

Abstract
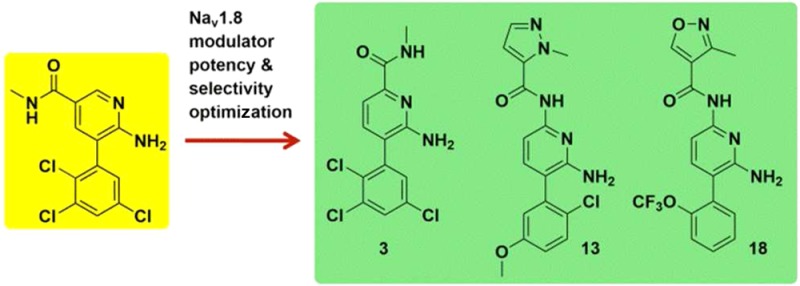
Voltage-gated sodium channels, in particular Nav1.8, can be targeted for the treatment of neuropathic and inflammatory pain. Herein, we described the optimization of Nav1.8 modulator series to deliver subtype selective, state, and use-dependent chemical matter that is efficacious in preclinical models of neuropathic and inflammatory pain.
Keywords: Voltage-gated sodium channels, sodium channel drugs, Nav1.8, SCN10A, TTX-R
Voltage-gated sodium channels (Nav) are a family of transmembrane (TM) ion channel proteins. Structurally, they are members of the 6-TM ion channel family and are composed of a TM α-subunit of approximately 260 kDa with associated transmembrane β-subunits of lower molecular weight. The family comprises nine members, Nav1.1–Nav1.9, which can be subdivided into tetrodotoxin-sensitive (TTX-S) and tetrodotoxin-resistant (TTX-R) subtypes. Navs play a key role in controlling excitability of neurons by regulating the threshold of firing, underlying the upstroke of the action potential and controlling the duration of interspike interval.1 Nonselective Nav blockers (e.g., lamotrigine, lacosamide, and mexilitine) have been successfully used in the clinic to treat pathological firing patterns of neurons that occur in a range of conditions such as chronic pain and epilepsy. However, such drugs have a narrow therapeutic window due to inhibition of sodium channels in the heart and throughout the central nervous system (CNS).
Selective block of Nav channels as pain targets gained traction with the recognition that some Nav subtypes showed preferential or exclusive expression in peripheral sensory neurons. A number of preclinical studies have implicated Nav1.3, 1.7, 1.8, and 1.9, which are expressed in DRG (dorsal root ganglion neurons) and trigeminal neurones, in nociceptive processing.2 Nav1.8 is highly (but not exclusively) expressed in nociceptors,3,4 and its expression and function is modulated by agents that cause pain5,6 Genetic ablation of NaV1.8 in rodents results in deficits in nociception following inflammation, but not neuropathic pain,7−10 while recent human genetic evidence suggest that gain of function mutations in NaV1.8 contributes to painful peripheral neuropathy.11 A-803467 is one of the first compounds in the public domain that demonstrated selectivity across human Nav subtypes and attenuated pain sensitivity in models of both nerve injury and inflammation induced pain (the latter with the exception of the formalin challenge paw withdrawal assay).12 This provided the first pharmacological evidence supporting a role for Nav1.8 in both inflammatory and neuropathic pain.
Existing subtype selective Nav1.8 inhibitors, for example, A-803467, exhibit poor oral pharmacokinetics in preclinical species.12 In this article, we discuss the discovery of subtype selective Nav1.8 modulators with good oral pharmacokinetics in preclinical studies.
Lamotrigine is a first generation sodium channel modulator and an anticonvulsant used in the treatment of epilepsy and bipolar disorder (Figure 1). Lamotrigine is a weak Nav1.8 inhibitor and shows no selectivity for Nav1.8 over TTX-S channels (measured in fluorescence based assays). Compound 1 was identified through file screening and suggested that trichloroaryl and aminopyridine units offered a potent Nav1.8 and TTX-S channel inhibition profile. Modification of the core to a diaminopyridine unit coupled with the introduction of 6-acetamide led to compound 2, which displayed a significant improvement in Nav1.8 potency together with approximately 20-fold selectivity over TTX-S channels.
Figure 1.
