

Abstract
Targeting the tumor microenvironment is critical toward improving the effectiveness of cancer therapeutics. Cancer-associated fibroblasts (CAFs) are one of the most abundant cell types of the tumor microenvironment, playing an important role in tumor progression. Multiple origins for CAFs have been proposed including resident fibroblasts, adipocytes, and bone marrow. Our laboratory previously identified a novel hematopoietic stem cell (HSC) origin for CAFs; however, the functional roles of HSC-derived CAFs (HSC-CAFs) in tumor progression have not yet been examined. To test the hypothesis that HSC-CAFs promote tumor progression through contribution to extracellular matrix (ECM) and paracrine production of pro-angiogenic factors, we developed a method to isolate HSC-CAFs. HSC-CAFs were profiled on the basis of their expression of hematopoietic and fibroblastic markers in two murine tumor models. Profiling revealed production of factors associated with ECM deposition and remodeling. Functional in vivo studies showed that co-injection of HSC-CAFs with tumor cells resulted in increased tumor growth rate and significantly larger tumors than tumor cells alone. Immunohistochemical studies revealed increased blood vessel density with co-injection, demonstrating a role for HSC-CAFs in tumor vascularization. Mechanistic in vitro studies indicated that HSC-CAFs play a role in producing vascular endothelial growth factor A and transforming growth factor–β1 in endothelial tube formation and patterning. In vitro and in vivo findings suggest that HSC-CAFs are a critical component of the tumor microenvironment and suggest that targeting the novel HSC-CAF may be a promising therapeutic strategy.
Abbreviations: HSC-CAFs, hematopoietic stem cell–derived cancer–associated fibroblasts; HSC-CAF-CM, hematopoietic stem cell–derived cancer–associated fibroblast conditioned medium
Introduction
Studies suggest that cancer-associated fibroblasts (CAFs) are associated with increased malignant potential in a variety of cancers. Women with denser breast tissue have an increased risk of developing breast cancer [1], and desmoplastic reaction was associated with poorer survival in a long-term breast cancer study [2]. The presence of a fibrous stroma was also associated with poor prognosis in lung squamous cell carcinoma [3], while stromal α-smooth muscle actin (α-SMA) expression was correlated with invasive squamous cell carcinoma of the cervix [4]. CAFs are significant contributors to desmoplasia and the composition of the tumor stroma, producing scaffolding for tumor growth through deposition of extracellular matrix (ECM) and supplying the microenvironment with a dense reservoir of tumor-promoting factors [5–7]. Tumor cell–CAF crosstalk has been shown to have profound effects on tumor angiogenesis [8], a critical step in tumor progression [9], both directly, through production of cytokines and chemokines, and indirectly, through production of matrix degrading molecules that act to remodel the ECM and release stored growth factors. Further, studies have demonstrated that this crosstalk is necessary for CAF phenotype and function [10,11]. Clinically, increased solid tumor vascular density has been associated with increased tumor grade [12], poor prognosis [13], increased metastatic potential [14,15], and shorter patient survival [16].
CAFs are heterogeneous [17–19] with respect to their phenotype, genetic diversity [18,20], and complex secretory profiles [21–23]. This has been demonstrated by altered gene expression profiles of CAFs in colon cancer [24], oral squamous cell carcinoma [25], non–small cell lung carcinoma [26], and breast cancer [27–29]. In prostate cancer models, heterogeneity in transforming growth factor–β (TGF-β) receptor II expression in CAFs was associated with increased tumorigenicity [30]. Because of the heterogeneity of the CAF population, CAFs cannot be defined by a single marker but are generally defined as a cell that expresses ECM molecules, matrix metalloproteinases (MMPs), and one or more markers such as vimentin, α-SMA, fibroblast activation protein (FAP), and/or fibroblast specific protein [31]. CAF heterogeneity is believed to be due, in part, to their proposed multiple origins (reviewed in [5,32–34]). Traditionally, it was thought that CAFs arose from resident fibroblasts [35,36]; however, additional sources have recently been suggested including cancer-associated adipocytes [37,38], endothelial cells [39,40], epithelial-to-mesenchymal transition (EMT) [41], and bone marrow cells [42]. Given that the CAF population is a significant driver of the pro-tumorigenic microenvironment in many solid tumors, therapeutic targeting of the CAF population may lead to better control of tumor progression. However, the multiple origins and thus heterogeneity of the CAF population suggest the challenge of identifying an appropriate anti-CAF therapeutic target and highlights the importance of understanding the biological contributions of CAFs from each origin.
Using a unique clonal cell transplantation method, our laboratory has previously identified a novel hematopoietic stem cell (HSC) origin for CAFs in multiple murine tumor models [43]. In this transplantation method, a clonal population from a single sorted enhanced green fluorescent protein (EGFP+) Lin−Sca-1+c-kithiCD34− HSC was transplanted into lethally irradiated mice. In these studies [43], clonally engrafted mice were injected subcutaneously with syngeneic Lewis lung carcinoma (LLC) or melanoma cells. Tumors extracted from these mice contained EGFP+ cells with a fibroblastic morphology and heterogeneous expression of collagen IαI (Col IαI) and α-SMA [43], hallmarks of activated fibroblasts [6,7]. Findings from these studies show that approximately 8% of HSC-derived CAFs (HSC-CAFs) expressed procollagen IαI [43]. In addition, in clonal transplantation studies, the HSC-CAF precursor was identified as a circulating fibroblast precursor population that is present in the peripheral blood, is elevated with increased tumor burden, and matures into a CAF phenotype (i.e., collagen and α-SMA expression) with exposure to tumor conditioned media [44]. While these previous studies demonstrated the contribution of HSC-derived cells to the CAF population, their functional roles in tumor progression have not yet been determined. In the present study, we sought to isolate and profile HSC-CAFs and test the hypothesis that HSC-CAFs promote tumor progression through deposition of ECM, ECM remodeling, and paracrine production of pro-angiogenic factors. A method for isolating HSC-CAFs from two murine solid tumor models (LLC and E0771 breast cancer) was developed in clonally engrafted animals. Profiling studies demonstrate that fibroblastic cells isolated by this method are derived from the HSC and have a CAF phenotype based on molecular and immunohistochemical analyses. Functionally, we have demonstrated that HSC-CAFs contribute to ECM and ECM remodeling. Co-injection studies show that HSC-CAFs promote tumor growth and vascularization in vivo. In vitro endothelial tube formation assays reveal production of vascular endothelial growth factor A (VEGF-A) and TGF-β1 as a mechanism by which HSC-CAFs promote vascularization and regulate vascular patterning. The studies herein represent, to our knowledge, the first isolation and profiling of CAFs of a specific HSC origin and reveal that HSC-CAFs promote tumor progression by contributing to ECM deposition, ECM remodeling, and tumor vascularization. These studies are essential toward understanding the functional contributions of CAFs from one source and may provide important insight into the therapeutic targeting of fibroblasts in the tumor microenvironment.
Materials and Methods
Ethics Statement
Research was conducted in strict accordance with guidelines set by the US Public Health Service Policy on Humane Care and Use of Laboratory Animals and the Veterans Affairs Medical Center (VAMC) Institutional Animal Care and Use Committee (IACUC), approved by the Ralph H. Johnson VAMC IACUC (Charleston, SC) under Protocol No. 541, VA AWA-A3137-01 (expiration 31 December 2017). All efforts were made to minimize suffering in animal studies. Human umbilical vein endothelial cells (HUVECs) were purchased from a commercially available source (Life Technologies, Carlsbad, CA).
Mice
C57Bl/6/CD45.1 breeders were from Jackson Laboratories, (Bar Harbor, Maine). EGFP breeders (C57Bl/6/CD45.2 background) were provided by Dr M. Okabe (Osaka University, Osaka, Japan) [45]. Mice were bred and maintained in the Animal Research Facility, VAMC. Research was conducted in accordance with guidelines set by the US Public Health Service Policy on Humane Care and Use of Laboratory Animals and the VAMC IACUC.
Antibodies
Fluorochrome-conjugated, biotinylated or purified versions of the following antibodies were used: anti–Sca-1 (anti–Ly-6A/E[D7]), anti–c-kit (anti-CD117[2B8]), anti–Gr-1 (anti–Ly-6G[RB6-8C5]), anti-CD45R/B220 (RA3-6B2), anti–Thy-1.2 (30-H12), anti–TER-119 (TER-119), anti-CD3e (145-2C11), anti-CD45 (leukocyte common antigen, Ly-5;30-F11), anti-CD8a (53-6.7), anti-CD4 (GK1.5), and anti-CD45.1 (A20) from BD Biosciences (San Jose, CA); anti-F4/80 (BM8) and anti-CD34 (RAM34) from eBioscience (San Diego, CA); anti–β-actin–HRP (5125 s) from Cell Signaling Technology (Danvers, MA); anti–Col I from Rockland (Limerick, PA); anti–α-SMA (ab5694), anti-vimentin, anti–wide spectrum cytokeratin (WS CyK), anti–Col I (ab21286), anti-CD45 (ab10558), anti-CD31 (ab13970), and anti-GFP (anti-green fluorescent protein, ab28364) from Abcam (Cambridge, MA); VEGF-A, TGF-β1 neutralizing antibodies from R&D Systems (Minneapolis, MN); isotype control antibodies from BD Biosciences; secondary antibodies from Jackson ImmunoResearch (West Grove, PA) or BD Pharmingen (San Diego, CA).
Clonal Cell Transplantation
Clonal cell transplantation was performed as previously described [43,44,46,47]. Briefly, lineage negative (Lin−) cells were isolated from bone marrow of C57Bl/6-EGFP/CD45.2 mice by negative selection following staining and DynaBead removal of B220, Gr-1, CD4, CD8a, and TER-119 positive cells. Lin− cells were stained with antibodies to Sca-1, c-kit, and CD34 and then incubated with Hoechst 33342 (Sigma, St. Louis, MO; 5 mg/ml). Single Lin−Sca-1+c-kithiCD34− side population cells were deposited into individual wells of 96-well culture plates (MoFlo CyClone System, Beckman Coulter, Inc., Indianapolis, IN). Eighteen hours post-deposition, wells containing single cells were identified and cultured for 7 days in α-modification of Eagle’s medium (αMEM; Life Technologies), 20% FBS (Atlanta Biologicals, Norcross, GA), 10% BSA (Life Technologies), 1 × 10− 2 M 2-mercaptoethanol (Sigma), 10 μg/ml stem cell factor, and 10 μg/ml interleukin-11 (R&D Systems). Wells containing ≤ 20 clonal cells were selected for transplantation. Recipient C57Bl/6/CD45.1 mice were lethally irradiated (total body irradiation, 950 cGy). Clonal cells were injected intravenously along with 500 CD45.1/EGFP−/Lin−c-kit+Sca-1+CD34+ radioprotective cells. These short-term repopulating cells allow the mouse to survive the post-irradiation pancytopenia period [43,44,46–50]. The two clonally engrafted mice herein showed total hematopoietic engraftment of 57.6% and 83.4% EGFP+ cells from a single sorted HSC, and multilineage engraftment of EGFP+ cells in the B cell, T cell, and granulocyte/macrophage lineages was confirmed.
