

Abstract
Intrarenal interleukin-15 (IL-15) plays a major role controlling epithelial survival and polarization both in physiological and pathologic conditions. Herein, we confirmed that human renal cell carcinomas (RCCs) express a membrane-bound IL-15 isoform displaying an unusual molecular weight of 27 kDa. Its stimulation with soluble IL-15 receptor α chain (s-IL-15Rα) triggers epithelial-mesenchymal transition (EMT) process as shown by the down-regulation of E-cadherin and zona occludens 1 and the up-regulation of vimentin and N-cadherin and promotes the migratory and invasive properties of RCC. S-IL-15Rα treatment triggered the Src/PI3K/Akt/GSK-3β pathway and promoted β-catenin nuclei translocation. Deactivation of this pathway by using Src-specific inhibitor PP2, PI3K inhibitor LY294002, and AKT inhibitor MK2206 hampered β-catenin nuclei translocation and suppressed EMT, migration, and invasion of RCC. S-IL-15Rα treatment also enhanced Src-dependent phosphorylation of focal adhesion kinase (FAK) and extracellular signal–regulated kinase (Erk1/2). FAK knockdown significantly decreased the migration and invasion of RCC, which suggest that Src-FAK signaling was involved in s-IL-15Rα–favored migration and invasion of RCC. At the same time, inhibitors of Erk1/2 also significantly decreased the migration and invasion of RCC but could not reverse s-IL-15Rα–induced EMT. Taken together, our results reveal that Src-dependent PI3K/Akt/GSK3b/β-catenin pathway is required for s-IL-15Ra–dependent induction of EMT in RCC, while Src-FAK and Src-Erk1/2 signaling were involved in s-IL-15Rα–promoted migration and invasion properties of RCC. Our study provides a better understanding of IL-15 signaling in RCC tumor progression, which may lead to novel targeted therapies and provide some suggestions when using IL-15 in clinic.
Abbreviations: tmb-IL-15, transmembrane-bound interleukin-15; RCC, renal cell carcinoma; EMT, epithelial-mesenchymal transition; s-IL-15Rα, soluble IL-15 receptor α chain; siRNA, small interfering RNA; TGF-β, transforming growth factor–β; PI3K, phosphatidylinositol 3-kinase; GSK-3β, glycogen synthase kinase-3β; FAK, focal adhesion kinase; Erk, extracellular signal–regulated kinase; ZO-1, zona occludens 1; Akt, protein kinase B
Introduction
Kidney cancer accounts for about 2% of all cancers, and worldwide, > 250,000 new cases of kidney cancer are diagnosed each year. Renal cell carcinoma (RCC) is the most common form of adult kidney cancer denoting a diverse set of neoplasias with unique genetic and histologic feature [1]. The RCCs emanate from the renal tubule, a highly heterogeneous epithelial structure, and the resulting cancer displays unique characteristics depending on which cell is malignified [2]. The tubular epithelium is normally mitotically quiescent but demonstrates a considerable regenerative capacity upon renal injury [2]. It is therefore reasonable to propose that altered behavior of factors controlling tubular epithelium homeostasis may be involved in renal cancer progression and that a better understanding of the underlying biologic mechanisms could result in improvements in the prevention and treatment of this disease. In this context, interleukin-15 (IL-15) that plays an important role in renal homeostasis in both physiological and pathologic conditions could be an interesting candidate [3–9].
IL-15 is a pleiotropic cytokine that links in vivo innate and adaptive immune responses and is characterized by different levels of complexity in its biology [10,11]. Indeed, IL-15 receptor (IL-15R) consists of a private α-chain and shares IL-2 receptor β- and γ-chains that are expressed individually or together to form various functional receptors with different affinities and signaling capabilities [12,13]. Moreover, different functional forms of IL-15 exist: 1) the soluble form that is secreted at very low concentration can only activate cells expressing high affinity receptor [10,11]; 2) the hyper-IL-15 is a functional complex formed by the association of the soluble IL-15 receptor α chain (s-IL-15Rα) with the soluble cytokine [14]; 3) the membrane-bound isoform can anchor at the cell membrane through IL-15Rα [membrane-bound IL-15 (mb-IL-15)] or through a membrane-binding domain located between residues 21 to 41 of the N-terminus in the IL-15 hydrophobic region [transmembrane-bound IL-15 (tmb-IL-15)]. The latter is the dominant physiological isoforms of the cytokine competent for specific interaction with bystander cells [10,15–17]. In addition, the transmembrane isoform (tmb-IL-15) can also deliver a reverse signaling to the presenting cells by interacting with its soluble receptor [16,17].
Experiments in IL-15−/− and IL-15Rα−/− mice show that binding with IL-15Rα, intrarenal mb-IL-15 behaves as an epithelial survival factor [3,4], while acting through the IL-2/15Rγ (CD132), IL-15 preserves E-cadherin expression and inhibits the epithelial-mesenchymal transition (EMT) process [10]. By contrast, in renal cancer, loss of IL-2/15Rγ by epithelial cells defines a tumoral microenvironment where IL-15 can trigger EMT [18,19].
As for now, the key pathways implicated in the IL-15–dependent progression of the disease are only partially explored. Our study here revealed that the tmb-IL-15 expressed in RCC could respond to the s-IL-15Rα and promote migration and EMT of RCC by stimulating Src/phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt)/β-catenin, Src–focal adhesion kinase (FAK), and Src–extracellular signal–regulated kinase (Erk) signaling cascades. This better cognition of the mb-IL-15–dependent signals may help to develop novel targeted therapies and lead to improvement in the treatment of RCC.
Materials and Methods
Chemicals and Reagents
Antibodies against IL-15 [monoclonal antibody (mAb) L-20], antibodies against N-cadherin, vimentin, and zona occludens 1 (ZO-1), protein A/G–agarose beads, control small interfering RNA (siRNA; sc-37007), and FAK-siRNA (sc-29310) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Primary antibodies against E-cadherin, p-Src, Src, p-Akt, Akt, p-Erk1/2, Erk1/2, p-FAK, FAK, p-PI3K, PI3K, p-glycogen synthase kinase-3β (GSK-3β), GSK-3β, β-catenin, p-Smad2, Smad2, p-Smad3, and Smad3 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies were purchased from Cell Signaling Technology (Beverly, MA). Anti–IL-15–phycoerythrin (mAb247-PE), anti-mouse IgG, anti-goat IgG, rhIL-15 Rα/Fc chimera soluble (s-IL-15Rα) chain, and transforming growth factor–β (TGF-β) were purchased from R&D Systems (Minneapolis, MN). Src inhibitor PP2, Akt inhibitor MK2206, and Erk inhibitor PD98059 were purchased from Sigma-Aldrich (St Louis, MO). HRP-conjugated secondary antibody, Alexa Fluor 488/594–conjugated secondary antibody, 4',6-diamidino-2-phenylindole (DAPI), and Lipofectamine 2000 were purchased from Invitrogen (Carlsbad, CA). All other chemicals were obtained from Sigma-Aldrich.
