

Abstract
Obesity is now widespread around the world. Obesity-associated chronic low-grade inflammation is responsible for the decrease of insulin sensitivity, which makes obesity a major risk factor for insulin resistance and related diseases such as type 2 diabetes mellitus and metabolic syndromes. The state of low-grade inflammation is caused by overnutrition which leads to lipid accumulation in adipocytes. Obesity might increase the expression of some inflammatory cytokines and activate several signaling pathways, both of which are involved in the pathogenesis of insulin resistance by interfering with insulin signaling and action. It has been suggested that specific factors and signaling pathways are often correlated with each other; therefore, both of the fluctuation of cytokines and the status of relevant signaling pathways should be considered during studies analyzing inflammation-related insulin resistance. In this paper, we discuss how these factors and signaling pathways contribute to insulin resistance and the therapeutic promise targeting inflammation in insulin resistance based on the latest experimental studies.
1. Introduction
Insulin resistance (IR) is a complicated condition in which three primary metabolic tissues that are sensitive to insulin, skeletal muscle, liver, and white adipose tissue (WAT) become less sensitive to insulin and its downstream metabolic actions under normal serum glucose concentrations [1]. IR is closely associated with obesity, hypertension, hyperglycaemia, polycystic ovary syndrome, and metabolic syndrome (see glossary) [2, 3]. As the key component of metabolic syndrome, IR is also closely associated with nonalcoholic fatty liver disease (NAFLD) [4]. The antilipolytic effect of insulin is decreased in insulin-resistant conditions, which may promote hepatic triglyceride synthesis. Another feature of insulin resistance is an increasing release of free fatty acid. As we know, FFA could be taken up by organs and accumulated as ectopic fat, such as hepatic and cardiac lipids [5]. And hepatic lipids including triglyceride deposition are involved in the pathogenesis and development of NAFLD. Several factors are implicated in the pathology of obesity-related NAFLD, including complex interactions between glucose and lipid metabolism, genetic predisposition, environmental conditions, and modulation of the intestinal microbiota [6]. IR encompasses a wide spectrum of disorders, such as defective insulin receptor signal transduction and mitochondrial function [7, 8], microvascular dysfunction [9, 10], and inflammation [11–13]. Obesity, characterized as a state of chronic low-grade inflammation caused by overnutrition, is a major cause of decreased insulin sensitivity, which makes obesity a major risk factor for IR [14–16]. Obesity, also manifested as excess adiposity, is a main cause of NAFLD [17]. NAFLD is recognized as a typical feature of metabolic syndrome and manifested as a series of hepatic injuries including steatosis, nonalcoholic steatohepatitis (NASH), and even hepatocellular carcinoma [18]. Obesity causes lipid accumulation in adipocytes, which activates c-Jun N-terminal kinase (JNK) and nuclear factor-kappa B (NF-κB) signaling pathways and might subsequently increase the production of proinflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6) [11, 19]. In most cases, adipose tissue (AT) is an important site of obesity-induced IR, and it can also affect the liver and muscle by releasing cytokines, including adipokines such as TNF-α [11, 18]. AT consists of several cell types. Among these, adipocytes and immune cells, such as macrophages and dendritic cells (DCs), have attracted significant attention as contributors that link inflammation to IR.
This review will focus on the relationship between inflammation and IR, and we analyze the mechanisms relating to how inflammatory cytokines, signaling pathways, and some other factors link inflammation to IR.
2. Cytokines That Link Inflammation to IR
2.1. TNF-α
Studies of TNF-α in the 1990s first analyzed the relationship between inflammation and IR [20]. TNF-α is an adipose tissue-derived proinflammatory cytokine that causes insulin resistance by enhancing adipocyte lipolysis and increasing the serine/threonine phosphorylation of IRS-1 (insulin receptor substrate-1) [11, 21]. Several signaling pathways, including the IKKβ/NF-κB pathway, are involved in the pathogenesis of IR (see Figure 1) [22, 23]. It was reported that TNF-α can increase glucose uptake in both visceral and subcutaneous adipocytes by activating the adenosine monophosphate activated protein kinase (AMPK) pathway, whereas it triggers insulin resistance in visceral adipocytes by activating JNK1/2. Because of the depot-specific effects of TNF-α on glucose uptake, approaches to treat IR by modulating TNF-α signaling are ongoing [24]. However, studies of therapies such as the TNF-α superfamily member sTWEAK (soluble tumour necrosis factor-like weak inducer of apoptosis), which aims to block TNF signaling to treat IR, have demonstrated that TNF-α plays a role in IR [25]. Interestingly, the plasma levels of TNF-α are higher in males than in females, as well as in obese individuals compared with lean ones. This suggests that obese males are more likely to suffer from IR and related diseases such as cardiovascular disease [26].
Figure 1.
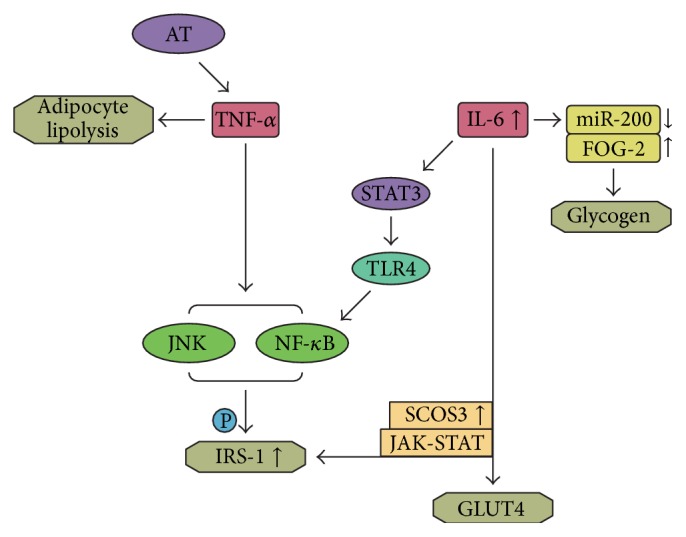
Influence of the inflammatory cytokines on the status of insulin resistance. TNF-α causes insulin resistance by enhancing adipocyte lipolysis stimulating JNK and IKKβ/NF-κB pathway which may increase serine/threonine phosphorylation of IRS1. IL-6 induces IR by reducing the expression of GLUT4 and IRS-1 by activating the JAK-STAT signaling pathway and increasing SOCS3 expression, and IL-6 can also lead to IR in skeletal muscle by inducing TLR-4 gene expression through activation of STAT3; besides, TLR4 is suggested to be major upstream molecules in the activation of NF-κB. Besides, IL-6 is also found to induce IR by impairing the synthesis of glycogen through downregulating the expression of miR-200s and upregulating that of FOG-2.
