

Abstract
The stroma of breast cancer can promote the disease’s progression, but whether its composition and functions are shared among different subtypes is poorly explored. We compared stromal components of a luminal [mouse mammary tumor virus (MMTV)–Neu] and a triple-negative/basal-like [C3(1)–Simian virus 40 large T antigen (Tag)] genetically engineered breast cancer mouse model. The types of cytokines and their expression levels were very different in the two models, as was the extent of innate immune cell infiltration; however, both models showed infiltration of innate immune cells that expressed matrix metalloproteinase 9 (MMP9), an extracellular protease linked to the progression of many types of cancer. By intercrossing with Mmp9 null mice, we found that the absence of MMP9 delayed tumor onset in the C3(1)-Tag model but had no effect on tumor onset in the MMTV-Neu model. We discovered that protein levels of insulin-like growth factor binding protein-1 (IGFBP-1), an MMP9 substrate, were increased in C3(1)-Tag;Mmp9−/− compared to C3(1)-Tag;Mmp9+/+ tumors. In contrast, IGFBP-1 protein expression was low in MMTV-Neu tumors regardless of Mmp9 status. IGFBP-1 binds and antagonizes IGFs, preventing them from activating their receptors to promote cell proliferation and survival. Tumors from C3(1)-Tag;Mmp9−/− mice had reduced IGF-1 receptor phosphorylation, consistent with slower tumor onset. Finally, gene expression analysis of human breast tumors showed that high expression of IGFBP mRNA was strongly correlated with good prognosis but not when MMP9 mRNA was also highly expressed. In conclusion, MMP9 has different effects on breast cancer progression depending on whether IGFBPs are expressed.
Abbreviations: αSMA, α smooth muscle actin; C3(1)-Tag, transgene mouse expressing Simian virus 40 large T antigen under a C3(1) promoter; HER2, human epidermal growth factor receptor 2; IGF, insulin-like growth factor; IGFBP, insulin-like growth factor binding protein; IL, interleukin; MMP, matrix metalloproteinase; MMTV, mouse mammary tumor virus; NF-κB, nuclear factor κB; PyMT, polyoma middle T antigen
Introduction
Breast cancer is classified on the basis of gene expression: About 20% of human breast cancers overexpress human epidermal growth factor receptor 2 (HER2/ErbB2) and these cancers have a worse prognosis than the more common estrogen and progesterone (hormone) receptor–positive subtypes [1]. Cases negative for hormone receptors and ErbB2 (triple negative subtypes) have the worst prognosis and fewest treatment options [2]. Besides the classification based on receptor expression status, breast cancer is also commonly classified into luminal and basal-like subtypes, based on global gene expression profiles [3]. Luminal breast cancers are often estrogen receptor– and/or HER2/ErbB2-positive, while the majority of basal-like breast cancers are triple negative [4].
In the mouse mammary tumor virus long terminal repeat driven-Neu (MMTV-Neu) model [5], the rodent homologue of HER2/ErbB2 is overexpressed. The MMTV-Neu model is a model of human luminal breast cancer as defined by expression profiling [6]. In another breast cancer model, expression of the Simian virus (SV) 40 large T antigen (C3(1)-Tag) [7] in the mammary epithelium induces estrogen receptor–, progesterone receptor–, and HER2-negative tumors [8], with expression profiles resembling human basal-like breast cancers [6]. In the MMTV-Neu model, the activation of the Ras/phosphoinositide 3-kinase pathway drives tumor progression, while in the C3(1)-Tag model, it is the inactivation of p53 and Rb that is the driving force.
Breast tumors contain stromal cells, such as immune, mesenchymal, and vascular cells, which communicate with cancer cells. Genetically engineered mouse models of cancer enable studies of how driving oncogenic changes influence tumor progression in the context of a stromal response [9–11]. The communication between cancer and stromal cells is mediated by, e.g., growth factors, cytokines, proteases, and extracellular matrix proteins [12–14]. These extracellular factors, together with the stromal cells, constitute the tumor microenvironment. Experimental studies have shown that the components of the tumor microenvironment possess functions that are vital for tumor growth, including support of angiogenesis (reviewed in [12]). However, it is still unclear whether specific stromal components have the same effects on tumor progression in different subtypes of cancer arising in the same organ.
Many approaches have been undertaken to block the influence of the microenvironment on cancer progression, including the pharmacological inhibition of matrix metalloproteinases (MMPs) [15–17]. MMPs belong to a large family of proteases involved in the degradation and modulation of extracellular proteins. The enzymatic activities of MMPs are important in the tumor microenvironment because proteolysis can regulate a range of different processes, such as angiogenesis and growth factor bioavailability [18].
Despite strong experimental proof that MMPs promote tumor initiation and progression, clinical trials using MMP inhibitors have failed (reviewed in [15,19]). In hindsight, the trials were not optimally designed: For example, they enrolled patients with late-stage cancer even though preclinical experiments showed that MMPs should be blocked in early-stage cancer [20]. Furthermore, no effort was made to ensure that MMPs were overexpressed in the cancer of the treated patients [19]. The design of the clinical trials also ignored the fact that MMPs can have different effects on tumor progression depending on the substrates they act on [21].
MMP9, one of the most studied MMPs in cancer, is mainly expressed by tumor-infiltrating myeloid cells [20,22–25]. Deletion of Mmp9, the gene encoding for MMP9, delays tumor onset or slows tumor progression in many genetically engineered mouse models of cancer [20,22–25]. Interestingly, in these models, the p53 and Rb tumor suppressors were inactivated, e.g., by interaction with the SV40 large T antigen or human papilloma virus early region oncogenes [20,22,23]. In contrast, the genetic deletion of Mmp9 in a model of mammary carcinoma driven by expression of tyrosine kinases [MMTV–polyoma middle T antigen (PyMT)] had no effect on tumor onset or primary tumor growth [24]. It is unclear whether this difference in the effects of MMP9 on tumor progression between the models was due to the different oncogenic events that drive the cancers or because the tumors originated from different tissues.
In this study, we compared expression levels of different stromal factors between the luminal MMTV-Neu and the basal-like C3(1)-Tag murine breast cancer models. Interestingly, we found that MMP9 was expressed by myeloid cells in both models, yet it only influenced tumor onset in the basal-like C3(1)-Tag model. We discovered that the protein levels of the MMP9 substrate insulin-like growth factor binding protein-1 (IGFBP-1) were increased in the absence of MMP9 only in the C3(1)-Tag model, the model that depended on MMP9 for tumor progression. Furthermore, in data sets of human breast cancer samples, high mRNA expression of IGFBPs correlated with a good prognosis, except when these tumors also expressed high levels of MMP9 mRNA. Collectively, our findings show that MMP9 and IGFBPs have different subtype-dependent effects on breast cancer and that a nuanced understanding of tumor biology is necessary to successfully target these stromal factors.
Materials and Methods
Mice
MMTV-Neu [5], C3(1)-Tag [7], and Mmp9−/− [26] mice have all been described previously. All three mice strains were used on the FVB/n background. MMTV-Neu and C3(1)-Tag mice were each crossed with Mmp9−/− mice, and the offspring were further intercrossed to generate MMTV-Neu;Mmp9+/+, MMTV-Neu;Mmp9+/−, MMTV-Neu;Mmp9−/−, C3(1)-Tag;Mmp9+/+, C3(1)-Tag;Mmp9+/−, and C3(1)-Tag;Mmp9−/− mice. Only mice hemizygous for the MMTV-Neu or C3(1)-Tag transgenes were used to compare tumor onset. All animal experiments were conducted in accordance with procedures approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco.
Tumor Growth
Tumor growth was monitored in all 10 mammary glands by weekly palpations in MMTV-Neu;Mmp9+/+ (n = 47), MMTV-Neu;Mmp9+/− (n = 48), MMTV-Neu;Mmp9−/− (n = 39), C(3)1-Tag;Mmp-9+/+ (n = 18), C(3)1-Tag;Mmp9+/− (n = 25), and C(3)1-Tag;Mmp9−/− (n = 13) mice. The length and width of all palpable tumors were measured by caliper, and the volume was calculated using the formula: volume = width2 × length/2.
Histology and Immunostaining
Dissected mammary carcinomas and lungs were fixed in 4% paraformaldehyde, processed in alcohol, embedded in paraffin, and cut into 5-μm-thick sections. Sections were stained with hematoxylin and eosin, Mayer's hematoxylin with Masson's trichrome, or Picrosirius Red using standard protocols. Lung metastatic burden was examined in tissue sections from lungs of C3(1)-Tag;Mmp9+/+ (n = 21), C3(1)-Tag;Mmp9+/− (n = 19), and C3(1)-Tag;Mmp9−/− (n = 14) mice, which were sacrificed at Institutional Animal Care and Use Committee–approved end stage (i.e., when the largest tumor reached 2 cm in diameter or a tumor had ulcerated). The percentage of the lung area occupied by metastasis, the number of metastatic foci, and the average size of each metastatic lesion were quantified on cross sections of lungs stained with hematoxylin and eosin using ImageScope software (Aperio Technologies, Vista, CA). Fibrillar collagen was visualized using Picrosirius Red-stained sections observed under cross-polarized light.
