green, TM5=Yellow, TM6=Orange, TM7=Red).
Figure 2.


A. Docking pose of 2 at the P2Y14R homology model. B. Crystal pose of 2MeSADP at the P2Y12R (TM1=Blue, TM2=Cyan, TM3= Cyan/Green, TM4=Green, TM5=Yellow, TM6=Orange, TM7=Red).
Figure 3.
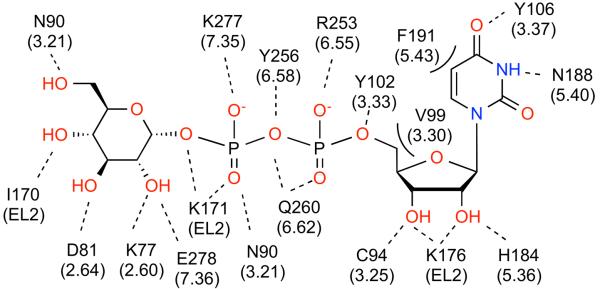

A. A 2D representation of the P2Y14R residues predicted to interact with 1.The dotted lines indicate hydrogen and ionic bonding interactions, the solid line represents π-π stacking, and the curved lines indicate steric hindrance observed with nucleotide modification at a particular position. The base and glucose moieties each dock to one-half of the bifurcated binding pocket. B. Summary of structure-activity relationships of 1 in the P2Y14R [20].
Figure 4.
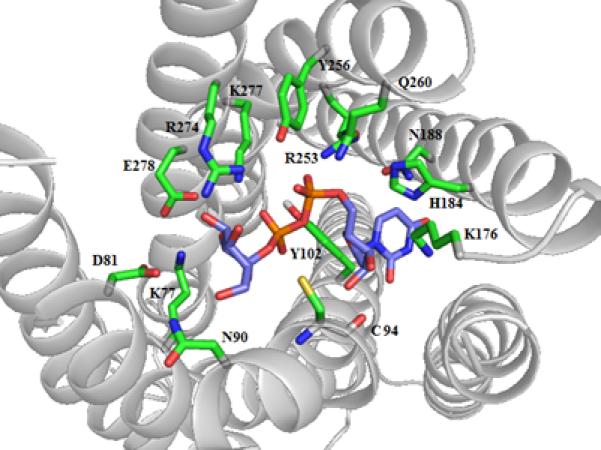
Key interactions in the P2Y14R homology model with 1 docked in the binding pocket.
Nucleobase
Docking results at the P2Y14R model showed that the binding conformation of the nucleobase of 2 around the glycosidic bond could dock in either a syn or anti conformation. If the base adopted a syn conformation, more H-bonding interactions were observed between the uracil base and residues lining the binding pocket. However, in an anti conformation less steric hindrance was observed, and 2 is in its natural energetically favorable torsional conformation around the glycosidic bond. In both conformations, the pyrimidine base is stabilized by π-π stacking with Y1023.33. A syn glycosyl conformation of the nucleobase provided for optimized H-bonding interactions in the receptor binding pocket. In fact, the carbonyl at the 2 position and the nitrogen at the 3 position would be involved in bidentate interactions with N1885.39. However, optimized hydrogen-bonding interactions in the orthosteric binding pocket may not be functionally relevant considering that the 2MeSADP-bound P2Y12R crystal structure suggests a preference for an anti glycosyl conformation in the orthosteric binding pocket.
In order to resolve the glycosyl conformation of the nucleotide compounds docked in the orthosteric binding pocket of the P2Y14R, we applied explicit molecular dynamics refinement to the docking pose of 2. When 2 was docked in a syn glycosyl conformation, we observed a base-flipping event during the first 500 fs of the equilibration and rearrangement of N1885.39 (Movie 1, Supporting information). Then, the docking pose remained stable in the binding pocket in an anti glycosyl conformation during a 10 ns production run. Thus, molecular dynamics refinement suggested an anti glycosyl conformation was more energetically favorable for 2 docked in the P2Y14R model binding pocket.