Tumor Models
Murine LLC-1 (American Type Culture Collection, Manassas, VA) cells were propagated using standard culture methods. LLC cells were grown to 95% confluency in RPMI 1640 containing l-glutamine (Life Technologies) with 10% FBS and penicillin/streptomycin (5% CO2, 37°C). LLC cells were resuspended [5 × 105 cells per 100 μl of phosphate-buffered saline (PBS)] and injected subcutaneously into anesthetized 14- to 16-week-old mice. Murine breast cancer E0771 cells were a kind gift to Dr Dennis Watson from Dr Ray B. Ratna (St Louis University, St Louis, MO). Cells were grown to 95% confluency in Dulbecco's modified Eagle's medium (Life Technologies) containing 20% FBS (5% CO2, 37°C). E0771 cells were resuspended (5 × 105 cells per 50 μl of PBS) and injected orthotopically into the fourth mammary fat pad of anesthetized 10- to 12-week-old female mice. LLC and E0771 tumors were harvested at maximal size allowed by local IACUC.
CAF Isolation
LLC or E0771 tumors were extracted, cleared of surrounding tissue, minced, and placed in collagenase solution [0.5 mg/ml collagenase (Sigma) in Dulbecco's modified Eagle's medium (Life Technologies)] for 35 minutes at 37°C. The suspension was vortexed, filtered through a 70 μM cell strainer, and centrifuged. The pellet was washed in 0.1% BSA/PBS and resuspended in 20% FBS/αMEM supplemented with penicillin/streptomycin and cultured (5% CO2, 37°C). After 3 to 5 days, cells were trypsinized with 0.25% trypsin-EDTA until tumor cells were released from the plate as visually assessed by microscopy, whereas stromal cells were retained. Plates were washed three times, and fresh culture medium was added. Cells were serially trypsinized to continually reduce tumor cell population before use for further studies. Primary CAFs were used below passage three.
Skin Fibroblast Isolation
Dorsal skin was harvested from euthanized EGFP+ mice, depilated, minced, and digested in 0.25% trypsin/EDTA for 4 hours at 37°C. Trypsin was inactivated, and cells were collected by centrifugation. The pellet was washed, triturated, resuspended in 20% FBS/αMEM containing penicillin/streptomycin, and cultured (5% CO2, 37°C). Primary skin fibroblasts were used below passage three.
Quantification of HSC-CAFs
Fluorescent and differential interference contrast (DIC) images of non-passaged isolated cells were taken at × 100 magnification (10 per sample). Numbers of EGFP+ and EGFP− cells, expressed as percent ± SD, were quantified by two independent observers from two animals.
Immunofluorescence
Cultured cells were fixed in 4% paraformaldehyde, permeabilized in 0.02% Triton X-100/PBS, serum blocked, and incubated with primary antibodies. Samples were washed in PBS and serum blocked before incubation with fluorochrome-conjugated secondary antibodies. Hoechst 33342 was used as a nuclear marker. For paraffin-embedded tumors, sections were deparaffinized in histoclear and dehydrated in alcohols, and heat-mediated antigen retrieval was performed in citrate buffer (Vector Laboratories, Burlingame, CA). Sections were then permeabilized in 0.02% Triton X-100/PBS, serum blocked, and incubated with anti-GFP antibodies followed by fluorochrome-conjugated secondary antibodies. Hoechst 33342 was used as a nuclear marker.
Quantitative Reverse Transcription–Polymerase Chain Reaction
Total RNA was extracted using the QIAshredder and RNeasyPlus Mini Kit (Qiagen). Total RNA concentration was measured using the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies). RNA was reverse transcribed in a 20-μl reaction using iScript (Bio-Rad). Real-time polymerase chain reaction (PCR) was performed in a 10-μl reaction with 2.5 μl of reverse transcribed cDNA (2.5 ng/μl), 5 μl of Bio-Rad SsoFast Probes Supermix, 0.1 μl of both forward and reverse primers (200 nM; Supplementary Table S1), and 0.1 μl of probe from the Universal Probe Library System (Roche) in a Roche LightCycler 480. Cycling conditions were given as follows: preincubation, 95°C (10 seconds); 50 cycles of denaturation at 95°C (5 seconds); annealing/extension at 60°C (30 seconds) with a single data acquisition taken at the end of each extension; and cooling at 40°C (30 seconds). Triplicate reactions were run for each sample. Relative gene expression was quantified on the basis of Ct value measured against an internal standard curve for each specific primer set using LightCycler 480–supplied software. Sample concentration was normalized to ribosomal protein encoding gene RPL13A. Isolated skin fibroblasts were used as a positive control for quantitative reverse transcription (qRT)–PCR to verify RT and PCR conditions given that these cells have an activated phenotype in vivo during wound healing and are activated by isolation and adherence in vitro (Supplementary Figure S1). In all experiments, no RT served as a negative control for DNA contamination and H2O served as an internal control for contamination.
Supplementary Figure S1.
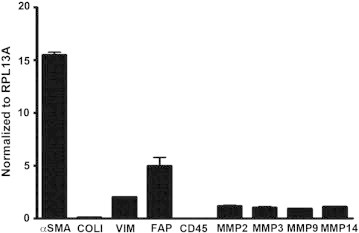
qRT-PCR: RT and PCR step controls. mRNA expression of α-SMA, Col I, vimentin, FAP, CD45, MMP-2, MMP-3, MMP-9, and MMP-14 in primary skin fibroblasts is shown. Relative expression is normalized to RPL13A as described in the Materials and methods section.
Total RNA was extracted using the QIAshredder and RNeasyPlus Mini Kit (Qiagen, Boston, MA). Total RNA concentration was measured using the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). RNA was reverse transcribed in a 20-μl reaction using iScript (Bio-Rad, Hercules, CA). Real-time polymerase chain reaction (PCR) was performed in a 10-μl reaction with 2.5 μl of reverse transcribed cDNA (2.5 ng/μl), 5 μl of Bio-Rad SsoFast Probes Supermix, 0.1 μl of both forward and reverse primers (200 nM; Supplementary Table S1), and 0.1 μl of probe from the Universal Probe Library System (Roche, Florence, SC) in a Roche LightCycler 480. Cycling conditions were given as follows: preincubation, 95°C (10 seconds); 50 cycles of denaturation at 95°C (5 seconds); annealing/extension at 60°C (30 seconds) with a single data acquisition taken at the end of each extension; and cooling at 40°C (30 seconds). Triplicate reactions were run for each sample. Relative gene expression was quantified on the basis of Ct value measured against an internal standard curve for each specific primer set using LightCycler 480–supplied software. Sample concentration was normalized to ribosomal protein encoding gene RPL13A. Isolated skin fibroblasts were used as a positive control for quantitative reverse transcription (qRT)–PCR to verify RT and PCR conditions given that these cells have an activated phenotype in vivo during wound healing and are activated by isolation and adherence in vitro (Supplementary Figure S1). In all experiments, no RT served as a negative control for DNA contamination and water served as an internal control for contamination.
Microscopy
Imaging was performed using a Nikon Eclipse Ti-U, 90i, or A1R/confocal microscope equipped with DIC optics and long band-pass GFP excitation cubes. Images were processed using NIS Elements (Nikon, Tokyo, Japan), ImageJ (NIH, Bethesda, MD), and Adobe Photoshop CS5 (Adobe Systems, San Jose, CA).
Flow Cytometry
Cells were trypsinized and recovered in culture media for a minimum of 30 minutes at 37°C in 5% CO2. Cells were resuspended in PBS and were blocked in 4% donkey serum (Jackson ImmunoResearch) and mouse Fc receptor block (Miltenyi Biotec, San Diego, CA) for 20 minutes. Antibodies to cell surface proteins were added for 15 minutes at 4°C in the dark. Cells were washed in PBS and fixed using fixation and permeabilization buffer according to the manufacturer’s instructions (BD Biosciences). Cells were stained in Perm/Wash buffer (BD Biosciences) with antibodies to intracellular proteins for 15 minutes at 4°C in the dark. Cells were washed in Perm/Wash buffer, and secondary antibody was added for 15 minutes at 4°C in the dark. Cells were washed in Perm/Wash buffer and resuspended in PBS and analyzed using an LSR Fortessa Cytometer (BD Biosciences). Fluorescence minus one controls were used to set gates. Data were analyzed using FlowJo v7.6.5 and v10 (TreeStar Inc, Ashland, OR).
MMP Activity Assay
MMP activity in HSC-CAF conditioned medium (HSC-CAF-CM/CAF-CM) or αMEM was determined using a pan-specific MMP quenched fluorogenic peptide hydrolysis assay (OmniMMP; Enzo Laboratories, Farmingdale, NY). Briefly, equal amounts (volume loaded) of CAF-CM or αMEM control medium were incubated for 8 hours at 37°C with a quenched fluorogenic peptide that contains a pan-specific MMP hydrolysis site (OmniMMP; Enzo Laboratories, Farmingdale, NY). Upon cleavage of the peptide by active MMPs (MMP-1, -2, -3, -7, -8, -9, -10, -12, -13, -14, -19, and -20), the quenching group is released and, once sufficiently separated from the fluorescent group, allows a fluorescent signal to be detected. Fluorescence was measured and recorded on a fluorescent microplate reader (SpectraMax-M3; Molecular Devices, Sunnyvale, CA) and compared to a standard curve.
Western Blot
Isolated HSC-CAFs were washed twice with ice-cold Hank’s buffered saline solution (HBSS, Life Technologies). Then, cells were lysed with RIPA with 1 mM PMSF (phenylmethylsulfonyl fluoride protease inhibitor) on ice for 15 minutes. The lysate was transferred into a prechilled tube and centrifuged at 12,000 rpm for 15 minutes at 4°C, and the supernatant was used for assay. Protein was estimated using bicinchoninic acid method according to the manufacturer’s protocol (Thermo Fisher Pierce BCA kit, Thermofisher, Waltham, MA). The lysate was then heated to 95°C for 5 minutes with loading buffer (Bio-Rad 161-0737) according to the manufacturer’s protocol. Equal amounts of the samples were loaded into each well, and electrophoresis was performed and transferred to a nitrocellulose membrane. The membranes were blocked with 5% milk for 30 minutes and incubated overnight in the primary antibodies. Anti–Col I antibody (1/1000 dilution), anti-SMA (1/1000 dilution), and anti–β-actin–HRP-conjugated antibody (1:3000) were used. This was followed by the appropriate secondary antibody for 1 hour at room temperature with rocking in milk. The blots were visualized using Bio-Rad Clarity Western ECL (170-5060).
Co-Injection Study
EGFP+ CAFs or EGFP− LLC cells were resuspended in sterile PBS and subcutaneously injected into 14- to 16-week-old C57Bl/6/CD45.1 mice as follows: 300,000 HSC-CAFs/0.1 ml, 100,000 LLCs with 300,000 HSC-CAFs/0.1 ml, or 100,000 LLCs/0.1 ml. Tumor length and width were measured with calipers (length × width). Following euthanasia, the tumor was excised, measured (length × width × height), and weighed.
Immunohistochemistry and Vessel Quantification
Paraffin sections (six per tumor) from LLC (n = 3) and LLC + CAF (n = 4) cohorts were deparaffinized in xylenes, dehydrated in alcohols, and stained with antibodies to CD31 using a rabbit IgG kit (Vector Laboratories) and NovaRed substrate (Vector Laboratories). Sections were taken at least four paraffin sections apart (~ 20 μm) to ensure the same vessels were not counted twice. Average number of blood vessels per field was quantified in three high power fields (200 ×) per section.