For immunostainings, antigen retrieval was performed by incubation with proteinase K (S3020; DAKO, Glostrup, Denmark) for 5 minutes at 37°C. Before adding primary antibodies, sections were blocked with a mixture of goat serum and 2.5% BSA (blocking buffer). Antibodies against Ly6B.2 (clone 7/4, rat anti-mouse mAb, #CL8993 AP-1, Cedarlane Laboratories, Burlington, NC) and F4/80 (rat anti-mouse mAb, MCA497R; Serotec, Raleigh, NC) were used at 1:400 dilutions in blocking buffer. Biotinylated anti-rat IgG Fab fragment (sc-3826; Santa Cruz Biotechnology, Dallas, TX) was diluted 1:400 in blocking buffer followed by incubation with VECTASTAIN ABC-Elite solution (Vector Labs, Burlingame, CA). To detect IGFBP-1 and phospho–IGF-1 receptor (IGF-1R), the tumor sections were heat-treated in Tris-EDTA (10 mM Tris, 1 mM EDTA, pH 9.0) buffer for 6 minutes before blocking and incubation with anti–IGFBP-1 (1:200 dilution, sc-13097; Santa Cruz Biotechnology) or anti-phospho (Y1161) IGF-1R (1:100 dilution, ab39398; Abcam, Cambridge, MA). Antibody binding was detected with biotin-conjugated anti-rabbit antibodies (1:250 dilution, sc-3840; Santa Cruz Biotechnology) and VECTASTAIN ABC-Elite solution and visualized by 3,3′-diaminobenzidine chromogen (Vector Labs). To quantify IGFBP-1 and phopho–IGF-1R proteins in tumor sections, we first stained for these antigens and then scanned tissue sections from C3(1)-Tag;Mmp9+/+ (n = 10) and C3(1)-Tag;Mmp9−/− (n = 10) tumors. We then used ImageScope software with the Color Deconvolution Algorithm to quantify the signal intensity of 3,3′-diaminobenzidine staining (using the H-score: the sum of three times the area with strong staining intensity and two times area with medium staining intensity) to compare signal intensity.
The number of cells positive for Ly6B.2 (neutrophils/monocytes) and F4/80 (macrophages) was quantified in tumors from MMTV-Neu;Mmp9+/+ (n = 3), MMTV-Neu;Mmp9−/− (n = 4), C(3)-1-Tag;Mmp9+/+ (n = 5), and C(3)-1-Tag;Mmp9−/− (n = 4) mice. Four representative images were acquired from the central parts of each tumor, and the number of positive cells was counted using NIH ImageJ software. For immunofluorescence staining of MMP9, a rabbit anti-mouse polyclonal MMP9 antibody [27] was used at 1 μg/ml together with the Ly6B.2 and F4/80 antibodies. Sections were incubated with the primary antibodies for 2 hours at room temperature before detection with Alexa Fluor 568–conjugated goat anti-rat IgG (Invitrogen, Carlsbad, CA) or Alexa Fluor 647–conjugated goat anti-rabbit IgG (Invitrogen). Secondary antibodies were diluted 1:150 in blocking buffer and incubated for 30 minutes at room temperature, followed by the addition of 4′,6-diamidino-2-phenylindole for labeling of cell nuclei. Sections were mounted with Fluorogel (Electron Microscopy Sciences, Hatfield, PA) and sealed with Cytoseal (Thermo Fisher Scientific Inc, Waltham, MA). The percentage of cells double positive for MMP9 and either Ly6B.2 (neutrophils/monocytes) or F4/80 (macrophages) was quantified in tumors from MMTV-Neu (n = 5 for neutrophils and n = 4 for macrophages) and C(3)-1-Tag mice (n = 5 for both neutrophils and macrophages) at the tumor-stroma interphase (where most inflammatory cells were found) using NIH ImageJ software.
Examination of Angiogenesis
Tumor vascularity and pericyte coverage of the vasculature were determined as previously described [28]. Briefly, tumor sections of MMTV-Neu;Mmp9+/+ (n = 8), C3(1)-Tag;Mmp9+/+ (n = 8), and C3(1)-Tag;Mmp9−/− (n = 8) mice were double immunofluorescence stained with anti-CD31 (1:25 dilution, ab28364; Abcam) and α smooth muscle actin (αSMA; 1:1500 dilution, A5228; Sigma-Aldrich, St Louis, MO). CD31-positive (endothelial cell marker) and αSMA-positive (pericyte marker) areas were defined as those with a fluorescence intensity above 150% of the mean of background intensity levels, as determined by Volocity software (PerkinElmer, Waltham, MA). Pericyte coverage was calculated by dividing the αSMA-positive (pericyte) area by the CD31-positive (endothelial cell) area. All images were analyzed without knowledge of mouse genotype.
Protein Arrays
Tumors were harvested, snap frozen, and stored in liquid nitrogen until use. The frozen tumor samples were homogenized on ice and transferred to lysis buffer (1% NP-40, 20 mM Tris-HCl, 137 mM NaCl, 10% glycerol, 2 mM EDTA, pH 8.0, containing EDTA-free proteinase inhibitor cocktail) for 30 minutes at 4°C on a rocking table. Lysates were centrifuged twice at 13,000g for 10 minutes at 4°C. Samples were analyzed using a Mouse Cytokine Antibody Array, Panel A and a Mouse Angiogenesis Antibody Array (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. Relative protein levels were determined by the intensity of spots representing a single protein on radiographic films. The intensity of spots was analyzed by Adobe Photoshop (version CS6) and normalized to internal standards on the arrays. Normalized data for individual tumors were plotted in a heat map and ranked according to the detected levels, with the highest at the top. For proteins analyzed by both the Cytokine and Angiogenesis arrays, highly reproducible expression levels were obtained when lysates from the same tumor were tested on the two different arrays. These proteins are therefore only represented in the figures for the cytokine/chemokine arrays. Statistical significance was determined using multiple t tests with the Holm-Šídák correction for multiple comparisons, with α = 0.05, using GraphPad Prism software (version 6; GraphPad Software, La Jolla, CA).
Necrosis Quantification
Micrographs of Masson’s trichrome–stained C3(1)-Tag tumor sections were captured using a Panoramic MIDI slide scanner (3DHISTECH Ltd, Budapest, Hungary), which provided overview pictures of the entire tissue section. Using 3DHISTECH software, necrosis was defined as areas with pale staining, containing dead cells and infiltrating leukocytes. The necrotic areas as percentage of the total tumor area were calculated for each tissue section.
Survival Analysis for Human Breast Cancer Patients
Four expression profile data sets from whole tumor tissue (including both cancer and stromal cells) of human breast cancer with clinical information were used: GSE1456 (overall survival, n = 159) [29], GSE2034 (metastasis-free survival, n = 286) [30], GSE2990 (relapse-free survival, n = 187) [31], and GSE3494 (overall survival, n = 236) [32]. Survival analysis based on expression levels of MMP9 and IGFBPs was conducted as previously described [33]. Briefly, signal intensities of each probe were acquired from the Gene Expression Omnibus database and analyzed by Expression Console software (Affymetrix, Santa Clara, CA). The cut-off level for dividing patients into the high or low expression groups was determined by the X-Tile software [34] and was followed by Kaplan-Meier survival analysis using GraphPad Prism (version 5; GraphPad Software).
Statistical Analysis of Data from Mouse Tumors
GraphPad Prism version 5 (GraphPad Software) was used to evaluate myeloid cell infiltration data, chemokine levels, and tumor-free survival. For all analyses, α = 0.05.
Results
Increased Infiltration of Myeloid-Derived Innate Immune Cells in C3(1)-Tag Compared to MMTV-Neu Tumors
We first compared the histology of mammary tumors in MMTV-Neu and C3(1)-Tag mice. There was a pronounced increase in stromal components, including collagen deposition, in C3(1)-Tag lesions versus MMTV-Neu lesions. This difference was already observable at the pre-invasive mammary intraepithelial neoplasia stage and became even more striking as the tumor progressed from low- to high-grade carcinomas (Figures 1A and S1). Collagen fibers were interspersed between the cancer cells in the C3(1)-Tag tumors, while they were almost exclusively found at the tumor-stroma border in MMTV-Neu tumors (Figure S1).
Figure 1.
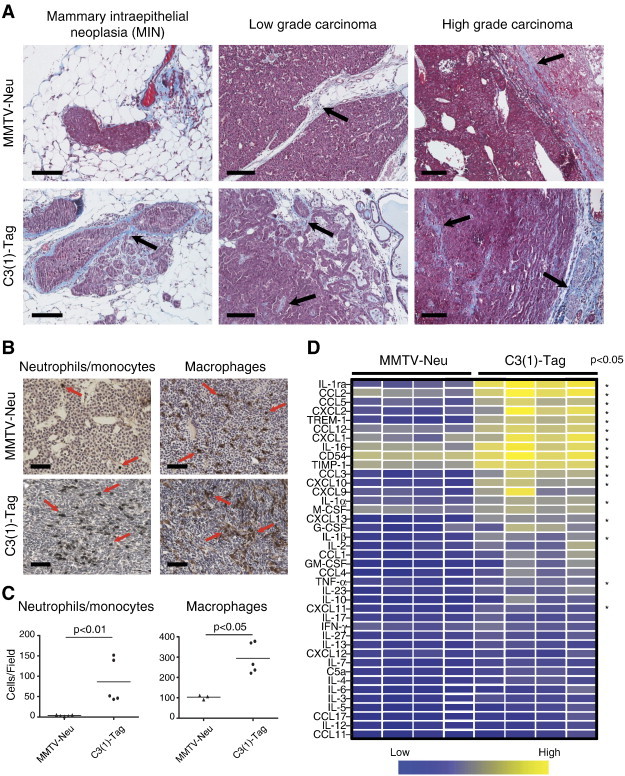
The tumor stroma of the MMTV-Neu and C3(1)-Tag mouse models of breast cancer is different. (A) Representative images of mammary tumors at different stages from MMTV-Neu and C3(1)-Tag mice. Tumors were stained with hematoxylin and Masson’s trichrome for visualization of collagen (which stains blue). Black arrows indicate deposition of collagen in the tumor microenvironment. Scale bars, 100 μm. (B) Myeloid cell infiltration in carcinomas from MMTV-Neu and C3(1)-Tag mice. The tumors were stained for reactivity against antigen Ly6B.2 (clone 7/4, expressed on neutrophils/monocytes) and F4/80 (expressed on macrophages). Red arrows indicate positively stained cells. Scale bars, 100 μm. (C) Significantly more neutrophils and macrophages were detected in tumors from C3(1)-Tag (n = 5) than MMTV-Neu (n = 3 for macrophage analysis and n = 5 for neutrophil analysis) mice (P = .008 and P = .04, respectively, Mann-Whitney, two-sided). (D) Relative protein levels of a panel of chemokines/cytokines measured on whole-tumor lysates from MMTV-Neu (n = 4) and C3(1)-Tag (n = 4) tumors. Each column represents a tumor from an individual mouse. The measured protein levels were normalized to an internal assay standard and plotted in a heat map. Statistical significance was determined by t tests using the Holm- Šídák method to correct for multiple comparisons, and the asterisk indicates P < .05.