Uracil nucleobase analogues modified at the 2 and 5 positions (compounds 3-7) were docked to the P2Y14R model. Analogues substituted at the 5 position are previously reported to be inactive at the P2Y14R. A possible explanation for the lack of activity of uracil derivatives substituted at the 5 position is steric hindrance with F1915.42, which reduced the size of the orthosteric binding pocket in the P2Y14R model compared to the P2Y12R crystal structure. Modification at the 2 position, as in 3, was previously associated with a high potency in activating the P2Y14R. The P2Y14R homology model predicted that there is enough space in the pocket to accommodate the bulkier thio group at the 2 position of the nucleobase and a better fit with the cavity can be an explanation for the increase in potency of 3 compared to 2 (Graphical abstract).
Ribose
The ribose H-bonding interactions included H1845.35, K176EL2, and the carbonyl backbone of C943.25. It is known that the ribose ring conformation is an important determinant of recognition in the P2YR family [17]. Analogues of 1 containing a conformationally constrained North (N) or South (S) methanocarba (bicyclo[3.1.0]hexane, mc) ring system in place of the ribose moiety (compounds 15 and 16) are reported to be inactive at the P2Y14R [11]. A structural explanation for the loss of activity of these analogues could be steric hindrance of the bicyclic ring system with V993.30, i.e. the presence of the fused cyclopropyl ring might interfere with ligand binding. However, when the ribose itself was constrained in either extreme conformation, the nucleotide 1 was able to dock to the homology model. Thus, we docked 1 with the ribose moiety frozen in a 2′-endo or 3′-endo pucker to further probe the selectivity of the model for a given ribose configuration. Figures 5A and 5B show both the frozen (S)- and (N)-conformations of 1 docked to the P2Y14R model. (S)-1 maintained the ability to dock in the binding pocket with the preferred pose, while (N)-1 did not dock with the preferred pose. The P2Y14R model suggests the (S)-ribose pucker may be preferred for receptor activation as a result of the relative positioning of the ribose hydroxyl groups for H-bonding interactions with C943.25 and K176EL2.
Figure 5.


A. UDPG 1 in the ribose 2′-endo conformation (purple carbons) docked to the P2Y14R model. This docking pose with the ribose ring frozen in a (S) pucker was consistent with the proposed binding mode. B. UDPG 1 in the ribose 3′-endo conformation (purple carbons) docked to the P2Y14R model. This docking pose with the ribose ring frozen in a (N) pucker was not consistent with the proposed binding mode.
Modifications of the hydroxyl groups of the ribose ring, such as in 2′-modified compounds 8 and 9, display a loss of in vitro activity. These analogues fit to the P2Y14R binding site with a similar orientation as 1, but were not able to form all of the H-bonding interactions observed in the native nucleotide structure. A possible explanation for the loss of activity in this series is the lack of favorable interactions with the highly conserved H-bond donor K176EL2.
Phosphates
Ionic and H-bonding interactions in the P2Y14R model were observed between the ligand phosphate groups and the following residues, which are conserved with the P2Y12R: R2536.55, Y2566.58, Q2606.62, and K2777.35. An additional ionic interaction is formed with R2747.32 that is not conserved in the P2Y12R.
Glucose
After inspecting the pose for the hexose moiety, we found that multiple conformations were possible for the pyranose ring in the second half of the binding pocket of the P2Y14R model (Figure 1B, pocket 2). Among those conformations, we selected the preferred glucose docking pose involving H-bonding interactions between residues in the P2Y14R model and each hydroxyl group of the glucose ring to maximize the stability of this moiety in the binding pocket (Figure S4, Supporting information). The residues lining the hexose binding pocket included R2747.32 E2787.36, K772.60, D812.64, the backbone carbonyl of I170EL2, and N903.21. The cyclohexyl derivative, compound 10, is a less potent analogue of compound 2. A lack of interactions to stabilize the pyranose ring in the receptor pocket due to the lack of hydroxyl groups may explain the loss of potency for 10 compared to 2.
