

Abstract
Heterozygous, germline nonsense mutations in AXIN2 have been reported in two families with oligodontia and colorectal cancer (CRC) predisposition, including an AXIN2 1989G>A mutation. Somatic AXIN2 mutations predicted to generate truncated AXIN2 (trAXIN2) proteins have been reported in some CRCs. Our studies of cells from an AXIN2 1989G>A mutation carrier showed that the mutant transcripts are not significantly susceptible to nonsense-mediated decay and, thus, could encode a trAXIN2 protein. In transient transfection assays, trAXIN2 was more abundant than wild-type AXIN2 protein, and in contrast to AXIN2, glycogen synthase kinase 3β inhibition did not increase trAXIN2 levels. Like AXIN2, the trAXIN2 protein interacts with β-catenin destruction complex proteins. When ectopically overexpressed, trAXIN2 inhibits β-catenin/T-cell factor–dependent reporter gene activity and SW480 CRC cell colony formation. These findings suggest the trAXIN2 protein may retain some wild-type functions when highly expressed. However, when stably expressed in rat intestinal IEC-6 cells, the trAXIN2 protein did not match AXIN2’s activity in inhibiting Wnt-mediated induction of Wnt-regulated target genes, and SW480 cells with stable expression of trAXIN2 but not AXIN2 could be generated. Our data suggest the AXIN2 1989G>A mutation may not have solely a loss-of-function role in CRC. Rather, its contribution may depend on context, with potential loss-of-function when AXIN2 levels are low, such as in the absence of Wnt pathway activation. However, given its apparent increased stability in some settings, the trAXIN2 protein might have gain-of-function in cells with substantially elevated AXIN2 expression, such as Wnt pathway–defective CRC cells.
Introduction
There are two AXIN proteins—AXIN1 and AXIN2. AXIN2 has been termed conductin or Axil in the case of the mouse or rat orthologs, respectively. A primary function of AXIN1 is in the assembly of the so-called “β-catenin destruction complex”, which plays a key role in regulating the pool of β-catenin that functions in Wnt pathway signaling and β-catenin/T-cell factor (TCF)–regulated gene expression. Activation of the Wnt signaling pathway and dysregulation of β-catenin protein levels and localization are commonly presumed to be critical events in colorectal cancer (CRC) development. Because AXIN1 is a negative regulator of β-catenin, AXIN1 has been classically thought of as a tumor suppressor protein. Studies in selected CRC and hepatocellular carcinoma cell lines showed that ectopic expression of AXIN1 can inhibit cell growth in vitro, reduce total β-catenin protein levels, and inhibit a TCF-responsive reporter gene construct [1]. Additionally, a mouse model in which Axin1 is conditionally inactivated in hepatocytes manifests an increased risk of hepatocellular cancer [2], further evidence of a role for AXIN1 as a tumor suppressor factor.
AXIN2 was initially identified through yeast two-hybrid experiments with β-catenin and glycogen synthase kinase 3β (GSK3β) and named for its homology to AXIN1 [3,4]. Like AXIN1, AXIN2 appears to act as a scaffold factor in the β-catenin destruction complex, and the two AXIN proteins have extensive similarity in several domains [4]. The two AXIN proteins are considered functionally equivalent, as an Axin2 cDNA inserted into the Axin1 mouse locus rescues the Axin1-null lethality [5]. However, while constitutional loss of Axin1 is lethal in the mouse during embryogenesis at e9.5 [6,7], mice carrying homozygous null mutations in Axin2 are viable and fertile, with a mild skull abnormality, indicating that the two genes are not fully redundant in vivo [8]. This difference in phenotypes likely reflects the differential expression patterns of the two genes; while AXIN1 is ubiquitous in various tissues [9], AXIN2 shows a more restricted developmental and cell-type–specific expression pattern [4]. Additionally, AXIN2, but not AXIN1, is a major transcriptional target of β-catenin–dependent Wnt signaling, and AXIN2 expression is significantly elevated in cancers with Wnt pathway mutations [10–12]. Because AXIN2 is positively regulated by upstream Wnt- and β-catenin–dependent signals and because the AXIN proteins are the least abundant members of the β-catenin destruction complex [13], control of AXIN2 protein levels could be a key negative feedback mechanism for the regulation of Wnt/β-catenin signaling in cells.
Mutations that lead to aberrant Wnt pathway activation are found in roughly 90% of sporadic CRCs [14]. Germline, inactivating mutations in the adenomatous polyposis coli (APC) gene underlie the inherited CRC predisposition syndrome familial adenomatous polyposis [15], and APC somatic mutations are present in about 70% to 80% of apparently sporadic CRCs [14]. Dysregulation of the Wnt pathway is believed to be an important first step in the genesis of CRCs [16], and this key role is also well supported by work in mouse models harboring mutations in Apc [17,18]. Akin to a key function of the AXIN proteins, APC is thought to have a crucial role in the assembly of the β-catenin destruction complex, and APC defects lead to increased levels of “free” β-catenin that drives cell survival and proliferation, in part through effects on β-catenin/TCF-regulated genes. While the most common mode of Wnt pathway dysregulation in CRC is abrogation of wild-type APC function, other Wnt pathway mutations are seen in CRCs [14]. Indeed, mutations in both AXIN1 and AXIN2 have been reported in association with CRC, although the functional significance in cancer of most reported AXIN1/2 mutations is uncertain [19]. In two unrelated kindreds with autosomal dominant oligodontia (the congenital absence of six or more adult teeth) and colorectal neoplasia (variable phenotypes, including oligo/attenuated polyposis and CRC), affected individuals have been found to carry heterozygous germline mutations predicted to prematurely truncate the AXIN2 open reading frame [20,21]. To better understand the role of AXIN2 defects in CRC, we have studied one of these germline AXIN2 mutations—the 1989G>A allele that leads to a premature stop at codon 663 (W663X)—and the truncated AXIN2 (trAXIN2) protein product predicted to be expressed from the allele. We present studies and data below that indicate that the trAXIN2 protein may have potentially complex and context-dependent effects that depend, in part, on the levels of trAXIN2 expression.
Materials and Methods
Expression Constructs and Cell Culture
A wild-type AXIN2 cDNA was generated by polymerase chain reaction (PCR)–based approaches, using total RNA isolated from the DLD-1 cell line (ATCC CCL-221). The identity of the AXIN2 wild-type cDNA sequence was confirmed by standard sequencing approaches. A trAXIN2 sequence, modeling that seen in the 1989G>A allele, was made by site-directed mutagenesis of the wild-type AXIN2 construct, and the wild-type AXIN2 and trAXIN2 sequences were subcloned into the following expression constructs: pPGS-CMV-Cite-Neo for retroviral expression, pCMV-3Tag for FLAG-tagged expression, pCS2+MT for myc-tagged expression, and pEYFP-C1 for EYFP-tagged expression. A human AXIN1 cDNA (isoform a) was generated previously by Janet Leung and cloned into pcDNA3.1 with an N-terminal FLAG-tag and C-terminal myc-tag. The sequence was confirmed by standard sequencing techniques. A β-catenin S33Y expression construct has been previously described [22]. All cells were cultured under sterile conditions at 37°C in 5% CO2. The HEK293T, RKO (ATCC CCL-2577), and SW480 (ATCC CCL-228) cell lines were cultured in Dulbecco's modified Eagle's medium (Gibco, Grand Island, NY) with the addition of 10% FBS (Fisher Pittsburgh, PA) and 1% penicillin/streptomycin (Gibco). IEC-6 cells (ATTC CRL-1592) were grown in Dulbecco's modified Eagle's medium (Gibco) with the addition of 10% FBS (Fisher), 1% penicillin/streptomycin (Gibco), and 0.1 U/ml recombinant human insulin (Gibco). For transfection-based studies, cells were transfected in 6-well plates or 12-well plates at 50% to 80% confluence, using 1 to 2 μg of total DNA per well with the TransIT-LT1 transfection reagent (Mirus, Madison, WI).