Figure S1.
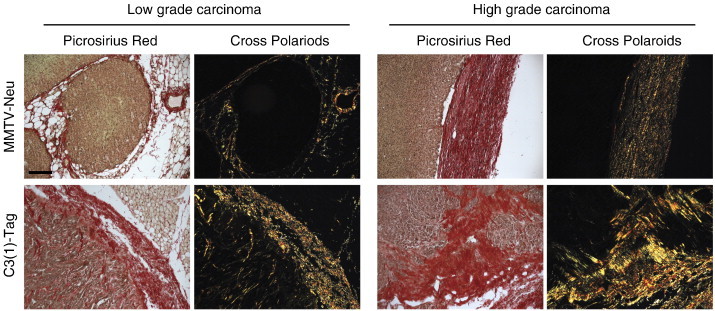
Difference in tumor stroma between MMTV-Neu and C3(1)-Tag mouse models. Picrosirius Red staining of low- and high-grade tumors from MMTV-Neu and C3(1)-Tag mice. The same fields are shown in bright-field (all collagens stain red) and under cross-polarized light (fibrillar collagens are birefringent, appearing yellow orange). Scale bar, 100 μm.
We first compared the histology of mammary tumors in MMTV-Neu and C3(1)-Tag mice. There was a pronounced increase in stromal components, including collagen deposition, in C3(1)-Tag lesions versus MMTV-Neu lesions. This difference was already observable at the pre-invasive mammary intraepithelial neoplasia stage and became even more striking as the tumor progressed from low- to high-grade carcinomas (Figures 1A and S1). Collagen fibers were interspersed between the cancer cells in the C3(1)-Tag tumors, while they were almost exclusively found at the tumor-stroma border in MMTV-Neu tumors (Figure S1).
We next compared the infiltration of two broad classes of myeloid cells, neutrophils/monocytes and macrophages, in the two cancer models by immunohistochemistry (Figure 1B). C3(1)-Tag tumors had 25-fold higher numbers of cells expressing the neutrophil/monocyte marker 7/4 (P = .008, two-sided Mann-Whitney) and a three-fold higher infiltration of cells expressing the macrophage marker F4/80 (P = .04, two-sided Mann-Whitney) than MMTV-Neu tumors (Figure 1C). The pronounced difference in the infiltration of innate immune cells between the two breast cancer models suggested different cytokine milieus. We therefore surveyed the relative protein levels of a panel of cytokines in whole-tumor lysates using an antibody-based protein array (Figure 1D). About half of the assayed cytokines, including CCL2, CCL5, CXCL2, CXCL9, and CXCL10, were present at significantly higher levels in the C3(1)-Tag tumors than in the MMTV-Neu tumors (P < .05, t tests and using the Holm-Šídák method to correct for multiple comparisons). These CC and CXC chemokine family members are chemotactic for, e.g., neutrophils, monocytes, and macrophages [35,36].
MMP9 Influences Tumor Onset of C3(1)-Tag, but Not MMTV-Neu, Mouse Mammary Carcinomas
Tumor-infiltrating myeloid cells are important contributors of tumor angiogenesis [37]. Therefore, the significantly higher levels of myeloid cells in the C3(1)-Tag tumors suggested that vascularity would be different between the two models. Yet, the number of tumor vessels was similar in the MMTV-Neu and C3(1)-Tag tumors (Figure 2A). The protein levels of a panel of angiogenic factors were also similar between the two models, except pentraxin-3, endothelin-1, and amphiregulin, which were all increased in the C3(1)-Tag compared to the MMTV-Neu tumors (Figure 2B; P < .05 after using the Holm-Šídák method to correct for multiple comparisons).
Figure 2.
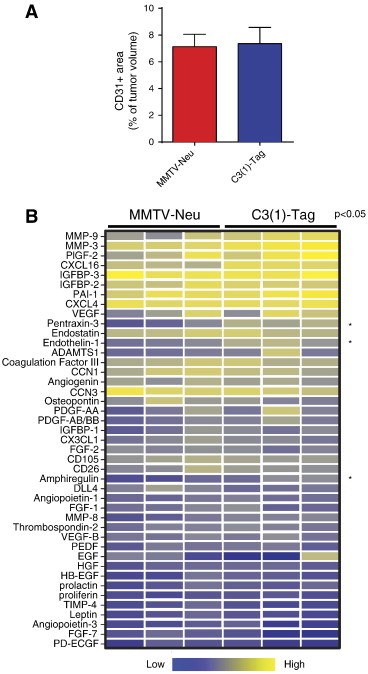
Similar angiogenic microenvironments in the MMTV-Neu and C3(1)-Tag mouse models of breast cancer. (A) The vascularity, as determined by the tumor area staining for the endothelial marker CD31, was similar in MMTV-Neu (n = 8) and C3(1)-Tag tumors (n = 8). (B) Relative protein levels of a panel of angiogenic factors measured on whole-tumor lysates from MMTV-Neu (n = 3) and C3(1)-Tag (n = 3) tumors. Each column represents a tumor from an individual mouse. The measured protein levels were normalized to an internal assay standard and plotted in a heat map. Statistical significance was determined by t tests using the Holm- Šídák method to correct for multiple comparisons, and the asterisk indicates P < .05.
MMP9 promotes tumor progression in several mouse models of cancer [20,22,23,25]. Therefore, we next compared the effect of MMP9 on tumor progression between our two models of different breast cancer subtypes, focusing on high-grade carcinomas. We found that in both subtype models, cells infiltrating the central, necrotic areas of the tumors and cells located in the stroma at the periphery of the tumors expressed MMP9. These MMP9-expressing cells co-expressed either the neutrophil/monocyte marker or the macrophage marker (Figure 3, A and B). The percentages of macrophages and neutrophils that expressed MMP9 were similar between the models (53-60% of neutrophils and 7% of macrophages; Figure 3, C and D). However, consistent with the very limited infiltration of neutrophils in the MMTV-Neu model (Figure 1C), most of the MMP9-expressing cells were macrophages in the MMTV-Neu model, while most of the MMP9-expressing cells were neutrophils in the C3(1)-Tag model (Figure 3, E and F).
Figure 3.
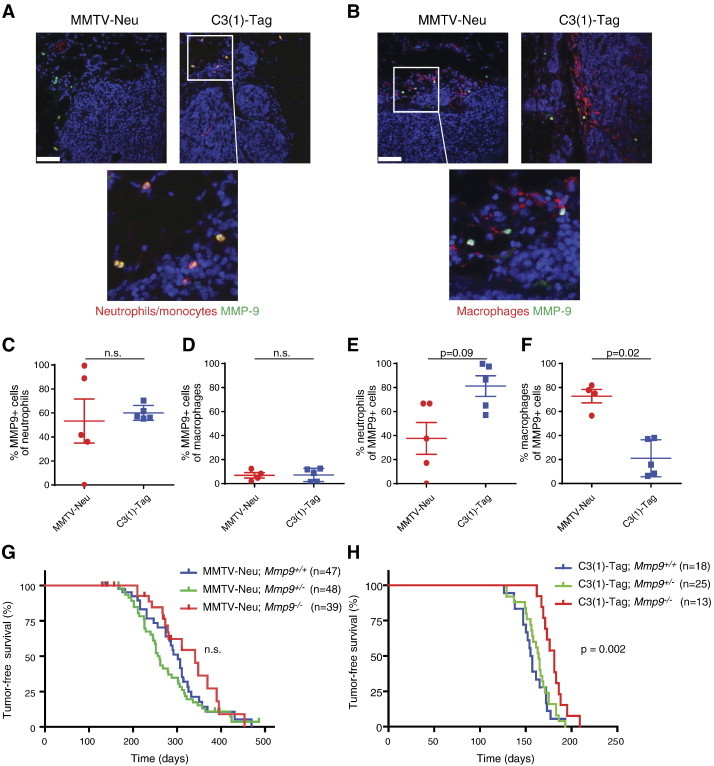
MMP9 promotes tumor onset in the C3(1)-Tag but not in the MMTV-Neu mouse model of breast cancer. (A) MMP9 is expressed in neutrophils/monocytes and (B) macrophages in tumors of both MMTV-Neu and C3(1)-Tag mice. Tumors were stained for reactivity against Ly6B.2 (clone 7/4, for neutrophils/monocytes), F4/80 (for macrophages), and MMP9. Lower panels are high magnifications of indicated areas. All sections were counterstained with 4′,6-diamidino-2-phenylindole for visualization of cell nuclei. Scale bars, 50 μm. (C and D) The percentages of neutrophils/monocytes and macrophages expressing MMP9 were the same in the MMTV-Neu and C3(1)-Tag models (n.s., not significant, Mann-Whitney, two-sided). (E and F) Neutrophils/monocytes constituted a higher percentage of MMP9-expressing cells in the C3(1)-Tag models than in the MMTV-Neu model (P = .09, Mann-Whitney), while macrophages constituted a higher percentage of MMP9-expressing cells in the MMTV-Neu model than in the C3(1)-Tag model (P = .02, Mann-Whitney, two-sided). (G) Kaplan-Meier curves showing tumor-free survival of MMTV-Neu;Mmp9+/+, MMTV-Neu;Mmp9+/−, and MMTV-Neu;Mmp9−/− mice. No significant differences among the curves were observed. (H) Kaplan-Meier curves showing tumor-free survival of C3(1)-Tag;Mmp9+/+, C3(1)-Tag;Mmp9+/−, and C3(1)-Tag;Mmp9−/− mice. A significant delay (P = .002, log rank test) in tumor onset was observed in C3(1)-Tag;Mmp9−/− compared to C3(1)-Tag;Mmp9+/+ mice.