Tube Formation Assay
HUVECs (purchased from Life Technologies) were cultured using standard culture methods in HUVEC medium [medium 200 supplemented with low serum growth supplement (Life Technologies)] and used below passage five. HUVECs were resuspended (18.75 × 103 cells per 200 μl) in HUVEC medium, αMEM (Life Technologies), or HSC-CAF-CM. Neutralizing antibodies to VEGF-A (0.06 μg/ml) or TGF-β (10 μg/ml) were added to HSC-CAF-CM for inhibition studies. Cells were plated onto 48-well tissue culture plates coated with 50 μl of Geltrex matrix (Life Technologies) and incubated for 16 to 18 hours (5% CO2, 37°C). Cells were stained [Calcein AM, Dead Red dye (LIVE/DEAD Cell Imaging Kit, Life Technologies)], and individual wells were imaged (40 ×). Nodes were defined as a branching point with two or more neighboring pixels and segments as a length adjoining two nodes. Vascular pattern was analyzed on the basis of methods adapted from [51]. The mean length of a bisecting line through an avascular space (Ma) and the mean diameter of the adjacent vascular area (Mv) of complete networks were calculated (μm). A minimum of three independent samples per group and two images per sample were quantified.
Collection of CM and ELISA
HSC-CAFs were starved in serum-free αMEM overnight (16-18 hours), and HSC-CAF-CM was centrifuged and used for ELISA and tube formation assays. TGF-β1 and VEGF-A ELISA (R&D Systems) results were normalized to protein levels using a BCA protein quantification kit (Thermo Fisher Pierce BCA kit). Data presented are pg/ml per 1000 μg of protein. Samples were analyzed with a SpectraMax-M2 microplate reader (Molecular Devices).
Statistics
Analysis was conducted using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA). Data were presented as the mean ± standard error of the mean. Student’s t test was used to compare groups. P ≤ .05 was regarded as statistically significant (indicated by * or ψ in figures).
Results
Isolation of HSC-CAFs
Our previous in vitro and in vivo studies identified the HSC as a novel source of CAFs and their circulating fibroblast precursors (CFPs) in solid tumor [43,44]. However, the functional roles of HSC-CAFs have not yet been investigated. To allow for lineage tracing of cells originating from a single HSC based on their EGFP expression, we first reconstituted the bone marrow of lethally irradiated mice with a clonal population of cells derived from a single EGFP+ HSC as described previously [43,44,46,47]. Total hematopoietic engraftment and multilineage hematopoietic engraftment in B cell, T cell, and granulocytes/macrophages were confirmed. Next, to generate HSC-CAFs, syngeneic EGFP− LLC cells were subcutaneously injected into these clonally engrafted mice, and tumors were allowed to progress to allow for in vivo recruitment and differentiation of mature HSC-CAFs. To isolate HSC-CAFs, resulting tumors were digested in collagenase and plated as a mixed population of EGFP+ and EGFP− cells, wherein EGFP expression indicates cells arising from the clonal EGFP population transplanted from a single HSC. EGFP− cells in culture are either tumor cells or fibroblasts from alternative sources, e.g., resident fibroblasts before transplantation. Cells underwent serial trypsinization to remove less adherent tumor cells, resulting in retention of a population of cells highly enriched for EGFP+ HSC-derived cells with a fibroblastic morphology. DIC images of cells were overlaid with fluorescent images to quantify EGFP+ (HSC-derived) and EGFP− cells (Figure 1A). Quantitative analysis as described in the Materials and Methods section revealed that EGFP+ HSC-derived fibroblast-like cells represented 79.41 ± 5.76% of the population isolated by this method.
Figure 1.
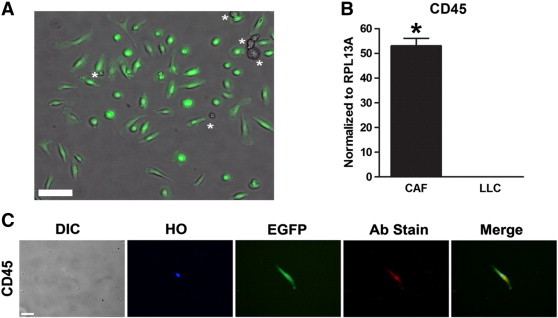
Isolation of HSC-CAFs from clonally engrafted mice. (A) Representative image of EGFP+ (HSC-derived) and EGFP− cells (asterisks) isolated from an LLC tumor grown in a clonally engrafted mouse. (B) qRT-PCR analysis of CD45 expression in HSC-CAFs (black bar) and LLC tumor cells (hatched bar; *P ≤ .0001). (C) DIC and immunofluorescence staining of representative isolated cells show morphology (DIC), nuclei (Hoechst dye, HO), expression of EGFP, and CD45 (Ab stain). Merged images (right panels) represent EGFP, HO, and antibody stain (Ab stain). Secondary only (no primary antibody) controls for immunofluorescence images are depicted in Supplementary Figure S2. Bars, 100 μm (B) and 25 μm (D).
Isolation of HSC-CAFs from clonally engrafted mice. (A) Representative image of EGFP+ (HSC-derived) and EGFP− cells (asterisks) isolated from an LLC tumor grown in a clonally engrafted mouse. (B) qRT-PCR analysis of CD45 expression in HSC-CAFs (black bar) and LLC tumor cells (hatched bar; *P ≤ .0001). (C) DIC and immunofluorescence staining of representative isolated cells show morphology (DIC), nuclei (Hoechst dye, HO), expression of EGFP, and CD45 (Ab stain). Merged images (right panels) represent EGFP, HO, and antibody stain (Ab stain). Secondary only (no primary antibody) controls for immunofluorescence images are depicted in Supplementary Figure S2. Bars, 100 μm (B) and 25 μm (D).
Molecular and Immunohistochemical Profiling of HSC-CAFs
Given that CAFs are a heterogeneous population and that there is no single marker for CAFs, a combination of several markers is used to identify CAFs [18,52]. Therefore, HSC-derived EGFP+ fibroblast-like cells from clonally engrafted mice were profiled by qRT-PCR and immunohistochemical analyses using a panel of standard fibroblast markers to confirm HSC origin and CAF identity. Molecular analysis by qRT-PCR of these cells showed mRNA expression of CD45, a pan-hematopoietic marker, indicating HSC origin (Figure 1B). LLC cells were negative for CD45 mRNA expression. Immunostaining confirmed that EGFP+ cells also expressed CD45 [53] (Figure 1C). Classic fibroblast/activated fibroblast markers including Col I, α-SMA, FAP, and vimentin were then profiled by mRNA analysis (Figure 2A). LLC cells did not express Col I or α-SMA, as assessed by mRNA analysis. LLC cells did express vimentin, a marker associated with an epithelial cell that has undergone EMT [54], and FAP but at significantly lower levels than that observed in the CAFs (P ≤ .001 and P = .0121, respectively). Production of MMPs has been associated with both the CAF phenotype and their function in critical components of tumor progression, including matrix remodeling and release of stored growth factors and vascularization [55–57]. Therefore, as an additional analysis of the activated fibroblastic nature of this population, mRNA expression of MMP-2, -3, -9, and -14 was demonstrated by qRT-PCR (Figure 2B). As further confirmation of the fibroblastic nature of the isolated HSC-derived population, immunostaining revealed expression of Col I, α-SMA, and vimentin protein (Figure 2C). Negative immunostaining with antibodies to F4/80, a macrophage marker [58], and WS CyK, an epithelial marker, demonstrated that EFGP+/HSC-derived cells were not macrophages or epithelial cells (Figure 2C). No primary antibody controls are shown in Supplementary Figure S2. Together, the fibroblastic morphology and gene and protein expression of a panel of fibroblastic markers confirm the CAF identity of EGFP+/HSC-derived cells obtained through our isolation method.
Figure 2.
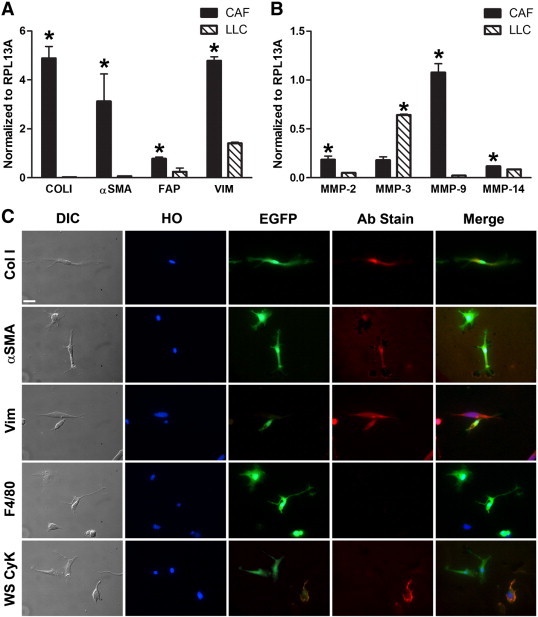
Characterization of HSC-CAFs from clonally engrafted mice. (A) qRT-PCR analysis of activated fibroblast markers Col I (*P ≤ .0001), α-SMA (*P = .0031), FAP (*P = .0121), and vimentin (*P ≤ .0001) in HSC-CAFs (black bars) and LLC tumor cells (hatched bars). (B) qRT-PCR analysis of MMP-2 (*P = .0035), MMP-3 (*P ≤ .0001), MMP-9 (*P ≤ .0001), and MMP-14 (*P = .0002) in HSC-CAFs (black bars) from clonally engrafted mice and LLC tumor cells (hatched bars). (C) DIC and immunofluorescence staining of representative isolated cells show morphology (DIC), nuclei (Hoechst dye, HO), and expression of EGFP, Col I, α-SMA, vimentin, F4/80, and WS CyK. Bars, 25 μm (C).
Supplementary Figure S2.
No primary antibody staining controls. Morphology (DIC), nuclei (HO), EGFP expression (green), and secondary only control stain for each antibody used. (A and B) Representative images of HSC-CAFs derived from LLC tumors stained with secondary antibodies without the addition of primary antibodies are presented for each antibody used in Figures 1, 2, and 3. (C and D) Depiction of staining controls for HSC-CAFs derived from E0771 tumors shown in Figure 8. Bars, 25 μm (A–D).
Given that CAFs are a heterogeneous population and that there is no single marker for CAFs, a combination of several markers is used to identify CAFs [18,52]. Therefore, HSC-derived EGFP+ fibroblast-like cells from clonally engrafted mice were profiled by qRT-PCR and immunohistochemical analyses using a panel of standard fibroblast markers to confirm HSC origin and CAF identity. Molecular analysis by qRT-PCR of these cells showed mRNA expression of CD45, a pan-hematopoietic marker, indicating HSC origin (Figure 1B). LLC cells were negative for CD45 mRNA expression. Immunostaining confirmed that EGFP+ cells also expressed CD45 [53] (Figure 1C). Classic fibroblast/activated fibroblast markers including Col I, α-SMA, FAP, and vimentin were then profiled by mRNA analysis (Figure 2A). LLC cells did not express Col I or α-SMA, as assessed by mRNA analysis. LLC cells did express vimentin, a marker associated with an epithelial cell that has undergone EMT [54], and FAP but at significantly lower levels than that observed in the CAFs (P ≤ .001 and P = .0121, respectively). Production of MMPs has been associated with both the CAF phenotype and their function in critical components of tumor progression, including matrix remodeling and release of stored growth factors and vascularization [55–57]. Therefore, as an additional analysis of the activated fibroblastic nature of this population, mRNA expression of MMP-2, -3, -9, and -14 was demonstrated by qRT-PCR (Figure 2B). As further confirmation of the fibroblastic nature of the isolated HSC-derived population, immunostaining revealed expression of Col I, α-SMA, and vimentin protein (Figure 2C). Negative immunostaining with antibodies to F4/80, a macrophage marker [58], and WS CyK, an epithelial marker, demonstrated that EGFP+/HSC-derived cells were not macrophages or epithelial cells (Figure 2C). No primary antibody controls are shown in Supplementary Figure S2. Together, the fibroblastic morphology and gene and protein expression of a panel of fibroblastic markers confirm the CAF identity of EGFP+/HSC-derived cells obtained through our isolation method.