2.2. IL-1β
Interleukin-1β (IL-1β) is a proinflammatory cytokine whose secretion is regulated by inflammasome activity. IL-1β contributes to IR by impairing insulin signaling in peripheral tissues and macrophages, which leads to the reduced insulin sensitivity of β-cells and possible impaired insulin secretion [27, 28]. The levels of IL-1β in various cells such as endothelial cells and monocytes are increased during hyperglycemia [29]. IL-1β also plays a vital role in initiating and maintaining inflammation-induced organ dysfunction in type 2 diabetes mellitus (T2DM) [30]. IL-1β might increase systemic inflammation and inhibit insulin action in the major insulin-target cells, such as macrophages [31].
2.3. IL-6
IL-6 is secreted by multiple tissues, particularly adipose tissue, and is recognized as an inflammatory mediator that causes IR by reducing the expression of glucose transporter-4 (GLUT-4) and insulin receptor substrate-1 (IRS-1). These effects are exerted by the activation of the Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling pathway (see Box 1) and increased the expression of suppressor of cytokine signaling 3 (SOCS3) [32, 33] (see Figure 1). Therefore, hybrid training can ameliorate insulin resistance by suppressing serum IL-6 in skeletal muscle [34]. IL-6 also induces IR by blocking the phosphoinositide 3-kinase (PI3K) pathway and impairing glycogen synthesis by downregulating the expression of microRNA-200s (miR-200s) and upregulating that of friend of GATA 2 (FOG-2) [35, 36]. It was suggested that IR in human skeletal muscle is related to IL-6 stimulation, which induces toll-like receptor-4 (TLR-4) gene expression by activating STAT3 [37] (see Figure 1).
2.4. Leptin
Leptin is a protein that is derived primarily from white adipose tissue (WAT) [38]. It suppresses appetite and increases energy expenditure by repressing anabolic neuronal circuits and activating catabolic neuronal circuits. In addition, leptin levels are affected by nutriture [39]. Leptin-mediated appetite and energy homeostasis are associated with the progression of IR [40]. Furthermore, a state called leptin resistance, which was disputed lately by the concept of hypothalamic leptin insufficiency, is often observed in the obese individuals, and weight loss simultaneously reduces serum leptin levels. This suggests that leptin might have a role in regulating IR. Consistent with this, the stimulation of PI3K signaling by leptin is essential for modulating glucose metabolism and the function of pancreatic β-cells [31–42]. It is likely that an increased concentration of leptin, an anti-inflammatory cytokine, during inflammation in AT is associated with leptin resistance in obese individuals. Interestingly, leptin was recommended as a biomarker for in utero insulin resistance based on the link between maternal and fetal leptin and IR [43, 44]. Leptin is a potential treatment for IR because it improves glycometabolism, insulin sensitivity, and lipometabolism [45, 46].
2.5. Adiponectin
Adiponectin is produced mainly by WAT. Its levels reduce in obesity, IR, or T2DM, where it acts as an anti-inflammatory cytokine, but increase in osteoarthritis (OA) and type 1 diabetes mellitus (T1DM), where it acts as a proinflammatory cytokine [39, 47]. Two receptors are involved in the glucose metabolism that links adiponectin to the amelioration of IR. Adiponectin receptor 1 (AdipoR1) is likely to reduce the expression of the genes that encode hepatic gluconeogenic enzymes and molecules involved in lipogenesis by activating AMPK. In contrast, adiponectin receptor 2 (AdipoR2) increases the expression of the genes that contribute to glucose consumption by activating peroxisome proliferator activated receptor-alpha (PPAR-α) signaling [48, 49]. AdipoR1 and AdipoR2 are expressed at high levels in skeletal muscle and the liver, respectively [28, 50]. In brief, adiponectin ameliorates hepatic insulin resistance by reducing glycogenesis and lipogenesis, as well as increasing glucose consumption.
2.6. Resistin
The production of resistin is complex. In rodents, it is generated from adipocytes, whereas it is produced mostly by macrophages in humans. Its concentrations increase concurrently with the levels of inflammatory mediators [51]. It was suggested that resistin participates in the pathogenesis of IR and that its levels might be elevated due to obesity and IR [52]. Resistin promotes IR by regulating the expression of proinflammatory cytokines, including TNF-α and IL-6, in macrophages via an NF-κB-dependent pathway. It also plays roles in inflammation and IR by binding directly to TLR4 receptors in the hypothalamus to activate JNK and mitogen-activated protein kinase (MAPK) signaling pathways [53].
2.7. MCP-1
Monocyte chemoattractant protein-1 (MCP-1) is a proinflammatory chemokine produced by adipocytes, macrophages, and endothelial cells, which might lead to the recruitment of macrophages, DCs, and memory T cells [11, 54]. Adipocytes and macrophages are the main source of proinflammatory cytokines. However, the expression of MCP-1 increases during adiposity, which might stimulate the recruitment of macrophages and DCs, which further increases the expression of cytokines to exacerbate inflammation-induced IR [22]. The expression of MCP-1 increases during obesity, particularly in visceral fat areas, which might contribute to the pathogenesis of IR, particularly in the liver [54, 55]. It plays a role in IR by regulating the inflammatory response, insulin sensitivity, lipid metabolism, macrophage polarization and infiltration, and the phosphorylation of extracellular signal-regulated kinase-1/2 (ERK-1/2) and p38 MAPK [56]. C-C motif chemokine receptor 2 (CCR2) is a vital MCP-1 receptor. In adipose tissue of CCR2 knockout mice, macrophage content and inflammatory profile were reduced. CCR2 deficiency also ameliorated hepatic steatosis and improved insulin sensitivity [57]. This suggests that MCP-1 plays a crucial role in the development of both inflammation and IR.
