

Abstract
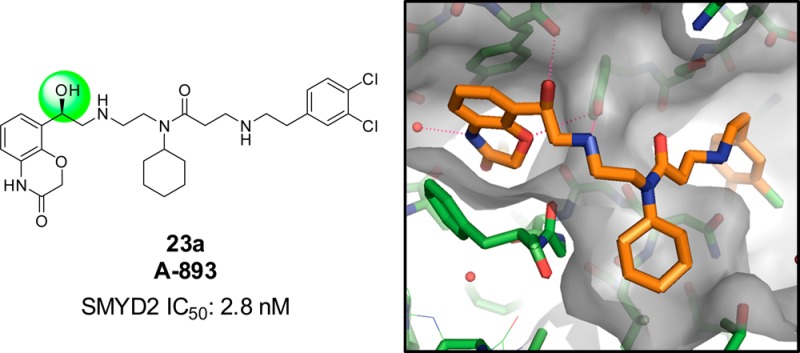
A lack of useful small molecule tools has precluded thorough interrogation of the biological function of SMYD2, a lysine methyltransferase with known tumor-suppressor substrates. Systematic exploration of the structure–activity relationships of a previously known benzoxazinone compound led to the synthesis of A-893, a potent and selective SMYD2 inhibitor (IC50: 2.8 nM). A cocrystal structure reveals the origin of enhanced potency, and effective suppression of p53K370 methylation is observed in a lung carcinoma (A549) cell line.
Keywords: Epigenetics, SMYD2, H3K36, p53, methyltransferase, lysine
SET and MYND domain-containing protein 2 (SMYD2) is a lysine methyltransferase that is highly expressed in esophageal squamous cell carcinoma and pediatric acute lymphoblastic leukemia.1,2 Elevated expression in both of these cancers correlates with a poor prognosis. Its function as a methyltransferase has been reported to span a variety of substrates including histone H3 (K363 and K44), retinoblastoma tumor suppressor (Rb) (K860),5 and tumor suppressor p53 (K370).6,7 It is through these processes that SMYD2 is believed to play a role in the network of post-translational modifications that regulates tumor growth. In particular, SMYD2 may serve as a putative oncogene by its methylation of lysine 370 of p53, which is repressive to p53 function.
Despite the potential role of SMYD2 methylation in epigenetic regulation of the aforementioned cancers, no small molecule inhibitors of this enzyme were known until the disclosure of AZ505 (1) a few years ago (Figure 1).8 This benzoxazinone was identified from a high-throughput screen (HTS) and shown via a cocrystal structure to bind to the lysine pocket of SMYD2. The potency of 1 toward SMYD2 was disclosed to be moderate (IC50: 120 nM) despite being quite selective (>700-fold) over six other methyltransferases. More recently, an additional SMYD2 inhibitor representing a new chemotype was published as a probe of the Structural Genomics Consortium (Figure 1).9 LLY-507 (2) exhibited much improved potency (IC50 < 15 nM) and inhibited p53K370 methylation in cells.
Figure 1.

Published inhibitors of SMYD2.
As compound 1 represented the only known inhibitor of SMYD2 until recently, we pursued an avenue of investigation that systematically interrogated all binding elements of this molecule in order to inspire development of a more potent inhibitor.
At the outset of this endeavor, we sought to address structural changes in three regions of the molecule (Figure 2). The first region comprises the benzoxazinone subunit. It occupies the lysine binding channel and contributes to overall binding via a water-bridged hydrogen bond of its hydroxyl and ring amine functionalities to the backbone of residues V202 and M205. In addition, its ether oxygen participates in another water-bridged hydrogen bond interaction with the amine linker of the molecule and N180, G183, and Y258 of the enzyme. The second and third regions of the molecule contain the cyclohexylamine and dichlorophenyl subunits, respectively. These moieties fill two hydrophobic pockets outside of the immediate lysine channel. Of note is a third water-bridged interaction between the amine-linker, flanked by regions 1 and 2, and side-chain N180.
Figure 2.

Published binding features of SMYD2 inhibitor 1 (AZ505).