To investigate the effect of MMP9 on tumor progression in our models, we crossed Mmp9−/− mice with MMTV-Neu and C3(1)-Tag mice. We found that the absence of Mmp9 significantly delayed tumor onset in the C3(1)-Tag model but had no effect on tumor onset in the MMTV-Neu model (Figure 3, G and H). MMP9 has been shown to promote lung metastasis in the MMTV-PyMT mouse model of breast cancer [24], but we did not find any difference in overall lung metastatic burden, in the number of metastatic foci, nor in the average size of the metastatic foci between tumors of Mmp9+/+, Mmp9+/−, and Mmp9−/− mice of the C3(1)-Tag strain (Figure S2).
Figure S2.
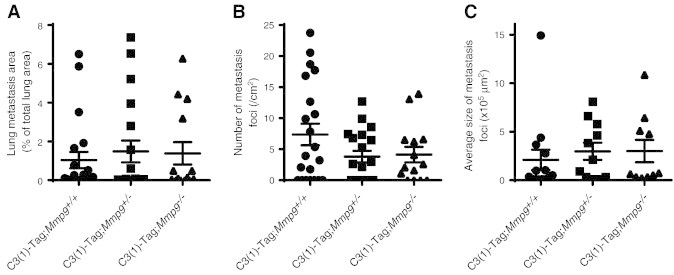
Lung metastasis in C3(1)-Tag mice was not influenced by Mmp9 status. The lung metastatic burden was examined by histology in C3(1)-Tag;Mmp9+/+ (n = 21), C3(1)-Tag;Mmp9+/− (n = 19), and C3(1)-Tag;Mmp9−/− (n = 14) mice at end stage. There were no significant differences in (A) total lung metastatic burden, (B) number of metastatic foci, nor (C) average size of metastatic foci when comparing C3(1)-Tag;Mmp9+/+, C3(1)-Tag;Mmp9+/−, and C3(1)-Tag;Mmp9−/− mice (one-way ANOVA).
To investigate the effect of MMP9 on tumor progression in our models, we crossed Mmp9−/− mice with MMTV-Neu and C3(1)-Tag mice. We found that the absence of Mmp9 significantly delayed tumor onset in the C3(1)-Tag model but had no effect on tumor onset in the MMTV-Neu model (Figure 3, G and H). MMP9 has been shown to promote lung metastasis in the MMTV-PyMT mouse model of breast cancer [24], but we did not find any difference in overall lung metastatic burden, in the number of metastatic foci, nor in the average size of the metastatic foci between tumors of Mmp9+/+, Mmp9+/−, and Mmp9−/− mice of the C3(1)-Tag strain (Figure S2).
Absence of MMP9 Increases IGFBP-1 Protein and Suppresses the IGF Signaling Pathway
The simplest explanation for the difference in effect of MMP9 on tumor onset between the two breast cancer models might be the slightly higher MMP9 expression levels in the C3(1)-Tag versus MMTV-Neu tumors. However, since MMP9 acts through proteolysis of its substrates, differences in expression levels of MMP9 substrates could be a contributing factor. MMPs can modulate cytokine bioactivity by proteolysis [18]. We therefore investigated whether cytokine protein levels increased in the absence of MMP9, when there was no MMP9-mediated proteolysis. No significant increases in cytokine protein levels were found in C3(1)-Tag;Mmp9−/− tumors compared to C3(1)-Tag;Mmp9+/+ tumors (Figure 4A), except for a decrease in interleukin-1α (IL-1α). Comparison of the MMTV-Neu;Mmp9+/+ and MMTV-Neu;Mmp9−/− tumors also revealed no significant changes (Figure 4B). Consistently, in the absence of MMP9, there were no significant changes in macrophage or neutrophil infiltration (Figure 4C).
Figure 4.
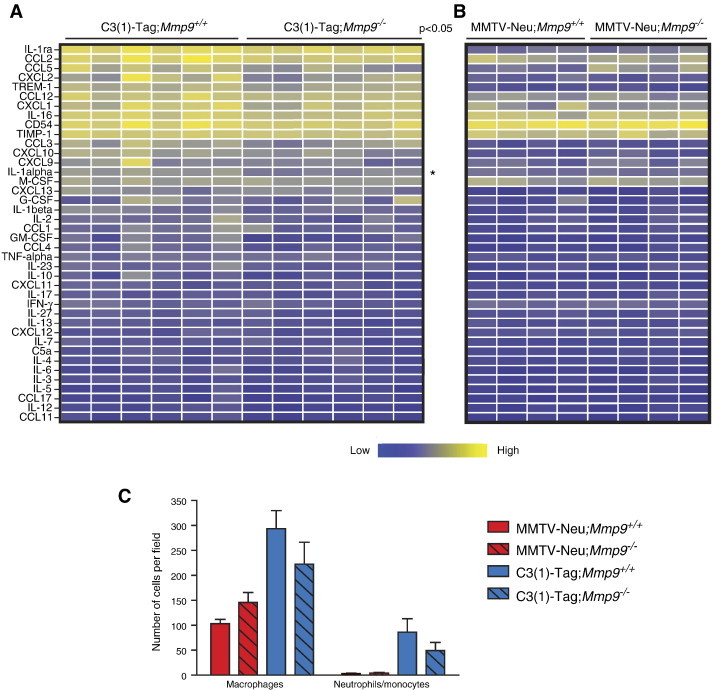
The absence of MMP9 leads to minimal changes in the cytokine environment in the C3(1)-Tag and MMTV-Neu mouse models of breast cancer. (A) Relative protein levels of a panel of cytokines in C3(1)-Tag;Mmp9+/+ versus C3(1)-Tag;Mmp9−/− and (B) MMTV-Neu;Mmp9+/+ versus MMTV-Neu;Mmp9−/− tumors. Each column represents a tumor from an individual mouse. The measured protein levels were normalized to an internal assay standard and plotted in a heat map. Statistical significance was determined by t tests using the Holm- Šídák method to correct for multiple comparisons, and the asterisk indicates P < .05. No differences were significant for B after correction for multiple comparisons. (C) The infiltration of macrophages and neutrophils/monocytes was not significantly influenced by Mmp9 gene expression in tumors from MMTV-Neu nor C3(1)-Tag mice. Infiltration was determined by immunohistochemistry of tumors from mice of the indicated genotypes.
MMP9 has complex effects on the tumor vasculature and can, for example, both promote the formation of tumor vessels and decrease vascular leakiness depending on tumor model and tumor stage ([20]; [25,28,38]). We previously reported that deletion of Mmp9 in MMTV-Neu tumors had no effect on the number of vessels but resulted in reduced coverage by pericytes [28]. Pericytes support endothelial cells, thereby reducing vascular leakiness. In the C3(1)-Tag model, deletion of Mmp9 instead led to a reduction in the number of tumor vessels, but the vessels had the same amount of vascular coverage by pericytes (Figure 5, A and B). Consistent with reduced vascularity, an analysis of histologic sections of C3(1)-Tag tumors revealed a non-significant trend toward increased necrosis in the absence of MMP9 (Figure 5, C and D; P = .1, two-sided Student’s t test, comparing size-matched tumors).
Figure 5.
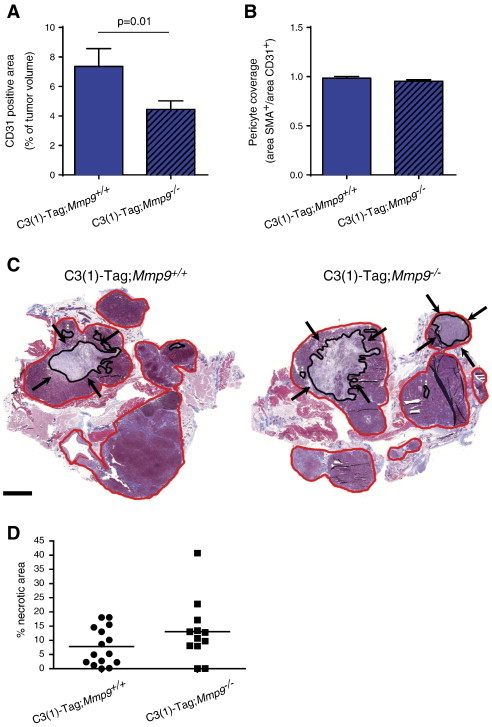
Tumor vascularity is reduced in the absence of MMP9 in the C3(1)-Tag model. (A) The CD31+ endothelial cell area was significantly decreased in C3(1)-Tag;Mmp9−/− tumors (P = .01, t test), while (B) the relative vascular coverage by αSMA-positive pericytes was not altered, as determined by double immunofluorescent staining using antibodies against CD31 and αSMA. For each genotype, eight tumors were stained and quantified. (C) Areas of necrosis were observed in Masson’s trichrome–stained tumor sections from C3-Tag;Mmp9+/+ and C3-Tag;Mmp9−/− mice. Representative examples of tumor sections are shown, with total tumor area (red line) and total necrotic area (black line and marked by black arrows) indicated. Scale bar, 2 mm. (D) A non-significant trend for increased necrosis area was observed in C3(1)-Tag;Mmp9−/− (n = 12) compared to C3(1)-Tag;Mmp9+/+ (n = 15) mice by quantification of necrotic areas (P = .1, t test).