3. Signaling Pathways Linking Inflammation to Insulin Resistance
3.1. IKKβ/NF-κB Pathway
NF-κB is a transcription factor comprised of Rel family proteins such as p65/RelA, RelB, c-Rel, p50/p105, and p52/p100. It is involved in a series of pathological processes such as inflammation and innate and adaptive immune responses [58, 59]. NF-κB is sequestered in the cytoplasm bound to IκB proteins in normal circumstances, which prevents the nuclear localization of NF-κB. After stimulation with various pathogenic stimuli, such as those in obese individuals, the IKK complex that contains two subunits (IKKα and IKKβ) is activated, which triggers the phosphorylation of IκBα on Ser32 and 36. This leads to the degradation of IκBα, exposes the nuclear localization sequence of NF-κB, and triggers its translocation to the nucleus and the upregulation of target genes that encode inflammatory mediators such as TNF-α, IL-1β, and IL-6 [20, 58] (see Figure 2). IKKβ deficiency in adipocytes completely prevented the free fatty acid- (FFA-) induced expression of TNF-α and IL-6, whereas the activation of IKKβ inhibited the expression of anti-inflammatory cytokines such as leptin and adiponectin [60]. According to this, the deletion of IKKβ improved glucose tolerance and insulin sensitivity [61]. In addition, treatments that inhibit NF-κB always improve IR, which suggests that the NF-κB pathway plays an important role in inflammation-associated IR [62]. NF-κB is also a vital intermediary that couples IR to the proinflammatory cytokine IL-1β in IR-related diseases such as obesity and T2DM [27].
Figure 2.
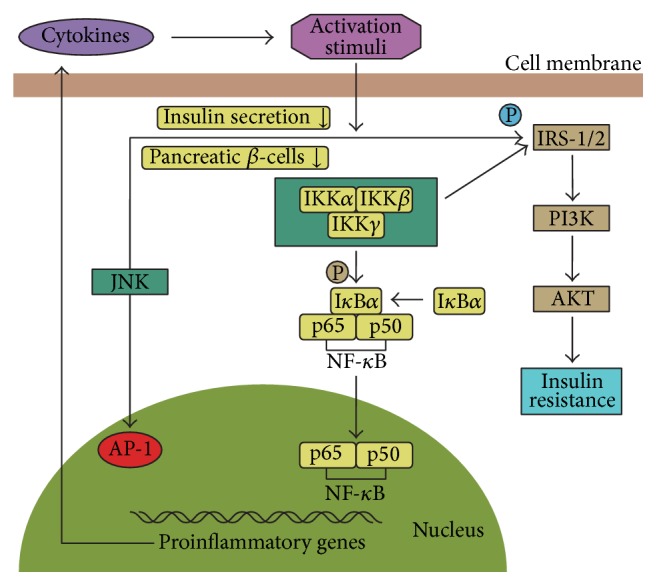
Inflammatory pathways linking inflammation to insulin resistance. Activation of JNK and NF-κB pathways causes serine kinase phosphorylation of IRS-1 or IRS-2, which may block insulin signaling and finally lead to the occurrence of IR. In addition, JNK and NF-κB pathways are involved in the production of proinflammatory cytokines which may in turn become activation stimuli of the pathways.
3.2. JNK Pathway
There are three different JNK isoforms (JNK-1, -2, and -3), which belong to MAPK family. JNK contributes to inflammation and metabolic syndrome (MS), obesity, and IR by regulating the production of proinflammatory cytokines, karyomitosis, and cellular apoptosis [63–65]. JNK can also be stimulated by endoplasmic reticulum (ER) stress, which leads to the serine phosphorylation of IRS-1 (see Figure 2). JNK plays a role in the phosphorylation of the c-Jun component of activator protein (AP-1) transcription factor, but there is no evidence of a direct relationship between this transcriptional pathway and JNK-reduced IR. The JNK pathway can be activated under diabetic conditions, which might increase IR. Conversely, suppressing the JNK pathway improves IR and glucose tolerance [66]. JNK plays an important role in IR by inhibiting insulin secretion from pancreatic β-cells via proinflammatory stimuli including IL-1. Moreover, the excessive activation of JNK in peripheral insulin-sensitive tissues promotes IR [67]. It was demonstrated that inhibiting JNK reduced the release of IR-related proinflammatory cytokines such as TNF-α and MCP-1 [68–70]. Interestingly, JNK-1 deficiency in adipose tissue protects against hepatic steatosis and promotes glucose intolerance, insulin clearance, IR, and hepatic steatosis. In skeletal muscle, JNK-1 does not affect the development of obesity and IR [65, 71]. However, JNK in isolated rat skeletal muscle plays a vital role during oxidant-induced IR because insulin-stimulated glucose transport activity was improved by the selective inhibition of JNK [72]. Taken together, these studies suggest that further studies are needed to analyze the effects of JNK in IR.
3.3. Inflammasome Pathway
The inflammasome consists of a large group of cytosolic protein complexes and plays roles in inflammation by regulating the secretion of IL-1β and IL-18. Therefore, it is important in innate immunity and metabolic syndromes such as obesity and IR [30, 73]. NOD-like receptor proteins (NLRPs), neutrophilic alkaline phosphatases (NALPs), apoptosis associated speck-like protein (ASC), and caspase-1 are the essential components of inflammasome complexes [20]. Inflammasome NLRP3 (nucleotide-binding domain, leucine-rich-containing family, and pyrin domain-containing-3), which links saturated FFAs to chronic inflammation, is being studied extensively because it is highly sensitive to nonmicrobial stress. It can be activated by mitochondrial dysfunction. In addition, the reduced expression of NLRP3 in obesity results in enhanced insulin signaling, decreased inflammation, and improved insulin sensitivity [73, 74] (see Figure 3). Caspase-1 is a cysteine protease that contributes to IR by counteracting the metabolic function of adipose tissue to impair insulin sensitivity and also mediates the infiltration of macrophages into adipose tissues [75, 76]. It was reported that the elimination of ASC and caspase-1 lowers the plasma levels of insulin, leptin, and resistin. Moreover, ASC deficiency might protect individuals against HFD-induced IR, hepatic steatosis, and adipocyte hypertrophy. In addition, caspase-1-deficient mice have high energy expenditure. Taken together, these studies suggest that the inflammasome plays a vital role in obesity-induced IR and that it is an important therapeutic target for the treatment of IR [76].
Figure 3.