Compounds 11 - 14, which are analogues of 1 that are fluoro-substituted on the glucose moiety, were docked to the model and their poses are shown in Figure 6. Importantly, the 2′′-fluoro modification was correlated with a loss of favorable interactions with three residues: R2747.32, E2787.36 and K772.60. Modification at the 3′′, 4′′ and 6′′ positions of the glucose moiety altered the docking conformation in the binding pocket and resulted in a loss of favorable interactions with R2747.32 and N902.64. The in vitro activity pattern of the fluoro-modification series indicates that loss of a hydroxyl group at the 2′′ position has a greater impact on altering receptor activity than modification at the 3′′, 4′′ and 6′′ positions. The loss of H-bonding interactions with R2747.32, E2787.36 and K772.60 through modification at the 2′′ position suggested these residues may be involved in a H-bonding network that could be important for binding and/or receptor activation.
Figure 6.




The fluoro-atom modification at each position prevented hydrogen bonding interactions with key residues in the hexose binding pocket. A. Compound 11 (purple carbons) lost interactions with three residues: R2747.32, E2787.36, and K772.60. B. Compound 12 (purplecarbons) lost interactions toR2747.32. C. Compound 13 (purple carbons) lost interactions withR2747.32. D. Compound 14 (purple carbons) lost interactions with N903.21.
DISCUSSION
The P2Y14R model has provided in silico validation for a previously predicted conserved binding mode in the P2Y12-like receptors. The P2Y14R model was initially refined to enhance the receptor conformation induced upon binding 1 and 2. We were able to induce a binding site conformation, which applied to both 1 and 2, that is consistent with the binding mode observed for 2MeSADP in the P2Y12R (Figure 2)The refined models were then used to dock several derivatives to explain the SAR patterns previously empirically observed for P2Y14R agonists.Interestingly, nucleotide-like antagonists of the P2Y12R are known, while nucleotide antagonists of the P2Y14R have not been reported. Molecular dynamics calculations of 2 docked in the P2Y14R homology model provide in silico evidence of the thermodynamic stability of the selected docking conformation in the binding pocket during a 6 ns equilibration and a 10 ns trajectory (Movie 1, Supporting information).
In the P2Y14R model we observed that the positively charged residues highly conserved in P2Y12-like receptors K2777.35 and R2536.55 were involved in ionic interactions with the negatively charged phosphates, consistent with a previous prediction based on sequence analysis of P2Y12-like receptors [34]. K176EL2 is also highly conserved in P2Y12-like receptors and was involved in stabilizing the ribose moiety by complementing the hydroxyl group at the 2′ position as a H-bond donor. The P2Y14R model suggested that the (S) pucker is the favored ribose conformation for receptor activation. This conformation in the P2Y14R model is also in agreement with the 2MeSADP (S) ribose conformation observed in the crystal structure of the P2Y12R.
The motif K-E-X-X-L in TM7 has been identified through sequence alignments as highly conserved in P2Y12-like receptors, which corresponded to the residues K2777.35, E2787.36, F2797.37, T2807.38 and L2817.39 in the P2Y14R. As already mentioned, K2777.35 stabilized the negatively charged β phosphate. The docking pose of the glucose moiety of 1 placed the hydroxyl group at the 2′′ position near E2787.36. It is interesting that fluoro modification at the 2′′ position introduced in compound 11 significantly reduced in vitro activity, which we attribute to a loss of favorable interactions with residues in the hexose binding pocket of the P2Y14R model – including the E2787.36 constituent of the K-E-X-X-L motif. The effect of the missing interaction with E2787.36 on the activity of compound 11 supported its functional role in mediating receptor activity.
The π-π stacking interaction involving Y1053.33 observed in the agonist-bound P2Y12R structure was conserved in the P2Y14R model to form the binding pocket for the nucleotide base. Molecular dynamics refinement of compound 2 docked at the P2Y14R model suggests an anti conformation may be favored in order to minimize steric hindrance in the orthosteric binding pocket. An anti glycosyl conformation has also been determined through spectroscopy as the favored conformation in solution for most nucleotides. However, structural modification of the nucleobase through the addition of substituents may alter the preference for a given glycosyl conformation since substitution of the nucleobase may affect the most energetically favorable torsional conformation [26,35].