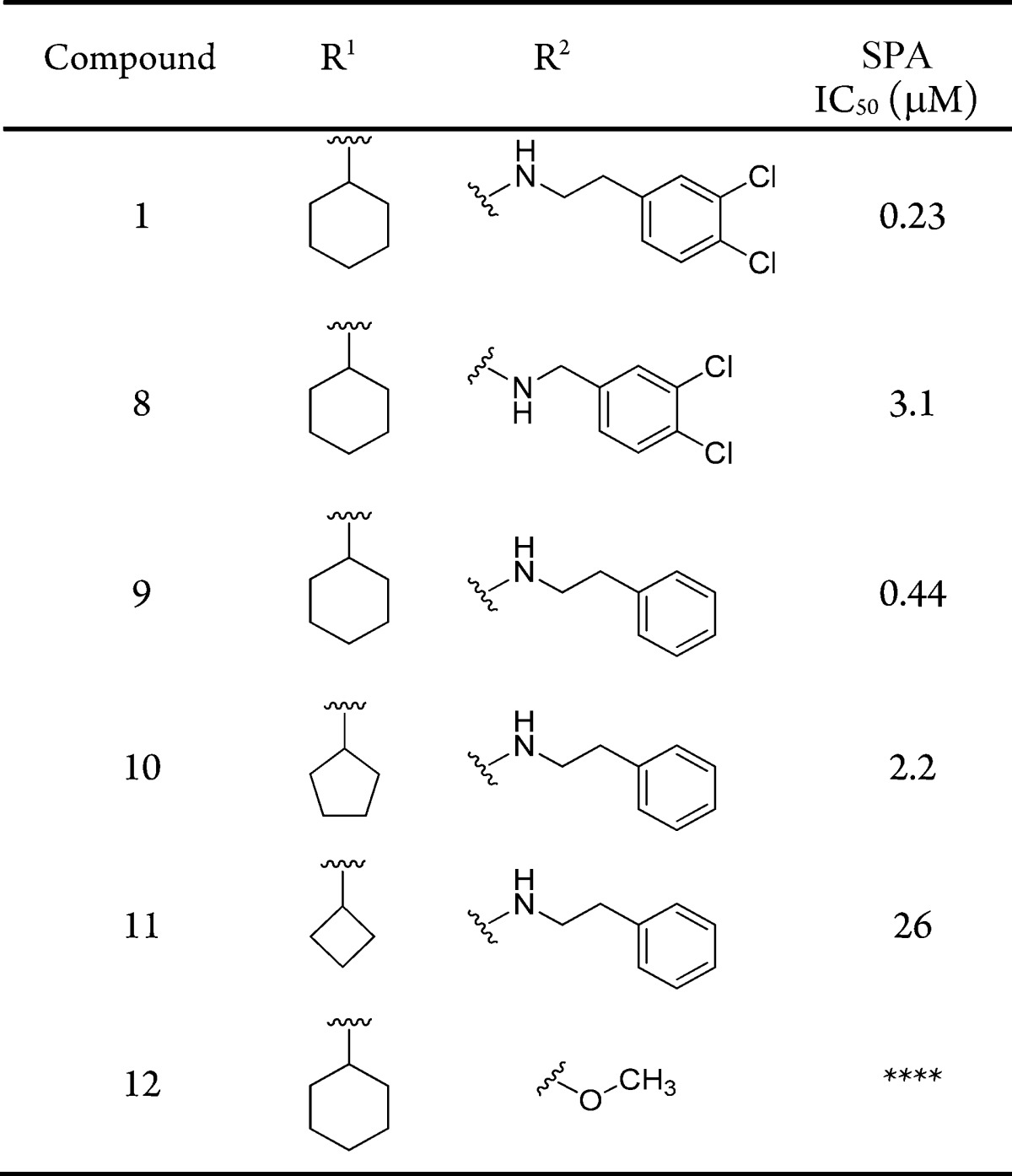
To establish structure–activity relationships (SAR) in these three regions, a scintillation proximity assay (SPA) was utilized, which measured methylation of the p53 peptide by SMYD2 (Table 1). At the outset, we postulated that the cyclohexyl and dichlorobenzyl subunits of compound 1 (occupying regions 2 and 3) did not significantly contribute much to the overall binding efficiency of the molecule. As such, the first targeted analogues were made to evaluate the effect of subtle changes, or omission, to these regions. Advanced intermediate 4 could be generated from 2-nitrobenzene-1,3-diol, following a published chemistry route, in order to enable SAR at these regions (Scheme 1).10 This benzoxazinone was then subjected to standard reductive amination conditions with a set of aldehydes, represented by 5, utilizing sodium triacetoxyborohydride. This installed variance at the R1 position on the central amine. Following temporary Boc-protection of the resultant secondary amine with di-tert-butyl dicarbonate, a 1,4-addition with a second set of amines enabled divergence at the R2 position as represented by compound 7.
Table 1. Structure–Activity Relationship of Benzoxazinone Derivatives.