Characterization of Endogenous AXIN2 Transcripts in a Heterozygous Carrier of the Mutant 1989G>A Allele
After obtaining informed consent, peripheral blood was drawn and peripheral blood lymphocytes (PBLs) were isolated using Ficoll-Hypaque separation. The buffy coat layer was removed and plated onto polystyrene tissue culture dishes for 2 hours at 37°C to allow for adherence depletion of monocytes. PBLs were cultured in RPMI 1640 (Gibco) supplemented with l-glutamine (Gibco), 10% FBS (Fisher), 1% penicillin/streptomycin (Gibco), 1% Hepes (Gibco), 0.5% β-mercaptoethanol (Gibco), 5 μg/ml PHA-M (Sigma, St. Louis, MO), and 7 ng/ml interleukin-2 (Sigma). PBLs were cultured for 48 hours and then treated with 5 μM BIO (No. 13123; Cayman Chemical, Ann Arbor, MI) or DMSO for 18 hours. Total RNA was isolated using TRIzol, and cDNA was generated with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA). Genomic DNA (gDNA) was isolated from PBLs by boiling the cells in 50 mM NaOH followed by neutralization with 1 M Tris (pH 8.0). PCR to specifically amplify gDNA or cDNA was done using Taq polymerase (Promega, Madison, WI). Bulk PCR products were gel-extracted and sequenced. PCR products from gDNA and cDNA were TOPO subcloned, using the pCR2.1 TOPO cloning vector (Invitrogen, Grand Island, NY). Single TOPO clones were then isolated and sequenced to confirm the 1989GA genotype and the presence of the AXIN2 3′untranslated region (UTR). The primers used to amplify AXIN2 gDNA are given as follows: forward primer located in intron 5 (CCGACTTGCTGAATTGTCTG) and reverse primer located in intron 7 (AAGCAGCAGCTTACTCATCCA). The primers used to amplify AXIN2 cDNA are given as follows: forward primer located in exon 6 (GGGAGGAAGGAGACAGGTCGC) and reverse primer located in the AXIN2 3′UTR (CAAAGCCAGACCCCAGGG).
TOPFlash Reporter Gene Assay
HEK293T cells were transfected with relevant plasmids. Twenty-four hours after transfection, cells were treated with 100 ng/ml Wnt3a (R&D Systems, Minneapolis, MN) for 8 hours. Cells were then lysed in Passive Lysis Buffer (Promega), and luciferase activity was analyzed using the Promega Luciferase Assay Reagent and a GloMax Luminometer (Promega). The TOPFlash construct has been described previously [23]. The luminescence of each sample was normalized to total protein by Bicinchoninic acid assay (Pierce, Grand Island, NY). Relative luminescence values were compared using a t test to determine statistical significance.
Quantitative PCR
For Wnt target gene analysis, IEC-6 cells were transduced with AXIN2 or trAXIN2 retroviral constructs and then grown under G418 selection to generate stable polyclonal cell lines. To assess effects of stable AXIN2 or trAXIN2 expression on Wnt-mediated induction of β-catenin/TCF target genes, 100,000 stably transduced IEC-6 cells were plated onto a 6-cm dish. Twenty-four hours after plating, the cells were treated with 200 ng/ml recombinant Wnt3a (ProSci, San Diego, CA) to induce target gene expression or with phosphate-buffered saline as a control. After 16 hours of induction, RNA was collected by TRIzol extraction, cDNA synthesized, and target genes analyzed by quantitative PCR (qPCR) using the primers shown in Table 1. For quantification of AXIN2 or trAXIN2 (1989G>A) transcripts in ectopic expression assays, HEK293T cells were transfected in six-well dishes with 2 μg of FLAG-tagged expression constructs. At 24 hours after transfection, RNA was isolated for analysis by qPCR. Ectopic transcripts were quantified by PCR-based detection with primers for the FLAG sequence in the AXIN2/trAXIN2 constructs.
Table 1.
qPCR Primers for Wnt Target Gene and AXIN2/trAXIN2 Transcript Quantification
| Gene | Primer Sequence |
|---|---|
| U6 | F—GTGCTCGCTTCGGCAGCACATAT R—AAAAATATGGAACGCTTCACGAA |
| Axin2 | F—CTCTAACGCTAGGCGGAATG R—CCAGAAGTCCAGGGTATCCA |
| Lgr5 | F—GCTGCCAAATTGTTGGTTTT R—CAGGCTAGAAAGGGGAGCTT |
| Irs1 | F—CCAGAAGCAACCAGAGGA R—CCATGAGTTAAAAAGGAGGAT |
| Nkd1 | F—AGGACGACTTCCCCCTAGAA R—TGCAGCAAGCTGGTAATGTC |
| β-actin | F—GCCTTCCTTCTTGGGTATGG R—GCCTGGGTACATGGTGGT |
| FLAG | F—AAGGACGATGATGACAAGGACTACA R—TCCGGGAGGCAAGTCACCAA |
Immunoprecipitation and Immunoblot Studies
Cells were transfected with plasmids using TransIT-LT1 (Mirus) or Fugene-HD (Promega). Total protein was extracted using RIPA buffer, followed by immunoprecipitation (IP) with the indicated antibody and protein A/G agarose and 200 μg of total cellular protein. Proteins were eluted from agarose by boiling in Laemmli buffer and analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblot (IB). β-Catenin IPs were performed following a 5-hour treatment with 10 μM of the proteasome inhibitor, MG132 (Cayman Chemical). The following antibodies were used for IB analysis: α-AXIN2 (Cell Signaling Technology, 76G6, Danvers, MA), α-FLAG (Sigma, M2), α-myc (Sigma, C3956), α-total β-catenin (BD Transduction Laboratories, Franklin Lakes, NJ), and α-β-actin (Sigma, AC-15), and for IP: α-FLAG (Sigma, F7425) and α-myc (as above). For protein stabilization studies, HEK293T cells were plated onto a six-well dish and transfected at 70% to 90% confluence with 2 μg of FLAG-tagged proteins. Six hours after transfection, each well was split into four wells of a six-well plate. Twenty-four hours post-transfection, the cells were treated with DMSO, 100 nM okadaic acid (OA; Cell Signaling Technology, No. 5934), 1 μM BIO, or both. Cells were collected for IB analysis after 6 hours of inhibitor treatment. XAV939 (Cayman Chemical) treatment was performed using the indicated concentrations, and cells were collected for IB analysis after 24 hours of inhibitor treatment. Protein quantification was estimated relative to β-actin using the ImageJ software [24].