The addition of an amino group at the 5 position of the nucleobase suggests improved π-π stacking interaction due to the presence of a strong electron donating group. Yet, synthetic analogues modified at the 5 position of the nucleobase are inactive at the P2Y14R. A question that remains unanswered with respect to the SAR of the nucleobase is whether the loss of activity of compounds 5, 6 and 7 is a result of an alteration in the favorable torsional conformation in the binding pocket or a result of steric effects with nearby residues in the nucleobase binding pocket. Ligand docking and molecular dynamics suggest an anti glycosyl conformation is thermodynamically favored for 2 in the orthosteric binding pocket of the P2Y14R model, similar to the torsional conformation of 2MeSADP in the P2Y12R crystal structure. Thus, the binding mode for nucleotides at the P2Y12-like subtype might require an anti glycosyl conformation for proper receptor activation behavior. We hypothesize that F1915.43 in the P2Y14R may provide a means for selectivity of pyrimidine nucleotides against purine nucleotides. Comparably, the distal residue C1945.43 in the base binding pocket of P2Y12R may permit binding of adenine nucleotides by expanding the size of the binding pocket to accommodate purine nucleotides. This provides additional rationale for a conserved agonist binding mode in P2Y12-like receptors by complementing the topology of endogenous uridine nucleotides selective for the P2Y14R over the adenine-binding P2Y12R and P2Y13R. Additionally, the high potency of the 2-thio modification of the nucleobase may be explained by a better fit of the region delimited by TM3 and TM4 that helps anchoring compound 3 in its anti conformation in the binding pocket.
We sought to compare the conformation of the P2Y14R model from this study to the previously reported P2Y14R model in which the final receptor conformation was induced by flexible docking of the highly selective P2Y14R antagonist 18 (MRS4174, Figure S2, Supporting information), which contains a fluorophore for receptor detection [13]. The P2Y14R RMSD between the two models is 4.3 Å; this high value reflects mainly the large conformational differences in the flexible loop domains. The conformation of residues in the trans-membrane region of the binding pocket is similar in both models The residues K171EL2, K772.60, and K176EL2 are involved in stabilizing 18 in the extracellular region of the receptor binding pocket. In our P2Y14R model, K176EL2 was oriented downward, and we propose that the orientation of this residue may function to stabilize nucleotide compounds in the orthosteric binding pocket of the P2Y14R by inducing a (S) pucker in the ribose moiety. Conversely, K176EL2 is oriented upward in the antagonist 18-bound P2Y14R model to stabilize the ligand through H-bonding interactions.
In conclusion, the P2Y14R model has provided an explanation for the structure-activity relationships of various previously reported synthetic nucleotide agonists. The results from this study have assigned roles to several residues highly conserved within the subfamily in mediating receptor-ligand interaction networks, which supports the validation of previous experimental work. The P2Y14R model has also predicted a previously unidentified non-conserved residue, F1915.42, as potentially allowing for structural selectivity of the nucleobase at the P2Y14R. This indicates that the P2Y12R agonist-bound structure is an adequate template for a P2Y14R model. Thus, P2Y14R homology modeling may allow detailed prediction of interactions to facilitate the design of high affinity and selective agonists as pharmacological tools to study the P2Y14R. This study also illustrates the potential of homology modeling, ligand docking and molecular dynamics as tools for understanding and predicting structural-functional relationships in G protein-coupled receptors.
EXPERIMENTAL SECTION
Homology modeling
The 2MeSADP-bound P2Y12R structure at 2.5 Å resolution was obtained from the Protein Data Bank (PDB ID: 4PXZ)[12] and used as a starting template. The BRIL protein fused in intracellular loop 3, 2MeSADP, cholesterols and waters were removed from the P2Y12R structure. Hydrogen atoms and missing side chains in unresolved regions were added, the hydrogen network was optimized, and minimization was performed using the OPLS-2005 forcefield with the Protein Preparation Wizard tool in the Schrödinger suite [36]. The FASTA sequence for P2Y14R was obtained through the Uniprot database and aligned against the P2Y12R sequence using the Prime tool in the Schrödinger suite. The alignment was visually inspected to ensure no gaps were present in predicted alpha-helical regions of the model. Then, the P2Y14R model was built using the Prime homology modeling tool (energy-based method) of the Schrödinger suite.