The lethal irradiation necessary to condition recipient mice for bone marrow transplantation with a clonal population of cells derived from a single sorted HSC may affect the behavior and function of HSC-CAFs. Therefore, to avoid any effects of irradiation, we applied the same HSC-CAF isolation method to non–bone marrow transplanted mice and confirmed HSC origin by CD45 expression and CAF identity by fibroblast-associated marker expression. EGFP/CD45.2 mice were injected subcutaneously with EGFP− LLC cells, and tumors were processed and cells subjected to serial trypsinization as described above. EGFP/CD45.2 mice were used to allow for distinction between EGFP+ stromal cells from EGFP− tumor cells. Isolated EGFP+ cells had a fibroblastic morphology and expressed CD45 mRNA at similar levels to those seen in CAFs isolated from clonally engrafted mice, indicating their HSC origin (compare Figures 3A and 1B). Expression of CD45 protein was confirmed by immunofluorescence staining (Figure 3B). qRT-PCR analysis of the same isolated cells demonstrated expression of mRNA for Col I, α-SMA, FAP, vimentin, and MMPs (Figure 3, C and D). Together with CD45 expression, this confirms isolation of a population of cells highly enriched for HSC-CAFs. As shown in representative images, expression of Col I, α-SMA, and vimentin protein by EGFP+ HSC-CAFs was confirmed by immunofluorescence staining (Figure 3E). No primary antibody controls are shown in Supplementary Figure S2. Together, findings using both clonally engrafted mice and non–bone marrow transplanted mice demonstrate an HSC origin for CAFs, profile the HSC-CAF phenotype, and confirm we have established an effective method of cell isolation to study the functional contributions of HSC-CAFs to tumor progression.
Figure 3.
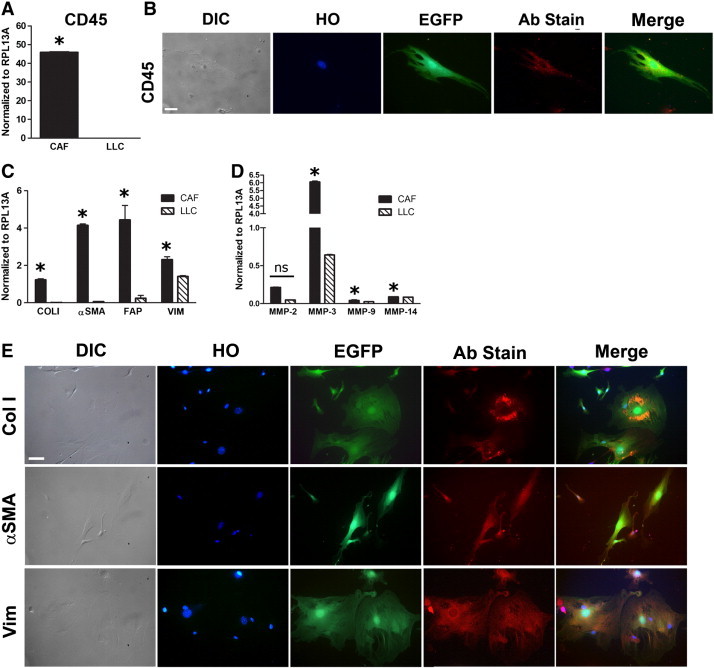
Expression profiles of HSC-CAFs. (A) qRT-PCR analysis of CD45 in HSC-CAFs isolated from non-transplanted mice injected with LLC tumor cells (black bar) and LLC tumor cells (hatched bar; *P ≤ .0001). (B) DIC and representative immunofluorescence staining of EGFP, nuclear (Hoescht dye, HO), and CD45 (Ab stain) expression in HSC-CAFs are depicted. (C) qRT-PCR analysis of activated fibroblast markers Col I (*P = .0001), α-SMA (*P ≤ .0001), FAP (*P = .0176), and vimentin (*P ≤ .0001) in HSC-CAFs (black bars) and LLC tumor cells (hatched bars). (D) qRT-PCR analysis of MMP-2 (*P = .5931), MMP-3 (*P ≤ .0001), MMP-9 (*P = .0012), and MMP-14 (*P = .0007) in HSC-CAFs (black bars) and LLC tumor cells (hatched bars). (E) DIC and representative immunofluorescence staining of nuclei (Hoescht dye, HO), EGFP, and Col I, α-SMA, and vimentin (Ab stain) expression in HSC-CAFs are depicted. Bars, 25 μm (B, E).
The lethal irradiation necessary to condition recipient mice for bone marrow transplantation with a clonal population of cells derived from a single sorted HSC may affect the behavior and function of HSC-CAFs. Therefore, to avoid any effects of irradiation, we applied the same HSC-CAF isolation method to non–bone marrow transplanted mice and confirmed HSC origin by CD45 expression and CAF identity by fibroblast-associated marker expression. EGFP/CD45.2 mice were injected subcutaneously with EGFP− LLC cells, and tumors were processed and cells subjected to serial trypsinization as described above. EGFP/CD45.2 mice were used to allow for distinction between EGFP+ stromal cells from EGFP− tumor cells. Isolated EGFP+ cells had a fibroblastic morphology and expressed CD45 mRNA at similar levels to those seen in CAFs isolated from clonally engrafted mice, indicating their HSC origin (compare Figures 3A and 1B). Expression of CD45 protein was confirmed by immunofluorescence staining (Figure 3B). qRT-PCR analysis of the same isolated cells demonstrated expression of mRNA for Col I, α-SMA, FAP, vimentin, and MMPs (Figure 3, C and D). Together with CD45 expression, this confirms isolation of a population of cells highly enriched for HSC-CAFs. As shown in representative images, expression of Col I, α-SMA, and vimentin protein by EGFP+ HSC-CAFs was confirmed by immunofluorescence staining (Figure 3E). No primary antibody controls are shown in Supplementary Figure S2. Together, findings using both clonally engrafted mice and non–bone marrow transplanted mice demonstrate an HSC origin for CAFs, profile the HSC-CAF phenotype, and confirm we have established an effective method of cell isolation to study the functional contributions of HSC-CAFs to tumor progression.
HSC-CAFs Contribute to a Pro-Tumorigenic Microenvironment
To quantitatively assess the contribution of HSC-derived cells to the α-SMA and Col I positive populations of isolated cells, flow cytometric analysis was used to determine the percentage of EGFP+ cells isolated from tumors of non–bone marrow transplanted mice that co-expressed CD45 with Col I or α-SMA (Figure 4A). Quantitative analysis from multiple mice demonstrated that 68% (± 0.5%) of EGFP+ cells co-expressed CD45 and Col I and 90% (± 0.1%) of EGFP+ cells co-expressed CD45 and α-SMA. ECM composition and stiffness are correlated with poor patient prognosis [1,59,60]. Two factors shown to play important roles in this are Col I ([61] and reviewed in [62]) and α-SMA [4,32]. To determine whether HSC-CAFs actively contribute to production of collagen and expression of α-SMA in the tumor microenvironment, Western blot analysis for Col I and α-SMA was also performed (Figure 4B). In addition to matrix deposition, CAFs have been shown to play a role in matrix remodeling through production of MMPs. Therefore, to determine whether CAFs originating from the HSC contribute to these processes, we examined HSC-CAFs for the production of active MMPs. CAF-CM was added to a fluorogenic OmniMMP substrate that emits fluorescence when cleaved by MMPs, and the resulting fluorescence is measured over time. CAF-CM resulted in increased fluorescence of the substrate over time, indicating that HSC-CAFs produce active MMPs (Figure 4C). HSC-CAF expression of active MMPs and production of Col I and α-SMA indicate that HSC-CAFs are important contributors to the pro-tumorigenic microenvironment.
Figure 4.
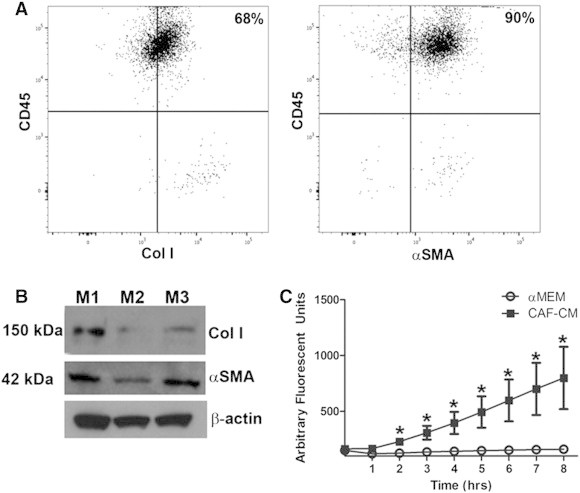
HSC-CAFs contribute to the pro-tumorigenic microenvironment. (A) Representative flow cytometric analysis shows percent of EGFP-expressing cells that co-express CD45 and Col I (68%, left panel) and those that co-express CD45 and α-SMA (90%, right panel). Gates were set on the basis of fluorescence minus one (FMO) controls. (B) Western blot analysis shows 150 and 42 kDa bands for Col I and α-SMA, respectively, from HSC-CAFs isolated from three biological replicates (M1, M2, and M3); β-actin loading control is shown below. (C) Functional MMP activity assay in HSC-CAF-CM (black squares) and αMEM (empty circles) shows that HSC-CAF-CM contains active MMPs (*P ≤ .05).
HSC-CAFs Enhance Tumor Growth In Vivo
To test the in vivo functional ability of HSC-CAFs to affect tumor progression, EGFP+ HSC-CAFs were isolated from LLC tumors from non-transplanted EGFP/CD45.2 mice. These EGFP+ HSC-CAFs were then co-injected with LLC cells (LLC + CAF) into C57Bl/6 mice in a ratio of 1:3 (LLCs:CAFs). Control cohorts of mice were injected with either LLC cells alone (LLC) or HSC-CAFs alone (CAF). The LLC cohort served as a control for tumor growth rate. Tumor size was measured in each cohort longitudinally (Figure 5A). Tumors were not observed in the CAF cohort, demonstrating that HSC-CAFs were not tumorigenic and confirming effective isolation of CAFs with minimal tumor cell contamination. Tumors from the LLC + CAF cohort grew faster than tumors in the LLC cohort (growth curve slope 0.19 ± 0.02 vs 0.07 ± 0.01, P ≤ .0001). In addition, tumors from co-injection cohort animals had a significantly larger size at each time point from day 14 to harvest (P = .0445, day 14; P = .009, day 16; P = .0046, day 18; P = .0075, day 20). At endpoint, tumors from the LLC + CAF cohort were about two-fold larger by weight than tumors in the LLC cohort (0.98 ± 0.33 g vs 0.45 ± 0.07 g, respectively, P = .0433; Figure 5, B and C). The pattern of EGFP+ cells in the LLC + CAF cohort tumors is shown by EGFP+ staining in tumor sections (Figure 5D), demonstrating that the co-injected HSC-CAF population persists as the tumors grow. The increased growth rate, size, and mass of tumors from co-injected mice indicate that HSC-CAFs play a significant role in promoting tumor growth in vivo.
Figure 5.