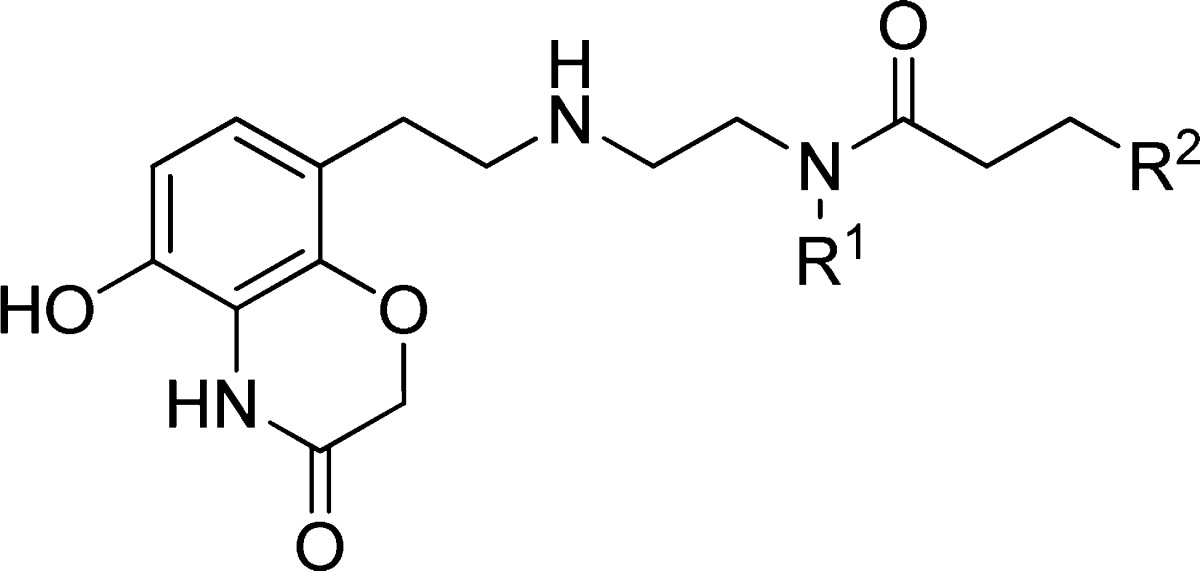
Scheme 1. Synthesis of Benzoxazinone Derivatives of Compound 1.

Reagents and conditions: (a) Na(OAc)3BH, NMP, rt; (b) di-tert-butyl dicarbonate, EtOAc, H2O, rt, 17–43% (over 2 steps); (c) R2OH or R2NH2, EtOH, 50 °C, 42–88%; (d) trifluoroacetic acid, EtOH, rt, 48–92%.
Surprising sensitivity was observed to subtle changes at positions R1 and R2. The potency of compound 1 (IC50: 230 nM) was in line with reported data (IC50: 120 nM). Shortening the linker length by one carbon atom provided benzylamine derivative 8. Over 10-fold potency was lost with this modification (IC50: 3.1 μM). A more conservative change by removing only the ortho-chlorine atoms resulted in compound 9, with slightly reduced but comparable potency (IC50: 440 nM). Using this as a template, the central region encompassing R1 was evaluated. Substituting the cyclohexane from 9 with a cyclopentane, as in 10, led to a significant loss in potency (IC50: 2.2 μM). This negative effect was further magnified by cyclobutyl derivative 11, which was over 10-fold less potent than 10. Finally, in the interest of reducing the molecular weight, complete omission of the dichlorophenyl subunit of R2 was evaluated by installing a methoxy-group in its place (12). This analogue was inactive against SMYD2.
This initial SAR made it apparent that reducing the size of the subunits that occupy the hydrophobic pockets of regions 2 and 3 was generally deleterious to activity. As such, the investigation shifted focus to changes at the benzoxazinone subunit that occupied the lysine channel. A slightly altered synthetic approach was employed to install diversity at the lysine channel region at the end of the synthesis (Scheme 2). This multistep sequence commenced with the addition of 2-(3,4-dichlorophenyl)ethanamine to tert-butyl acrylate followed by protection of the resultant secondary amine with benzylchloroformate. Subsequent removal of the tert-butyl ester with trifluoroacetic acid furnished carboxylic acid 14. This was then coupled to N-(2,2-dimethoxyethyl)cyclohexylamine using hydroxybenzotriazole (HOBt)/1-ethyl-3-(3-(dimethylamino)propyl)carbo-diimide (EDC), followed by acetal cleavage to yield aldehyde 15. This advanced intermediate was then used for installing benzoxazinone replacements (as in 16) via a two-step sequence: reductive amination with sodium triacetoxyborohydride and deprotection of the secondary amine with boron tribromide.
Scheme 2. Synthesis of Analogues with Alternative Subunits to the Benzoxazinone of 1.

Reagents and conditions: (a) 2-(3,4-dichlorophenyl)ethanamine, ethanol, rt 85%; (b) Et3N, benzylchloroformate, CH2Cl2, rt, 71%; (c) CF3CO2H, CH2Cl2, rt, 80%; (d) HOBt, EDC, (i-Pr)2NEt, N-(2,2-dimethoxyethyl)-cyclohexanamine, DMF, rt, 61%; (e) p-toluenesulfonic acid, CH2Cl2, rt, 84%; (f) RNH2, Na(OAc)3BH, CH2Cl2, 0 °C–rt, 68–87%; (g) BBr3, CH2Cl2, 0 °C–rt, 53–64%.