Cell Culture
The 786-O renal carcinoma cell line was obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The ACHN renal carcinoma cell line was kindly given by Prof. Bruno Azzarone from France. 786-O cells and ACHN cells were, respectively, cultured in RPMI-1640 (Gibco, NY, USA) and Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% FBS (Gibco), 100 U/ml penicillin, and 100 mg/ml streptomycin under a humidified 5% CO2 atmosphere at 37°C in an incubator.
Flow Cytometry Analysis
Membrane expression of IL-15 (mb-IL-15) was revealed by flow cytometry using the anti–IL-15 phycoerythrin-conjugated mAb (mAb247-PE). Anti-mouse IgG1 PE was used as control. For each sample, 10,000 cells were acquired for data analysis using FACSCalibur and analyzed with CellQuest software (BD Biosciences, San Jose, CA).
Migration and Invasion Assay
Migration and invasion assays were performed in Boyden chambers. The polycarbonate filters (8-mm pore size; Corning, NY, USA) pre-coated with Matrigel Matrix (BD Biosciences) were used for invasion assay, and uncoated filters were used for migration assay. Cells (3 × 105) in 300 ml of medium (containing 0.1% FBS) with or without 100 ng/ml s-IL-15Rα were seeded in the upper chamber. Then, 600 ml of medium with 10% FBS was added to the lower chamber and served as a chemotactic agent. After 4-day incubation, for migration, the cells that migrated and adhered onto the lower chamber were fixed in 4% paraformaldehyde for 30 minutes, stained with crystal violet, and counted under an upright microscope (five fields per chamber). For invasion, the cells in the upper chamber were fixed in 4% paraformaldehyde for 30 minutes. Then, the Matrigel was mechanically removed from the filter with a cotton swab. The cells adhering to the underside of the filter were stained with crystal violet and counted under an upright microscope (five fields per chamber).
Western Blot
The cells were washed three times with ice-cold phosphate-buffered saline (PBS) and then lysed in lysis buffer containing 50 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.5% Na-deoxycholate, 5 m g/ml aprotinin, 5 mg/ml leupeptin, and 1 mM phenylmethylsulfonyl fluoride. Lysates were cleared by centrifugation and denatured by boiling in Laemmli buffer. Equal amounts of protein samples were loaded per well and separated on sodium dodecyl sulfate–polyacrylamide gels and then electrophoretically transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, MA, USA). Following blocking with 5% non-fat milk at room temperature for 1 hour, membranes were incubated with primary antibodies (1:500) at 4°C overnight and then incubated with HRP-conjugated secondary antibodies (1:5000) for 2 hours at room temperature. Detection was carried out using a LumiGLO chemiluminescent substrate system (KPL, Guildford, United Kingdom).
Immunofluorescence
The cells were cultured on chamber slides, serum starved for 12 hours, and then exposed to s-IL-15Rα for the indicated time. Cells were washed three times with PBS, fixed with 4% paraformaldehyde for 30 minutes, and permeabilized with 0.3% Triton X-100 for 30 minutes. After blocking with 3% BSA for 1 hour, cells were incubated with antibodies against vimentin, ZO-1, and β-catenin (1:50 dilution) at room temperature for 2 hours. Slides were washed three times with PBS and incubated with Alexa Fluor 488– or Alexa Fluor 594–conjugated secondary antibodies (1:200) for 1 hour at room temperature. Nuclei were stained with DAPI (10 mg/ml) for 1 minute. Images were acquired using fluorescence microscopy (BX51TRF; Olympus, Japan).
Immunoprecipitation
Cells were lysed in 1% digitonin cell extraction buffer [1 mM EDTA, 150 mM NaCl, 20 mM Tris (pH 8.0), and 10% glycerol] and protease inhibitor cocktail tablets (complete; Roche, Switerland). For immunoprecipitation, lysates and plasma membrane fraction were first pre-cleared with the protein A/G and then incubated overnight at 4°C with 2 μg of antibody against GSK-3β/AKT bound to protein A/G or anti-goat IgG bound to protein A/G as control. After washing, the immunocomplexes were boiled for 5 minutes in sodium dodecyl sulfate sample buffer and analyzed by Western blot.
RNA Interference
RNA interference was performed as per the manufacturer’s protocols. Briefly, 2 × 105 cells were seeded in six-well plates and allowed to grow to 50% confluence. Then, cells were transfected with siRNA. The cells were allowed to grow for another 18 hours before they were collected for the following experiments.
Statistical Analysis
Data are expressed as mean ± SD. Student’s t test and one-way analysis of variance test were used for statistical analyses of the data. All statistical analyses were conducted using GraphPad Prism Software Version 5.0 (GraphPad Software Inc, La Jolla, CA). Cases in which P values are < .05 were considered statistically significant.
Results
Stimulation of tmb-IL-15 with s-IL-15Rα Promoted Cellular Migration and Invasion of RCC
In Figure 1, A and B, flow cytometric and Western blot analyses revealed that the RCC cell lines 786-O and ACHN express membrane-bound IL-15 isoform, characterized by an unusual molecular weight (MW) of 27 kDa (Figure 1B). To examine whether membrane-bound IL-15 isoform has any functions, we stimulated RCC with s-IL-15Rα. As shown in Figure 1C, stimulation of RCC with s-IL-15Rα strongly enhanced their invasive potential in Matrigel (two-fold increase) and their migratory property in migration assay (five-fold increase).
Figure 1.
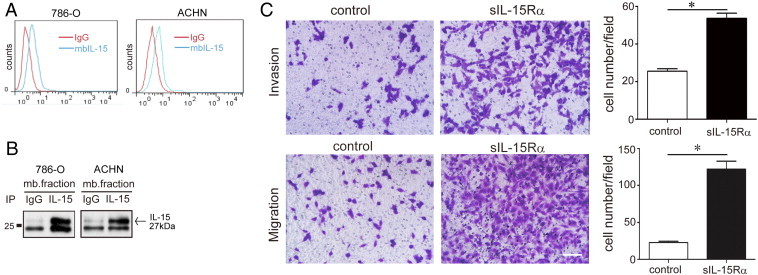
Stimulation of tmb-IL-15 with s-IL-15Rα promoted cellular migration and invasion in RCC cells. (A) Mb-IL-15 expression in human primary RCCs (786-O, ACHN) was detected by flow cytometry (mAb247-PE, blue peaks). Isotype-matched antibody IgG1 was used as control (red peaks). (B) The plasma membrane fraction of 786-O cells was immunoprecipitated with anti–IL-15 goat antibody L-20 and subsequently probed with the same L-20 antibody. A single 27 kDa specific band was detected, whereas the isotype control (goat IgG) detected no band. (C) The number of invasive and migrated cells after treatment with or without s-IL-15Rα (100 ng/ml) for 4 days. Data represent mean ± SEM of five fields. *P < .05 compared with control. Scale bar, 20 μm.