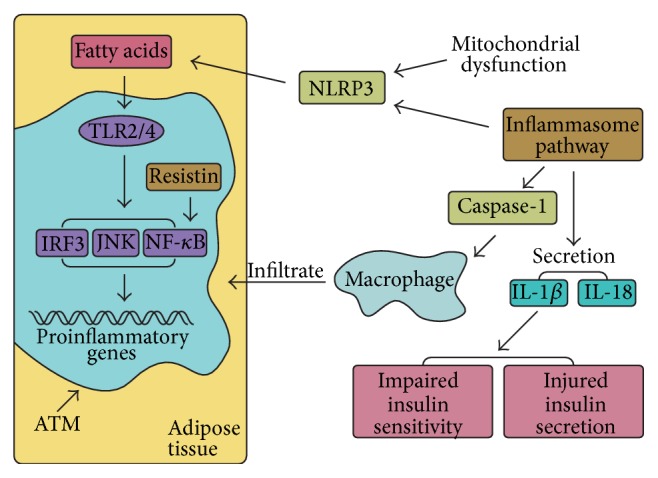
Inflammasome pathway and macrophages are involved in development of insulin resistance. The secretion of IL-β and IL-18 can be regulated by inflammasome pathway. Inflammasome consists of a large group of cytosolic protein complexes including NLRP3 and caspase-1. NLRP3 can be activated by mitochondrial dysfunction through causing ROS accumulation, and NLRP3 is also a novel molecular link between saturated FFA and chronic inflammation. Caspase-1 mediates macrophages that infiltrate into adipose tissues. Dietary saturated fatty acids lead to activation of TLR2 and TLR4 in ATMs, giving rise to the activation of IRF3, JNK, and NF-κB.
4. Other Factors Linking Inflammation to IR
4.1. Macrophages
Macrophages infiltrate and reside in adipose tissue, named ATMs, and usually play an important role in obesity-induced IR. There are two types of ATM: classically activated (M1) in obese animals and alternatively activated (M2) in lean species [77]. ATMs have an important role in the development of chronic inflammation, including obesity-induced inflammation, because they are the primary source of cytokine production. In addition, obesity might change the number of ATMs by increasing the triple positive CD11b + F4/80 + CD11 + ATM subpopulation [20]. As well as using CD11c as an M1 marker, Fujisaka et al. used CD206 rather than CD209 and CD301 as M2 markers by flow cytometry to demonstrate that IR might be regulated by the number of M1 ATMs and the M1 : M2 ratio. In addition, intervention with pioglitazone could reduce inflammation and ameliorate IR by upregulating the expression of IL-10, which might contribute to the reduction of M2 quantity [77]. In another study, it was suggested that the MCP-1/CCR2 axis might contribute to a shift from M2 to M1 polarization, which is an important cause of IR as it leads to the production of inflammatory factors such as TNF-α and IL-6 [78]. During obesity, dietary saturated fatty acids lead to the activation of TLR2 and TLR4 in ATMs, which is followed by the activation of interferon regulatory factor-3 (IRF3), JNK, and NF-κB and subsequent inflammatory signaling (see Figure 3) [1].
4.2. hs-CRP
C-reactive protein (CRP) is an acute-phase protein synthesized by the liver. It is an inflammatory marker whose expression is increased significantly during inflammation, mainly due to its regulation by proinflammatory cytokines such as IL-6 and TNF-α [79, 80]. In most clinical and scientific studies, CRP is measured using high-sensitivity assays and is known as high-sensitivity CRP (hs-CRP) [81]. It was suggested that increased hs-CRP levels might be caused by an insufficient insulin-induced suppression of CRP synthesis. Moreover, CRP might contribute to vascular inflammation by activating complement proteins and increasing the production of thrombogenic components bound to the membranes of injured vascular cells, which contributes to the development of IR [80]. In addition, elevated CRP expression is a potential risk factor and indicator for T2DM. However, there is no apparent causality between serum CRP, IR, and diabetes, which suggests that CRP is more likely to be a downstream marker rather than an upstream effector that links inflammation to IR [82]. Nevertheless, hs-CRP is closely associated with IR, and thus its expression should be assessed during investigations of IR.
5. Concluding Remarks
Inflammation plays an important role in the development of IR via various cytokines and molecular pathways, and so inflammation should be targeted with appropriate interventions to prevent IR. Because dietary fat might play a role in the production of inflammatory molecules by modifying the intestinal microbiota, which might result in an inappropriate immune reaction [83], it is important for individuals to develop good dietary and living habits. Specific factors and signaling pathways are often correlated with each other. For example, the activation of IKKβ/NF-κB signaling might increase the secretion of proinflammatory cytokines such as TNF-α and IL-1β, which might in turn stimulate IKKβ/NF-κB signaling. Therefore, both of the fluctuation of cytokines and the status of relevant signaling pathways should be taken into account during studies analyzing inflammation-related IR. Most current studies of inflammation-related IR are performed in animals, which makes it challenging to apply these methods to humans to exert a curative effect of IR in the clinic. Because the mechanisms that link inflammation to IR are not understood completely, additional well-designed clinical and laboratory studies are in demand to elaborate their relationship.
6. Box 1
6.1. The JAK-STAT Signaling Pathway
The Janus kinase-signal transducers and activators of transcription (JAK-STAT) signaling pathway are a cytokines-activated cascade involved in many important biological processes including the proliferation, differentiation, and apoptosis of the cells [84]. This signaling pathway contains three components: tyrosine kinase associated receptor, Janus kinase and signal transducer, and activator of transcription [85]. To date, four members of JAK kinase family have been identified including JAK1, JAK2, JAK3, and TYK2, and the STAT family consists of seven proteins (STATs 1, 2, 3, 4, 5A, 5B, and 6) [86]. The signaling pathway is initiated through binding of ligands to membrane-bound receptors which may lead to receptor dimerization and then activate the JAK kinases; in turn, the activation of JAK kinases phosphorylates the tyrosine residues with the receptors [87]. As a result, STAT proteins are phosphorylated by JAK, then dimerize via their src-homology 2 (SH2) domains, and translocate to the nucleus where they regulate transcription of specific target genes involved in multiple diseases including leukemia, rheumatoid arthritis, cancer, and diabetic nephropathy [88, 89].
Acknowledgments
The authors thank all study participants for valuable contributions to the study. The study was supported by the National Natural Science Foundation of China (no. 81001557 and no. 81473787).
Glossary
Metabolic Syndrome. A pathophysiological disorder characterized by a cluster of risk factors for cardiovascular disease, type 2 diabetes, and renal disease.
Adenosine Monophosphate Activated Protein Kinase. A key molecule implicated in metabolic modulation as it increases O2 consumption, glucose metabolism, and fatty acid oxidation.