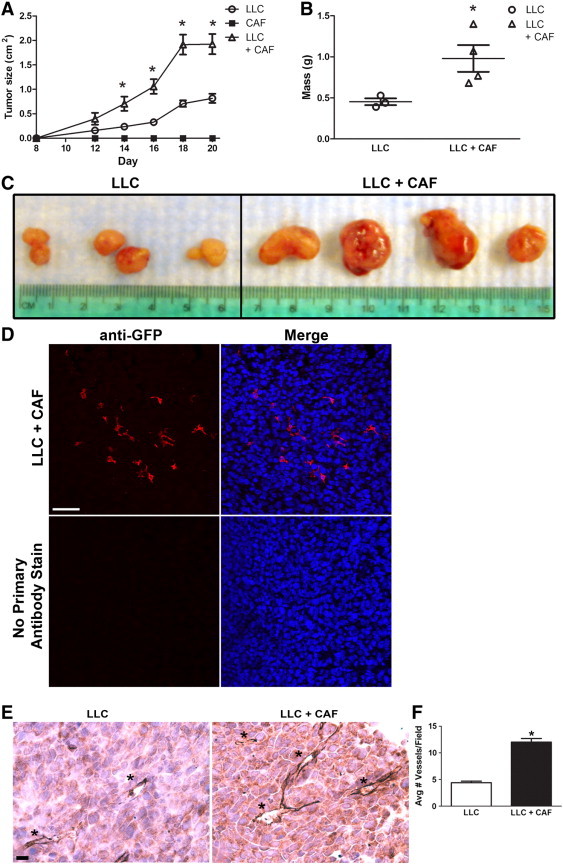
Effect of HSC-CAFs on tumor growth and vascularization. Measurement of size (A) and mass (B) of tumors showed that LLC + CAF co-injection (empty triangles) resulted in larger tumors versus LLC (empty circles) injected cohort (*P < .05). No tumor was observed in the CAF (black squares) cohort. (C) Extracted tumors from LLC and LLC + CAF cohorts; LLC + CAF cohort tumors show increased size and gross vascularity. (D) Immunofluorescence staining for anti-GFP (red) shows that the EGFP+ HSC-CAF population persists in the LLC + CAF cohort of tumors at endpoint (upper panels). No primary antibody staining is shown in the lower panels. Hoechst nuclear stain is represented in blue in merged images. (E) Immunohistochemical staining for CD31 with hematoxylin counterstain demonstrates increased vascularization (blood vessels indicated by asterisks) in the LLC + CAF cohort. (F) Quantification of blood vessels in LLC (white bar) and LLC + CAF (black bar) from immunohistochemical staining images (*P < .05). Bar, 25 μm (D).
HSC-CAFs Promote Tumor Vascularization In Vivo
Angiogenesis is critical toward tumor growth, and because the tumors from the co-injected mice (LLC + CAF cohort) appeared to be more vascularized on excision than those from LLC cohort mice (Figure 5C), we sought to determine whether the observed increased growth was because of the ability of HSC-CAFs to promote blood vessel formation in vivo. Paraffin sections of tumor from LLC + CAF and LLC cohorts harvested at endpoint were stained using antibodies to CD31, an endothelial cell marker. Quantitative analysis showed that tumors from co-injected mice were highly vascularized (Figure 5E), with an about three-fold increase (P ≤ .0001) in the average number of blood vessels per field (12.0 ± 0.7 vessels per field) over that of LLC-only cohort tumors (4.4 ± 0.3 vessels per field; Figure 5F). These findings suggest that HSC-CAFs enhanced tumor growth, in part, by promoting tumor vascularization as reflected by increased blood vessel density in vivo.
HSC-CAFs Promote Angiogenesis In Vitro
To determine the mechanism by which HSC-CAFs promote tumor vascularization and to establish whether HSC-CAFs promote blood vessel formation through paracrine factors, HUVECs were plated onto a Geltrex matrix in serum-free αMEM conditioned medium from HSC-CAFs (HSC-CAF-CM) isolated from LLC tumors. Serum-free αMEM served as a negative control, and complete HUVEC medium was used as a positive control. HUVECs were stained with Calcein AM dye to allow for better visualization of networks by fluorescence microscopy. Tube formation assay showed that HSC-CAF-CM promoted tube formation over that of αMEM and to a similar extent as HUVEC medium positive control (Figure 6A).
Figure 6.
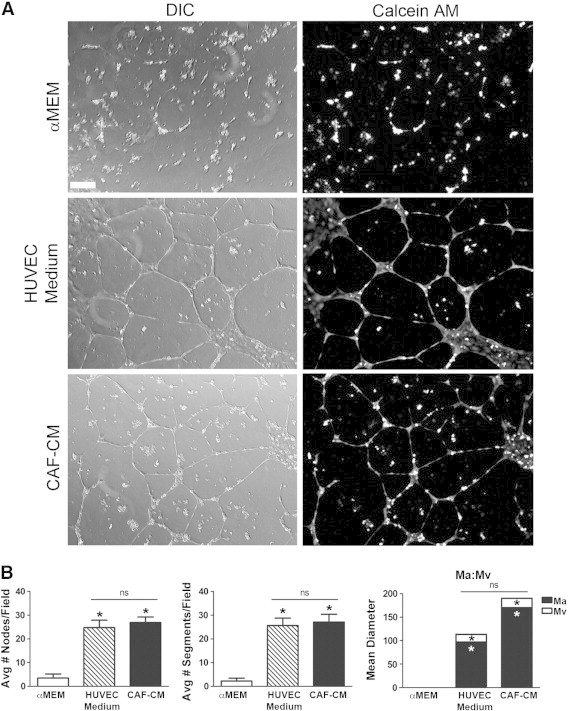
HSC-CAF-CM supports HUVEC tube formation. (A) HUVEC tube formation assay of cells treated with αMEM (negative control), HUVEC medium (positive control), or HSC-CAF-CM. Tubes and vascular pattern were visualized with DIC microscopy (left panels) and Calcein AM dye (right panels). (B) Number of nodes, segments and Ma:Mv were quantified. In B, *P < .05 compared to αMEM; ns, not significant. Bar, 250 μm (A).
Images of the tube formation assay were quantitatively evaluated for numbers of nodes, numbers of endothelial segments, and overall vascular pattern (Figure 6B). Analysis revealed a statistically significant increase (P < .0001) in the number of nodes resulting from treatment with CAF-CM (26.9 ± 2.3) over control αMEM (3.5 ± 1.7) and a significant increase in the number of endothelial segments (P < .0001) with CAF-CM (27.1 ± 3.3) over αMEM (2.3 ± 1.2). Numbers of nodes and segments were statistically similar between CAF-CM and HUVEC medium treatment (P = .5958 and P = .7469, respectively). Vascular pattern can be reflected in the ratio of vascular to avascular space, as calculated by measurements of Ma and Mv diameters [51]. For example, a fine reticular vascular network has a large Ma to small Mv, while a larger, more sinusoidal network has a small Ma to large Mv ratio [51]. On the basis of this algorithm [51], HUVEC medium cultures revealed a fine vascular network indicated by increased Ma to Mv ratio (Figure 6B, Ma:Mv). HUVECs cultured in HSC-CAF-CM resulted in a network pattern similar to HUVEC control medium with Mv (35.2 ± 4.8 vs 53.1 ± 8.3, respectively, P = .0507) and Ma (362.0 ± 16.9 vs 331.3 ± 20.7, P = .2497). Negative control (αMEM) did not support vascular patterning, and there was no significant difference in cell death between HUVECs cultured in αMEM and HSC-CAF-CM (data not shown, P = .1690). These data indicate that HSC-CAFs promote endothelial tube formation through production of paracrine factors.
HSC-CAF Promotion of Vascular Patterning Is VEGF-A and TGF-β1 Dependent
VEGF and TGF-β are known to play a role in angiogenesis and vascular patterning; thus, we sought to determine whether HSC-CAF promotion of endothelial tube formation is supported through these factors. ELISA analysis of HSC-CAF-CM as used in tube formation assays demonstrated that HSC-CAFs produce both VEGF-A (289.60 ± 37.38 pg/ml) and TGF-β1 (89.50 ± 2.47 pg/ml), factors commonly associated with CAF populations (Figure 7A). This expression was confirmed by qRT-PCR (Figure 7B). As a positive control, production of VEGF-A and TGF-β1 by cultured primary skin fibroblasts was measured by ELISA (VEGF-A, 263.30 ± 5.03 pg/ml; TGF-β1, 30.64 ± 4.46 pg/ml) and qRT-PCR to allow for comparison of CAF-produced factors to an activated cell type known to produce biologically relevant levels of angiogenic factors, including VEGF and TGF-β [63,64]. To assess whether production of these factors by HSC-CAFs contributed to HUVEC tube formation, neutralizing antibodies to VEGF-A or TGF-β1 were added to HSC-CAF-CM in HUVEC tube formation assays. Morphologic comparison of HUVECs cultured in HSC-CAF-CM versus HSC-CAF-CM with VEGF-A neutralizing antibody revealed decreased network formation, fewer complete vascular networks or “meshes”, and presence of cell aggregates (Figure 7C). Quantitative comparison of networks cultured in HSC-CAF-CM versus those cultured in HSC-CAF-CM with VEGF-A neutralization (Figure 7D) showed a significant decrease in the number of nodes (26.9 ± 2.3 vs 15.1 ± 3.2, respectively, P = .0107) and segments (27.1 ± 3.3 vs 11.8 ± 3.9, respectively, P = .0094). Analysis of vascular pattern indicated similar Ma in HSC-CAF-CM with VEGF-A neutralization (327.5 ± 32.5) versus CAF-CM (362.0 ± 16.9, P = .3150). An approximate 31% reduction in Mv was seen with VEGF-A neutralization; however, this difference was not found to be statistically significant (24.2 ± 2.8 in HSC-CAF-CM with inhibitor vs 35.2 ± 4.8 in HSC-CAF-CM; P = .1758). Together, morphologic and quantitative analyses suggest that neutralization of VEGF in HSC-CAF-CM skews the vascular patterning toward a finer vascular network with larger avascular spaces, increased cell aggregates, and decreased nodes and segments, overall resulting in inhibited tube formation.
Figure 7.
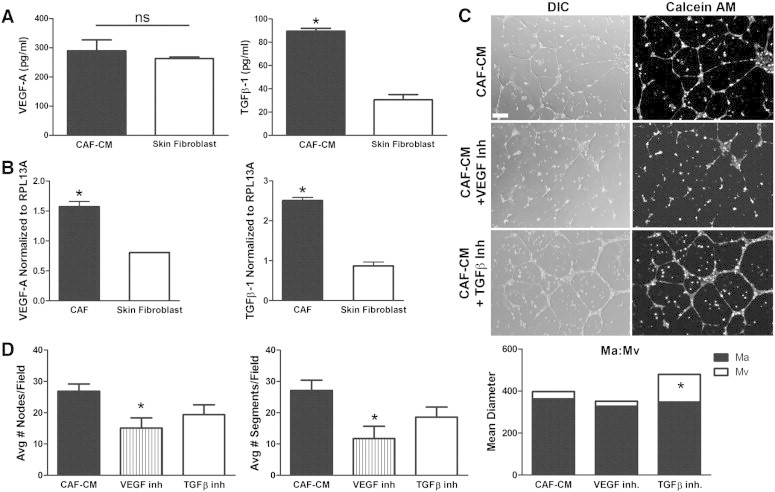
VEGF-A and TGF-β from HSC-CAFs promote HUVEC tube formation. (A) ELISA of VEGF-A (left panel) and TGF-β1 (right panel) in HSC-CAF-CM (black bars) and skin fibroblast–CM (white bars; positive control; *P < .05). (B) qRT-PCR analysis of VEGF-A (left panel) and TGF-β1 (right panel) mRNA expression in HSC-CAFs (black bars) and skin fibroblasts (white bars; positive control). (C) DIC (left panels) and Calcein AM dye (right panels) representative images of HUVEC tube formation assay with the addition of VEGF-A or TGF-β neutralizing antibodies in HSC-CAF-CM. (D) Quantification of nodes (left panel) and segments (right panel) with neutralization of VEGF-A (hatched bars) or TGF-β (white bars) in HSC-CAF-CM (black bars; *P < .05). The ratios of Ma area, Mv area, and Ma:Mv are quantified. In D, *P < .05 compared to HSC-CAF-CM. Bar, 250 μm (C).