S-IL-15Rα Favored EMT of RCC
The role of tmb-IL-15 signaling in cell migration led us to examine if it has any effect on the process of EMT in RCC. In Figure 2A, immunofluorescence analysis showed that stimulation of tmb-IL-15 with 100 ng/ml s-IL-15Rα is as efficient as 5 ng/ml TGF-β in inducing the up-regulation of the mesenchymal marker vimentin and the down-regulation of the epithelial marker ZO-1. Interestingly, analysis of the magnified inset showed that untreated RCC displayed epithelial morphology and a very scant expression of vimentin, while s-IL-15Rα treatment induced a fibroblast-like morphology with a strong expression of a network of vimentin filaments. In Figure 2B, Western blot analysis confirmed that s-IL-15Rα efficiently induced the down-regulation of epithelial markers (ZO-1 and E-cadherin) and the up-regulation of mesenchymal markers (vimentin and N-cadherin) as that of TGF-β. In summary, these data suggest that mb-IL-15Rα signaling regulates the process of EMT, resulting in the enhanced migratory ability of RCC in vitro.
Figure 2.
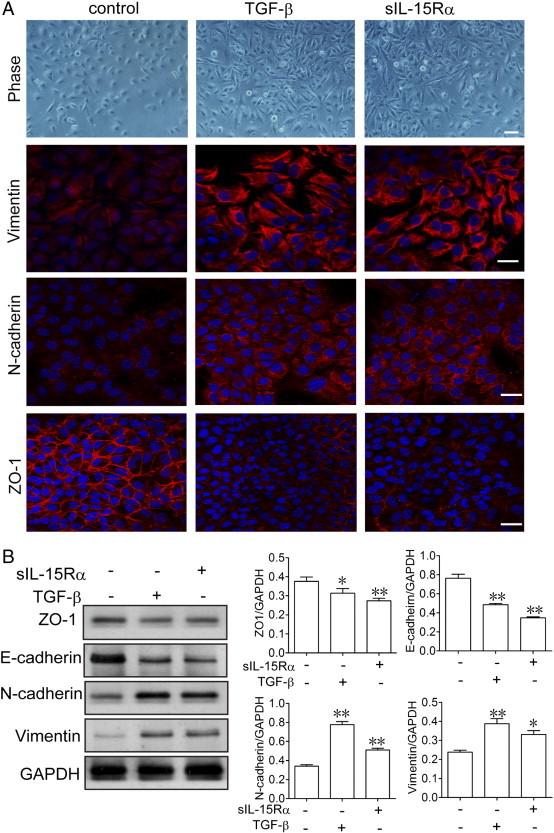
Stimulation of tmb-IL-15 with s-IL-15Rα triggered EMT in RCC cells.786-O cells were stimulated with 10 ng/ml TGF-β or 100 ng/ml s-IL-15Rα for 4 days. Morphology of cells was observed by phase-contrast microscopy (A). The epithelial marker ZO-1 and the mesenchymal markers vimentin and N-cadherin were analyzed by immunofluorescence staining (B) and Western blot (C). Scale bar, 20 μm. Relative band intensities were shown as mean ± SEM of three different experiments. *P < .05, **P < .01 compared with control.
Src/PI3K/Akt/GSK-3β/β-Catenin Signaling Cascade Is Responsible for s-IL-15Rα–Favored EMT
Next, we tried to clarify the signaling cascades that are responsible for the s-IL-15Rα–favored EMT process. First, we examined the typical TGF-β targets Smad2 and Smad3. As shown in Figure S1, s-IL-15Rα treatment did not significantly modify the phosphorylation levels of Smad2 and Smad3, which may suggest that this pathway was not involved in this process. At the same time, we found that under stimulation of s-IL-15Rα (100 ng/ml), phosphorylations of Src, PI3K, Akt, and GSK-3β were observed to have started from 30 up to 120 minutes (Figure 3A). S-IL-15Rα also dose-dependently promoted phosphorylation of Src and Akt (Figure 3B). Subsequently, the interaction between Akt and GSK-3β was enhanced after s-IL-15Rα stimulation, as shown by immunoprecipitation assay. These data all demonstrated that s-IL-15Rα activated the Src/PI3K/Akt/GSK-3β signaling cascade. As we know, the EMT process starts with the disruption of cell-cell adherent junctions and destabilization of cadherin-catenin complex. Subsequently, the release of β-catenin from E-cadherin favors nuclear translocation of β-catenin and renders it available for transcriptional regulation [20]. This latter step is under the control of Src/PI3K/Akt/GSK-3β signal cascade [21]. Immunofluorescence analysis shows that β-catenin was localized in cell-cell adherent junctions, while s-IL-15Rα treatment induces the nuclear localization of β-catenin (Figure 3D). To further confirm the relationship between the Src/PI3K/Akt/GSK-3β/β-catenin signal cascade and RCC EMT favored by s-IL-15Rα, Src inhibitor PP2, PI3K inhibitor LY4294002, and Akt inhibitor MK2206 were employed (Figures S2 and S3). As expected, Src inhibitor PP2 decreased Src and Akt phosphorylation, suggesting that Akt is the downstream of Src (Figure 4C). PI3K inhibitor LY4294002 treatment significantly inhibited s-IL-15Rα–triggered β-catenin nuclear translocation (Figure 4, A and B) and migration of RCC (Figure 4E). At the same time, Akt inhibitor MK2206 blocked tmb-IL-15–mediated down-regulation of the epithelial marker E-cadherin and up-regulation of the mesenchymal marker vimentin (Figure 4D) and finally reduced the migration of RCC (Figure 4E). All these data proved a previously unrecognized cross-talk between tmb-IL-15 and Src/PI3K/Akt/GSK-3β/β-catenin signaling in EMT of RCC.
Figure 3.