The reduced vascularity of the C3(1)-Tag;Mmp9−/− compared to the C3(1)-Tag;Mmp9+/+ tumors led us to survey for differences in the levels of angiogenic factors. IGFBP-1 is a known MMP9 substrate [39,40]. Protein levels of IGFBP-1 were increased in the absence of Mmp9 in the C3(1)-Tag tumors (Figure 6A; P = .02, t test; the difference was not significant when corrected for multiple comparisons using the Holm-Šídák method). Immunostaining confirmed that there was more IGFBP-1 protein in C3(1)-Tag;Mmp9−/− tumors compared to C3(1)-Tag;Mmp9+/+ tumors (Figure 6, C and D). In the MMTV tumors, IGFBP-1 protein levels were very low and not influenced by Mmp9 status (Figure 6B; P = .73, t test, also not significant using the Holm-Šídák method).
Figure 6.
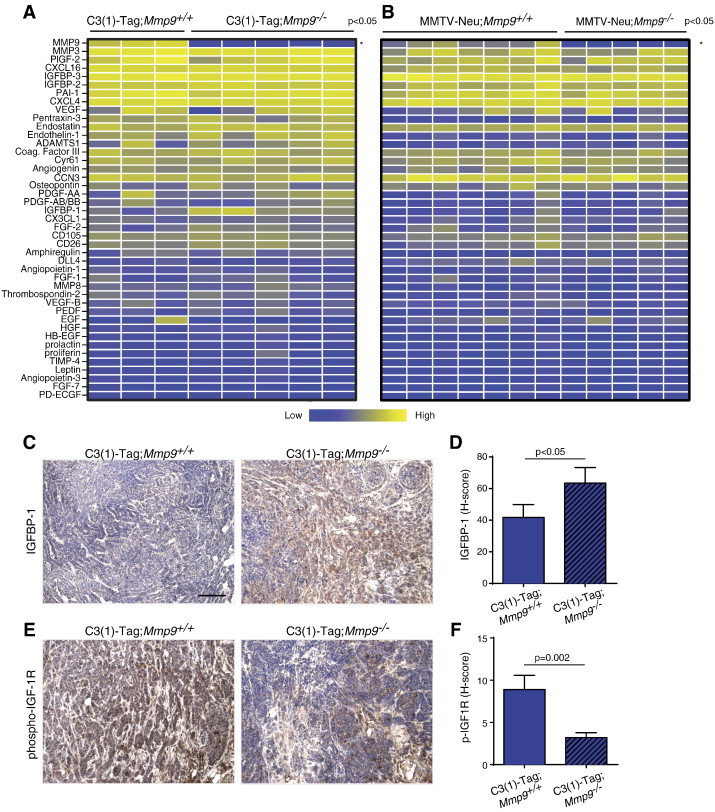
The absence of MMP9 increases IGFBP-1 protein and suppresses the IGF signaling pathway. (A) Relative protein levels of a panel of angiogenic factors in C3(1)-Tag;Mmp9+/+ versus C3(1)-Tag;Mmp9−/− and (B) MMTV-Neu;Mmp9+/+ versus MMTV-Neu;Mmp9−/− tumors. Each column represents a tumor from an individual mouse. The measured protein levels were normalized to an internal assay standard and plotted in a heat map. Statistical significance was determined by t tests using the Holm-Šídák method to correct for multiple comparisons, and the asterisk indicates P < .05. Only MMP9 protein levels were significantly different after correction for multiple comparisons. (C) Representative images of immunohistochemistry with antibodies against IGFBP-1 and (E) phospho IGF-1R on tumor sections from C3(1)-Tag;Mmp9+/+ (n = 10) and C3(1)-Tag;Mmp9−/− (n = 10) mice. Positive staining is shown in brown; slides were counterstained with hematoxylin. Quantification of the (D) increase in IGFBP-1 protein (P = .05, t test) and (F) decrease in phosphorylated IGF-1R protein (P = .002, t test) in tumors of C3(1)-Tag;Mmp9−/− mice. The H-score was determined by ImageScope software after scanning the stained slides and was calculated as the sum of three times the area of strong staining intensity and two times the area of medium staining intensity. Scale bar, 100 μm.
IGFBP-1 binds to IGF, preventing it from binding to and activating its receptors. We used immunohistochemistry to examine the levels of the phosphorylated (activated) IGF receptor (IGF-1R) and found decreased IGF-1R activation in C3(1)-Tag;Mmp9−/− tumors compared to C3(1)-Tag;Mmp9+/+ tumors (Figure 6, E and F).
High IGFBP Expression Is Associated with Good Prognosis in Human Breast Cancer Patients Only When MMP9 Is Expressed at Low Levels
High levels of IGFBP-1 can reduce cell proliferation and increase cell death through sequestration of IGFs [41,42]. Our data from mouse models suggested that IGFBPs are important substrates for MMP9 in breast cancer and that interaction between the protease and its substrate influences tumor progression. To investigate whether this might have clinical relevance, we looked for correlations between IGFBP and MMP9 mRNA expression and patient outcome in publicly available gene expression data sets from human breast tumors. IGFBP-1 was expressed at very low levels in human breast cancer. However, the related IGFBP-2, -3, -4, -5, -6, and -7 mRNA were all expressed (Figure 7A), and of these, IGFBP-2, -3, -4, and -6 proteins have been shown to be substrates of MMP9 [41,43–46]. We found that high expression of IGFBP-2, -3, -4, -6, and -7 mRNA was associated with good prognosis (Figure S3). On the contrary, high expression of MMP9 mRNA was associated with poor prognosis (Figure S4), consistent with prior reports [47,48]. High expression of MMP9 mRNA was significantly correlated with the Elston grade of breast cancer in the GSE3494 data set, which contained information on tumor grade [Figure S4; P = .016, one-way analysis of variance (ANOVA)]. Interestingly, in both the GSE3494 and GSE1456 data sets, for patients that expressed high levels of MMP9 mRNA, high expression of IGFBP-2, -3, -4, -6, or -7 mRNA was no longer associated with good, but instead with poor, prognosis. In contrast, low expression levels of these IGFBP mRNAs were associated with poor outcome regardless of MMP9 expression levels (Figures 7, B–D, F, and G, and S5). IGFBP-5 protein has not been shown to be an MMP9 substrate. We found that high IGFBP-5 mRNA expression was not associated with good, but instead with poor, prognosis. Notably, high versus low MMP9 expression did not affect outcome for patients with high IGFBP-5 expression levels (Figure 7E); however, the number of patients with high IGFBP-5 levels was small (n = 31) and it is therefore possible that a true association would be missed. MMP2 is another protease that can digest IGFBPs [45], but we found that high mRNA expression level of MMP2 was associated with good prognosis in breast cancer (Figure S6A), also for patients with high mRNA expression levels of IGFBP-2 or IGFBP-4 (Figure S6, B–D). Together, our data suggest that MMP9 cleaves IGFBPs, which in turn leads to activation of pro-tumorigenic IGF signaling pathways in breast cancer.
Figure 7.
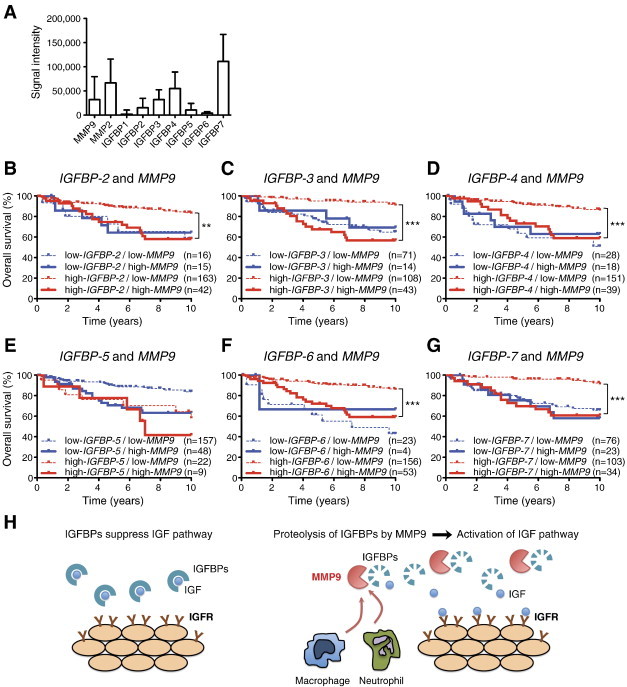
High MMP9 expression levels overcome the association of high IGFBP expression with good prognosis in human breast cancer patients. (A) Relative expression of IGFBPs (IGFBP-1–7), MMP2, and MMP9 genes in human breast cancer data set GSE3494. (B–G) Kaplan-Meier survival analysis was performed for breast cancer patient data, divided into four groups with high and low IGFBPs and MMP9 expression levels (using the GSE3494 data set, n = 236). High expression levels of IGFBPs, except (E) IGFBP-5, were significantly associated with better overall survival for patients with low expression of MMP9. (H) Model of how MMP9 and IGFBPs interact to promote tumor progression: When MMP9 is not present, IGFBPs sequester IGF, preventing it from acting on its receptors. However, tumor-infiltrating innate immune cells can provide MMP9, which digests IGFBPs, resulting in the release of IGF and the activation of the IGF receptor pathway to promote tumor growth. Asterisks indicate statistical significance: **P < .01 or ***P < .001.