On the basis of morphology, vascular pattern was also affected by the addition of TGF-β neutralizing antibody to HSC-CAF-CM. Vascular networks appeared complete, with similar mesh pattern to those observed with CAF-CM; however, the diameter of the tubes formed with TGF-β inhibition appeared to be larger (Figure 7C). While the numbers of nodes and segments did not reflect significant differences (comparison of CAF-CM to CAF-CM with inhibitor: nodes, 26.9 ± 2.3 vs 19.4 ± 3.1, P = .0749; segments, 27.1 ± 3.3 vs 18.6 ± 3.2, P = .0851), there was a trend toward decreased numbers of nodes (27.90%) and segments (31.33%) with TGF-β inhibition (Figure 7D). Analysis of vascular pattern reflected significant morphologic changes on inhibition of TGF-β. While Ma remained the same compared to HUVEC cultures treated with CAF-CM (P = .6279), Mv was significantly increased with TGF-β neutralization (131.8 ± 27.1 vs 35.2 ± 4.8 in CAF-CM alone, P ≤ .0001), suggesting that neutralization of TGF-β altered vascular patterning, resulting in increased diameter of endothelial tubes. Together, these studies demonstrate that HSC-CAFs promote tumor vascularization and vascular patterning through paracrine production of VEGF-A and TGF-β1.
HSC-CAFs from a Murine Model of Breast Cancer Promote In Vitro Angiogenesis
To determine whether HSC-CAFs and their tumor-promoting functions were tumor type specific, we profiled HSC-CAFs and examined their impact on angiogenesis in a syngeneic E0771 breast cancer model. E0771 tumor cells were injected orthotopically into non–bone marrow transplanted EGFP/CD45.2 female mice, and CAFs were isolated as described above. EGFP+ cells isolated from E0771 tumors expressed CD45 mRNA and protein (Figure 8, A and B, respectively), supporting their HSC origin, as well as markers of activated fibroblasts including Col I, α-SMA, FAP, vimentin, and MMPs (Figure 8, C–E). As with LLC cells, E0771 cells did not express CD45 or profiled fibroblast markers, with the exception of the EMT marker, vimentin. For all immunofluorescence staining, no primary staining controls are shown in Supplementary Figure S2. Together, gene and protein expression of CD45, as well as markers of activated fibroblasts, indicate an HSC origin for the isolated population of CAFs from an E0771 murine breast cancer model.
Figure 8.
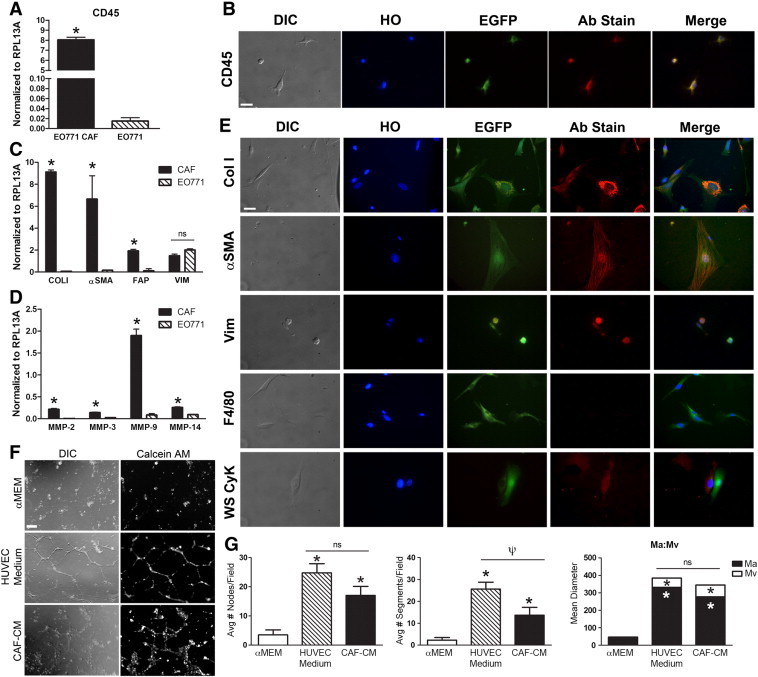
HSC-CAFs contribute to murine breast cancer. (A) qRT-PCR analysis of CD45 expression in HSC-CAFs from E0771 tumors (black bar) and E0771 tumor cells (hatched bar; *P ≤ .0001). (B) DIC and immunofluorescence staining of representative E0771 HSC-CAFs show morphology (DIC), nuclei (Hoechst dye, HO), expression of EGFP, and CD45 (Ab stain). (C, D) qRT-PCR analysis (C) for fibroblast markers Col I (*P ≤ .0001), α-SMA (*P = .0104), FAP (*P = .0016), and vimentin (P = .0082) and (D) MMP-2 (*P ≤ .0001), MMP-3 (*P = .0013), MMP-9 (*P ≤ .0001), and MMP-14 (*P ≤ .0001) in E0771 HSC-CAFs (black bars) and E0771 tumor cells (hatched bars). (E) DIC and immunofluorescence staining of representative E0771 HSC-CAFs show morphology (DIC), nuclei (HO), EGFP expression, antibody (Col I, α-SMA, vimentin, F4/80, WS CyK) stain (Ab stain), and merged images from representative cells. (F) HUVEC tube formation assay of cells treated with αMEM (negative control), HUVEC medium (positive control), or E0771 HSC-CAF-CM. Tubes and vascular pattern were visualized with DIC microscopy (left panels) and Calcein AM dye (right panels). (G) Number of nodes (left panel), segments (middle panel), and Ma:Mv (right panel) from F were quantified. In G, *P < .05 compared to αMEM and ψP = .0280 compared to HUVEC medium. Bars, 25 μm (A, C) and 250 μm (F).
To determine whether HSC-CAFs and their tumor-promoting functions were tumor type specific, we profiled HSC-CAFs and examined their impact on angiogenesis in a syngeneic E0771 breast cancer model. E0771 tumor cells were injected orthotopically into non–bone marrow transplanted EGFP/CD45.2 female mice, and CAFs were isolated as described above. EGFP+ cells isolated from E0771 tumors expressed CD45 mRNA and protein (Figure 8, A and B, respectively), supporting their HSC origin, as well as markers of activated fibroblasts including Col I, α-SMA, FAP, vimentin, and MMPs (Figure 8, C–E). As with LLC cells, E0771 cells did not express CD45 or profiled fibroblast markers, with the exception of the EMT marker, vimentin. For all immunofluorescence staining, no primary staining controls are shown in Supplementary Figure S2. Together, gene and protein expression of CD45, as well as markers of activated fibroblasts, indicate an HSC origin for the isolated population of CAFs from an E0771 murine breast cancer model.
Finally, functional analysis of HSC-CAF-CM in HUVEC tube formation assays showed that CAFs from E0771 tumors also supported endothelial cell network formation (Figure 8, F and G). HSC-CAF-CM from E0771 tumors compared to αMEM showed an increase in the number of nodes (17.0 ± 3.1 vs 3.5 ± 1.7, respectively, P = .0016) and segments (13.7 ± 3.6 vs 2.3 ± 1.2, respectively, P = .0053). While there was no significant difference between HSC-CAF-CM and HUVEC medium treatment with respect to nodes (P = .1142), there was a decrease in number of segments (P = .0280). In addition, HSC-CAF-CM from E0771 tumors promoted network patterning where HSC-CAF-CM Ma (277.3 ± 36.7) and Mv (67.6 ± 13.4) were similar to HUVEC medium Ma (331.3 ± 20.7, P = .1762) and Mv (53.1 ± 8.3, P = .3398). As with the LLC model, these data demonstrate an HSC origin for CAFs in a murine model of breast cancer and together demonstrate a role for HSC-CAFs in ECM production, ECM remodeling, and tumor vascularization in multiple tumor models.
Discussion
Multiple origins for CAFs have been suggested (reviewed in [5,32–34]), and understanding the roles of CAFs in various aspects of tumor development, progression, and metastasis is expanding. Many of the studies demonstrating these roles do not directly address the origin of the CAFs studied as they rely on laser capture microscopy for CAF isolation, negative selection excluding CD45-expressing cells, cell lines, and in vitro activation or induction of the CAF phenotype. Thus, the functional impact of CAFs from each specific origin on tumor progression remains unclear. This may be important for the development of strategic therapeutic targets, as it is undetermined whether CAFs of differing origins have distinct roles, potentially requiring different therapeutic targeting strategies (as reviewed in [65] and [52]). In the current study, we have taken steps toward addressing these questions with respect to the function of HSC-CAFs.
The method of CAF isolation developed in the current study is advantageous in that CAFs are recruited to the primary tumor in vivo and selected for in vitro to achieve a highly enriched primary CAF population that can be traced back to an HSC origin. Through the use of clonal engraftment and confirmation through expression of the pan-hematopoietic marker CD45, we were able to isolate a population of CAFs that are highly enriched for cells of a specific HSC origin to conduct in vitro and in vivo analyses of their function. Given that there is no single marker to identify a CAF, their identification relies on morphology and expression of multiple markers associated with fibroblasts/activated fibroblasts [31,32]. Thus, in this study, isolated cells from two murine tumor models were profiled for mRNA and protein expression of CD45 and a panel of activated fibroblast markers. Expression of the combination of markers used herein suggested this isolation method enriched for the CAF phenotype. As further confirmation, HSC-CAFs were also shown to be negative for expression of macrophage and epithelial markers, suggesting they were not the result of cell fusion, macrophage engulfment, or EMT.
Data indicate that HSC-CAFs express Col I, α-SMA, FAP, and MMPs, factors known to affect the ECM environment, stiffness, and composition. The expression of these factors by HSC-CAFs provides further insight into their potential functions in vivo, suggesting that this HSC-derived subset of CAFs directly contributes to changes seen during the development of tumor-associated matrix. Expression of Col I and α-SMA were demonstrated by Western blot, and flow cytometric analysis of HSC-CAFs demonstrated that ~ 68% of isolated HSC-CAFs co-express CD45 with Col I and ~ 90% co-express CD45 and α-SMA. This confirms that the isolated population is highly enriched for HSC-CAFs and that these cells are significant contributors to matrix composition in the tumor microenvironment. In addition to matrix deposition, matrix remodeling is an important aspect of a pro-tumorigenic microenvironment, acting to promote growth factor release, tumor vascularization, and migratory and invasive capacities of tumor cells, all of which can promote tumor cell metastasis. Factors known to play a role in this process include MMPs [5,31] and FAP, a membrane-bound serine protease that is thought to be selectively expressed by CAFs from tumors of epithelial origin [66]. In the present study, data show mRNA expression of FAP and production of active MMPs by HSC-CAFs. Together, these findings demonstrate contributions by HSC-CAFs to matrix deposition, composition, and remodeling in the tumor stroma.