CRC Cell Line Colony Formation Assays and Stable Cell Lines
CRC cells were transfected with vector, AXIN2, or trAXIN2 pCMV-3Tag expression constructs, which contain a Neomycin resistance cassette. Twenty-four hours post-transfection, cells were counted, plated in triplicate onto 24-well dishes, and grown under selection in 250 μg/ml G418. RKO cells were plated at a density of 5000 cells per well. SW480 cells were plated at a density of 1000 cells per well. After 21 days, colonies were fixed in 4% paraformaldehyde, stained with crystal violet, and counted. Colony formation was performed in triplicate and repeated in three independent transfections. Colony numbers were analyzed using a t test to compare AXIN2 or trAXIN2 transfections to vector-only colony number. In parallel studies, G418-resistant SW480 colonies were harvested after 14 days of selection following transfection with empty vector, AXIN2, or trAXIN2 pCMV3Tag expression constructs, and protein lysates were prepared for IB studies, as above.
Results
The Sequence of the 1989G>A AXIN2 Mutant Allele Does Not Generate Novel Splicing Motifs
Our initial efforts sought to determine if the AXIN2 1989G>A mutant allele contained a novel sequence motif that might promote altered splicing of AXIN2 transcripts. The 1989G>A sequence change does not introduce a novel consensus splice donor or acceptor element into the exon where it is located (exon 7 of AXIN2). Further studies were undertaken using SplicePort [25] (spliceport.cbcb.umd.edu/) to analyze the gDNA sequence of the 1989G>A and wild-type AXIN2 alleles from intron 5 to exon 8 for potential splice recognition elements. SplicePort analysis found no changes in the predicted splice sites for the 1989A allele (Figure 1A). Exploration of the 1989G>A and wild-type AXIN2 alleles with an exonic splicing enhancer prediction program, ESEfinder [26,27] (rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi), found that the A allele does not disrupt any predicted endogenous exonic splicing enhancers (ESEs) but does encode an additional splicing factor 1 (SF1) ESE (Figure 1B), which, if of any consequence, might be expected to favor inclusion of exon 7 in transcripts, consistent with the notion that the 1989G>A AXIN2 variant allele likely does not generate variant AXIN2 transcripts.
Figure 1.
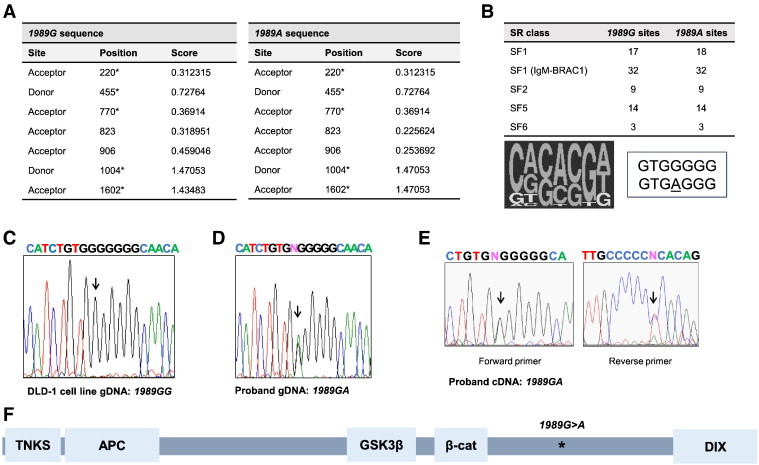
Analysis of AXIN2 gDNA and transcripts from a heterozygous 1989G>A allele carrier and the predicted AXIN2 truncated protein expressed. (A) The 1989G>A allele does not generate new splice elements. The sequences of the AXIN2 1989G (wild-type) and 1989A (mutant) alleles were analyzed using the SplicePort prediction algorithm. On the basis of the DNA sequence analysis, the positions of the predicted splice acceptor and donor positions in the wild-type and mutant AXIN2 alleles are indicated, with no new splice sites predicted in the mutant AXIN2 allele. The actual splice sites surrounding exon 7 are denoted with an asterisk. (B) The exonic splicing enhancer factor finder (ESEfinder) prediction identified one new predicted exon splicing enhancer factor binding site for SF1 binding in the 1989A allele. The number of each type of potential serine/arginine-rich (SR) factor binding motifs found in the AXIN2 1989G and 1989A alleles is indicated at the left. At the right, the SF1 consensus binding sequence motif is shown, with the corresponding sequences of the 1989G and A alleles, indicating the new consensus site match for the “A” allele. (C) Sequencing analysis of PCR products for the relevant region of AXIN2 exon 7 gDNA (gDNA) sequences in control DLD-1 CRC cells and (D) proband PBL samples. (E) Sequencing analysis of bulk PCR products from proband PBL cDNA identifies transcripts from both the wild-type “G” and mutant “A” AXIN2 alleles. (F) A schematic diagram of the location of the presumptive AXIN2 protein interaction domains [AXIN2 domains predicted to interact with TNKS (tankyrase), APC, GSK3, β-catenin] and DIX dimerization motif, as well as the location of the 1989G>A (W663X) nonsense mutation.
Mutant AXIN2 1989G>A Transcripts are Detected in a Heterozygous Carrier
To address the possibility that nonsense-mediated degradation (NMD) might reduce the levels of the 1989G>A transcripts, we examined lymphocytes from the proband of Marvin et al. [21]. PBLs were isolated from whole blood and briefly cultured in vitro before RNA isolation. NMD of transcripts can result from the presence of a stop codon in an exon that is upstream of the exon containing the polyadenylation signal for a given mRNA. NMD is triggered during the pioneering round of translation, when the exon junction proteins are removed from an mRNA transcript. To enhance the levels of AXIN2 transcripts in the cultured PBLs, the cells were treated with BIO, a GSK3β inhibitor that mimics to some degree the effects of activating Wnt ligands and increases the expression of Wnt target genes, including AXIN2 [28]. Total RNA and gDNA were then isolated from cells, cDNA was generated from total RNA, and the sequences immediately surrounding the 1989G>A allele were PCR amplified from both cDNA and gDNA. These bulk PCR products were directly sequenced as well as subcloned to detect the presence of wild-type and mutant alleles. The 1989G>A allele was confirmed in gDNA from the proband and was absent in gDNA from the control DLD-1 CRC cell line (Figure 1, C and D). Sequencing of the bulk PCR products obtained from cDNA templates demonstrated that the 1989G>A allele was present at roughly equivalent levels to that of the wild-type allele (Figure 1E). To confirm these findings, individually subcloned PCR products from gDNA and cDNA were sequenced, confirming that both wild-type and mutant AXIN2 transcripts were present in PBLs. These findings indicate that AXIN2 transcripts in PBLs of a 1989G>A mutation carrier do escape NMD, and the transcripts would be expected to generate a truncated (trAXIN2) protein product of 662 amino acids (Figure 1F). The studies of mutation carrier cDNAs did not uncover alternatively spliced transcripts arising from the 1989G>A mutation.
The trAXIN2 Protein is More Abundant than Wild-Type AXIN2 Protein in Transient Transfection-Based Studies
In transient transfection-based studies in HEK293T cells, the ectopically expressed trAXIN2 protein was present at higher levels than the wild-type AXIN2 protein (Figure 2, A and B). These findings were true not only for the FLAG–epitope-tagged version of trAXIN2 but also for ectopically expressed myc–epitope-tagged and untagged versions of trAXIN2 (below and data not shown). Reverse transcription (RT)–PCR–based studies revealed no significant differences in AXIN2 transcript levels between HEK293T cells transfected with the wild-type AXIN2 or trAXIN2 expression constructs (Figure 2B).