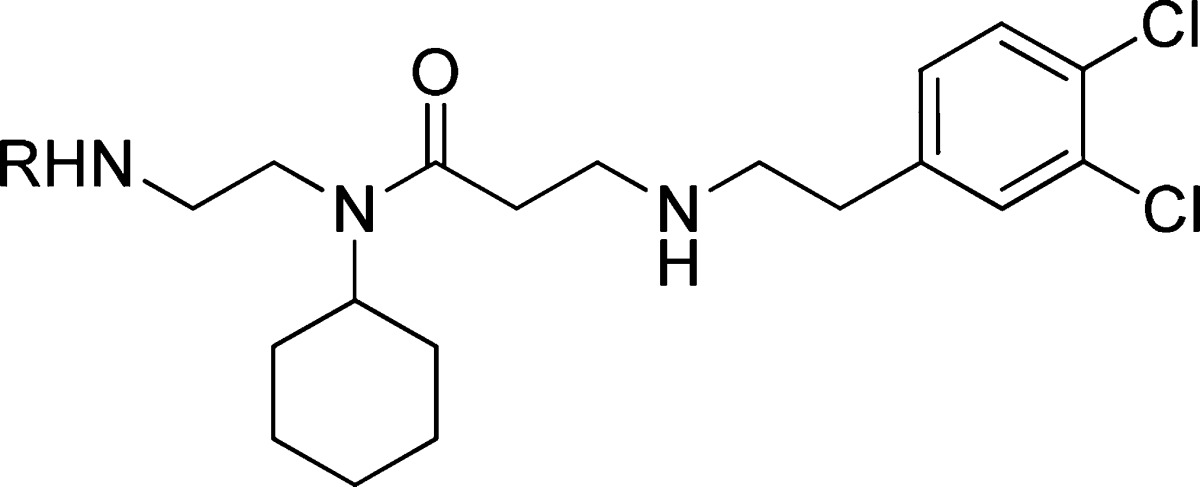
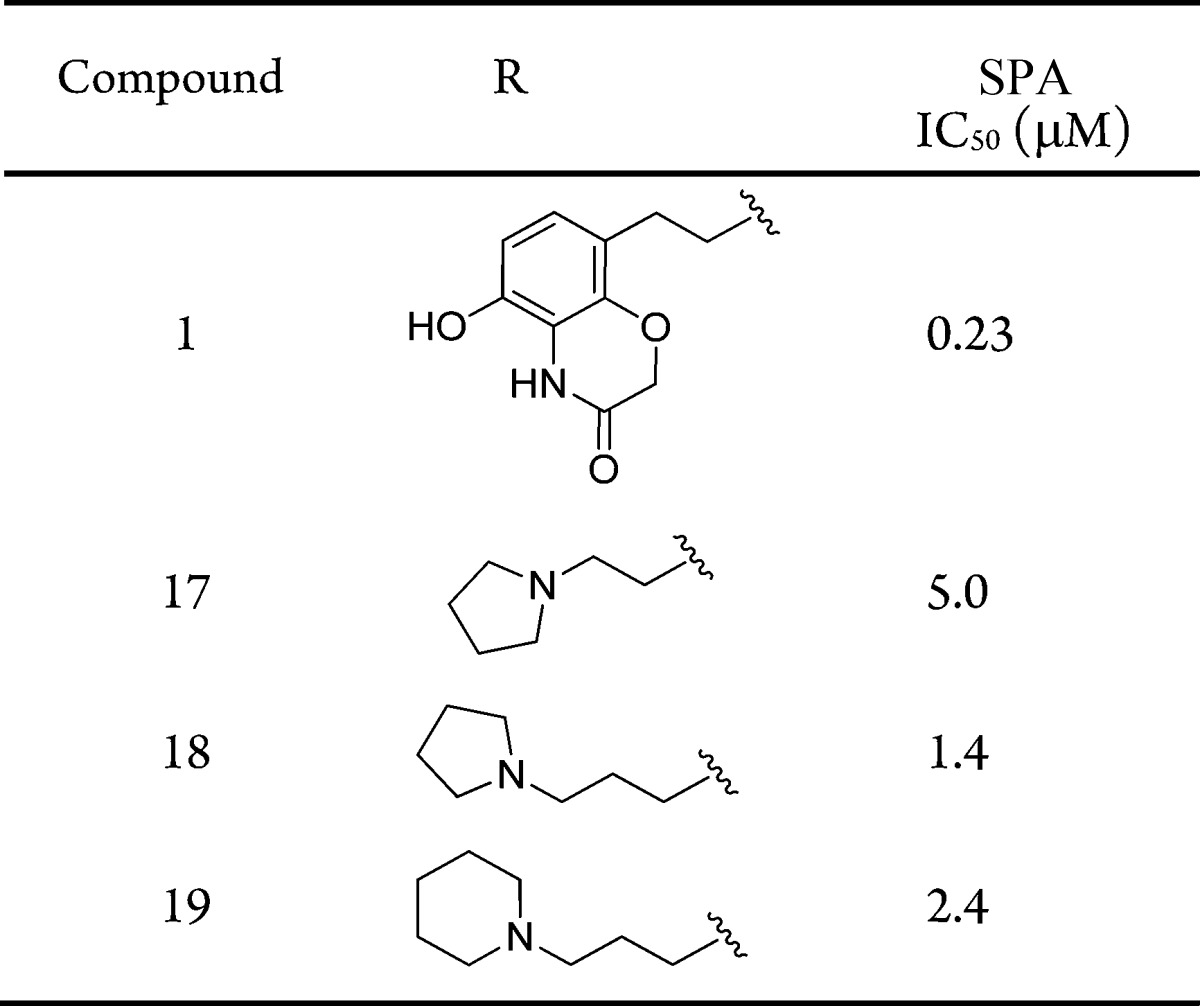
The initial replacements of the benzoxazinone subunit of compound 1 were envisioned to possess a simpler secondary amine mimic of methyl lysine (Table 2).11 The first example of this was pyrrolidine 17, as it represents a common motif of methyltransferase inhibitors that effectively binds to the lysine-peptide site.12−14 Unfortunately, a >20-fold loss in activity was observed (IC50: 5.0 μM). The ethyl-linked pyrrolidine was likely too short to properly occupy the lysine channel. Accordingly, propyl-pyrrolidine 18 was evaluated and found to have more favorable IC50 of 1.4 μM. The slightly larger piperidine replacement, 19, was less effective (IC50: 2.4 μM). This survey revealed that replacements to the benzoxazinone subunit of 1 were capable of maintaining SMYD2 inhibition, albeit with less potency.
Table 2. Structure–Activity Relationship of Benzoaxazinone Replacements.
Before embarking on a more extensive analysis of benzoxazinone replacements, we first sought to address the relative importance of each heteroatom in the benzoxazinone itself in order to inspire a more promising structural change. The first facet of this evaluation involved removing the hydroxyl-group, which was reported to participate in a water-bridged hydrogen bond with the backbone of residues V202 and M205 (Figure 1). Similar chemistry to that employed for the previous benzoxazinone analogues was utilized, with the exception that the hydroxyl-group in question was removed (Scheme 3). This route led to intermediate azide 20, which we attempted to reduce to the primary amine en route to the des-hydroxy analogue 21.
Scheme 3. Synthesis of A-893 Arising from Exclusive Formation of Intermediate 22.