Soluble Tumour Necrosis Factor-Like Weak Inducer of Apoptosis. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) is a member of the tumor necrosis factor (TNF) superfamily. Soluble tumour necrosis factor-like weak inducer of apoptosis (sTWEAK) is a soluble variant of TWEAK. sTWEAK plays a role in a series of biological processes including cellular proliferation, differentiation, apoptosis, and inflammation.
Glucose Transporter. It is a wide group of membrane proteins that facilitate the transport of glucose.
Phosphoinositide 3-Kinase. It is an enzyme that generates lipid second messenger molecules, resulting in the activation of multiple intracellular signalling cascades.
Friend of GATA. The GATA family refers to a kind of transcription factors that recognizes and binds to the GATA motifs. GATA proteins play an essential role in hematopoiesis and tissue specific gene expression through functional interactions with friend of GATA (FOG-) proteins. There are two FOG proteins, FOG-1 and FOG-2. FOG-1 is mainly expressed in hematopoietic tissues and FOG-2 in the heart, brain, and gonads.
Toll-Like Receptors. Toll-like receptors are pattern recognition receptors that play an important role in recognizing the conserved molecular structure of pathogens and triggering of the innate immune response.
Peroxisome Proliferator Activated Receptors. Peroxisome proliferator activated receptors are a group of ligand-activated nuclear receptors involved in the gene expressions associated with the metabolic processes.
Mitogen-Activated Protein Kinase. It is an important signal transducer acting as a regulator of physiology and immune responses.
Disclosure
Li Chen and Rui Chen are co-first authors.
Conflict of Interests
The authors declare that they have no conflict of interests to this work.
Authors' Contribution
Li Chen and Rui Chen contributed equally to the paper.
References
- 1.Chawla A., Nguyen K. D., Goh Y. P. S. Macrophage-mediated inflammation in metabolic disease. Nature Reviews Immunology. 2011;11(11):738–749. doi: 10.1038/nri3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liang F. X., Koya D. Acupuncture: is it effective for treatment of Insulin resistance? Diabetes, Obesity and Metabolism. 2010;12(7):555–569. doi: 10.1111/j.1463-1326.2009.01192.x. [DOI] [PubMed] [Google Scholar]
- 3.Laakso M., Kuusisto J. Insulin resistance and hyperglycaemia in cardiovascular disease development. Nature Reviews Endocrinology. 2014;10(5):293–302. doi: 10.1038/nrendo.2014.29. [DOI] [PubMed] [Google Scholar]
- 4.Mavrogiannaki A. N., Migdalis I. N. Nonalcoholic fatty liver disease, diabetes mellitus and cardiovascular disease: newer data. International Journal of Endocrinology. 2013;2013:8. doi: 10.1155/2013/450639.450639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaggini M., Morelli M., Buzzigoli E., DeFronzo R. A., Bugianesi E., Gastaldelli A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients. 2013;5(5):1544–1560. doi: 10.3390/nu5051544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karlas T., Wiegand J., Berg T. Gastrointestinal complications of obesity: non-alcoholic fatty liver disease (NAFLD) and its sequelae. Best Practice and Research: Clinical Endocrinology and Metabolism. 2013;27(2):195–208. doi: 10.1016/j.beem.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Abdul-Ghani M. A., DeFronzo R. A. Pathogenesis of insulin resistance in skeletal muscle. Journal of Biomedicine and Biotechnology. 2010;2010:19. doi: 10.1155/2010/476279.476279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahn J., Lee H., Im S. W., Jung C. H., Ha T. Y. Allyl isothiocyanate ameliorates insulin resistance through the regulation of mitochondrial function. The Journal of Nutritional Biochemistry. 2014;25(10):1026–1034. doi: 10.1016/j.jnutbio.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 9.Muris D. M. J., Houben A. J. H. M., Schram M. T., Stehouwer C. D. A. Microvascular dysfunction: an emerging pathway in the pathogenesis of obesity-related insulin resistance. Reviews in Endocrine and Metabolic Disorders. 2013;14(1):29–38. doi: 10.1007/s11154-012-9231-7. [DOI] [PubMed] [Google Scholar]
- 10.Karaca Ü., Schram M. T., Houben A. J. H. M., Muris D. M. J., Stehouwer C. D. A. Microvascular dysfunction as a link between obesity, insulin resistance and hypertension. Diabetes Research and Clinical Practice. 2014;103(3):382–387. doi: 10.1016/j.diabres.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 11.Shoelson S. E., Lee J., Goldfine A. B. Inflammation and insulin resistance. The Journal of Clinical Investigation. 2006;116(7):1793–1801. doi: 10.1172/jci29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Könner A. C., Brüning J. C. Toll-like receptors: linking inflammation to metabolism. Trends in Endocrinology and Metabolism. 2011;22(1):16–23. doi: 10.1016/j.tem.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Cai D. S. Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends in Endocrinology and Metabolism. 2013;24(1):40–47. doi: 10.1016/j.tem.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohanraj L., Kim H.-S., Li W., et al. IGFBP-3 inhibits cytokine-induced insulin resistance and early manifestations of atherosclerosis. PLoS ONE. 2013;8(1) doi: 10.1371/journal.pone.0055084.e55084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daniele G., Mendoza R. G., Winnier D., et al. The inflammatory status score including IL-6, TNF-α, osteopontin, fractalkine, MCP-1 and adiponectin underlies whole-body insulin resistance and hyperglycemia in type 2 diabetes mellitus. Acta Diabetologica. 2014;51(1):123–131. doi: 10.1007/s00592-013-0543-1. [DOI] [PubMed] [Google Scholar]
- 16.Osborn O., Olefsky J. M. The cellular and signaling networks linking the immune system and metabolism in disease. Nature Medicine. 2012;18(3):363–374. doi: 10.1038/nm.2627. [DOI] [PubMed] [Google Scholar]
- 17.Martins C., Aires L., Júnior I. F., et al. Physical activity is related to fatty liver marker in obese youth, independently of central obesity or cardiorespiratory fitness. Journal of Sports Science and Medicine. 2015;14(1):103–109. [PMC free article] [PubMed] [Google Scholar]