Ligand docking
Ligands were built using Maestro and prepared using the Ligprep module and OPLS-2005 forcefield in the Schrödinger suite. The sitemap tool was used to identify probable binding site pockets in the receptor. Molecular docking of ligands to the P2Y14R model was performed by means of the Glide package from the Schrödinger suite. In particular, a Glide grid was positioned on the centroid of residues located within 5 Å from the previously identified cavity. The Glide grid was built using an inner box (ligand diameter midpoint box) of 10 Å × 10 Å × 10 Å and an outer box (box within which all the ligand atoms must be contained) that extended 20 Å in each direction from the inner box. Induced fit was used to refine the binding site by separately docking 1 and 2 to provide optimized receptor models for the base and hexose binding pockets. The poses that best fit the coordinates of superimposed 2MeSADP-bound P2Y12R were used for further inspection of ligand-receptor interactions. The Glide package was used for SP (standard precision) docking to the optimized models of all previously reported synthetic nucleotide agonist compounds. The conformational poses were selected based on similarity to the docking pose of 1 and agreement with available experimental data.
Molecular dynamics
P2Y14R models were uploaded to the “Orientations of Proteins in Membranes (OPM)” database and a suggested orientation for each structure was provided based on the 2MeSADP-bound P2Y12R orientation (PDB: 4PXZ) [37]. Each receptor model was then positioned in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipid bilayer (70 Å × 70 Å) generated by a grid-based method using the VMD Membrane Plugin tool, and overlapping lipids within 0.6 Å were removed upon combining the protein-membrane system [38,39]. Each protein-membrane system was then solvated with TIP3P water using the Solvate 1.0 VMD Plugin tool and neutralized by 0.154 M Na+/Cl− counterions [40].
This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, Md (http://biowulf.nih.gov). Molecular dynamics simulations with periodic boundary conditions were carried out using Nanoscale Molecular Dynamics (NAMD) software and the CHARMM36 Force Field [41,42]. The ligands were parameterized by analogy using the ParamChem service (0.9.7.1) and implementing the CHARMM General Force Field for organic molecules (2b8) [43-46]. A 3000-step conjugate gradient minimization was initially performed to minimize steric clashes. The protein and ligand atoms were kept fixed during an initial 8 ns equilibration of the lipid and water molecules. Atom constraints were then removed and the entire system was allowed to equilibrate. The UDP-bound P2Y14R structure was allowed to equilibrate for 6 ns (first plateau reached at around 1 ns, Figure S4), and the orthosteric binding pocket was visually inspected to ensure solvation of the binding pocket. An aporeceptor P2Y14R model was allowed to equilibrate for 8 ns. A 10 ns production run was then carried out on the equilibrated UDP-bound P2Y14R and apo-P2Y14R models. The temperature was maintained at 300 K using a Langevin thermostat with a damping constant of 3 ps-1. The pressure was maintained at 1 atm using a Berendsen barostat. An integration time step of 1 fs was used, while hydrogen-oxygen bond lengths and hydrogen-hydrogen angles in water molecules were constrained using the SHAKE algorithm [47]. VMD 1.9 was used for trajectory visualization and movie making. The PyMOL Molecular Graphics System, Version 1.6.0 Schrödinger, LLC was used for making figures.
Supplementary Material
Acknowledgements
Funding from the NIH Undergraduate Scholarship Program (UGSP) to KT, the NIGMS Postdoctoral Research Associate (PRAT) program to EK and the Intramural Research Program of NIDDK (Z01 DK031126) is gratefully acknowledged.