Not unexpectedly, hydroxyethyl amine 22 was the major product observed from this step. The lack of a benzoxazinone hydroxyl group removed the stabilization of a quinone-methide intermediate during the reduction,15 rendering the newly formed benzyl hydroxyl group, 22, with a lower propensity toward further reduction. As a result, we chose to take forward 22, following chiral separation, via the same sequence as shown in Scheme 1 to furnish separated enantiomers 23a and 23b.
The resultant potency of these compounds was remarkable. The hydroxyl-substituted derivative 23a inhibited SMYD2 with an IC50 of 2.8 nM. Its enantiomer was less efficacious, but nonetheless far more potent than any analogue observed in this series (IC50: 5.6 nM). The >80-fold enhancement in potency was fortuitous, yet unexpected. We next investigated the mechanism of action to assess whether this change in structure affected its ability to be a peptide competitive inhibitor (Figure 3). The dependence of IC50 on peptide and SAM concentrations suggests that 23a retains the same mechanism of inhibition (peptide-competitive) as inhibitor 1.
Figure 3.

Peptide-competitive and SAM-uncompetitive inhibition of SMYD2 by 23a.
The dramatic increase in potency resulting from a modest change in structure highlighted the possibility of a critical hydrogen bond interaction accessed by the newly installed secondary alcohol. To illuminate the key features of binding with this new compound, we obtained a cocrystal structure of SMYD2 with inhibitor 23a (Figure 4). Overall, the three regions of SMYD2 that are occupied with 23a are generally unchanged from compound 1. The central cyclohexylamine and the terminal dichlorophenyl group occupy the two hydrophobic pockets of aforementioned regions 2 and 3, respectively (from Figure 2).
Figure 4.