Figure 2.
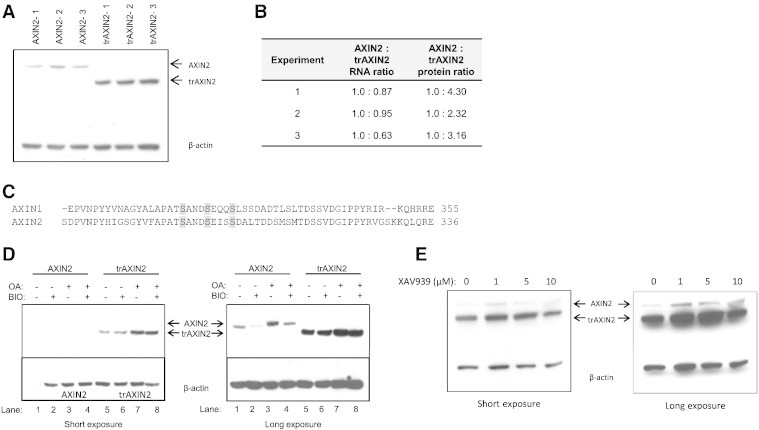
Analysis of AXIN2 and trAXIN2 protein abundance and regulation in transient transfection assays in HEK293T and effects of inhibitors of GSK3β, PP2A, and tankyrase on AXIN2 expression. (A) The trAXIN2 protein is expressed at increased levels in HEK293T cells relative to wild-type AXIN2. HEK293T cells were transfected with equal molar amounts of the AXIN2 or trAXIN2 expression vectors. Protein and RNA were collected for analysis. An IB from protein lysates was prepared from three separate transfection experiments for each of the two constructs, with AXIN2 and trAXIN2 proteins detected by electrochemiluminescence (ECL)-based IBs with an antibody against the FLAG-epitope. (B) Quantification of ectopically expressed AXIN2 transcripts in the transfected HEK293T cells by quantitative RT-PCR using primers in the FLAG sequence and protein levels of AXIN2 protein, based on IB analyses relative to β-actin levels, using ImageJ software. (C) A relevant portion of the AXIN1 and AXIN2 protein sequences are shown, with potential GSK3β phosphorylation sites in the proteins highlighted. (D) IB analysis of AXIN2 and trAXIN2 expression in transiently transfected HEK293T cells ectopically expressing the full-length or trAXIN2 proteins, following a 6-hour BIO and/or OA treatment, with “−” indicating no treatment and “+” indicating treatment. Shorter and longer exposures of the IBs are shown. (E) HEK293T cells were co-transfected with FLAG-tagged AXIN2 and trAXIN2 expression constructs and then treated with increasing doses of XAV939, a tankyrase inhibitor, for 24 hours after transfection, before preparing protein lysates. IB analysis to detect the ectopically expressed AXIN2 and trAXIN2 proteins was carried out with an anti-FLAG antibody.
The stability of the AXIN1 protein has been reported to be regulated by GSK3β phosphorylation and protein phosphatase 2A (PP2A) dephosphorylation [29,30]. The predicted GSK3β phosphorylation sites in AXIN1 are conserved in AXIN2 (Figure 2C). Hence, AXIN2 may be subject to GSK3β phosphorylation-mediated stabilization. The PP2A interaction domain in AXIN1 has been mapped to a region of approximately 200 amino acids in the carboxyl-terminal half of the protein [31]. Assuming PP2A might also interact directly with AXIN2, the trAXIN2 protein might be predicted to lack the PP2A interaction motif, or it may fold in a way as to preclude PP2A interaction. To test the effects of GSK3β and PP2A activity on the wild-type AXIN2 and trAXIN2 proteins, we employed the GSK3β inhibitor, BIO [28], and the PP2A inhibitor, OA [32], for studies of effects on the ectopically expressed proteins in cells. As might be predicted from prior studies of the effects of GSK3β or PP2A inhibition on AXIN1 levels, treatment of transfected HEK293T cells with BIO reduced the levels of the AXIN2 protein, and OA treatment increased AXIN2 protein levels (Figure 2D). The levels of the trAXIN2 were unchanged by BIO treatment, suggesting that the stability of the trAXIN2 protein is not regulated by a GSK3β-dependent mechanism (Figure 2D). However, like wild-type AXIN2, the trAXIN2 protein is stabilized by OA treatment. Another mechanism regulating the AXIN2 protein levels is polyADP-ribosylation (PARsylation), with PARsylation leading to AXIN2 degradation through ubiquitin ligation- and proteasome-dependent mechanisms [33]. Treatment of HEK293T cells ectopically expressing both AXIN2 and trAXIN2 with XAV939, a small molecule inhibitor of tankyrase-mediated PARsylation of AXIN proteins, yielded data suggesting that the wild-type AXIN2 protein was not significantly more sensitive to PARsylation-mediated degradation than trAXIN2 (Figure 2E).
The trAXIN2 Protein Retains Interactions with Components of the β-Catenin Destruction Complex
The trAXIN2 protein, arising from the 1989G>A allele, lacks the protein-encoding sequences from the last three AXIN2 exons, including the so-called disheveled and AXIN (DIX) domain. The DIX domain is a dimerization motif found in the carboxyl-terminal regions of both AXIN1 and AXIN2 [9,34,35]. Lacking a DIX domain, the trAXIN2 protein would be predicted to be unable to dimerize with AXIN proteins, which might affect the formation of the β-catenin destruction complex [36–38]. Expression constructs encoding FLAG- or myc-epitope–tagged AXIN2 and trAXIN2 cDNAs were co-transfected into HEK293T cells, and the interaction between the AXIN2 and trAXIN2 proteins was assessed by IP followed by IB studies. The expression of the epitope-tagged AXIN2 wild-type and trAXIN2 proteins was studied, and the levels of trAXIN2 proteins with either the FLAG- or the myc-tag were higher than those for the corresponding epitope-tagged wild-type AXIN2 proteins (Figure 3A). Unexpectedly, the trAXIN2 protein was found to immunoprecipitate with both AXIN2 and trAXIN2 (Figure 3B, upper), in ratios largely representative of protein expression levels in the cells (Figure 3A). However, when the IP was carried out with the anti-myc antibody and the trAXIN2-myc protein, some reduction was seen in the amount of AXIN2-FLAG protein recovered as compared to the IP with anti-myc antibody and AXIN2-myc (Figure 3B). Overall, the experiments imply that because trAXIN2 does not contain the DIX domain, the trAXIN2 protein interactions with trAXIN2 or full-length AXIN2 are likely not direct but are instead facilitated by another protein in the destruction complex that contains AXIN binding sites, such as APC [3]. Interactions of trAXIN2 and full-length AXIN2 with AXIN1 were also tested, because AXIN1 is a component of the β-catenin destruction complex, and AXIN1 contains a DIX domain that may dimerize with AXIN2 and other AXIN1 molecules. Interestingly, IP of AXIN1 led to the recovery of a greater amount of wild-type AXIN2 protein than with the trAXIN2 protein (Figure 3C), despite lower expression of AXIN2 in HEK293T cells (Figure 3A). This observation suggests that much of the AXIN1-AXIN2 protein interactions detected in the IP assay may reflect direct interaction (i.e., heterodimerization), while the AXIN1-trAXIN2 interaction may be an indirect interaction mediated by APC and/or other proteins. Finally, both AXIN2 and trAXIN2 proteins were found to complex with β-catenin, demonstrating that the trAXIN2 protein is present in β-catenin protein-containing complexes (Figure 3D).