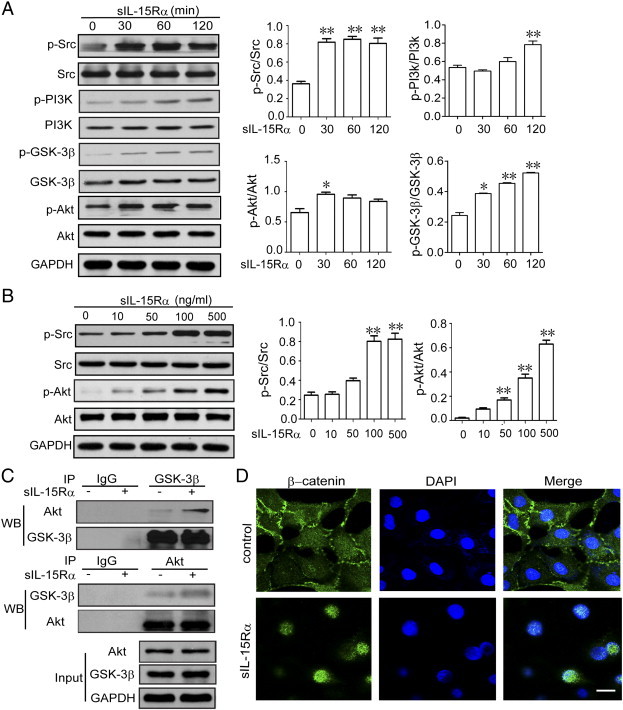
S-IL-15Rα stimulated Src/PI3K/Akt signaling pathway and β-catenin nuclei translocation in RCC cells. (A) 786-O cells were treated with 100 ng/ml s-IL-15Rα for the indicated time or with increasing concentrations (1-500 ng/ml) for 30 minutes (B). The expressions of p-Src, p-PI3K, p-GSK-3β, and p-Akt were examined by Western blot. GAPDH serves as the loading control, and relative band intensities were shown as mean ± SEM of three different experiments. *P < .05, **P < .01 versus control group. (C) 786-O cells were treated with or without s-IL-15Rα (100 ng/ml) for 1 hour. GSK-3β and Akt interaction was detected by immunoprecipitation. IgG was performed as a negative control. (D) 786-O cells were treated with or without s-IL-15Rα (100 ng/ml) for 6 hours. After fixation, the cellular location of β-catenin was examined by immunofluorescence staining. Scale bar, 20 μm.
Figure 4.
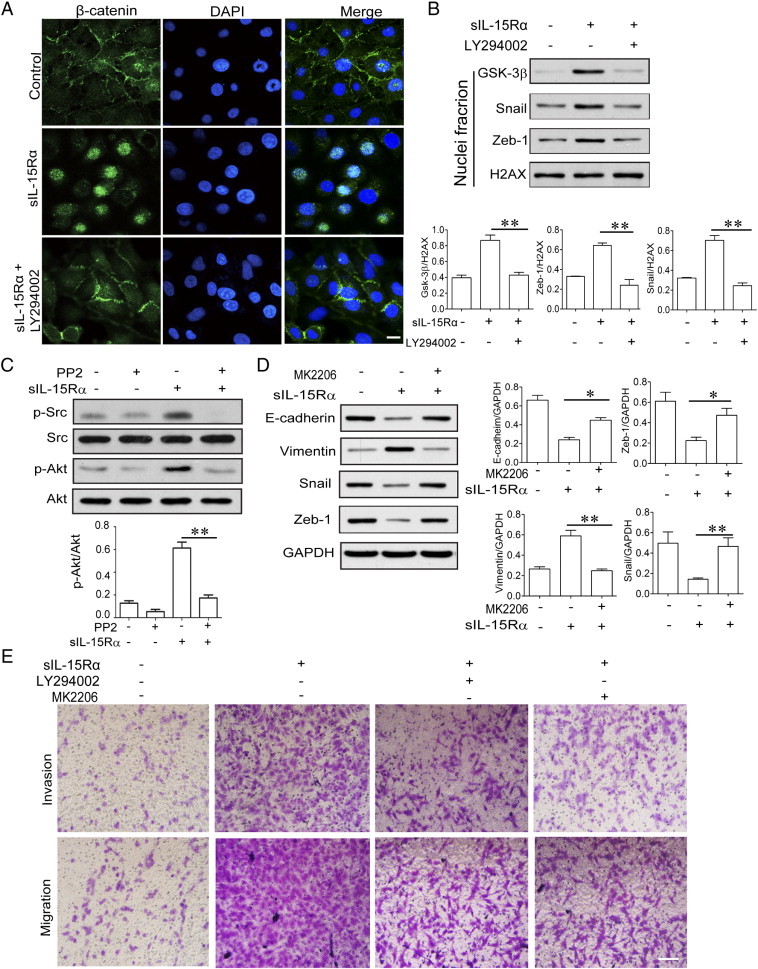
Src/PI3K/Akt/β-catenin activation was responsible for s-IL-15Rα–induced EMT and metastasis of RCC cells. 786-O cells were pretreated with or without 10 μM PI3K LY4294002 for 1 hour, followed by stimulation with s-IL-15Rα (100 ng/ml) for 6 hours. Cellular location of β-catenin was examined by immunofluorescence staining (A) and Western blot (B). Scale bar, 20 μm. (C) 786-O cells were pretreated with or without 10 μM SRC inhibitor PP2 for 1 hour, followed by stimulation with s-IL-15Rα (100 ng/ml) for 30 minutes. Phosphorylation and protein levels of Src and Akt in total cell lysate were detected by Western blot. (D and E) 786-O cells were treated with or without PI3K inhibitor LY4294002 (10 μM) or AKT inhibitor MK2206 (2 μM) accompanied by s-IL-15Ra (100 ng/ml) for 4 days. EMT, migration, and invasion of cells were examined.
Src-Dependent FAK Phosphorylation Contributed to s-IL-15Rα–Promoted Migration and Invasion of RCC
In cancer cells, Src activation plays a major role not only favoring EMT process through the Src/Akt signal but may also promoting cellular migration and invasion through the Src/FAK pathway [22]. These latter activities are triggered by the engagement of integrins with extracellular matrix proteins, which cause the activation of the Src/FAK signal [23]. In Figure 5A, stimulation of RCC tmb-IL-15 with the s-IL-15Rα chain strongly enhances FAK phosphorylation, while Src inhibitor PP2 can significantly inhibit FAK phosphorylation. To find out whether s-IL-15Rα–promoted cell migration was associated with Src-FAK signaling, FAK-specific siRNA (si-FAK) and negative control siRNA (si-NC) were transfected into 786-O cells. We observed that si-FAK but not si-NC inhibits FAK expression and Src phosphorylation in the cells stimulated with s-IL-15Rα is not affected because it is the upstream of FAK (Figure 5B). We then further explored the influence of FAK in the regulation of cell mobility: si-FAK strongly inhibited both migratory and invasive activities of 786-O cells treated for 4 days with s-IL-15Rα chain (Figure 5C). These data suggested that Src-dependent FAK phosphorylation contributed to s-IL-15Rα–promoted migration and invasion of RCC.