Abbreviations
- EL
extracellular loop
- GPCR
G protein-coupled receptor
- 2MeSADP
2-methylthioadenosine 5′-diphosphate
- TM
transmembrane helical domain
- UDP
uridine 5′-diphosphate
- UDPG
uridine 5′-diphosphoglucose
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting information available:
Alignment of P2Y12R and P2Y14R; structures of compounds 15, 16 and 17; ; docking pose of 1 in P2Y14R model; roles of corresponding residues in hP2Y12R and hP2Y14R; 1.5ps movie of 2 in P2Y14R model; RMSD of 6 ns UDP-bound P2Y14R .
References
- 1.Moro S, Jacobson KA. Curr.. Pharm. Des. 2002;8:2401. doi: 10.2174/1381612023392892. [DOI] [PubMed] [Google Scholar]
- 2.Jacobson KA, Jayasekara MP, Costanzi S. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2012;1 doi: 10.1002/wmts.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao ZG, Ding Y, Jacobson KA. Biochem. Pharmacol. 2010;79:873. doi: 10.1016/j.bcp.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao Z-G, Wei Q, Jayasekara MPS, Jacobson KA. Purinergic Signalling. 2013;9:31. doi: 10.1007/s11302-012-9325-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunschweiger A, Muller CE. Curr. Med. Chem. 2006;13:289. doi: 10.2174/092986706775476052. [DOI] [PubMed] [Google Scholar]
- 6.Dovlatova N, Wijeyeratne YD, Fox SC, Manolopoulos P, Johnson AJ, White AE, Latif ML, Ralevic V, Heptinstall S. Thromb. Haemost. 2008;100:261. [PubMed] [Google Scholar]
- 7.Abbracchio MP, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Miras-Portugal MT, King BF, Gachet C, Jacobson KA, Weisman GA, Burnstock G. Trends Pharmacol. Sci. 2003;24:52. doi: 10.1016/S0165-6147(02)00038-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carter RL, Fricks IP, Barrett MO, Burianek LE, Zhou Y, Ko H, Das A, Jacobson KA, Lazarowski ER, Harden TK. Mol. Pharmacol. 2009;76:1341. doi: 10.1124/mol.109.058578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ko H, Fricks I, Ivanov AA, Harden TK, Jacobson KA. J. Med. Chem. 2007;50:2030. doi: 10.1021/jm061222w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ko H, Das A, Carter RL, Fricks IP, Zhou Y, Ivanov AA, Melman A, Joshi BV, Kováč P, Hajduch J, Kirk KL, Harden TK, Jacobson KA. Bioorg. Med. Chem. 2009;17:5298. doi: 10.1016/j.bmc.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das A, Ko H, Burianek LE, Barrett MO, Harden TK, Jacobson KA. J. Med. Chem. 2009;53:471. doi: 10.1021/jm901432g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, Zhang K, Gao ZG, Paoletta S, Zhang D, Han GW, Li T, Ma L, Zhang W, Muller CE, Yang H, Jiang H, Cherezov V, Katritch V, Jacobson KA, Stevens RC, Wu B, Zhao Q. Nature. 2014;509:119. doi: 10.1038/nature13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiselev E, Barrett M, Katritch V, Paoletta S, Weitzer CD, Hammes E, Yin AL, Zhao Q, Stevens RC, Harden TK, Jacobson KA. ACS Chem. Biol. 2014;9:2833. doi: 10.1021/cb500614p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moro S, Hoffmann C, Jacobson KA. Biochemistry. 1999;38:3498. doi: 10.1021/bi982369v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim HS, Barak D, Harden TK, Boyer JL, Jacobson KA. J. Med. Chem. 2001;44:3092. doi: 10.1021/jm010082h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yeagle PL, Choi G, Albert AD. Biochemistry. 2001;40:11932. doi: 10.1021/bi015543f. [DOI] [PubMed] [Google Scholar]
- 17.Costanzi S, Joshi BV, Maddileti S, Mamedova L, Gonzalez-Moa MJ, Marquez VE, Harden TK, Jacobson KA. J. Med. Chem. 2005;48:8108. doi: 10.1021/jm050911p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ivanov AA, Fricks I, Harden TK, Jacobson KA. Bioorg. Med. Chem. Lett. 2007;17:761. doi: 10.1016/j.bmcl.2006.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmidt P, Ritscher L, Dong EN, Hermsdorf T, Cöster M, Wittkopf D, Meiler J, Schöneberg T. Mol. Pharmacol. 2013;83:256. doi: 10.1124/mol.112.082198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. J. Mol. Biol. 2004;342:571. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 21.Hibert MF, Trumpp-Kallmeyer S, Bruinvels A, Hoflack J. Mol. Pharmacol. 1991;40:8. [PubMed] [Google Scholar]