- 18.Ramadori P., Kroy D., Streetz K. L. Immunoregulation by lipids during the development of non-alcoholic steatohepatitis. Hepatobiliary Surgery and Nutrition. 2015;4(1):11–23. doi: 10.3978/j.issn.2304-3881.2015.01.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma M., Vikram N. K., Misra A., et al. Assessment of 11-β hydroxysteroid dehydrogenase (11-βHSD1) 4478T>G and tumor necrosis factor-α (TNF-α)-308 G>A polymorphisms with obesity and insulin resistance in Asian Indians in North India. Molecular Biology Reports. 2013;40(11):6261–6270. doi: 10.1007/s11033-013-2738-5. [DOI] [PubMed] [Google Scholar]
- 20.Lee B.-C., Lee J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochimica et Biophysica Acta—Molecular Basis of Disease. 2014;1842(3):446–462. doi: 10.1016/j.bbadis.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hotamisligil G. S. Molecular mechanisms of insulin resistance and the role of the adipocyte. International Journal of Obesity and Related Metabolic Disorders. 2000;24(4):S23–S27. doi: 10.1038/sj/ijo/0801497. [DOI] [PubMed] [Google Scholar]
- 22.Kahn S. E., Hull R. L., Utzschneider K. M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 23.Zhou X., You S. Rosiglitazone inhibits hepatic insulin resistance induced by chronic pancreatitis and IKK-β/NF-κB expression in liver. Pancreas. 2014;43(8):1291–1298. doi: 10.1097/mpa.0000000000000173. [DOI] [PubMed] [Google Scholar]
- 24.Fernández-Veledo S., Vila-Bedmar R., Nieto-Vazquez I., Lorenzo M. c-Jun N-terminal kinase 1/2 activation by tumor necrosis factor-α induces insulin resistance in human visceral but not subcutaneous adipocytes: reversal by liver X receptor agonists. Journal of Clinical Endocrinology and Metabolism. 2009;94(9):3583–3593. doi: 10.1210/jc.2009-0558. [DOI] [PubMed] [Google Scholar]
- 25.Vázquez-Carballo A., Ceperuelo-Mallafré V., Chacón M. R., et al. TWEAK prevents TNF-α-induced insulin resistance through PP2A activation in human adipocytes. American Journal of Physiology: Endocrinology and Metabolism. 2013;305(1):E101–E112. doi: 10.1152/ajpendo.00589.2012. [DOI] [PubMed] [Google Scholar]
- 26.El-Haggar S. M., Mostafa T. M. Adipokines and biochemical changes in Egyptian obese subjects: possible variation with sex and degree of obesity. Endocrine. 2015;48(3):878–885. doi: 10.1007/s12020-014-0390-z. [DOI] [PubMed] [Google Scholar]
- 27.Su D., Coudriet G. M., Kim D. H., et al. FoxO1 links insulin resistance to proinflammatory cytokine IL-1β production in macrophages. Diabetes. 2009;58(11):2624–2633. doi: 10.2337/db09-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Böni-Schnetzler M., Donath M. Y. How biologics targeting the IL-1 system are being considered for the treatment of type 2 diabetes. British Journal of Clinical Pharmacology. 2013;76(2):263–268. doi: 10.1111/j.1365-2125.2012.04297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koenen T. B., Stienstra R., van Tits L. J., et al. Hyperglycemia activates caspase-1 and TXNIP-mediated IL-1β transcription in human adipose tissue. Diabetes. 2011;60(2):517–524. doi: 10.2337/db10-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grant R. W., Dixit V. D. Mechanisms of disease: inflammasome activation and the development of type 2 diabetes. Frontiers in Immunology. 2013;4, article 50 doi: 10.3389/fimmu.2013.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hardaway A. L., Podgorski I. IL-1β, RAGE and FABP4: targeting the dynamic trio in metabolic inflammation and related pathologies. Future Medicinal Chemistry. 2013;5(10):1089–1108. doi: 10.4155/fmc.13.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lukic L., Lalic N. M., Rajkovic N., et al. Hypertension in obese type 2 diabetes patients is associated with increases in insulin resistance and IL-6 cytokine levels: potential targets for an efficient preventive intervention. International Journal of Environmental Research and Public Health. 2014;11(4):3586–3598. doi: 10.3390/ijerph110403586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Serrano-Marco L., Barroso E., El Kochairi I., et al. The peroxisome proliferator-activated receptor (PPAR) β/δ agonist GW501516 inhibits IL-6-induced signal transducer and activator of transcription 3 (STAT3) activation and insulin resistance in human liver cells. Diabetologia. 2012;55(3):743–751. doi: 10.1007/s00125-011-2401-4. [DOI] [PubMed] [Google Scholar]
- 34.Kawaguchi T., Shiba N., Maeda T., et al. Hybrid training of voluntary and electrical muscle contractions reduces steatosis, insulin resistance, and IL-6 levels in patients with NAFLD: a pilot study. Journal of Gastroenterology. 2011;46(6):746–757. doi: 10.1007/s00535-011-0378-x. [DOI] [PubMed] [Google Scholar]
- 35.Dou L., Zhao T., Wang L., et al. MiR-200s contribute to interleukin-6 (IL-6)-induced insulin resistance in hepatocytes. The Journal of Biological Chemistry. 2013;288(31):22596–22606. doi: 10.1074/jbc.m112.423145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin J., Hao Z., Ma Y., et al. Concomitant activation of the PI3K/Akt and ERK1/2 signalling is involved in cyclic compressive force-induced IL-6 secretion in MLO-Y4 cells. Cell Biology International. 2014;38(5):591–598. doi: 10.1002/cbin.10235. [DOI] [PubMed] [Google Scholar]
- 37.Kim T. H., Choi S. E., Ha E. S., et al. IL-6 induction of TLR-4 gene expression via STAT3 has an effect on insulin resistance in human skeletal muscle. Acta Diabetologica. 2013;50(2):189–200. doi: 10.1007/s00592-011-0259-z. [DOI] [PubMed] [Google Scholar]
- 38.Pedroso J. A., Buonfiglio D. C., Cardinali L. I., et al. Inactivation of SOCS3 in leptin receptor-expressing cells protects mice from diet-induced insulin resistance but does not prevent obesity. Molecular Metabolism. 2014;3(6):608–618. doi: 10.1016/j.molmet.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stofkova A. Leptin and adiponectin: from energy and metabolic dysbalance to inflammation and autoimmunity. Endocrine Regulations. 2009;43(4):157–168. doi: 10.4149/endo_2009_04_157. [DOI] [PubMed] [Google Scholar]
- 40.van der Wijden C. L., Delemarre-van de Waal H. A., van Mechelen W., van Poppel M. N. The relationship between moderate-to-vigorous intensity physical activity and insulin resistance, insulin-like growth factor (IGF-1)-system 1, leptin and weight change in healthy women during pregnancy and after delivery. Clinical Endocrinology. 2015;82(1):68–75. doi: 10.1111/cen.12593. [DOI] [PubMed] [Google Scholar]