Figure 5.
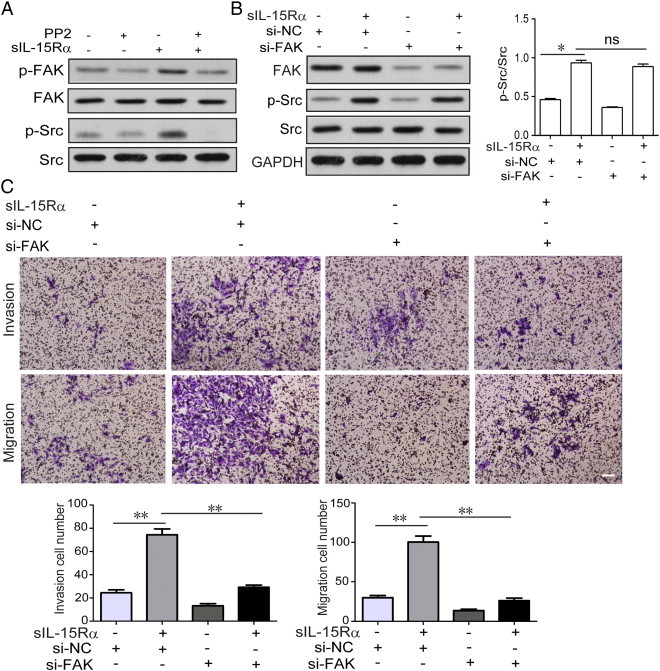
FAK signaling accounted for s-IL-15Rα–promoted RCC migration and invasion. (A) 786-O cells were pretreated with or without 10 μM Src inhibitor PP2 for 1 hour, followed by stimulation with/without s-IL-15Rα (100 ng/ml) for 30 minutes; p-SRC and p-FAK expressions were examined by Western blot. (B) 786-O cells transfected with si-FAK or si-NC were stimulated with or without s-IL-15Rα (100 ng/ml). The expressions of FAK and p-Src were detected by Western blot. (C) The number of invasive cells and migrated cells transfected with si-FAK or si-NC after treatment with or without s-IL-15 Rα (100 ng/ml) for 4 days was quantified. Data represent mean ± SEM of five fields. *P < .05 compared with control. Scale bar, 20 μm.
Src-Dependent Erk1/2 Phosphorylation Was Involved in s-IL-15Rα–Promoted Migration and Invasion of RCC
In Figure 6, A and B, Western blot analysis shows that s-IL-15Rα dose and time-dependently promoted Erk1/2 phosphorylation, while Src kinase inhibitor PP2 blocked its phosphorylation (Figure 6C), which confirmed that s-IL-15Rα–induced Erk1/2 phosphorylation depends on Src. Further, we found out that Erk1/2 inhibitor PD98059 can block tmb-IL-15–promoted migration of RCC (Figure 6E) but has no influence on tmb-IL-15–mediated down-regulation of the epithelial marker E-cadherin and up-regulation of the mesenchymal marker vimentin (Figure 6D). These results show that the Src/Erk1/2 pathway may not be involved in the EMT process but contribute to RCC migration and invasion favored by tmb-IL-15 with its soluble-specific ligand.
Figure 6.
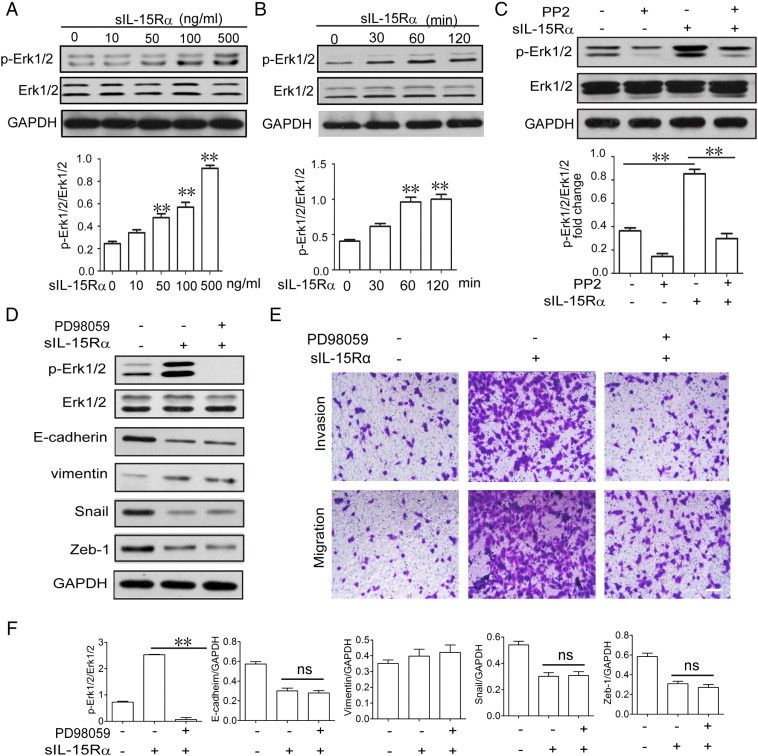
Erk1/2 signaling was involved in s-IL-15Rα–promoted RCC migration and invasion. (A) 786-O cells were treated with 100 ng/ml s-IL-15Rα for the indicated time or with increasing concentrations (1-500 ng/ml) for 30 minutes (B). The expressions of p-Erk1/2 and Erk1/2 were examined by Western blot. (C) 786-O cells were pretreated with or without 10 μM Src inhibitor PP2 for 1 hour, followed by stimulation with/without s-IL-15Rα (100 ng/ml) for 30 minutes; p-Erk1/2 and Erk1/2 expressions were examined by Western blot. (D) 786-O cells were pretreated with or without Erk1/2 inhibitor PD98059 (10 μM) for 1 hour, followed by stimulation with/without s-IL-15Rα (100 ng/ml) for 30 minutes; p-Erk1/2, Erk1/2, E-cadherin, vimentin, Snail, and Zeb-1 expressions were examined by Western blot. Relative band densities were calculated and shown as mean ± SEM of three different experiments. *P < .05, **P < .01 versus control group (F). (E) 786-O cells were treated with or without Erk1/2 inhibitor PD98059 (20 μM) accompanied by s-IL-15Ra (100 ng/ml) for 4 days. Migration and invasion of cells were examined. Scale bar, 20 μm.