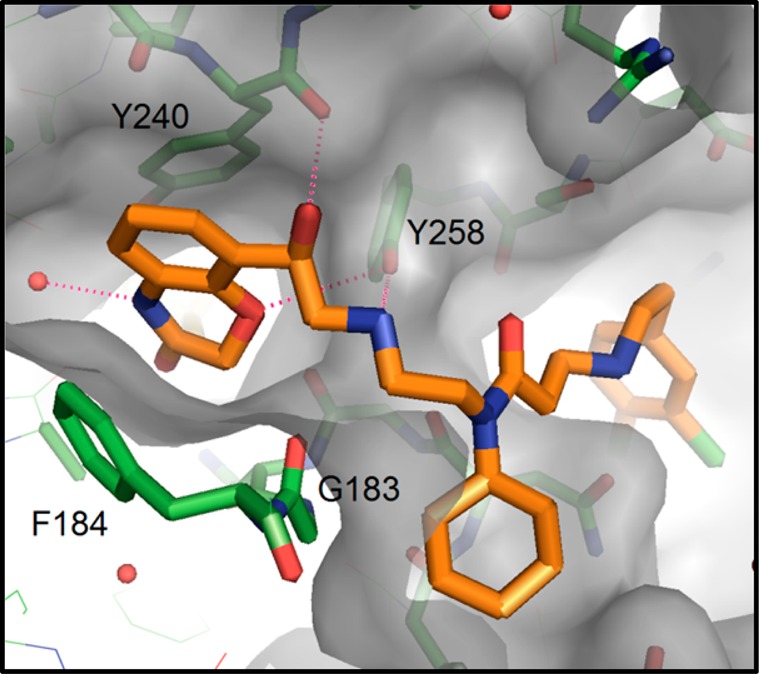
X-ray cocrystal structure of 23a and SMYD2, revealing the orientation of key residues around the hydroxyethylamine linker and the benzoxazinone moieties.
In the lysine channel, the ether oxygen of the benzoxazinone accepts a hydrogen bond from the side-chain hydroxyl of Y258, and the ring amine maintains a water-bridged interaction, albeit without a nearby ring-hydroxyl group that was present in 1. The secondary amine of the linker region also participates in an electrostatic interaction with Y258. The newly installed benzylic alcohol accesses a hydrogen bond with the backbone carbonyl group of Y240. In evaluating this network of binding, the enhanced potency of 23a over 1 reflects the more coordinated nature of the flexible linker region, attained by additional hydrogen bond of the alcohol. Conversely, the loss of the benzoxazinone hydroxyl group that was present in 1 does not have a deleterious impact on binding, perhaps due to its redundancy in the water-bridged interaction engaged by the amine segment of the benzoxazinone ring. Irrespective of this, its relative importance to overall binding is eclipsed by the enhanced coordination brought by the secondary alcohol.
In order to assess the ability of 23a to serve as a meaningful probe of SMYD2 biology, we evaluated its selectivity against a panel of 31 other methyltransferases (Figure 5). Three concentrations were chosen: 1, 10, and 50 μM. No significant (>50%) inhibition was observed for any other methyltransferase at 1 μM. At 10 and 50 μM, inhibition values between 65% and 80% were observed for PRMT3, PRMT7, SUV39H2, and SMYD3. The exceptional selectivity observed with A-893 (23a) rendered it as a suitable compound to evaluate inhibition of SMYD2 in cells.
Figure 5.
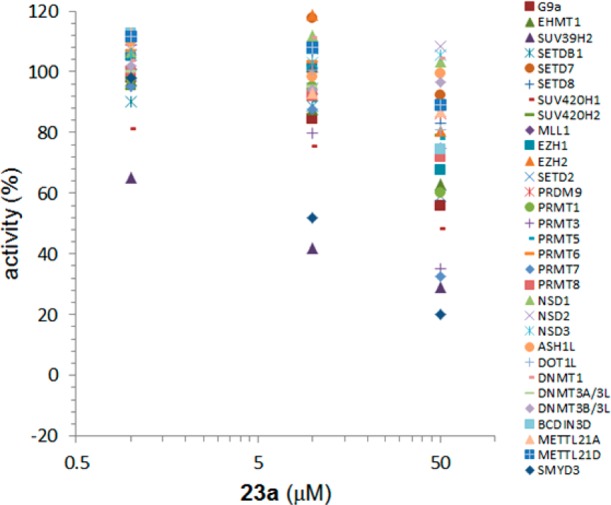
Selectivity of A-893 (23a). The inhibition of 30 methyltransferases at various concentrations of compound.