Figure S3.
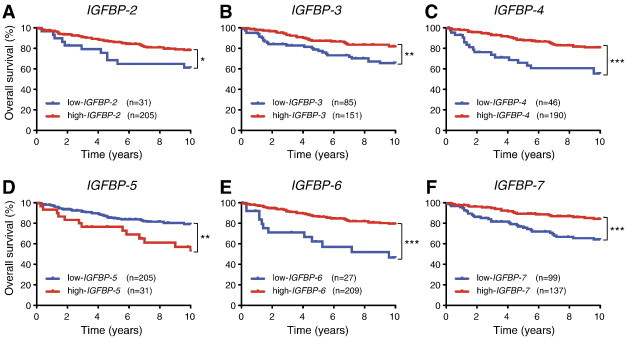
High IGFBP expression correlates with better prognosis of human breast cancer patients. The human breast cancer data set of expression profiles (GSE3494, n = 236) was analyzed for correlation of IGFBP expression with patient outcome. (A–F) Longer overall survival was observed in cancer patients with high expression of IGFBPs, except IGFBP-5 (D). Asterisks indicate statistical significance: *P < .05, **P < .01, or ***P < .001.
Figure S4.
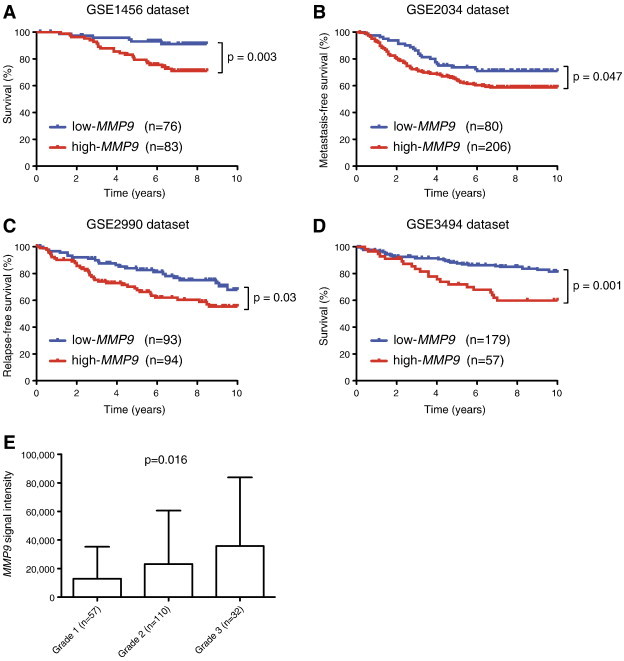
High MMP9 expression correlates with poor prognosis of human breast cancer patients. Four human breast cancer data sets of expression profiles (GSE1456, n = 159; GSE2034, n = 286; GSE2990, n = 187; and GSE3494, n = 236) were analyzed for correlation of MMP9 expression with patient outcome. (A and D) Shorter overall survival was observed in cancer patients with high expression of MMP9, and high MMP9 expression also correlated with (B) decreased metastasis-free and (C) relapse-free survival. (E) High expression of MMP9 was significantly correlated (P = .016, one-way ANOVA) with the Elston grade of breast cancer in the GSE3494 data set.
Figure S6.
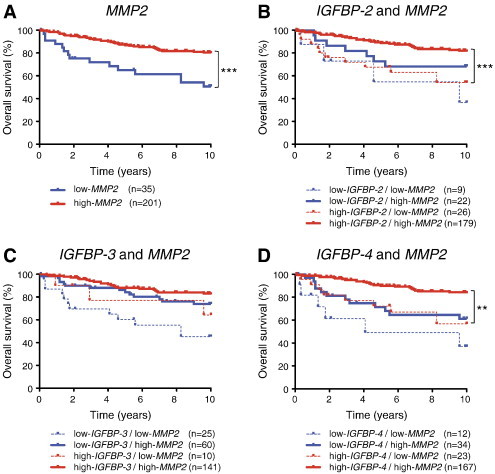
High IGFBP expression is associated with good prognosis in human breast cancer patients with high MMP2 expression. (A) The human breast cancer data set of expression profiles (GSE3494, n = 236) was analyzed for correlation of MMP2 expression with patient outcome. Longer overall survival was observed in cancer patients with high expression of MMP2. (B–D) Breast cancer patients were divided into four groups with high and low IGFBP and MMP2 expression levels. High expression of IGFBP-2 and -4 and high expression of MMP2 were significantly associated with better overall survival compared to high expression of IGFBP-2 and -4 and low MMP2 expression. Asterisks indicate statistical significance: **P < .01 or ***P < .001.
High levels of IGFBP-1 can reduce cell proliferation and increase cell death through sequestration of IGFs [41,42]. Our data from mouse models suggested that IGFBPs are important substrates for MMP9 in breast cancer and that interaction between the protease and its substrate influences tumor progression. To investigate whether this might have clinical relevance, we looked for correlations between IGFBP and MMP9 mRNA expression and patient outcome in publicly available gene expression data sets from human breast tumors. IGFBP-1 was expressed at very low levels in human breast cancer. However, the related IGFBP-2, -3, -4, -5, -6, and -7 mRNA were all expressed (Figure 7A), and of these, IGFBP-2, -3, -4, and -6 proteins have been shown to be substrates of MMP9 [41,43–46]. We found that high expression of IGFBP-2, -3, -4, -6, and -7 mRNA was associated with good prognosis (Figure S3). On the contrary, high expression of MMP9 mRNA was associated with poor prognosis (Figure S4), consistent with prior reports [47,48]. High expression of MMP9 mRNA was significantly correlated with the Elston grade of breast cancer in the GSE3494 data set, which contained information on tumor grade [Figure S4; P = .016, one-way analysis of variance (ANOVA)]. Interestingly, in both the GSE3494 and GSE1456 data sets, for patients that expressed high levels of MMP9 mRNA, high expression of IGFBP-2, -3, -4, -6, or -7 mRNA was no longer associated with good, but instead with poor, prognosis. In contrast, low expression levels of these IGFBP mRNAs were associated with poor outcome regardless of MMP9 expression levels (Figures 7, B–D, F, and G, and S5). IGFBP-5 protein has not been shown to be an MMP9 substrate. We found that high IGFBP-5 mRNA expression was not associated with good, but instead with poor, prognosis. Notably, high versus low MMP9 expression did not affect outcome for patients with high IGFBP-5 expression levels (Figure 7E); however, the number of patients with high IGFBP-5 levels was small (n = 31) and it is therefore possible that a true association would be missed. MMP2 is another protease that can digest IGFBPs [45], but we found that high mRNA expression level of MMP2 was associated with good prognosis in breast cancer (Figure S6A), also for patients with high mRNA expression levels of IGFBP-2 or IGFBP-4 (Figure S6, B–D). Together, our data suggest that MMP9 cleaves IGFBPs, which in turn leads to activation of pro-tumorigenic IGF signaling pathways in breast cancer.
Discussion
Relatively little is known about how stromal responses differ between different subtypes of breast cancer. Here, we compared the stromal responses of the luminal MMTV-Neu and basal-like C3(1)-Tag mouse models of breast cancer and found that C3(1)-Tag tumors had higher levels of collagen, more inflammatory myeloid cells, and higher levels of several chemokines (including CCL5, CXCL9, and CXCL10) than the MMTV-Neu tumors. We further report that the protease MMP9 only influenced tumor progression in the C3(1)-Tag mouse model of breast cancer and that protein levels of a known MMP9 substrate, IGFBP-1, correlated with the tumor-promoting effect of MMP9 in the models. Finally, analysis of the correlation between expression levels of MMP9 and IGFBPs versus patient outcome suggested that these two genes interact to promote human breast cancer.
Pro-angiogenic effects of MMP9 might be the cause for the delay in tumor onset observed in the C3(1)-Tag;Mmp9−/− mice, and we did indeed observe a reduced number of blood vessels in the C3(1)-Tag;Mmp9−/− tumors compared to the C3(1)-Tag;Mmp9+/+ tumors. However, there was no significant difference in the level of necrosis between tumors with and without Mmp9. Instead, we detected increased levels of IGFBP-1, an MMP9 substrate [39], in the C3(1)-Tag;Mmp9−/− compared to the C3(1)-Tag;Mmp9+/+ tumors. Deletion of Mmp9 would be expected to result in decreased proteolysis of IGFBP-1, consistent with this finding. The major function of IGFBP-1 is to bind and sequester IGF-1, thereby antagonizing IGFs’ ability to stimulate the proliferation of breast cancer cells [41,49]. A slower cancer cell growth rate would increase the time until tumors become detectable, consistent with our observations of extended tumor-free survival in the C3(1)-Tag;Mmp9−/− mice. On the basis of our preclinical data, we propose a model in which high levels of IGFBPs are associated with good prognosis because they sequester IGFs to reduce IGFR signaling and cancer proliferation. However, in the presence of high levels of MMP9, the IGFBPs are digested, IGFs are released, and cancer progression accelerates (Figure 7H). One way to test whether MMP9-mediated proteolysis of IGFBP-1 is critical for tumor progression in the C3(1)-Tag model would be to generate mice expressing IGFBP-1 with a mutated MMP9 cleavage site, so that it no longer can be processed by MMP9, and intercross such mice with C3(1)-Tag.