Judah Folkman established that tumors could not achieve growth beyond 1 to 2 mm without the formation of a new blood supply [9], thus demonstrating the critical importance of vascularization to growth and progression of solid tumor. Angiogenesis provides both a nutrient-rich blood flow to the developing tumor and a route for metastatic dissemination of tumor cells. Herein, we identify EGFP+ cells in the LLC + CAF cohort tumors demonstrating that the co-injected HSC-CAF population persists as the tumors grow. A role for HSC-CAFs in tumor vascularization was evidenced by increased tumor size, enhanced vascularization on gross inspection, and augmented CD31 staining of tumors from mice co-injected with tumor cells and HSC-CAFs. Mechanistic in vitro studies showed HSC-CAF–derived VEGF-A and TGF-β directly regulate vascular patterning in vitro, suggesting that HSC-CAFs promote vascularization in vivo by similar mechanisms. Increased levels of VEGF-A and TGF-β have been clinically correlated with poor prognosis and disease progression in several cancer models [67–70]. The studies herein demonstrated that HSC-CAFs exhibit expression, release, and activation of MMPs, suggesting that HSC-CAFs may also contribute to tumor vascularization by degrading ECM to allow endothelial cell movement, endothelial cell organization, and release of stored pro-angiogenic growth factors. Given that both aberrant vascularization and MMP production have been associated with increased invasiveness of tumor cells and dissemination of aggressive tumor initiating cells, these data suggest the potential for HSC-CAFs to impact early pro-metastatic processes.
Through their contributions to ECM deposition and remodeling as well as the ability of HSC-CAFs to regulate vascular patterning and to produce active MMPs, these findings demonstrate that HSC-CAFs are a critical component of the pro-tumorigenic microenvironment. Thus, with our studies and others, resulting in an increased appreciation for the roles of the tumor microenvironment in tumor progression and metastasis, current therapeutic efforts are being aimed at targeting both tumor cells and stromal cells (reviewed in [6,71,72]). Devising optimal anti-stromal or anti-CAF therapies requires identification of suitable targets as well as the development of reliable biomarkers that indicate the type and composition of the tumor microenvironment in a given tumor. Elucidation of the contribution of CAFs from different sources may inform both of these aims.
Our studies show that HSC-CAFs, a previously underappreciated source of CAFs, play an integral role in the development of the tumor microenvironment and biologically impact tumor growth and vascularization in vitro and in vivo in multiple models. HSC-CAFs, once incorporated into the tumor, share phenotypic and functional similarities with classically identified CAFs and those derived from bone marrow mesenchymal stem cells [42,73] in that they display an activated phenotype, produce and remodel matrix, and contribute to tumor growth and angiogenesis. However, the HSC-CAF is unique in that this population may be continually recruited from the bone marrow into circulation as a circulating fibroblast precursor [44], then to the developing tumor mass. This recruitment potentially provides a unique opportunity to target HSC-CAFs before their incorporation into the tumor stroma, in addition to targeting their effects on the pro-tumorigenic microenvironment once established in the tumor stroma. Further, the ability to identify and trace the HSC-CAF before its incorporation into the tumor stroma may serve as a powerful biomarker for assessing efficacy of anti-tumor therapies.
The following are the supplementary data related to this article.
Primers and Probes Used for qRT-PCR
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2015.04.004.
Acknowledgements
The authors thank Haiqun Zeng and Danielle Woodford, HCC Flow Cytometry and Cell Sorting Shared Resource, for technical assistance. The authors also thank the staff of Radiation Oncology Department for assistance in irradiation of mice, and the staff of the Ralph H. Johnson VAMC Animal Facility for care of mice.
Footnotes
This work is supported in part by the NIH (National Institutes of Health)/NCI (National Institutes of Health) (R01 CA148772, A.C.L.), the Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs (Merit Awards, A.C.L.), and the Hollings Cancer Center (Translational Research Pilot Project, P30 CA138313, A.C.L.).
This article refers to supplementary materials, which are designated by Supplementary Table S1 and Supplementary Figures S1 and S2 and are available online at www.neoplasia.com.
References
- 1.Boyd N.F., Guo H., Martin L.J., Sun L., Stone J., Fishell E., Jong R.A., Hislop G., Chiarelli A., Minkin S. Mammographic density and the risk and detection of breast cancer. N Engl J Med. 2007;356:227–236. doi: 10.1056/NEJMoa062790. [DOI] [PubMed] [Google Scholar]
- 2.Cardone A., Tolino A., Zarcone R., Borruto Caracciolo G., Tartaglia E. Prognostic value of desmoplastic reaction and lymphocytic infiltration in the management of breast cancer. Panminerva Med. 1997;39:174–177. [PubMed] [Google Scholar]
- 3.Takahashi Y., Ishii G., Taira T., Fujii S., Yanagi S., Hishida T., Yoshida J., Nishimura M., Nomori H., Nagai K. Fibrous stroma is associated with poorer prognosis in lung squamous cell carcinoma patients. J Thorac Oncol. 2011;6:1460–1467. doi: 10.1097/JTO.0b013e318229189d. [DOI] [PubMed] [Google Scholar]
- 4.Barth P.J., Ebrahimsade S., Ramaswamy A., Moll R. CD34 + fibrocytes in invasive ductal carcinoma, ductal carcinoma in situ, and benign breast lesions. Virchows Arch. 2002;440:298–303. doi: 10.1007/s004280100530. [DOI] [PubMed] [Google Scholar]
- 5.McDonald L.T., LaRue A.C. Hematopoietic stem cell derived carcinoma-associated fibroblasts: a novel origin. Int J Clin Exp Pathol. 2012;5:863–873. [PMC free article] [PubMed] [Google Scholar]
- 6.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 8.Ito T.K., Ishii G., Chiba H., Ochiai A. The VEGF angiogenic switch of fibroblasts is regulated by MMP-7 from cancer cells. Oncogene. 2007;26:7194–7203. doi: 10.1038/sj.onc.1210535. [DOI] [PubMed] [Google Scholar]
- 9.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 10.Spaeth E.L., Dembinski J.L., Sasser A.K., Watson K., Klopp A., Hall B., Andreeff M., Marini F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One. 2009;4:e4992. doi: 10.1371/journal.pone.0004992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mishra P.J., Humeniuk R., Medina D.J., Alexe G., Mesirov J.P., Ganesan S., Glod J.W., Banerjee D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008;68:4331–4339. doi: 10.1158/0008-5472.CAN-08-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gyftopoulos K., Vourda K., Sakellaropoulos G., Perimenis P., Athanasopoulos A., Papadaki E. The angiogenic switch for vascular endothelial growth factor-A and cyclooxygenase-2 in prostate carcinoma: correlation with microvessel density, androgen receptor content and Gleason grade. Urol Int. 2011;87:464–469. doi: 10.1159/000329289. [DOI] [PubMed] [Google Scholar]
- 13.Weidner N., Folkman J., Pozza F., Bevilacqua P., Allred E.N., Moore D.H., Meli S., Gasparini G. Tumor angiogenesis: a new significant and independent prognostic indicator in early-stage breast carcinoma. J Natl Cancer Inst. 1992;84:1875–1887. doi: 10.1093/jnci/84.24.1875. [DOI] [PubMed] [Google Scholar]
- 14.Rofstad E.K., Mathiesen B. Metastasis in melanoma xenografts is associated with tumor microvascular density rather than extent of hypoxia. Neoplasia. 2010;12:889–898. doi: 10.1593/neo.10712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weidner N. Intratumor microvessel density as a prognostic factor in cancer. Am J Pathol. 1995;147:9–19. [PMC free article] [PubMed] [Google Scholar]
- 16.Barresi V., Di Gregorio C., Regiani-Bonetti L., Ponz-De Leon M., Barresi G., Vitarelli E. Stage I colorectal carcinoma: VEGF immunohistochemical expression, microvessel density, and their correlation with clinical outcome. Virchows Arch. 2011;457:11–19. doi: 10.1007/s00428-010-0933-5. [DOI] [PubMed] [Google Scholar]
- 17.Orimo A., Gupta P.B., Sgroi D.C., Arenzana-Seisdedos F., Delaunay T., Naeem R., Carey V.J., Richardson A.L., Weinberg R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 18.Sugimoto H., Mundel T.M., Kieran M.W., Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther. 2006;5:1640–1646. doi: 10.4161/cbt.5.12.3354. [DOI] [PubMed] [Google Scholar]
- 19.Kiskowski M.A., Jackson R.S., II, Banerjee J., Li X., Kang M., Iturregui J.M., Franco O.E., Hayward S.W., Bhowmick N.A. Role for stromal heterogeneity in prostate tumorigenesis. Cancer Res. 2011;71:3459–3470. doi: 10.1158/0008-5472.CAN-10-2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tchou J., Kossenkov A.V., Chang L., Satija C., Herlyn M., Showe L.C., Pure E. Human breast cancer associated fibroblasts exhibit subtype specific gene expression profiles. BMC Med Genomics. 2012;5:39. doi: 10.1186/1755-8794-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis M.P., Lygoe K.A., Nystrom M.L., Anderson W.P., Speight P.M., Marshall J.F., Thomas G.J. Tumour-derived TGF-beta1 modulates myofibroblast differentiation and promotes HGF/SF-dependent invasion of squamous carcinoma cells. Br J Cancer. 2004;90:822–832. doi: 10.1038/sj.bjc.6601611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davalos A.R., Coppe J.P., Campisi J., Desprez P.Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010;29:273–283. doi: 10.1007/s10555-010-9220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenthal E.L., McCrory A., Talbert M., Carroll W., Magnuson J.S., Peters G.E. Expression of proteolytic enzymes in head and neck cancer-associated fibroblasts. Arch Otolaryngol Head Neck Surg. 2004;130:943–947. doi: 10.1001/archotol.130.8.943. [DOI] [PubMed] [Google Scholar]
- 24.Herrera M., Islam A.B., Herrera A., Martin P., Garcia V., Silva J., Garcia J.M., Salas C., Casal I., de Herreros A.G. Functional heterogeneity of cancer-associated fibroblasts from human colon tumors shows specific prognostic gene expression signature. Clin Cancer Res. 2013;19:5914–5926. doi: 10.1158/1078-0432.CCR-13-0694. [DOI] [PubMed] [Google Scholar]
- 25.Lim K.P., Cirillo N., Hassona Y., Wei W., Thurlow J.K., Cheong S.C., Pitiyage G., Parkinson E.K., Prime S.S. Fibroblast gene expression profile reflects the stage of tumour progression in oral squamous cell carcinoma. J Pathol. 2011;223:459–469. doi: 10.1002/path.2841. [DOI] [PubMed] [Google Scholar]
- 26.Navab R., Strumpf D., Bandarchi B., Zhu C.Q., Pintilie M., Ramnarine V.R., Ibrahimov E., Radulovich N., Leung L., Barczyk M. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc Natl Acad Sci U S A. 2011;108:7160–7165. doi: 10.1073/pnas.1014506108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finak G., Bertos N., Pepin F., Sadekova S., Souleimanova M., Zhao H., Chen H., Omeroglu G., Meterissian S., Omeroglu A. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 28.Bauer M., Su G., Casper C., He R., Rehrauer W., Friedl A. Heterogeneity of gene expression in stromal fibroblasts of human breast carcinomas and normal breast. Oncogene. 2010;29:1732–1740. doi: 10.1038/onc.2009.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casey T., Bond J., Tighe S., Hunter T., Lintault L., Patel O., Eneman J., Crocker A., White J., Tessitore J. Molecular signatures suggest a major role for stromal cells in development of invasive breast cancer. Breast Cancer Res Treat. 2009;114:47–62. doi: 10.1007/s10549-008-9982-8. [DOI] [PubMed] [Google Scholar]