Discussion
IL-15 is a cytokine displaying pleiotropic immune-enhancing activities, as it stimulates natural killer (NK), T and natural killer T (NKT) cell proliferation, survival, and functions. In view of these properties, IL-15 is regarded as a good candidate for cancer immunotherapy [11]. This possibility is reinforced by its low toxicity and efficacy in preclinical tumor models [24]. In this context, IL-15 has been recently proposed in clinical trials for the treatment of kidney cancer (NCT01021059 Protocol). However, it is important to be aware of the potential side effects of IL-15 that plays a major regulatory role in renal physiopathology. Indeed, renal cancer cells do not secrete IL-15, do not express IL-15Rγ, and express a tmb-IL-15 [18,19], while renal cancer stem cells express, as normal tubular cells, the three subunits of the IL-15R, creating a complex intratumoral interplay where IL-15 could favor (RCC cells) or inhibit cancer stem cells (CSC) tumor progression [8]. Therefore, much more attention should be paid to when using IL-15 in renal tumor therapy.
Herein, we confirmed that human RCC express an unusual transmembrane IL-15 isoform of 27 kDa. Its stimulation with the s-IL-15Rα chain triggered a complex reverse signal leading to the development of EMT and the acquisition of increased migratory and invasive properties, clearly contributing to cancer progression.
Mb-IL-15 exists under two different isoforms: The former is anchored at the cell membrane through the IL-15Rα chain and activates surrounding cells expressing the IL-15R through juxtacrine loops (transpresentation) [14,15], whereas the latter one (tm- IL-15 isoform) is anchored through an IL-15R–independent mechanism competent not only for transpresentation to opposing cells but also for the activation of a reverse signal in response to its soluble ligand s-IL-15Rα (Figure 7) [16,17].
Figure 7.
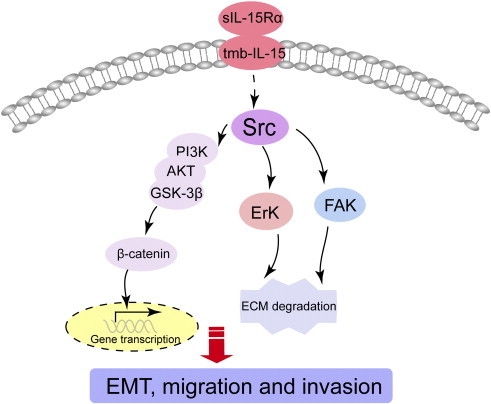
Illustration for the mechanism underlying s-IL-15Rα–induced EMT, migration, and invasion of human RCCs. When s-IL-15Rα binds to tmb-IL-15 in RCC, Src-dependent PI3K/Akt/GSK-3β/β-catenin pathway is activated and responsible for EMT induction. However, Src-FAK and Src-Erk1/2 signaling are also triggered by s-IL-15Rα/tmb-IL-15 interaction and involved in s-IL-15Rα–induced migration and invasion properties of RCC.
Tmb-IL-15–induced EMT in RCC cells is documented by the loss of epithelial markers (E-cadherin, ZO-1), the acquisition of mesenchymal ones (vimentin and N-cadherin), and the nuclear translocation of β-catenin. E-cadherin is considered as a tumor suppressor gene [25] and its down-regulation is a critical event in tumor invasion and a “master” programmer of EMT. Indeed, disruption of E-cadherin/β-catenin complexes leads to the loss of epithelial polarization and allows nuclear translocation of β-catenin. This multifunctional protein in normal tissues maintains with E-cadherin integrity and polarization of epithelia, while its aberrant nuclear expression can induce malignant pathways in normal cells and, through its abnormal activity, behaves as an oncogene and modulates transcription of genes competent to drive cancer initiation, progression, survival, and relapse [26].
Several factors have been suggested as potential initiators of renal EMT. TGF-β is also abundantly produced in the intratumoral RCC environment where it is likely involved in the induction of the neoplastic EMT [27,28]. With a few key exceptions, EMT induction depends on TGF-β and its downstream mediators, including SMADs. Our data show that RCC stimulation with s-IL-15Rα induces EMT, triggering events similar to those induced by TGF-β, but independently on SMAD activation, showing the existence of an alternative EMT program controlled by tmb-IL-15/s-IL-15Rα interplay totally independent from TGF-β signaling.
The tmb-IL-15–induced reverse signal involves first of all the activation of the tyrosine kinase Src, which in turn induces the phosphorylation of the Akt and Erk1/2 pathways. Both signals may favor EMT [29]. However, the use of specific inhibitors clearly shows that in RCC cells stimulated with s-IL-15Rα, only the Akt pathway is involved in the induction of EMT. Subsequently, Akt phosphorylation is associated to a cascade of signals leading to the activation of PI3K/Akt pathway and/or the phosphorylation of GSK-3β and finally to the nuclear translocation of β-catenin. The reverse signal induced by tmb-IL-15 causes in addition the phosphorylation of the FAK that is associated to the acquisition of increased migratory and invasive properties by RCC cells.
In conclusion, the results of this study clarify the signaling pathway triggered by s-IL-15Rα with tmb-IL-15 and its role during EMT and migration process of RCC. The detection in RCC of a novel tmb-IL-15–dependent program controlling EMT activation and the careful dissection of the reverse signals involved could help to develop novel targeted therapies for a more efficient treatment of RCC. Indeed, one could consider the intratumoral silencing of the tmb-IL-15 and of its ligand that is probably presented to tmb-IL-15 as a soluble molecule cleaved by ADAM metalloproteases [7,30] or alternatively transported as a functional molecule by exosomes [31] or transpresented by bystander cells [32]. These alternatives could offer different levels of intervention for interfering in the activation of the reverse signal. However, silencing of the IL-15Rα could be dangerous because its loss causes severe impairment of NK, NKT, CD8, and intraepithelial lymphocyte (IEL) lymphocyte homeostasis [33], and it is therefore mandatory to develop alternative strategies. Actually, specific inhibitors for Src, Akt, PI3K, GSK-3β, and FAK have been developed [34–37], and some of them are currently proposed in clinical trials for RCC combination therapy. It is promising that they may represent potential therapeutic targets for renal cancer metastasis in the near future.
Footnotes
This work was supported by the National Natural Science Foundation of China (Nos. 91129728, 81101563, 81422050, and 91429308), Key Personnel of Jiangsu Province (RC2011170), Jiangsu Province Clinical Science and Technology Projects (Clinical Research Center, BL2012008), and Priority Academic Program Development of Jiangsu Higher Education Institutions. Conflict of interest: The authors declared no conflict of interest.
This article refers to supplementary materials, which are designated by Figures S1 to S3 and are available online at www.neoplasia.com.
Contributor Information
Yang Sun, Email: yangsun@nju.edu.cn.
Qiang Xu, Email: molpharm@163.com.
Yanhong Gu, Email: guluer@163.com.
Appendix A. Supplementary Materials
Figure S1. S-IL-15Rα has no effect on Smad phosphorylation. 786-O cells were stimulated with s-IL-15Rα (100 ng/ml) for different times, and Smad expressions were examined by Western blot.