The functional consequence of SMYD2 inhibition was interrogated using both A-893 (23a) and AZ505 (1) in a p53 cellular assay.16 Human A549 lung carcinoma cells were chosen due to their high SMYD2 expression levels and wild-type p53 status.17 After 18 h of treatment with 10 μM of compound, changes in p53K370me1 were measured along with changes to overall p53 levels (Figure 6). The ratio of p53K370me1 to overall p53 levels was reduced, as expected, by treatment with either A-893 or AZ505 (Figure 6, part A). While this unexpectedly depicts a more robust response with AZ505, further dissection of the data provides clarity into the origin of this. While overall p53 levels were unaffected by A-893, a surprising >3-fold increase was observed with AZ505 (Figure 6B). Analysis of p53K370me1 levels reveals that inhibitor A-893 exhibited 42% reduction in the methyl mark, while AZ-505 was slightly less effective at 28% reduction (Figure 6C).
Figure 6.

Changes to p53K370me1 and overall p53 levels in A549 cells after treatment with SMYD2 inhibitors.
Accordingly, while the p53K370me1/p53 ratio looks far lower with AZ505 (Figure 6A), this is predominantly a result of an overall increase in p53. This may reflect an off-target phenotype that fortuitously is not present in A-893. The results of this experiment suggest that A-893 cleanly targets p53K370 methylation, without convolution, and highlight its usefulness as a cellular probe of SMYD2 function.
In summary, an analysis of the critical binding elements of the first reported SMYD2 inhibitor, AZ505 (1), via structure–activity relationships, has been conducted. Three regions of the molecule were systematically varied (Figure 2), and the results show large changes in activity from relatively modest changes in structure for regions 2 and 3, which predominantly function by occupying hydrophobic pockets in the enzyme. Analysis of region 1, the lysine channel binding site, reveals the importance of several key hydrogen bond interactions provided by the benzoxazinone subunit. Installation of a secondary alcohol in the linker region of the molecule led to A-893 (23a), a compound with an unexpectedly notable improvement in potency (>80-fold). This molecule was found to function as a peptide-competitive inhibitor and exhibited a high degree of selectivity over a panel of 30 additional methyltransferases. An analysis of the cocrystal structure of A-893 highlights the contribution of the newly–installed hydroxyl group to an intricate network of hydrogen bonds around the lysine pocket. Most notably, A-893 represents a viable cell-active probe to explore SMYD2 function. It reduced p53 methylation levels by 42% in A549 cells and is devoid of the confounding effect of total p53 protein elevation as observed with AZ505. The insights gained in the discovery of inhibitor A-893 (23a) provide the foundation for continued refinement and development of the next-generation of SMYD2 inhibitors
Supporting Information Available
Experimental details are provided pertaining to X-ray crystallography, biological assays, and the synthesis and characterization of all compounds. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00124.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The design, study conduct, and financial support for this study were provided by AbbVie and the SGC. AbbVie and the SGC participated in the data generation, interpretation of data, review, and approval of this publication. The SGC is a registered charity (no. 1097737) that receives funds from AbbVie, Bayer, Boehringer Ingelheim, Genome Canada through Ontario Genomics Institute Grant OGI- 055, GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and Wellcome Trust Grant 092809/Z/10/Z.
The authors declare the following competing financial interest: R.F.S., Z.W., M.A., G.G.C., J.G., C.G.J., D.M., B.S., N.B.S., and W.N.P. are employees of AbbVie, Inc. C.H.A., P.J.B., S.K., F.L., and M.V. receive funding from the Structural Genomics Consortium, University of Toronto, Toronto, Canada.
Supplementary Material
References
- Komatsu S.; Imoto I.; Tsuda H.; Kozaki K.; Muramatsu T.; Shimada Y.; Aiko S.; Yoshizumi Y.; Ichikawa D.; Otsuji E.; Inazawa J. Overexpression of SMYD2 relates to tumor cell proliferation and malignant outcome of esophageal squamous cell carcinoma. Carcinogenesis 2009, 30, 1139–1146. [DOI] [PubMed] [Google Scholar]
- Sakamoto L. H. T.; de Andrade R. V.; Felipe M. S. S.; Motoyama A. B.; Silva F. P. SMYD2 is highly expressed in pediatric acute lymphoblastic leukemia and constitutes a bad prognostic factor. Leuk. Res. 2014, 38, 496–502. [DOI] [PubMed] [Google Scholar]