Figure 3.
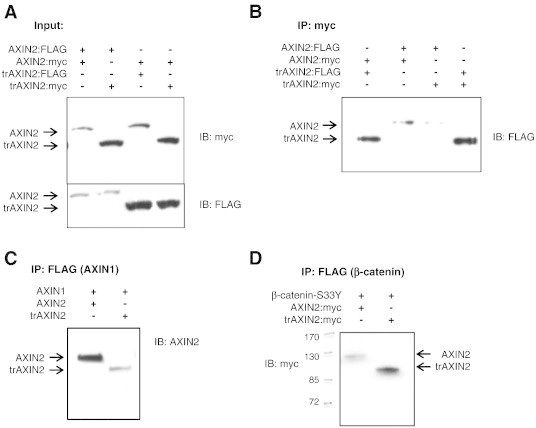
The trAXIN2 protein is present in complexes with AXIN2, AXIN1, and β-catenin. (A) HEK293T cells were transfected with the indicated FLAG- or myc-tagged expression constructs. The expression of the FLAG- and myc-epitope–tagged AXIN2 and trAXIN2 proteins was assessed in IB assays to address the levels of the proteins in the input material used for IP. (B–D) HEK293T cells were transfected with the indicated plasmids. IPs were carried out with the indicated antibodies: anti–myc-epitope antibody in B and anti-FLAG-epitope antibody in C and D (for AXIN1-FLAG and β-catenin–FLAG, respectively), with the studies showing that trAXIN2 can form complexes with trAXIN2, AXIN2, AXIN1, and β-catenin. FLAG–β-catenin IPs (D) were performed following treatment with MG132 to stabilize β-catenin destruction complexes.
Expression of trAXIN2 Inhibits Colony Formation in an APC-Mutant CRC Cell Line
It has previously been shown that small molecules that stabilize the AXIN1 and AXIN2 proteins, such as the XAV939 tankyrase inhibitor, can inhibit the in vitro proliferation and/or survival of some CRC cell lines with APC mutations [33,39]. We sought to determine if the trAXIN2 protein retains this ability to inhibit the growth and/or survival of the SW480 CRC cell line, which has constitutively activated Wnt signaling due to APC mutational inactivation [40]. We also studied the effects of the trAXIN2 protein in the RKO CRC cell line, as it lacks mutations in APC, CTNNB1, or other Wnt pathway factors and it displays no evidence of activated Wnt pathway signaling [41]. These two cell lines were transfected with empty (control) vector, AXIN2, or trAXIN2 expression constructs that also encode resistance to geneticin (G418). After 21 days of G418 selection, the resultant colonies were stained and counted. Similar to the effects seen with wild-type AXIN2, trAXIN2 inhibited colony formation in the SW480 cell line (Figure 4, A and B). The somewhat more potent activity of trAXIN2 in inhibiting SW480 colony formation may result from the higher expression levels of the trAXIN2 protein relative to AXIN2 at early time points after transfection, based on the observed higher expression of trAXIN2 relative to wild-type AXIN2 in the transient transfection assays in HEK293 cells, as described above. In the RKO cell line, we did not observe inhibition of G418-resistant colony formation with wild-type AXIN2 or trAXIN2 (Figure 4, C and D), consistent with the absence of inhibitory effects on RKO cell proliferation and/or survival in published studies with the XAV939 tankyrase inhibitor that stabilizes AXIN1/2 [33]. Interestingly, when selected resultant G418-resistant colonies of SW480 cells were harvested at day 14 after G418 selection and analyzed by IB, detectable expression of trAXIN2 but not wild-type AXIN2 was seen in harvested colonies (Figure 4E and data not shown). These findings suggest that when acutely overexpressed, the trAXIN2 and wild-type AXIN2 proteins may have similar inhibitory effects in APC-mutant colon cancer cells. However, when stably and more modestly expressed in APC-mutant CRC cells, a potential loss-of-function of the trAXIN2 protein in CRC growth suppression may be revealed.
Figure 4.
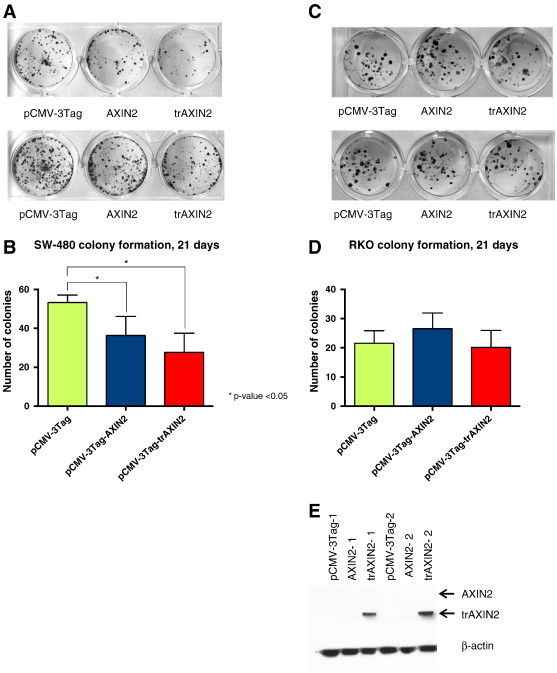
AXIN2 and trAXIN2 inhibit G418-resistant colony formation in a Wnt pathway mutant CRC cell line. G418-resistant colony formation assays were undertaken in the APC mutant SW480 CRC cell line (A and B) and in the RKO CRC cell line that lacks mutations in APC or CTNNB1 (C and D). SW480 and RKO cells were transfected with the indicated plasmids and plated in triplicate at low density under G418 drug selection for 21 days, and then the cells were fixed and stained with crystal violet, so that colonies could be visualized (A and C); experiments were undertaken three separate times to quantify colony numbers (B and D). (E) SW480 cells were transfected with the indicated expression plasmids and grown under G418 selection for 14 days and then lysed for IB analysis with the anti–FLAG-epitope antibody to assess AXIN2 and trAXIN2 protein expression.