Figure S2. LY294002 inhibited PI3K and GSK-3β phosphorylation. 786-O cells were pretreated with or without 10 μM LY294002 for 1 hour, followed by stimulation with/without s-IL-15Rα (100 ng/ml) for 30 minutes; p-PI3K and p-GSK-3 expressions were examined by Western blot.
Figure S3. MK2206 inhibited AKT phosphorylation. 786-O cells were pretreated with or without 2 μM MK2206 for 1 hour, followed by stimulation with/without s-IL-15Rα (100 ng/ml) for 30 minutes; p-AKT expression was examined by Western blot.
References
- 1.Maher E.R. Genomics and epigenomics of renal cell carcinoma. Semin Cancer Biol. 2013;23:10–17. doi: 10.1016/j.semcancer.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 2.Axelson H., Johansson M.E. Renal stem cells and their implications for kidney cancer. Semin Cancer Biol. 2013;23:56–61. doi: 10.1016/j.semcancer.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 3.Shinozaki M., Hirahashi J., Lebedeva T., Liew F.Y., Salant D.J., Maron R., Kelley V.R. IL-15, a survival factor for kidney epithelial cells, counteracts apoptosis and inflammation during nephritis. J Clin Invest. 2002;109:951–960. doi: 10.1172/JCI14574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eini H., Tejman-Yarden N., Lewis E.C., Chaimovitz C., Zlotnik M., Douvdevani A. Association between renal injury and reduced interleukin-15 and interleukin-15 receptor levels in acute kidney injury. J Interferon Cytokine Res. 2010;30:1–8. doi: 10.1089/jir.2009.0005. [DOI] [PubMed] [Google Scholar]
- 5.Pavlakis M., Strehlau J., Lipman M., Shapiro M., Maslinski W., Strom T.B. Intragraft IL-15 transcripts are increased in human renal allograft rejection. Transplantation. 1996;62:543–545. doi: 10.1097/00007890-199608270-00020. [DOI] [PubMed] [Google Scholar]
- 6.Trinder P., Seitzer U., Gerdes J., Seliger B., Maeurer M. Constitutive and IFN-gamma regulated expression of IL-7 and IL-15 in human renal cell cancer. Int J Oncol. 1999;14:23–31. doi: 10.3892/ijo.14.1.23. [DOI] [PubMed] [Google Scholar]
- 7.Badoual C., Bouchaud G., Agueznay Nel H., Mortier E., Hans S., Gey A., Fernani F., Peyrard S., Puig P.L., Bruneval P. The soluble α chain of interleukin-15 receptor: a proinflammatory molecule associated with tumor progression in head and neck cancer. Cancer Res. 2008;68:3907–3914. doi: 10.1158/0008-5472.CAN-07-6842. [DOI] [PubMed] [Google Scholar]
- 8.Azzi S., Bruno S., Giron-Michel J., Clay D., Devocelle A., Croce M., Ferrini S., Chouaib S., Vazquez A., Charpentier B. Differentiation therapy: targeting human renal cancer stem cells with interleukin 15. J Natl Cancer Inst. 2011;103:1884–1898. doi: 10.1093/jnci/djr451. [DOI] [PubMed] [Google Scholar]
- 9.Budagian V., Bulanova E., Paus R., Bulfone-Paus S. IL-15/IL-15 receptor biology: a guided tour through an expanding universe. Cytokine Growth Factor Rev. 2006;17:259–280. doi: 10.1016/j.cytogfr.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Giron-Michel J., Azzi S., Ferrini S., Chouaib S., Camussi G., Eid P., Azzarone B. Interleukin-15 is a major regulator of the cell-microenvironment interactions in human renal homeostasis. Cytokine Growth Factor Rev. 2013;24:13–22. doi: 10.1016/j.cytogfr.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 11.Waldmann T.A. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol. 2006;6:595–601. doi: 10.1038/nri1901. [DOI] [PubMed] [Google Scholar]
- 12.Anderson D.M., Kumaki S., Ahdieh M., Bertles J., Tometsko M., Loomis A., Giri J., Copeland N.G., Gilbert D.J., Jenkins N.A. Functional characterization of the human interleukin-15 receptor α chain and close linkage of IL15RA and IL2RA genes. J Biol Chem. 1995;270:29862–29869. doi: 10.1074/jbc.270.50.29862. [DOI] [PubMed] [Google Scholar]
- 13.Giri J.G., Ahdieh M., Eisenman J., Shanebeck K., Grabstein K., Kumaki S., Namen A., Park L.S., Cosman D., Anderson D. Utilization of the beta and gamma chains of the IL-2 receptor by the novel cytokine IL-15. EMBO J. 1994;13:2822–2830. doi: 10.1002/j.1460-2075.1994.tb06576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giron-Michel J., Giuliani M., Fogli M., Brouty-Boye D., Ferrini S., Baychelier F., Eid P., Lebousse-Kerdiles C., Durali D., Biassoni R. Membrane-bound and soluble IL-15/IL-15Rα complexes display differential signaling and functions on human hematopoietic progenitors. Blood. 2005;106:2302–2310. doi: 10.1182/blood-2005-01-0064. [DOI] [PubMed] [Google Scholar]
- 15.Dubois S., Mariner J., Waldmann T.A., Tagaya Y. IL-15Rα recycles and presents IL-15 in trans to neighboring cells. Immunity. 2002;17:537–547. doi: 10.1016/s1074-7613(02)00429-6. [DOI] [PubMed] [Google Scholar]
- 16.Budagian V., Bulanova E., Orinska Z., Pohl T., Borden E.C., Silverman R., Bulfone-Paus S. Reverse signaling through membrane-bound interleukin-15. J Biol Chem. 2004;279:42192–42201. doi: 10.1074/jbc.M403182200. [DOI] [PubMed] [Google Scholar]
- 17.Neely G.G., Epelman S., Ma L.L., Colarusso P., Howlett C.J., Amankwah E.K., McIntyre A.C., Robbins S.M., Mody C.H. Monocyte surface-bound IL-15 can function as an activating receptor and participate in reverse signaling. J Immunol. 2004;172:4225–4234. doi: 10.4049/jimmunol.172.7.4225. [DOI] [PubMed] [Google Scholar]
- 18.Khawam K., Giron-Michel J., Gu Y., Perier A., Giuliani M., Caignard A., Devocelle A., Ferrini S., Fabbi M., Charpentier B. Human renal cancer cells express a novel membrane-bound interleukin-15 that induces, in response to the soluble interleukin-15 receptor α chain, epithelial-to-mesenchymal transition. Cancer Res. 2009;69:1561–1569. doi: 10.1158/0008-5472.CAN-08-3198. [DOI] [PubMed] [Google Scholar]