- Brown M. A.; Sims R. J. III; Gottlieb P. D.; Tucker P. W. Identification and characterization of Smyd2: a split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol. Cancer 2006, 5, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Farha M.; Lambert J.-P.; Al-Madhoun A. S.; Elisma F.; Skerjanc I. S.; Figeys D. The tale of two domains: proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol. Cell. Proteomics 2008, 7, 560–572. [DOI] [PubMed] [Google Scholar]
- Saddic L. A.; West L. E.; Aslanian A.; Yates J. R. III; Rubin S. M.; Gozani O.; Sage J. Methylation of the retinoblastoma tumor suppressor by SMYD2. J. Biol. Chem. 2010, 285, 37733–37740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Perez-Burgos L.; Placek B. J.; Sengupta R.; Richter M.; Dorsey J. A.; Kubicek S.; Opravil S.; Jenuwein T.; Berger S. L. Repression of p53 activity by Smyd2-mediated methylation. Nature 2006, 444, 629–632. [DOI] [PubMed] [Google Scholar]
- Other histones (H2B and H4) have also been reported as substrates of SMYD2 methylation:Wu J.; Cheung T.; Grande C.; Ferguson A. D.; Zhu X.; Theriault K.; Code E.; Birr C.; Keen N.; Chen H. Biochemical characterization of human SET and MYND domain-containing protein 2 methyltransferase. Biochemistry 2011, 50, 6488–6497. [DOI] [PubMed] [Google Scholar]
- Ferguson A. D.; Larsen N. A.; Howard T.; Pollard H.; Green I.; Grande C.; Cheung T.; Garcia-Arenas R.; Cowen S.; Wu J.; Godin R.; Chen H.; Keen N. Structural basis of substrate methylation and inhibition of SMYD2. Structure 2011, 19, 1262–1273. [DOI] [PubMed] [Google Scholar]
- Nguyen H.; Allali-Hassani A.; Antonysamy S.; Chang S.; Chen L. H.; Curtis C.; Emtage S.; Fan L.; Gheyi T.; Li F.; Liu S.; Martin J. R.; Mendel D.; Olsen J. B.; Pelletier L.; Shatseva T.; Wu S.; Zhang F. F.; Arrowsmith C. H.; Brown P. J.; Campbell R. M.; Garcia B. A.; Barsyte-Lovejoy D.; Mader M.; Vedadi M. LLY-507, a cell-active, potent and selective inhibitor of protein lysine methyltransferase SMYD2. J. Biol. Chem. 2015, 10.1074/jbc.M114.626861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcaraz L.; Bonnert R. V.; Connolly S.; Cook A. R.; Fisher A.; Humphries A.; Raubo P.. Amine derivatives and their use in beta-2-adrenoreceptor mediated diseases. WO 2008/075025 A1, June 26, 2008.
- Herold J. M.; Wigle T. J.; Norris J. L.; Lam R.; Korboukh V. K.; Gao C.; Ingerman L. A.; Kireev D. B.; Senisterra G.; Vedadi M.; Tripathy A.; Brown P. J.; Arrowsmith C. H.; Jin J.; Janzen W. P.; Frye S. V. Small-molecule ligands of methyl-lysine binding proteins. J. Med. Chem. 2011, 54, 2504–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedadi M.; Barsyte-Lovejoy D.; Liu F.; Rival-Gervier S.; Allali-Hassani A.; Labrie V.; Wigle T. J.; DiMaggio P. A.; Wasney G. A.; Siarheyeva A.; Dong A.; Tempel W.; Wang S.-C.; Chen X.; Chau I.; Mangano T. J.; Huang X.; Simpson C. D.; Pattenden S. G.; Norris J. L.; Kireev D. B.; Tripathy A.; Edwards A.; Roth B. L.; Janzen W. P.; Garcia B. A.; Petronis A.; Ellis J.; Brown P. J.; Frye S. V.; Arrowsmith C. H.; Jin J. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 2011, 7, 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweis R. F.; Pliushchev M.; Brown P. J.; Guo J.; Li F.; Maag D.; Petros A. M.; Soni N. B.; Tse C.; Vedadi M.; Michaelides M. R.; Chiang G. G.; Pappano W. N. Discovery and development of potent and selective inhibitors of histone methyltransferase G9a. ACS Med. Chem. Lett. 2014, 5, 205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Barsyte-Lovejoy D.; Li F.; Xiong Y.; Korboukh V.; Huang X.; Allali-Hassani A.; Janzen W. P.; Roth B. L.; Frye S. V.; Arrowsmith C. H.; Brown P. J.; Vedadi M.; Jin J. Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J. Med. Chem. 2013, 56, 8931–8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toteva M. M.; Moran M.; Amyes T. L.; Richard J. P. Substituent effects on carbocation stability: The pKR for p-quinone methide. J. Am. Chem. Soc. 2003, 125, 8814–8819. [DOI] [PubMed] [Google Scholar]
- Total p53 Assay Whole Cell Lysate Kit (MSD). https://www.mesoscale.com/en/products/k150dbd-3/.
- O’Connor P. M.; Jackman J.; Bae I.; Myers T. G.; Fan S.; Mutoh M.; Scudiero D. A.; Monks A.; Sausville E. A.; Weinstein J. N.; Friend S.; Fornace A. J. Jr.; Kohn K. W. Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute Anticancer Drug Screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 1997, 56, 4285–4300. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.