trAXIN2 Effects on β-Catenin/TCF-Dependent Reporter Gene and Endogenous Target Gene Activities
One of the primary functions of the AXIN proteins is to promote the turnover of β-catenin, thereby inhibiting the expression of downstream Wnt/β-catenin/TCF-regulated target genes. As expected, Wnt3a treatment of HEK293T cells was found to increase the activity of the well-characterized TCF-dependent luciferase reporter construct, TOPFlash, by roughly nine-fold (Figure 5A). Transient transfection and ectopic overexpression of the wild-type AXIN2 and trAXIN2 proteins potently inhibited TOPFlash reporter gene activity, reducing the Wnt3a-induced reporter gene activity back to the baseline level seen in the non–Wnt3a-treated cells. To assess the effect of the wild-type and trAXIN2 proteins on endogenous target genes when the proteins were expressed at potentially more physiological levels, we generated polyclonal rat intestinal IEC-6 cell lines that were stably transduced with an empty retroviral expression vector or with retroviral constructs encoding wild-type AXIN2 or trAXIN2 and assessed the levels of the transduced AXIN2 proteins using an anti-FLAG epitope antibody (Figure 5B). We then assessed the effects of Wnt3a treatment on expression of multiple candidate Wnt/β-catenin/TCF target genes, including Axin2, Lgr5, Nkd1, Bmp4, Irs1, and Edn1 [42]. The genes most potently induced by Wnt3a treatment were Axin2 and Lgr5, with roughly 20-fold and 10-fold induction, respectively, whereas the other four genes studied were much more modestly induced by Wnt treatment. AXIN2 overexpression was moderately to significantly active in inhibiting the Wnt3a-induced expression of all six target genes (Figure 5C). In contrast, trAXIN2 showed negligible, if any, ability to inhibit Wnt-mediated induction of any of the six target genes (Figure 5C). Curiously, IEC-6 cells stably overexpressing the trAXIN2 protein showed enhanced Lgr5 gene expression in response to Wnt3a treatment (Figure 5C). The basis for the apparent ability of the trAXIN2 protein to enhance Lgr5 expression in the IEC-6 cells in response to Wnt3a treatment is not known but is consistent with the notion that the trAXIN2 mutant protein may have more complex functions than solely a loss-of-function mutation.
Figure 5.
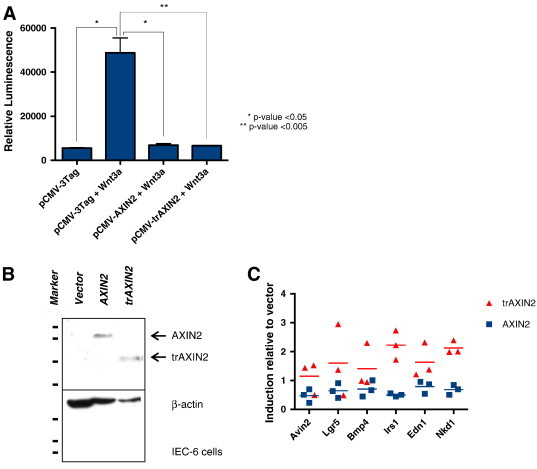
AXIN2- and trAXIN2-mediated inhibition of Wnt/β-catenin/TCF transcriptional targets is context dependent. (A) Transient ectopic expression of AXIN2 and trAXIN2 suppress Wnt3a-mediated TCF reporter gene activity. HEK293T cells were transiently transfected with the pCMV-3Tag vector, AXIN2, or trAXIN2 expression constructs as well as TOPFlash reporter vector. Twenty-four hours after transfection, cells were treated with Wnt3a for 8 hours before harvesting for luciferase assays. Luciferase assays were performed in triplicate and mean and SDs are indicated. (B and C) Stable expression of AXIN2 but not trAXIN2 inhibits Wnt3a-mediated induction of endogenous Wnt/β-catenin/TCF target genes in rat intestinal IEC-6 cells. IEC-6 cells were transduced with empty retroviral expression vector construct or constructs for AXIN2 or trAXIN2, and drug selection was undertaken to create stable polyclonal cell lines. IB studies of the resultant IEC-6 cell lines show stable expression of AXIN2 or trAXIN2, as detected with anti-AXIN2 antibody (B). Stable IEC-6 transductants were treated for 16 hours with Wnt3a. The cells were then harvested, total RNA was collected, and expression of the indicated Wnt/β-catenin/TCF target genes was assessed in three separate quantitative RT-PCR experiments. The individual data points for three independent qPCR experiments with the mean of each group designated by a horizontal line are shown in C.
Discussion
We studied the consequences of the AXIN2 1989G>A (W663X) mutation on AXIN2 protein function using the predicted trAXIN2 protein in cell culture–based assays to assess how its function might contribute to CRC development. The 1989G>A AXIN2 mutation segregates with oligodontia and colorectal neoplasia in an autosomal dominant manner [21]. Whether the mutation influences cancer development through a gain-of-function or a loss-of-function mechanism, or perhaps through both mechanisms, is not yet understood. If the AXIN2 1989G>A allele mRNA was susceptible to NMD, then it would point to a loss-of-function mechanism in colon neoplasia, consistent with the current, more widely held notion that AXIN proteins act as tumor suppressors [19]. However, transcripts from both the wild-type and 1989G>A alleles were detected in PBLs from a heterozygous 1989G>A carrier, suggesting that the 1989G>A allele can encode a trAXIN2 protein product in patient cells. IP studies found that, like wild-type AXIN2, the trAXIN2 protein was present in complexes containing β-catenin as well as AXIN2, trAXIN2, and AXIN1. However, while the trAXIN2 protein retains these interactions, unlike wild-type AXIN2, trAXIN2 likely does not dimerize with the AXIN1 and AXIN2 proteins, because of the absence of the C-terminal DIX domain in trAXIN2. Our transient transfection studies revealed that the trAXIN2 protein was expressed at higher levels than wild-type AXIN2. The expression difference may reflect differences in the mechanisms regulating trAXIN2 stability, as evidenced by the reduction in wild-type AXIN2 but not trAXIN2 protein in transiently transfected HEK293T cells treated with the GSK3β inhibitor, BIO. Like wild-type AXIN2, trAXIN2 potently inhibited the Wnt3a-mediated induction of the TCF reporter, TOPFlash, when highly expressed in transient transfection assays in HEK293T cells. However, when stably expressed at more moderate levels in IEC-6 cell lines, the trAXIN2 protein lacked the ability to inhibit Wnt3a-mediated activation of endogenous Wnt/β-catenin/TCF target genes. Similarly, trAXIN2 was just as effective as wild-type AXIN2 in inhibiting colony formation in APC-mutant SW480 CRC cells, with AXIN2-mediated growth inhibition and/or apoptosis likely resulting from acute effects of overexpression. However, when selected drug-resistant SW480 colonies resulting from transfection were expanded over 14 days, stable expression of trAXIN2 but not wild-type AXIN2 could be seen.
Taken together, our studies and data suggest that the AXIN2 1989G>A mutation is unlikely to have a simple loss-of-function role in colon neoplasia. Rather, the trAXIN2 protein appears to have reduced or absent function in regulating the canonical Wnt pathway when AXIN2 levels are low to moderate, which might be akin to the situation in cells lacking mutations in the canonical Wnt pathway, such as APC mutations. When the 1989G>A allele is highly expressed, such as in CRC cells with constitutive Wnt pathway activation, the trAXIN2 protein might be highly expressed and might otherwise have a function akin to those of wild-type AXIN2 in the canonical Wnt signaling pathway. However, because of its higher levels of expression than wild-type AXIN2 in certain contexts, the trAXIN2 protein could have potential gain-of-function effects, such as in CRC cells with APC mutations or other downstream defects (e.g., mutations affecting β-catenin). These gain-of-function effects for trAXIN2 might be expected to predominantly affect the non-Wnt pathways in which the AXIN1 protein has been shown to function, such as the extracellular signal-regulated kinase (ERK) [43] or c-Jun N-terminal kinase (JNK) pathways [44] or in regulation of GSK3β [45]. In fact, some studies have suggested that high levels of AXIN2 can promote tumor invasion and progression in certain contexts, including in CRC cells [45,46]. A possible means by which high levels of AXIN2 may promote cancer cell invasion is by the ability of AXIN2 to inhibit GSK3β, leading to stabilization of the Snail transcription factor and the resultant induction of epithelial-mesenchymal transition by Snail’s role in transcriptional repression [45]. Hence, it is possible that the trAXIN2 protein could contribute to colon neoplasia through a gain-of-function effect in initiated colon neoplastic cells, perhaps due to elevated expression of a trAXIN2 protein that inhibits GSK3β and stabilizes Snail and/or mediates effects on other known AXIN-regulated pathways, such as ERK or JNK signaling.