- 19.Giron-Michel J., Azzi S., Khawam K., Mortier E., Caignard A., Devocelle A., Ferrini S., Croce M., Francois H., Lecru L. Interleukin-15 plays a central role in human kidney physiology and cancer through the γc signaling pathway. PLoS One. 2012;7:e31624. doi: 10.1371/journal.pone.0031624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmalhofer O., Brabletz S., Brabletz T. E-cadherin, β-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28:151–166. doi: 10.1007/s10555-008-9179-y. [DOI] [PubMed] [Google Scholar]
- 21.Yan D., Avtanski D., Saxena N.K., Sharma D. Leptin-induced epithelial-mesenchymal transition in breast cancer cells requires β-catenin activation via Akt/GSK3- and MTA1/Wnt1 protein-dependent pathways. J Biol Chem. 2012;287:8598–8612. doi: 10.1074/jbc.M111.322800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macha M.A., Rachagani S., Gupta S., Pai P., Ponnusamy M.P., Batra S.K., Jain M. Guggulsterone decreases proliferation and metastatic behavior of pancreatic cancer cells by modulating JAK/STAT and Src/FAK signaling. Cancer Lett. 2013;341:166–177. doi: 10.1016/j.canlet.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taherian A., Li X., Liu Y., Haas T.A. Differences in integrin expression and signaling within human breast cancer cells. BMC Cancer. 2011;11:293. doi: 10.1186/1471-2407-11-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waldmann T.A., Lugli E., Roederer M., Perera L.P., Smedley J.V., Macallister R.P., Goldman C.K., Bryant B.R., Decker J.M., Fleisher T.A. Safety (toxicity), pharmacokinetics, immunogenicity, and impact on elements of the normal immune system of recombinant human IL-15 in rhesus macaques. Blood. 2011;117:4787–4795. doi: 10.1182/blood-2010-10-311456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez F.J., Lewis-Tuffin L.J., Anastasiadis P.Z. E-cadherin's dark side: possible role in tumor progression. Biochim Biophys Acta. 2012;1826:23–31. doi: 10.1016/j.bbcan.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thakur R., Mishra D.P. Pharmacological modulation of beta-catenin and its applications in cancer therapy. J Cell Mol Med. 2013;17:449–456. doi: 10.1111/jcmm.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burns W.C., Thomas M.C. The molecular mediators of type 2 epithelial to mesenchymal transition (EMT) and their role in renal pathophysiology. Expert Rev Mol Med. 2010;12:e17. doi: 10.1017/S1462399410001481. [DOI] [PubMed] [Google Scholar]
- 28.Gregory P.A., Bracken C.P., Smith E., Bert A.G., Wright J.A., Roslan S., Morris M., Wyatt L., Farshid G., Lim Y.Y. An autocrine TGF-β/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell. 2011;22:1686–1698. doi: 10.1091/mbc.E11-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Godoy P., Hengstler J.G., Ilkavets I., Meyer C., Bachmann A., Muller A., Tuschl G., Mueller S.O., Dooley S. Extracellular matrix modulates sensitivity of hepatocytes to fibroblastoid dedifferentiation and transforming growth factor β–induced apoptosis. Hepatology. 2009;49:2031–2043. doi: 10.1002/hep.22880. [DOI] [PubMed] [Google Scholar]
- 30.Mortier E., Bernard J., Plet A., Jacques Y. Natural, proteolytic release of a soluble form of human IL-15 receptor α-chain that behaves as a specific, high affinity IL-15 antagonist. J Immunol. 2004;173:1681–1688. doi: 10.4049/jimmunol.173.3.1681. [DOI] [PubMed] [Google Scholar]
- 31.Viaud S., Terme M., Flament C., Taieb J., Andre F., Novault S., Escudier B., Robert C., Caillat-Zucman S., Tursz T. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: a role for NKG2D ligands and IL-15Rα. PLoS One. 2009;4:e4942. doi: 10.1371/journal.pone.0004942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bulfone-Paus S., Bulanova E., Budagian V., Paus R. The interleukin-15/interleukin-15 receptor system as a model for juxtacrine and reverse signaling. Bioessays. 2006;28:362–377. doi: 10.1002/bies.20380. [DOI] [PubMed] [Google Scholar]
- 33.Lodolce J.P., Boone D.L., Chai S., Swain R.E., Dassopoulos T., Trettin S., Ma A. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity. 1998;9:669–676. doi: 10.1016/s1074-7613(00)80664-0. [DOI] [PubMed] [Google Scholar]
- 34.Sampath D., Malik A., Plunkett W., Nowak B., Williams B., Burton M., Verstovsek S., Faderl S., Garcia-Manero G., List A.F. Phase I clinical, pharmacokinetic, and pharmacodynamic study of the Akt-inhibitor triciribine phosphate monohydrate in patients with advanced hematologic malignancies. Leuk Res. 2013;37:1461–1467. doi: 10.1016/j.leukres.2013.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ravaud A., Gross-Goupil M., Bellmunt J. Combination therapy in metastatic renal cell cancer. Semin Oncol. 2013;40:472–481. doi: 10.1053/j.seminoncol.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 36.Bilim V., Ougolkov A., Yuuki K., Naito S., Kawazoe H., Muto A., Oya M., Billadeau D., Motoyama T., Tomita Y. Glycogen synthase kinase-3: a new therapeutic target in renal cell carcinoma. Br J Cancer. 2009;101:2005–2014. doi: 10.1038/sj.bjc.6605437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Golubovskaya V.M., Huang G., Ho B., Yemma M., Morrison C.D., Lee J., Eliceiri B.P., Cance W.G. Pharmacologic blockade of FAK autophosphorylation decreases human glioblastoma tumor growth and synergizes with temozolomide. Mol Cancer Ther. 2013;12:162–172. doi: 10.1158/1535-7163.MCT-12-0701. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. S-IL-15Rα has no effect on Smad phosphorylation. 786-O cells were stimulated with s-IL-15Rα (100 ng/ml) for different times, and Smad expressions were examined by Western blot.
Figure S2. LY294002 inhibited PI3K and GSK-3β phosphorylation. 786-O cells were pretreated with or without 10 μM LY294002 for 1 hour, followed by stimulation with/without s-IL-15Rα (100 ng/ml) for 30 minutes; p-PI3K and p-GSK-3 expressions were examined by Western blot.
Figure S3. MK2206 inhibited AKT phosphorylation. 786-O cells were pretreated with or without 2 μM MK2206 for 1 hour, followed by stimulation with/without s-IL-15Rα (100 ng/ml) for 30 minutes; p-AKT expression was examined by Western blot.