It is interesting that in the C3(1)-Tag model, Mmp9 deletion only resulted in increased IGFBP-1 levels, while IGFBP-2 and IGFBP-3 levels were not altered. Mouse IGFBP-3 is an MMP9 substrate [44], but it can be cleaved by multiple other proteases. It is therefore possible that other proteases compensate for the depletion of MMP9 in the C3(1)-Tag mouse model. Our analysis of the association of MMP9 and IGFBP levels with prognosis in human data sets is intriguing, as it suggests a population of breast cancer patients that could benefit from treatment with MMP9 inhibitors. Further studies are needed to confirm this possibility.
After crossing our models with Mmp9−/− mice, we found that tumor onset was significantly delayed in the absence of MMP9 in the C3(1)-Tag model, while there was no change in tumor onset in the MMTV-Neu model. It is unclear why deletion of MMP9 had no effect on tumor onset in the MMTV-Neu model, although it could simply be that expression levels are somewhat lower to begin with or that MMP9 is not activated to the same extent as in the C3(1)-Tag tumors. However, the low level of IGFBP-1 in the MMTV-Neu tumors (both with and without MMP9) may also be a contributing factor. Lack of MMP9 also delays tumor onset in the pancreatic islet cell carcinoma model (RIP1-Tag2) [20], a cancer model that, similar to C3(1)-Tag, is driven by expression of the SV40 large T antigen with p53 and Rb inactivation. In contrast, lack of MMP9 does not affect tumor onset in the luminal MMTV-PyMT model, a model that, like the MMTV-Neu model, is driven by activation of the Ras pathway [24]. Together, these reports suggest that MMP9 has a tumor-promoting effect in tumors driven by p53/Rb inactivation and a minimal effect when Ras activation is driving the cancer.
We found a substantial difference in the levels of cytokines between tumors of the MMTV-Neu and C3(1)-Tag models. Both types of tumors originate from luminal mammary epithelial cells [5,7], suggesting that the significant difference in stromal response between the models is not due to the cell type of origin. Activated Ras can transcriptionally induce chemokines involved in stromal cell recruitment, including IL-8/CXCL8 [50], IL-6, and CXCL1 [51,52]. In contrast, p53 can antagonize nuclear factor κB (NF-κB), thereby suppressing the expression of its target genes [53]. Increased expression of the NF-κB–regulated CXCL9 and CXCL10 chemokines has been reported in the thymuses of p53-deficient mice compared to their wild-type littermates [54]. We did indeed find higher levels of these two chemokines in the C3(1)-Tag tumors compared to the MMTV-Neu tumors, suggesting that the SV40 large T antigen, through inactivation of p53, induced an inflammatory response through NF-κB. Thus, oncogenesis driven by either Ras activation or p53 inactivation likely elicits transcription of different cytokines by the cancer cells, and these cytokine differences may drive the difference in the stromal responses.
The different inflammatory environment in the two mouse models might also contribute to the difference in the effect of MMP9: In the MMTV-Neu model, the majority of MMP9-expressing cells were macrophages, while in the C3(1)-Tag model, MMP9 was mostly supplied by neutrophils. Interestingly, it has previously been shown that MMP9 supplied by neutrophils better supports tumor angiogenesis than MMP9 from macrophages [55], likely because neutrophils secrete MMP9 in a form not bound to its endogenous inhibitor, tissue inhibitor of metalloproteinase-1 [56,57]. In human breast cancer, expression of MMP9 protein as detected by immunohistochemistry has been reported in cancer cells as well as stromal cells, including macrophages, neutrophils, and pericytes (reviewed in [21]). An interesting study compared MMP9 protein (detected by immunohistochemistry) with MMP9 mRNA expression (detected by in situ hybridization) and found that whereas macrophages and vascular cells expressed both MMP9 mRNA and MMP9 protein, breast cancer cells expressed only the protein, suggesting that it was supplied by the stromal cells [58]. Neutrophils were similarly found to express MMP9 protein and not MMP9 mRNA, which is consistent with MMP9 protein being already stored inside the neutrophils for rapid release rather than being actively synthesized by neutrophils that infiltrate the tumors.
In conclusion, our study highlights the fact that oncogenic alterations in cancer cells drive specific stromal responses and the function of proteases depends on the presence of their specific substrates. Importantly, we show that an extracellularly acting protease, MMP9, despite being present in both models, only promotes tumor progression in the C3(1)-Tag model. We provide correlative data suggesting that IGFBPs, both in murine and human breast tumors, interact with MMP9 to promote tumorigenesis. However, we had previously found that the absence of MMP9 results in better responses to the chemotherapeutic drug doxorubicin in the MMTV-Neu and MMTV-PyMT models but has no effect in the C3(1)-Tag model [28]. Together, these findings highlight that MMP9 has multiple context-dependent functions in the tumor tissue. Our data suggest that a thorough characterization of the altered expression of oncogenes and tumor suppressors, as well as the stromal components of human breast tumors, is needed to inform diagnosis, prognosis, and treatment regimens of breast cancer patients.
The following are the supplementary data related to this article.
Figure S5.
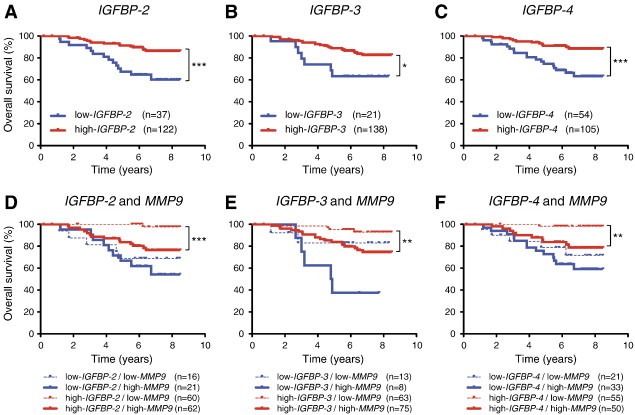
High IGFBP expression is associated with poor prognosis in human breast cancer patients who also have high MMP9 expression. (A–C) The human breast cancer data set of expression profiles (GSE1456, n = 159) was analyzed for correlation of IGFBP-2, -3, and -4 expression and patient outcome. Longer overall survival was observed in cancer patients with high expression of these IGFBPs. (D–F) Breast cancer patients were divided into four groups with high and low IGFBP and MMP9 expression levels. High expression of IGFBPs and low expression of MMP9 were significantly associated with better overall survival compared to high expression of both IGFBPs and MMP9. Asterisks indicate statistical significance: *P < .05, **P < .01, or ***P < .001.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2015.04.003.
Acknowledgements
We thank Elena Atamaniuc, Ying Yu, and Lidiya Korets for excellent help with animal handling; Öznur Turan for technical assistance with histology; Johannes Waage for construction of the heat maps; Ib J. Christensen for statistical evaluation; Janie Karp and Marcela Barros for cataloging tissue samples; and Lisa Coussens and Zena Werb for discussions about the project.
Footnotes
This work was supported by funds from the National Institutes of Health (R01CA057621). M.E. was supported by the Breast Cancer Alliance, the Long Island 2 Day Walk to Fight Breast Cancer, and the Manhasset Women’s Coalition Against Breast Cancer. M.G.R. was supported by Rigshospitalet, the University of Copenhagen, the Augustinus Fonden, the Dagmar Marshalls Fond, and the European Association for Cancer Research. J.-H.P. was supported by a postdoctoral fellowship from the US Department of Defense Breast Cancer Research Program.
This article refers to supplementary materials, which are designated by Figures S1 to S6 and are available online at www.neoplasia.com.