In spite of the ability of trAXIN2 to function in an analogous fashion to wild-type AXIN2 in some assays when overexpressed, our findings clearly highlight differences in the regulation and function of the two proteins. A possible contributing mechanism that could alter the stability and/or function of trAXIN2 is that loss of the AXIN2 C-terminus creates a functionally significant change in its protein conformation. A recent study suggested the N- and C-terminal regions of AXIN1 interact to form a protected, “closed” conformation that prevents association of AXIN1 with LRP5/6 [47]. This “closed” conformation of AXIN1 acts to inhibit Wnt signal transduction. This interaction between the N- and C-termini, if conserved in the AXIN2 protein, would be predicted to be abolished for the trAXIN2 protein, perhaps leading to an increased likelihood that a trAXIN2 protein molecule is in an “open” conformation and inactive in the inhibition of Wnt signaling.
As negative regulators of Wnt signaling, the AXIN proteins promote the assembly of complexes that target β-catenin for degradation. As such, the classic view of the AXIN proteins is that they function as tumor suppressors through inhibition of Wnt signaling [19]. However, the mutational evidence to support their role as tumor suppressor genes in cancer in general and CRC specifically is limited [19]. While the Axin2-null mouse has no reported cancer phenotype, studies with mammary stem cells have shown that Axin2-null cells have a more sustained response to Wnt ligands and that Axin2-null mammary stem cells outcompete wild-type cells to repopulate the mammary fat pad [48]. Additionally, stabilization of AXIN1 and AXIN2 proteins in breast cancer cell lines inhibited Wnt signaling, migration, and under low serum conditions, reduced colony formation [49]. Moreover, using a wound-healing model, it was shown that Axin2-null mice display increased Wnt signaling and cell proliferation and decreased cell death, when compared to heterozygous mice [50]. As such, the loss-of-AXIN2 function remains a potential contributing mechanism for the AXIN2G>A allele in predisposition to neoplasia. However, increased stability and effects of trAXIN2 on non-Wnt factors and pathways, such as GSK3β-dependent phosphorylation of targets, such as Snail, or ERK and JNK signaling, may have profound effects on promoting the progression of initiated colon lesions that harbor APC or other Wnt pathway defects that lead to elevated AXIN2 gene expression. The data in this paper highlight the potential role of complex and context-dependent trAXIN2 function in tumorigenesis. Additional studies, perhaps using mouse models, will likely be needed to advance further our understanding of how the trAXIN2 protein contributes to colon neoplasia.
Acknowledgements
We thank Monica Marvin for her continued communication with the proband and Janet Leung for cloning the Axin1 cDNA.
Footnotes
S.M.M. was supported by DOD Award W81XWH-11-1-0147.
References
- 1.Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, Kawasoe T, Ishiguro H, Fujita M, Tokino T. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet. 2000;24:245–250. doi: 10.1038/73448. [DOI] [PubMed] [Google Scholar]
- 2.Feng GJ, Cotta W, Wei XQ, Poetz O, Evans R, Jarde T, Reed K, Meniel V, Williams GT, Clarke AR. Conditional disruption of Axin1 leads to development of liver tumors in mice. Gastroenterology. 2012;143:1650–1659. doi: 10.1053/j.gastro.2012.08.047. [DOI] [PubMed] [Google Scholar]
- 3.Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, Kuhl M, Wedlich D, Birchmeier W. Functional interaction of an axin homolog, conductin, with β-catenin, APC, and GSK3β. Science. 1998;280:596–599. doi: 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- 4.Yamamoto H, Kishida S, Uochi T, Ikeda S, Koyama S, Asashima M, Kikuchi A. Axil, a member of the Axin family, interacts with both glycogen synthase kinase 3β and β-catenin and inhibits axis formation of Xenopus embryos. Mol Cell Biol. 1998;18:2867–2875. doi: 10.1128/mcb.18.5.2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chia IV, Costantini F. Mouse axin and axin2/conductin proteins are functionally equivalent in vivo. Mol Cell Biol. 2005;25:4371–4376. doi: 10.1128/MCB.25.11.4371-4376.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gluecksohn-Schoenheimer S. The effects of a lethal mutation responsible for duplications and twinning in mouse embryos. J Exp Zool. 1949;110:47–76. doi: 10.1002/jez.1401100105. [DOI] [PubMed] [Google Scholar]
- 7.Perry WL, III, Vasicek TJ, Lee JJ, Rossi JM, Zeng L, Zhang T, Tilghman SM, Costantini F. Phenotypic and molecular analysis of a transgenic insertional allele of the mouse Fused locus. Genetics. 1995;141:321–332. doi: 10.1093/genetics/141.1.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu HM, Jerchow B, Sheu TJ, Liu B, Costantini F, Puzas JE, Birchmeier W, Hsu W. The role of Axin2 in calvarial morphogenesis and craniosynostosis. Development. 2005;132:1995–2005. doi: 10.1242/dev.01786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeng L, Fagotto F, Zhang T, Hsu W, Vasicek TJ, Perry WL, III, Lee JJ, Tilghman SM, Gumbiner BM, Costantini F. The mouse Fused locus encodes Axin, an inhibitor of the Wnt signaling pathway that regulates embryonic axis formation. Cell. 1997;90:181–192. doi: 10.1016/s0092-8674(00)80324-4. [DOI] [PubMed] [Google Scholar]
- 10.Leung JY, Kolligs FT, Wu R, Zhai Y, Kuick R, Hanash S, Cho KR, Fearon ER. Activation of AXIN2 expression by β-catenin-T cell factor. A feedback repressor pathway regulating Wnt signaling. J Biol Chem. 2002;277:21657–21665. doi: 10.1074/jbc.M200139200. [DOI] [PubMed] [Google Scholar]
- 11.Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/β-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aulehla A, Wehrle C, Brand-Saberi B, Kemler R, Gossler A, Kanzler B, Herrmann BG. Wnt3a plays a major role in the segmentation clock controlling somitogenesis. Dev Cell. 2003;4:395–406. doi: 10.1016/s1534-5807(03)00055-8. [DOI] [PubMed] [Google Scholar]