- 41.Wang T.-N., Chang W.-T., Chiu Y.-W., et al. Relationships between changes in leptin and insulin resistance levels in obese individuals following weight loss. Kaohsiung Journal of Medical Sciences. 2013;29(8):436–443. doi: 10.1016/j.kjms.2012.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yadav A., Kataria M. A., Saini V. Role of leptin and adiponectin in insulin resistance. Clinica Chimica Acta. 2013;417:80–84. doi: 10.1016/j.cca.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 43.Walsh J. M., Byrne J., Mahony R. M., Foley M. E., McAuliffe F. M. Leptin, fetal growth and insulin resistance in non-diabetic pregnancies. Early Human Development. 2014;90(6):271–274. doi: 10.1016/j.earlhumdev.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 44.Jialal I., Adams-Huet B., Duong F., Smith G. Relationship between retinol-binding protein-4/adiponectin and leptin/adiponectin ratios with insulin resistance and inflammation. Metabolic Syndrome and Related Disorders. 2014;12(4):227–230. doi: 10.1089/met.2014.0013. [DOI] [PubMed] [Google Scholar]
- 45.Toyoshima Y., Gavrilova O., Yakar S., et al. Leptin improves insulin resistance and hyperglycemia in a mouse model of type 2 diabetes. Endocrinology. 2005;146(9):4024–4035. doi: 10.1210/en.2005-0087. [DOI] [PubMed] [Google Scholar]
- 46.Yau S. W., Henry B. A., Russo V. C., et al. Leptin enhances insulin sensitivity by direct and sympathetic nervous system regulation of muscle IGFBP-2 expression: evidence from nonrodent models. Endocrinology. 2014;155(6):2133–2143. doi: 10.1210/en.2013-2099. [DOI] [PubMed] [Google Scholar]
- 47.Passos M., Gonçalves M. Regulation of insulin sensitivity by adiponectin and its receptors in response to physical exercise. Hormone and Metabolic Research. 2014;46(09):603–608. doi: 10.1055/s-0034-1377026. [DOI] [PubMed] [Google Scholar]
- 48.Yamauchi T., Nio Y., Maki T., et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nature Medicine. 2007;13(3):332–339. doi: 10.1038/nm1557. [DOI] [PubMed] [Google Scholar]
- 49.Crimmins N. A., Martin L. J. Polymorphisms in adiponectin receptor genes ADIPOR1 and ADIPOR2 and insulin resistance. Obesity Reviews. 2007;8(5):419–423. doi: 10.1111/j.1467-789x.2007.00348.x. [DOI] [PubMed] [Google Scholar]
- 50.Bermúdez V. J., Rojas E., Toledo A., et al. Single-nucleotide polymorphisms in adiponectin, AdipoR1, and AdipoR2 genes: insulin resistance and type 2 diabetes mellitus candidate genes. American Journal of Therapeutics. 2013;20(4):414–421. doi: 10.1097/mjt.0b013e318235f206. [DOI] [PubMed] [Google Scholar]
- 51.Reilly M. P., Lehrke M., Wolfe M. L., Rohatgi A., Lazar M. A., Rader D. J. Resistin is an inflammatory marker of atherosclerosis in humans. Circulation. 2005;111(7):932–939. doi: 10.1161/01.cir.0000155620.10387.43. [DOI] [PubMed] [Google Scholar]
- 52.Szulinska M., Musialik K., Suliburska J., Lis I., Bogdanski P. The effect of L-arginine supplementation on serum resistin concentration in insulin resistance in animal models. European Review for Medical and Pharmacological Sciences. 2014;18(4):575–580. [PubMed] [Google Scholar]
- 53.Benomar Y., Gertler A., De Lacy P., et al. Central resistin overexposure induces insulin resistance through toll-like receptor 4. Diabetes. 2013;62(1):102–144. doi: 10.2337/db12-0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanda H., Tateya S., Tamori Y., et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. Journal of Clinical Investigation. 2006;116(6):1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kouyama K., Miyake K., Zenibayashi M., et al. Association of serum MCP-1 concentration and MCP-1 polymorphism with insulin resistance in Japanese individuals with obese type 2 diabetes. Kobe Journal of Medical Sciences. 2007;53(6):345–354. [PubMed] [Google Scholar]
- 56.Nio Y., Yamauchi T., Iwabu M., et al. Monocyte chemoattractant protein-1 (MCP-1) deficiency enhances alternatively activated M2 macrophages and ameliorates insulin resistance and fatty liver in lipoatrophic diabetic A-ZIP transgenic mice. Diabetologia. 2012;55(12):3350–3358. doi: 10.1007/s00125-012-2710-2. [DOI] [PubMed] [Google Scholar]
- 57.Weisberg S. P., Hunter D., Huber R., et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. Journal of Clinical Investigation. 2006;116(1):115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rahman M. M., McFadden G. Modulation of NF-κB signalling by microbial pathogens. Nature Reviews Microbiology. 2011;9(4):291–306. doi: 10.1038/nrmicro2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sakurai H., Suzuki S., Kawasaki N., et al. Tumor necrosis factor-α-induced IKK phosphorylation of NF-κB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. The Journal of Biological Chemistry. 2003;278(38):36916–36923. doi: 10.1074/jbc.m301598200. [DOI] [PubMed] [Google Scholar]
- 60.Jiao P., Ma J., Feng B., et al. FFA-induced adipocyte inflammation and insulin resistance: involvement of ER stress and IKKβ pathways. Obesity. 2011;19(3):483–491. doi: 10.1038/oby.2010.200. [DOI] [PubMed] [Google Scholar]
- 61.Arkan M. C., Hevener A. L., Greten F. R., et al. IKK-β links inflammation to obesity-induced insulin resistance. Nature Medicine. 2005;11(2):191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 62.Yekollu S. K., Thomas R., O'Sullivan B. Targeting curcusomes to inflammatory dendritic cells inhibits NF-κB and improves insulin resistance in obese mice. Diabetes. 2011;60(11):2928–2938. doi: 10.2337/db11-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee M.-Y., Yuk J.-E., Kwon O.-K., Oh S.-R., Lee H.-K., Ahn K.-S. Zuonin B inhibits lipopolysaccharide-induced inflammation via downregulation of the ERK1/2 and JNK pathways in RAW264.7 macrophages. Evidence-Based Complementary and Alternative Medicine. 2012;2012:8. doi: 10.1155/2012/728196.728196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Han M. S., Jung D. Y., Morel C., et al. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science. 2013;339(6116):218–222. doi: 10.1126/science.1227568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pal M., Wunderlich C. M., Spohn G., Brönneke H. S., Schmidt-Supprian M., Wunderlich F. T. Alteration of JNK-1 signaling in skeletal muscle fails to affect glucose homeostasis and obesity-associated insulin resistance in mice. PLoS ONE. 2013;8(1) doi: 10.1371/journal.pone.0054247.e54247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakatani Y., Kaneto H., Kawamori D., et al. Modulation of the JNK pathway in liver affects insulin resistance status. The Journal of Biological Chemistry. 2004;279(44):45803–45809. doi: 10.1074/jbc.m406963200. [DOI] [PubMed] [Google Scholar]