References
- 1.Davoli A, Hocevar BA, Brown TL. Progression and treatment of HER2-positive breast cancer. Cancer Chemother Pharmacol. 2010;65:611–623. doi: 10.1007/s00280-009-1208-1. [DOI] [PubMed] [Google Scholar]
- 2.Gluz O, Liedtke C, Gottschalk N, Pusztai L, Nitz U, Harbeck N. Triple-negative breast cancer—current status and future directions. Ann Oncol. 2009;20:1913–1927. doi: 10.1093/annonc/mdp492. [DOI] [PubMed] [Google Scholar]
- 3.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 4.Weigelt B, Reis-Filho JS. Histological and molecular types of breast cancer: is there a unifying taxonomy? Nat Rev Clin Oncol. 2009;6:718–730. doi: 10.1038/nrclinonc.2009.166. [DOI] [PubMed] [Google Scholar]
- 5.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, Rasmussen KE, Jones LP, Assefnia S, Chandrasekharan S. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maroulakou IG, Anver M, Garrett L, Green JE. Prostate and mammary adenocarcinoma in transgenic mice carrying a rat C3(1) simian virus 40 large tumor antigen fusion gene. Proc Natl Acad Sci U S A. 1994;91:11236–11240. doi: 10.1073/pnas.91.23.11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett CN, Green JE. Genomic analyses as a guide to target identification and preclinical testing of mouse models of breast cancer. Toxicol Pathol. 2010;38:88–95. doi: 10.1177/0192623309357074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cardiff RD. Mouse models of human breast cancer. Comp Med. 2003;53:250–253. [PubMed] [Google Scholar]
- 10.Egeblad M, Ewald AJ, Askautrud HA, Truitt ML, Welm BE, Bainbridge E, Peeters G, Krummel MF, Werb Z. Visualizing stromal cell dynamics in different tumor microenvironments by spinning disk confocal microscopy. Dis Model Mech. 2008;1:155–167. doi: 10.1242/dmm.000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN, Pollard JW. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006;66:11238–11246. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- 12.Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell. 2010;18:884–901. doi: 10.1016/j.devcel.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol. 2010;22:697–706. doi: 10.1016/j.ceb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajaram M, Li J, Egeblad M, Powers RS. System-wide analysis reveals a complex network of tumor-fibroblast interactions involved in tumorigenicity. PLoS Genet. 2013;9:e1003789. doi: 10.1371/journal.pgen.1003789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown PD. Ongoing trials with matrix metalloproteinase inhibitors. Expert Opin Investig Drugs. 2000;9:2167–2177. doi: 10.1517/13543784.9.9.2167. [DOI] [PubMed] [Google Scholar]
- 16.Kruger A, Soeltl R, Sopov I, Kopitz C, Arlt M, Magdolen V, Harbeck N, Gansbacher B, Schmitt M. Hydroxamate-type matrix metalloproteinase inhibitor batimastat promotes liver metastasis. Cancer Res. 2001;61:1272–1275. [PubMed] [Google Scholar]
- 17.Whittaker M, Floyd CD, Brown P, Gearing AJ. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem Rev. 1999;99:2735–2776. doi: 10.1021/cr9804543. [DOI] [PubMed] [Google Scholar]
- 18.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: proteolytic regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 20.Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 22.Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell. 2000;103:481–490. doi: 10.1016/s0092-8674(00)00139-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest. 2004;114:623–633. doi: 10.1172/JCI22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin MD, Carter KJ, Jean-Philippe SR, Chang M, Mobashery S, Thiolloy S, Lynch CC, Matrisian LM, Fingleton B. Effect of ablation or inhibition of stromal matrix metalloproteinase-9 on lung metastasis in a breast cancer model is dependent on genetic background. Cancer Res. 2008;68:6251–6259. doi: 10.1158/0008-5472.CAN-08-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006;103:12493–12498. doi: 10.1073/pnas.0601807103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rasch MG, Lund IK, Illemann M, Hoyer-Hansen G, Gardsvoll H. Purification and characterization of recombinant full-length and protease domain of murine MMP-9 expressed in Drosophila S2 cells. Protein Expr Purif. 2010;72:87–94. doi: 10.1016/j.pep.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 28.Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, Fein M, Rasch MG, Tan YX, Qiu J. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell. 2012;21:488–503. doi: 10.1016/j.ccr.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pawitan Y, Bjohle J, Amler L, Borg AL, Egyhazi S, Hall P, Han X, Holmberg L, Huang F, Klaar S. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast Cancer Res. 2005;7:R953–R964. doi: 10.1186/bcr1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 31.Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, Smeds J, Nordgren H, Farmer P, Praz V, Haibe-Kains B. Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst. 2006;98:262–272. doi: 10.1093/jnci/djj052. [DOI] [PubMed] [Google Scholar]
- 32.Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, Pawitan Y, Hall P, Klaar S, Liu ET. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci U S A. 2005;102:13550–13555. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Littlepage LE, Adler AS, Kouros-Mehr H, Huang G, Chou J, Krig SR, Griffith OL, Korkola JE, Qu K, Lawson DA. The transcription factor ZNF217 is a prognostic biomarker and therapeutic target during breast cancer progression. Cancer Discov. 2012;2:638–651. doi: 10.1158/2159-8290.CD-12-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Camp RL, Dolled-Filhart M, Rimm DL. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res. 2004;10:7252–7259. doi: 10.1158/1078-0432.CCR-04-0713. [DOI] [PubMed] [Google Scholar]
- 35.Allavena P, Germano G, Marchesi F, Mantovani A. Chemokines in cancer related inflammation. Exp Cell Res. 2011;317:664–673. doi: 10.1016/j.yexcr.2010.11.013. [DOI] [PubMed] [Google Scholar]
- 36.Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9:259–270. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–631. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- 38.Ahn GO, Brown JM. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell. 2008;13:193–205. doi: 10.1016/j.ccr.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coppock HA, White A, Aplin JD, Westwood M. Matrix metalloprotease-3 and -9 proteolyze insulin-like growth factor-binding protein-1. Biol Reprod. 2004;71:438–443. doi: 10.1095/biolreprod.103.023101. [DOI] [PubMed] [Google Scholar]
- 40.Lee SE, Han BD, Park IS, Romero R, Yoon BH. Evidence supporting proteolytic cleavage of insulin-like growth factor binding protein-1 (IGFBP-1) protein in amniotic fluid. J Perinat Med. 2008;36:316–323. doi: 10.1515/JPM.2008.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Firth SM, Baxter RC. Cellular actions of the insulin-like growth factor binding proteins. Endocr Rev. 2002;23:824–854. doi: 10.1210/er.2001-0033. [DOI] [PubMed] [Google Scholar]
- 42.Oh JS, Kucab JE, Bushel PR, Martin K, Bennett L, Collins J, DiAugustine RP, Barrett JC, Afshari CA, Dunn SE. Insulin-like growth factor-1 inscribes a gene expression profile for angiogenic factors and cancer progression in breast epithelial cells. Neoplasia. 2002;4:204–217. doi: 10.1038/sj.neo.7900229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Larsen PH, DaSilva AG, Conant K, Yong VW. Myelin formation during development of the CNS is delayed in matrix metalloproteinase-9 and -12 null mice. J Neurosci. 2006;26:2207–2214. doi: 10.1523/JNEUROSCI.1880-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishijima T, Piriz J, Duflot S, Fernandez AM, Gaitan G, Gomez-Pinedo U, Verdugo JM, Leroy F, Soya H, Nunez A. Neuronal activity drives localized blood-brain-barrier transport of serum insulin-like growth factor-I into the CNS. Neuron. 2010;67:834–846. doi: 10.1016/j.neuron.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 45.Prudova A, auf dem Keller U, Butler GS, Overall CM. Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol Cell Proteomics. 2010;9:894–911. doi: 10.1074/mcp.M000050-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rorive S, Berton A, D'Haene N, Takacs CN, Debeir O, Decaestecker C, Salmon I. Matrix metalloproteinase-9 interplays with the IGFBP2-IGFII complex to promote cell growth and motility in astrocytomas. Glia. 2008;56:1679–1690. doi: 10.1002/glia.20719. [DOI] [PubMed] [Google Scholar]
- 47.Li HC, Cao DC, Liu Y, Hou YF, Wu J, Lu JS, Di GH, Liu G, Li FM, Ou ZL. Prognostic value of matrix metalloproteinases (MMP-2 and MMP-9) in patients with lymph node-negative breast carcinoma. Breast Cancer Res Treat. 2004;88:75–85. doi: 10.1007/s10549-004-1200-8. [DOI] [PubMed] [Google Scholar]
- 48.Song J, Su H, Zhou YY, Guo LL. Prognostic value of matrix metalloproteinase 9 expression in breast cancer patients: a meta-analysis. Asian Pac J Cancer Prev. 2013;14:1615–1621. doi: 10.7314/apjcp.2013.14.3.1615. [DOI] [PubMed] [Google Scholar]
- 49.Huff KK, Kaufman D, Gabbay KH, Spencer EM, Lippman ME, Dickson RB. Secretion of an insulin-like growth factor-I-related protein by human breast cancer cells. Cancer Res. 1986;46:4613–4619. [PubMed] [Google Scholar]
- 50.Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6:447–458. doi: 10.1016/j.ccr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 51.Ancrile B, Lim KH, Counter CM. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007;21:1714–1719. doi: 10.1101/gad.1549407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang G, Rosen DG, Zhang Z, Bast RC, Jr., Mills GB, Colacino JA, Mercado-Uribe I, Liu J. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc Natl Acad Sci U S A. 2006;103:16472–16477. doi: 10.1073/pnas.0605752103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol. 1999;19:3485–3495. doi: 10.1128/mcb.19.5.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Komarova EA, Krivokrysenko V, Wang K, Neznanov N, Chernov MV, Komarov PG, Brennan ML, Golovkina TV, Rokhlin OW, Kuprash DV. p53 is a suppressor of inflammatory response in mice. FASEB J. 2005;19:1030–1032. doi: 10.1096/fj.04-3213fje. [DOI] [PubMed] [Google Scholar]
- 55.Deryugina EI, Zajac E, Juncker-Jensen A, Kupriyanova TA, Welter L, Quigley JP. Tissue-infiltrating neutrophils constitute the major in vivo source of angiogenesis-inducing MMP-9 in the tumor microenvironment. Neoplasia. 2014;16:771–788. doi: 10.1016/j.neo.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2007;104:20262–20267. doi: 10.1073/pnas.0706438104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ardi VC, Van den Steen PE, Opdenakker G, Schweighofer B, Deryugina EI, Quigley JP. Neutrophil MMP-9 proenzyme, unencumbered by TIMP-1, undergoes efficient activation in vivo and catalytically induces angiogenesis via a basic fibroblast growth factor (FGF-2)/FGFR-2 pathway. J Biol Chem. 2009;284:25854–25866. doi: 10.1074/jbc.M109.033472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nielsen BS, Sehested M, Kjeldsen L, Borregaard N, Rygaard J, Dano K. Expression of matrix metalloprotease-9 in vascular pericytes in human breast cancer. Lab Invest. 1997;77:345–355. [PubMed] [Google Scholar]