- 13.Lee E, Salic A, Kruger R, Heinrich R, Kirschner MW. The roles of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol. 2003;1:E10. doi: 10.1371/journal.pbio.0000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas N Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 16.Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 17.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 18.Fodde R, Edelmann W, Yang K, van Leeuwen C, Carlson C, Renault B, Breukel C, Alt E, Lipkin M, Khan PM. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci U S A. 1994;91:8969–8973. doi: 10.1073/pnas.91.19.8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mazzoni SM, Fearon ER. AXIN1 and AXIN2 variants in gastrointestinal cancers. Cancer Lett. 2014;355:1–8. doi: 10.1016/j.canlet.2014.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I, Pirinen S, Nieminen P. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet. 2004;74:1043–1050. doi: 10.1086/386293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marvin ML, Mazzoni SM, Herron CM, Edwards S, Gruber SB, Petty EM. AXIN2-associated autosomal dominant ectodermal dysplasia and neoplastic syndrome. Am J Med Genet A. 2011;155A:898–902. doi: 10.1002/ajmg.a.33927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolligs FT, Hu G, Dang CV, Fearon ER. Neoplastic transformation of RK3E by mutant β-catenin requires deregulation of Tcf/Lef transcription but not activation of c-myc expression. Mol Cell Biol. 1999;19:5696–5706. doi: 10.1128/mcb.19.8.5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 24.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dogan RI, Getoor L, Wilbur WJ, Mount SM. SplicePort–an interactive splice-site analysis tool. Nucleic Acids Res. 2007;35:W285–W291. doi: 10.1093/nar/gkm407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, Krainer AR. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum Mol Genet. 2006;15:2490–2508. doi: 10.1093/hmg/ddl171. [DOI] [PubMed] [Google Scholar]
- 28.Polychronopoulos P, Magiatis P, Skaltsounis AL, Myrianthopoulos V, Mikros E, Tarricone A, Musacchio A, Roe SM, Pearl L, Leost M. Structural basis for the synthesis of indirubins as potent and selective inhibitors of glycogen synthase kinase-3 and cyclin-dependent kinases. J Med Chem. 2004;47:935–946. doi: 10.1021/jm031016d. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto H, Kishida S, Kishida M, Ikeda S, Takada S, Kikuchi A. Phosphorylation of axin, a Wnt signal negative regulator, by glycogen synthase kinase-3β regulates its stability. J Biol Chem. 1999;274:10681–10684. doi: 10.1074/jbc.274.16.10681. [DOI] [PubMed] [Google Scholar]
- 30.Strovel ET, Wu D, Sussman DJ. Protein phosphatase 2Cα dephosphorylates axin and activates LEF-1-dependent transcription. J Biol Chem. 2000;275:2399–2403. doi: 10.1074/jbc.275.4.2399. [DOI] [PubMed] [Google Scholar]
- 31.Hsu W, Zeng L, Costantini F. Identification of a domain of Axin that binds to the serine/threonine protein phosphatase 2A and a self-binding domain. J Biol Chem. 1999;274:3439–3445. doi: 10.1074/jbc.274.6.3439. [DOI] [PubMed] [Google Scholar]
- 32.Cohen P, Klumpp S, Schelling DL. An improved procedure for identifying and quantitating protein phosphatases in mammalian tissues. FEBS Lett. 1989;250:596–600. doi: 10.1016/0014-5793(89)80803-8. [DOI] [PubMed] [Google Scholar]
- 33.Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–620. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 34.Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3β and β-catenin and promotes GSK-3β-dependent phosphorylation of β-catenin. EMBO J. 1998;17:1371–1384. doi: 10.1093/emboj/17.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mai M, Qian C, Yokomizo A, Smith DI, Liu W. Cloning of the human homolog of conductin (AXIN2), a gene mapping to chromosome 17q23-q24. Genomics. 1999;55:341–344. doi: 10.1006/geno.1998.5650. [DOI] [PubMed] [Google Scholar]
- 36.Sakanaka C, Williams LT. Functional domains of axin. Importance of the C terminus as an oligomerization domain. J Biol Chem. 1999;274:14090–14093. doi: 10.1074/jbc.274.20.14090. [DOI] [PubMed] [Google Scholar]
- 37.Fagotto F, Jho E, Zeng L, Kurth T, Joos T, Kaufmann C, Costantini F. Domains of axin involved in protein-protein interactions, Wnt pathway inhibition, and intracellular localization. J Cell Biol. 1999;145:741–756. doi: 10.1083/jcb.145.4.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kishida S, Yamamoto H, Hino S, Ikeda S, Kishida M, Kikuchi A. DIX domains of Dvl and axin are necessary for protein interactions and their ability to regulate β-catenin stability. Mol Cell Biol. 1999;19:4414–4422. doi: 10.1128/mcb.19.6.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waaler J, Machon O, Tumova L, Dinh H, Korinek V, Wilson SR, Paulsen JE, Pedersen NM, Eide TJ, Machonova O. A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012;72:2822–2832. doi: 10.1158/0008-5472.CAN-11-3336. [DOI] [PubMed] [Google Scholar]
- 40.Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, Koyama K, Utsunomiya J, Baba S, Hedge P. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253:665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 41.da Costa LT, He TC, Yu J, Sparks AB, Morin PJ, Polyak K, Laken S, Vogelstein B, Kinzler KW. CDX2 is mutated in a colorectal cancer with normal APC/β-catenin signaling. Oncogene. 1999;18:5010–5014. doi: 10.1038/sj.onc.1202872. [DOI] [PubMed] [Google Scholar]
- 42.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 43.Jeon SH, Yoon JY, Park YN, Jeong WJ, Kim S, Jho EH, Surh YJ, Choi KY. Axin inhibits extracellular signal-regulated kinase pathway by Ras degradation via β-catenin. J Biol Chem. 2007;282:14482–14492. doi: 10.1074/jbc.M611129200. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y, Neo SY, Wang X, Han J, Lin SC. Axin forms a complex with MEKK1 and activates c-Jun NH(2)-terminal kinase/stress-activated protein kinase through domains distinct from Wnt signaling. J Biol Chem. 1999;274:35247–35254. doi: 10.1074/jbc.274.49.35247. [DOI] [PubMed] [Google Scholar]
- 45.Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, Cha SY, Ryu JK, Choi YJ, Kim J. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8:1398–1406. doi: 10.1038/ncb1508. [DOI] [PubMed] [Google Scholar]
- 46.Wu ZQ, Brabletz T, Fearon E, Willis AL, Hu CY, Li XY, Weiss SJ. Canonical Wnt suppressor, Axin2, promotes colon carcinoma oncogenic activity. Proc Natl Acad Sci U S A. 2012;109:11312–11317. doi: 10.1073/pnas.1203015109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang S, Yin J, Chen D, Nie F, Song X, Fei C, Miao H, Jing C, Ma W, Wang L. Small-molecule modulation of Wnt signaling via modulating the Axin-LRP5/6 interaction. Nat Chem Biol. 2013;9:579–585. doi: 10.1038/nchembio.1309. [DOI] [PubMed] [Google Scholar]
- 48.Zeng YA, Nusse R. Wnt proteins are self-renewal factors for mammary stem cells and promote their long-term expansion in culture. Cell Stem Cell. 2010;6:568–577. doi: 10.1016/j.stem.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bao R, Christova T, Song S, Angers S, Yan X, Attisano L. Inhibition of tankyrases induces Axin stabilization and blocks Wnt signalling in breast cancer cells. PLoS One. 2012;7:e48670. doi: 10.1371/journal.pone.0048670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whyte JL, Smith AA, Liu B, Manzano WR, Evans ND, Dhamdhere GR, Fang MY, Chang HY, Oro AE, Helms JA. Augmenting endogenous Wnt signaling improves skin wound healing. PLoS One. 2013;8:e76883. doi: 10.1371/journal.pone.0076883. [DOI] [PMC free article] [PubMed] [Google Scholar]