- 30.Franco O.E., Jiang M., Strand D.W., Peacock J., Fernandez S., Jackson R.S., II, Revelo M.P., Bhowmick N.A., Hayward S.W. Altered TGF-beta signaling in a subpopulation of human stromal cells promotes prostatic carcinogenesis. Cancer Res. 2011;71:1272–1281. doi: 10.1158/0008-5472.CAN-10-3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J., Liu J. Tumor stroma as targets for cancer therapy. Pharmacol Ther. 2013;137:200–215. doi: 10.1016/j.pharmthera.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Polanska U.M., Orimo A. Carcinoma-associated fibroblasts: non-neoplastic tumour-promoting mesenchymal cells. J Cell Physiol. 2013;228:1651–1657. doi: 10.1002/jcp.24347. [DOI] [PubMed] [Google Scholar]
- 33.Madar S., Goldstein I., Rotter V. 'Cancer associated fibroblasts'–more than meets the eye. Trends Mol Med. 2013;19:447–453. doi: 10.1016/j.molmed.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 34.Augsten M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front Oncol. 2014;4:62. doi: 10.3389/fonc.2014.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu J., Clark R.A. Extracellular matrix alters PDGF regulation of fibroblast integrins. J Cell Biol. 1996;132:239–249. doi: 10.1083/jcb.132.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dunphy J.E. The Fibroblast — A Ubiquitous Ally for the Surgeon. N Engl J Med. 1963;268:1367–1377. [Google Scholar]
- 37.Jotzu C., Alt E., Welte G., Li J., Hennessy B.T., Devarajan E., Krishnappa S., Pinilla S., Droll L., Song Y.H. Adipose tissue-derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor-derived factors. Anal Cell Pathol (Amst) 2010;33:61–79. doi: 10.3233/ACP-CLO-2010-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan J., Buache E., Chenard M.P., Dali-Youcef N., Rio M.C. Adipocyte is a non-trivial, dynamic partner of breast cancer cells. Int J Dev Biol. 2011;55:851–859. doi: 10.1387/ijdb.113365jt. [DOI] [PubMed] [Google Scholar]
- 39.Zeisberg E.M., Potenta S., Xie L., Zeisberg M., Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67:10123–10128. doi: 10.1158/0008-5472.CAN-07-3127. [DOI] [PubMed] [Google Scholar]
- 40.Potenta S., Zeisberg E., Kalluri R. The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer. 2008;99:1375–1379. doi: 10.1038/sj.bjc.6604662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petersen O.W., Nielsen H.L., Gudjonsson T., Villadsen R., Rank F., Niebuhr E., Bissell M.J., Ronnov-Jessen L. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am J Pathol. 2003;162:391–402. doi: 10.1016/S0002-9440(10)63834-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quante M., Tu S.P., Tomita H., Gonda T., Wang S.S., Takashi S., Baik G.H., Shibata W., Diprete B., Betz K.S. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19:257–272. doi: 10.1016/j.ccr.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.LaRue A.C., Masuya M., Ebihara Y., Fleming P.A., Visconti R.P., Minamiguchi H., Ogawa M., Drake C.J. Hematopoietic origins of fibroblasts: I. In vivo studies of fibroblasts associated with solid tumors. Exp Hematol. 2006;34:208–218. doi: 10.1016/j.exphem.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 44.Abangan R.S., Jr., Williams C.R., Mehrotra M., Duncan J.D., Larue A.C. MCP1 directs trafficking of hematopoietic stem cell-derived fibroblast precursors in solid tumor. Am J Pathol. 2010;176:1914–1926. doi: 10.2353/ajpath.2010.080839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakanishi T., Kuroiwa A., Yamada S., Isotani A., Yamashita A., Tairaka A., Hayashi T., Takagi T., Ikawa M., Matsuda Y. FISH analysis of 142 EGFP transgene integration sites into the mouse genome. Genomics. 2002;80:564–574. doi: 10.1006/geno.2002.7008. [DOI] [PubMed] [Google Scholar]
- 46.Ebihara Y., Masuya M., Larue A.C., Fleming P.A., Visconti R.P., Minamiguchi H., Drake C.J., Ogawa M. Hematopoietic origins of fibroblasts: II. In vitro studies of fibroblasts, CFU-F, and fibrocytes. Exp Hematol. 2006;34:219–229. doi: 10.1016/j.exphem.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 47.Visconti R.P., Ebihara Y., LaRue A.C., Fleming P.A., McQuinn T.C., Masuya M., Minamiguchi H., Markwald R.R., Ogawa M., Drake C.J. An in vivo analysis of hematopoietic stem cell potential: hematopoietic origin of cardiac valve interstitial cells. Circ Res. 2006;98:690–696. doi: 10.1161/01.RES.0000207384.81818.d4. [DOI] [PubMed] [Google Scholar]
- 48.Osawa M., Hanada K., Hamada H., Nakauchi H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 1996;273:242–245. doi: 10.1126/science.273.5272.242. [DOI] [PubMed] [Google Scholar]
- 49.Mehrotra M., Williams C.R., Ogawa M., LaRue A.C. Hematopoietic stem cells give rise to osteo-chondrogenic cells. Blood Cells Mol Dis. 2013;50:41–49. doi: 10.1016/j.bcmd.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sato T., Laver J.H., Ogawa M. Reversible expression of CD34 by murine hematopoietic stem cells. Blood. 1999;94:2548–2554. [PubMed] [Google Scholar]
- 51.LaRue A.C., Mironov V.A., Argraves W.S., Czirok A., Fleming P.A., Drake C.J. Patterning of embryonic blood vessels. Dev Dyn. 2003;228:21–29. doi: 10.1002/dvdy.10339. [DOI] [PubMed] [Google Scholar]
- 52.Marsh T., Pietras K., McAllister S.S. Fibroblasts as architects of cancer pathogenesis. Biochim Biophys Acta. 2013;1832:1070–1078. doi: 10.1016/j.bbadis.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trowbridge I.S., Thomas M.L. CD45: an emerging role as a protein tyrosine phosphatase required for lymphocyte activation and development. Annu Rev Immunol. 1994;12:85–116. doi: 10.1146/annurev.iy.12.040194.000505. [DOI] [PubMed] [Google Scholar]
- 54.Ivaska J. Vimentin: Central hub in EMT induction? Small GTPases. 2011;2:51–53. doi: 10.4161/sgtp.2.1.15114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heppner K.J., Matrisian L.M., Jensen R.A., Rodgers W.H. Expression of most matrix metalloproteinase family members in breast cancer represents a tumor-induced host response. Am J Pathol. 1996;149:273–282. [PMC free article] [PubMed] [Google Scholar]
- 56.Singer C.F., Kronsteiner N., Marton E., Kubista M., Cullen K.J., Hirtenlehner K., Seifert M., Kubista E. MMP-2 and MMP-9 expression in breast cancer-derived human fibroblasts is differentially regulated by stromal-epithelial interactions. Breast Cancer Res Treat. 2002;72:69–77. doi: 10.1023/a:1014918512569. [DOI] [PubMed] [Google Scholar]
- 57.Bisson C., Blacher S., Polette M., Blanc J.F., Kebers F., Desreux J., Tetu B., Rosenbaum J., Foidart J.M., Birembaut P. Restricted expression of membrane type 1-matrix metalloproteinase by myofibroblasts adjacent to human breast cancer cells. Int J Cancer. 2003;105:7–13. doi: 10.1002/ijc.11012. [DOI] [PubMed] [Google Scholar]
- 58.Jin G., Kawsar H.I., Hirsch S.A., Zeng C., Jia X., Feng Z., Ghosh S.K., Zheng Q.Y., Zhou A., McIntyre T.M. An antimicrobial peptide regulates tumor-associated macrophage trafficking via the chemokine receptor CCR2, a model for tumorigenesis. PLoS One. 2010;5:e10993. doi: 10.1371/journal.pone.0010993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neglia J.P., FitzSimmons S.C., Maisonneuve P., Schoni M.H., Schoni-Affolter F., Corey M., Lowenfels A.B. The risk of cancer among patients with cystic fibrosis. Cystic Fibrosis and Cancer Study Group. N Engl J Med. 1995;332:494–499. doi: 10.1056/NEJM199502233320803. [DOI] [PubMed] [Google Scholar]
- 60.Pandol S., Edderkaoui M., Gukovsky I., Lugea A., Gukovskaya A. Desmoplasia of pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol. 2009;7:S44–S47. doi: 10.1016/j.cgh.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Provenzano P.P., Inman D.R., Eliceiri K.W., Knittel J.G., Yan L., Rueden C.T., White J.G., Keely P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seewaldt V. ECM stiffness paves the way for tumor cells. Nat Med. 2014;20:332–333. doi: 10.1038/nm.3523. [DOI] [PubMed] [Google Scholar]
- 63.Nissen N.N., Polverini P.J., Koch A.E., Volin M.V., Gamelli R.L., DiPietro L.A. Vascular endothelial growth factor mediates angiogenic activity during the proliferative phase of wound healing. Am J Pathol. 1998;152:1445–1452. [PMC free article] [PubMed] [Google Scholar]
- 64.Mori Y., Chen S.J., Varga J. Expression and regulation of intracellular SMAD signaling in scleroderma skin fibroblasts. Arthritis Rheum. 2003;48:1964–1978. doi: 10.1002/art.11157. [DOI] [PubMed] [Google Scholar]
- 65.Ohlund D., Elyada E., Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med. 2014;211:1503–1523. doi: 10.1084/jem.20140692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu R., Li H., Liu L., Yu J., Ren X. Fibroblast activation protein: A potential therapeutic target in cancer. Cancer Biol Ther. 2012;13:123–129. doi: 10.4161/cbt.13.3.18696. [DOI] [PubMed] [Google Scholar]
- 67.Saito H., Tsujitani S., Oka S., Kondo A., Ikeguchi M., Maeta M., Kaibara N. An elevated serum level of transforming growth factor-beta 1 (TGF-beta 1) significantly correlated with lymph node metastasis and poor prognosis in patients with gastric carcinoma. Anticancer Res. 2000;20:4489–4493. [PubMed] [Google Scholar]
- 68.Reis S.T., Pontes-Junior J., Antunes A.A., Sousa-Canavez J.M., Abe D.K., Cruz J.A., Dall'oglio M.F., Crippa A., Passerotti C.C., Ribeiro-Filho L.A. Tgf-beta1 expression as a biomarker of poor prognosis in prostate cancer. Clinics. 2011;66:1143–1147. doi: 10.1590/S1807-59322011000700004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seo Y., Baba H., Fukuda T., Takashima M., Sugimachi K. High expression of vascular endothelial growth factor is associated with liver metastasis and a poor prognosis for patients with ductal pancreatic adenocarcinoma. Cancer. 2000;88:2239–2245. doi: 10.1002/(sici)1097-0142(20000515)88:10<2239::aid-cncr6>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 70.Karayiannakis A.J., Syrigos K.N., Polychronidis A., Zbar A., Kouraklis G., Simopoulos C., Karatzas G. Circulating VEGF levels in the serum of gastric cancer patients: correlation with pathological variables, patient survival, and tumor surgery. Ann Surg. 2002;236:37–42. doi: 10.1097/00000658-200207000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hanahan D., Coussens L.M. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 72.Togo S., Polanska U.M., Horimoto Y., Orimo A. Carcinoma-associated fibroblasts are a promising therapeutic target. Cancers. 2013;5:149–169. doi: 10.3390/cancers5010149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhao Q., Gregory C.A., Lee R.H., Reger R.L., Qin L., Hai B., Park M.S., Yoon N., Clough B., McNeill E. MSCs derived from iPSCs with a modified protocol are tumor-tropic but have much less potential to promote tumors than bone marrow MSCs. Proc Natl Acad Sci U S A. 2015;112:530–535. doi: 10.1073/pnas.1423008112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers and Probes Used for qRT-PCR