- 67.Lanuza-Masdeu J., Isabel Arévalo M., Vila C., Barberà A., Gomis R., Caelles C. In vivo jnk activation in pancreatic β-cells leads to glucose intolerance caused by insulin resistance in pancreas. Diabetes. 2013;62(7):2308–2317. doi: 10.2337/db12-1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu X., Mi Y., Yang H., Hu A., Zhang Q., Shang C. The activation of HMGB1 as a progression factor on inflammation response in normal human bronchial epithelial cells through RAGE/JNK/NF-κB pathway. Molecular and Cellular Biochemistry. 2013;380(1-2):249–257. doi: 10.1007/s11010-013-1680-0. [DOI] [PubMed] [Google Scholar]
- 69.Hsu W.-H., Lee B.-H., Liao T.-H., Hsu Y.-W., Pan T.-M. Monascus-fermented metabolite monascin suppresses inflammation via PPAR-γ regulation and JNK inactivation in THP-1 monocytes. Food and Chemical Toxicology. 2012;50(5):1178–1186. doi: 10.1016/j.fct.2012.02.029. [DOI] [PubMed] [Google Scholar]
- 70.Jin S.-M., Kim K. S., Lee S.-Y., et al. The sequential combination of a JNK inhibitor and simvastatin protects porcine islets from peritransplant apoptosis and inflammation. Cell Transplantation. 2011;20(7):1139–1151. doi: 10.3727/096368910X550170. [DOI] [PubMed] [Google Scholar]
- 71.Sabio G., Cavanagh-Kyros J., Ko H. J., et al. Prevention of steatosis by hepatic JNK1. Cell Metabolism. 2009;10(6):491–498. doi: 10.1016/j.cmet.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Santos F. R., Diamond-Stanic M. K., Prasannarong M., Henriksen E. J. Contribution of the serine kinase c-Jun N-terminal kinase (JNK) to oxidant-induced insulin resistance in isolated rat skeletal muscle. Archives of Physiology and Biochemistry. 2012;118(5):231–236. doi: 10.3109/13813455.2012.713366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miao H., Ou J., Ma Y., et al. Macrophage CGI-58 deficiency activates ros-inflammasome pathway to promote insulin resistance in mice. Cell Reports. 2014;7(1):223–235. doi: 10.1016/j.celrep.2014.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vandanmagsar B., Youm Y. H., Ravussin A., et al. The NALP3/NLRP3 inflammasome instigates obesity-induced autoinflammation and insulin resistance. Nature Medicine. 2011;17(2):179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stienstra R., Joosten L. A. B., Koenen T., et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metabolism. 2010;12(6):593–605. doi: 10.1016/j.cmet.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stienstra R., Van Diepen J. A., Tack C. J., et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(37):15324–15329. doi: 10.1073/pnas.1100255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fujisaka S., Usui I., Bukhari A., et al. Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes. 2009;58(11):2574–2582. doi: 10.2337/db08-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lumeng C. N., Bodzin J. L., Saltiel A. R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. The Journal of Clinical Investigation. 2007;117(1):175–184. doi: 10.1172/jci29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spruijt-Metz D., Adar Emken B., Spruijt M. R., et al. CRP is related to higher leptin levels in minority peripubertal females regardless of adiposity levels. Obesity. 2012;20(3):512–516. doi: 10.1038/oby.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hanyu O., Yoshida J., Abe E., et al. High-sensitivity CRP reflects insulin resistance in smokers. Journal of Atherosclerosis and Thrombosis. 2009;16(5):560–567. doi: 10.5551/jat.869. [DOI] [PubMed] [Google Scholar]
- 81.Alemzadeh R., Kichler J. Gender differences in the association of insulin resistance and high-sensitivity c-reactive protein in obese adolescents. Journal of Diabetes & Metabolic Disorders. 2014;13, article 35:9. doi: 10.1186/2251-6581-13-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brunner E. J., Kivimäki M., Witte D. R., et al. Inflammation, insulin resistance, and diabetes—mendelian randomization using CRP haplotypes points upstream. PLoS Medicine. 2008;5(8):1278–1286. doi: 10.1371/journal.pmed.0050155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tanti J.-F., Ceppo F., Jager J., Berthou F. Implication of inflammatory signaling pathways in obesity-induced insulin resistance. Frontiers in Endocrinology. 2013;3, article 181 doi: 10.3389/fendo.2012.00181.Article 181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xu D., Yin C., Wang S., Xiao Y. JAK-STAT in lipid metabolism of adipocytes. JAK-STAT. 2014;2(4) doi: 10.4161/jkst.27203.e27203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Harrison D. A. The JAK/STAT pathway. Cold Spring Harbor Perspectives in Biology. 2012;4(3) doi: 10.1101/cshperspect.a011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Richard A. J., Stephens J. M. Emerging roles of JAK-STAT signaling pathways in adipocytes. Trends in Endocrinology & Metabolism. 2011;22(8):325–332. doi: 10.1016/j.tem.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Croker B. A., Kiu H., Nicholson S. E. SOCS regulation of the JAK/STAT signalling pathway. Seminars in Cell and Developmental Biology. 2008;19(4):414–422. doi: 10.1016/j.semcdb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kiu H., Nicholson S. E. Biology and significance of the JAK/STAT signalling pathways. Growth Factors. 2012;30(2):88–106. doi: 10.3109/08977194.2012.660936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li W. X. Canonical and non-canonical JAK-STAT signaling. Trends in Cell Biology. 2008;18(11):545–551. doi: 10.1016/j.tcb.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]