- 22.Jaakola V-P, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, IJzerman AP, Stevens RC. Science (New York, N.Y.) 2008;322:1211. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu B, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Science (New York, N.Y.) 2010;330:1066. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deflorian F, Jacobson KA. J. Comput. Aided Mol. Des. 2011;25:329. doi: 10.1007/s10822-011-9423-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang C, Srinivasan Y, Arlow DH, Fung JJ, Palmer D, Zheng Y, Green HF, Pandey A, Dror RO, Shaw DE, Weis WI, Coughlin SR, Kobilka BK. Nature. 2012;492:387. doi: 10.1038/nature11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schweizer MP, Banta EB, Witkowski JT, Robins RK. J. Am. Chem. Soc. 1973;95:3770. doi: 10.1021/ja00792a049. [DOI] [PubMed] [Google Scholar]
- 27.Jacobson KA, Ivanov AA, de Castro S, Harden TK, Ko H. Purinergic Signalling. 2009;5:75. doi: 10.1007/s11302-008-9106-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Platania CBM, Salomone S, Leggio GM, Drago F, Bucolo C. PLoS ONE. 2012;7:e44316. doi: 10.1371/journal.pone.0044316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sabbadin D, Ciancetta A, Moro S. Journal of Chemical Information and Modeling. 2013;54:169. doi: 10.1021/ci400532b. [DOI] [PubMed] [Google Scholar]
- 30.Kufareva I, Katritch V, Stevens Raymond C., Abagyan R. Structure. 2013;22:1120. doi: 10.1016/j.str.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dror RO, Jensen M, Borhani DW, Shaw DE. The Journal of General Physiology. 2010;135:555. doi: 10.1085/jgp.200910373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buch I, Harvey MJ, Giorgino T, Anderson DP, De Fabritiis G. J Chem Inf Model. 2010;50:397. doi: 10.1021/ci900455r. [DOI] [PubMed] [Google Scholar]
- 33.Ballesteros JA, Weinstein H. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 34.Costanzi S, Joshi BV, Maddileti S, Mamedova L, Gonzalez-Moa MJ, Marquez VE, Harden TK, Jacobson KA. J. Med. Chem. 2005;48:8108. doi: 10.1021/jm050911p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ginsburg-Shmuel T, Haas M, Schumann M, Reiser G, Kalid O, Stern N, Fischer B. J. Med. Chem. 2010;53:1673. doi: 10.1021/jm901450d. [DOI] [PubMed] [Google Scholar]
- 36.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. J. Comput. Aided Mol. Des. 2013;27:221. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 37.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. Bioinformatics. 2006;22:623. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 38.Sommer B. Computational and Structural Biotechnology Journal. 2013;5:e201302014. doi: 10.5936/csbj.201302014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Humphrey W, Dalke A, Schulten K. J Mol Graph. 1996;14:33. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 40.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. The Journal of Chemical Physics. 1983;79:926. [Google Scholar]
- 41.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J Comput Chem. 2005;26:1781. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, MacKerell AD. Journal of Chemical Theory and Computation. 2012;8:3257. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vanommeslaeghe K, MacKerell AD., Jr. J Chem Inf Model. 2012;52:3144. doi: 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vanommeslaeghe K, Raman EP, MacKerell AD., Jr. J Chem Inf Model. 2012;52:3155. doi: 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD., Jr. J Comput Chem. 2010;31:671. doi: 10.1002/jcc.21367. 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu W, He X, Vanommeslaeghe K, MacKerell AD., Jr. J Comput Chem. 2012;33:2451. doi: 10.1002/jcc.23067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ryckaert J-P, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–341. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.