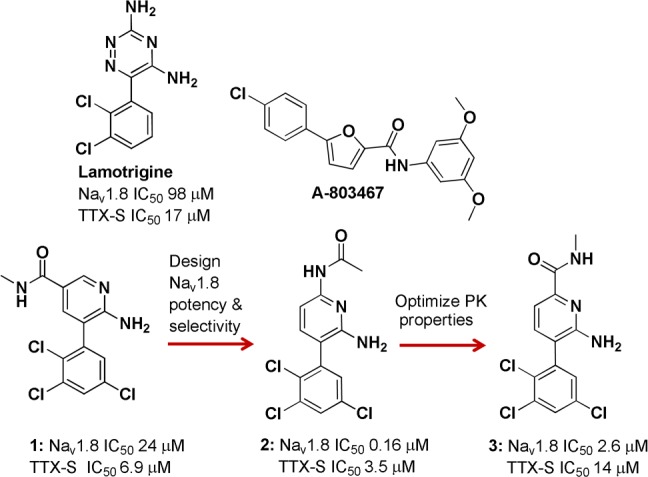
Discovery of candidate compound 3. IC50 data generated in VSP-FRET at hNav1.8 in HEK293 cells (in house cell line) with at least three tests on three different assay runs. TTX-S data generated in VSP-FRET in SHSY5Y neuroblastoma cell line expressing hNav1.2, hNav1.3, and hNav1.7.16
Compound 2 was assessed in an oral pharmacokinetic (PK) study in rat where it demonstrated high in vivo clearance (CL) (Table 1). The high CL was likely to be mediated by amide deacetylation as evidenced by rapid formation of the corresponding diaminopyridine metabolite in vivo. Reversal of the amide produced 3, which was more stable toward amide hydrolysis based on in vitro ADME data. Compound 2 readily underwent metabolism in rat and human liver microsomes (RLM and HLM), with intrinsic clearance (CLint) values of 29 and 41 μL/min/mg protein in RLM and HLM, respectively, while 3 displayed very little turnover (CLint < 9 and 7.1 μL/min/mg protein in RLM and HLM, respectively).
Table 1. Compounds 2, 3, 13, and 18: Pharmacokinetic Studies in Rat (R) and Dog (D).
| Cmpd | Dose (mg/kg) route | T1/2 (h) | Tmax (h) | Plasma CLa | Vd (L/kg) | Oral F (%) |
|---|---|---|---|---|---|---|
| 2 (R) | 3, p.o. | 4.1 | 0.75 | 204 | N.D. | |
| 3 (R) | 2, i.v. | 4.0 | 11.7 | 3.0 | ||
| 5, p.o. | 4.6 | 1.0 | 13.3 | 91 | ||
| 13 (R) | 1, i.v. | 3.9 | 6.7 | 2.25 | ||
| 2, p.o. | N.D. | 1.3 | 11.4 | 59 | ||
| 18 (D) | 0.1, i.v. | 9.7 | 6.2 | 5.3 | ||
| 0.25, p.o. | N.D. | 3.5 | 10.0 | 63 |
Plasma CL (i.v.) or CL/F (p.o.) in mL/min/kg.
Moreover, this analogue retained Nav1.8 activity (albeit weaker activity when compared with 2) and was Nav1.8 selective. Oral and i.v. rat PK studies with 3 exhibited low CL with high oral bioavailability of 91% (Table 1). Allometric scaling from rat data predicted low CL in human, oral bioavailability >90%, and half-life 8–28 h.13 Compound 3 was profiled through manual electrophysiology in order to assess potency and selectivity across multiple ion channels. The IC50 value for 3 at hNav1.8, was 0.19 μM with ≥50-fold selectivity for Nav1.8 over the other ion channels studied, including hERG. Furthermore, 3 inhibited native TTX-R currents in both human and rat DRG neurons (Table 3). The block of both human recombinant Nav1.8 and TTX-R currents from rat DRG neurons was found to be frequency and state dependent.14 State dependence of NaV modulators results from different affinities for each channel state, whereby binding to the inactivated state is often preferred. Many NaV modulators also demonstrate use dependence, which occurs when potency increases with NaV channel firing frequency. As 3 was both state and frequency dependent, it may be possible to achieve functional selectivity by preferentially targeting high frequency firing rates associated with neuroma ectopic activity and sparing the low frequency firing rates of the normal somatosensory system leading to an improved therapeutic index.15
Table 3. hNav1.8 Potency (IC50), Selectivity (IC50), and Antiallodynic Effects in Rodent Models of Neuropathic Pain for 3, 13, and 18a.
| hNav1.8 | hNav subtype selectivity | TTX-r Rat DRG | TTX-R human DRG | hERG | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | IC50 (μM) | n | IC50 (μM) | n | IC50 (μM) | n | IC50 (μM) | n | IC50 (μM) | Model | Effect significance | Unbound exposure (μM) |
| 3 | 0.19 | 5 | Nav1.1 = 13 | 5 | 0.44 | 4 | 0.31 | 4 | 30 | SNL hypersensitivity | P < 0.05 (equal to 100 mg/kg gabapentin) | 0.25 |
| Nav1.2 = 12.8 | 5 | |||||||||||
| Nav1.5 = 9.0 | 5 | |||||||||||
| Nav1.7 = 19 | 5 | |||||||||||
| 13 | 0.19 | 2 | Nav1.1 = 37 | 2 | 0.54 | 4 | 0.20 | 3 | >30 | TNT mechanical allodynia | SP < 0.05 (comparable to 10 mg/kg pregabalin) | 0.20 |
| Nav1.5 = 37 | 2 | |||||||||||
| Nav1.7 = 36 | 2 | |||||||||||
| 18 | 0.26 | 4 | SHSY5Yb = 10 Nav1.5 = 12 |
5 | 0.33 | 6 | ND | >30 | TNT mechanical allodynia | P < 0.05 (comparable to 10 mg/kg pregabalin) | 0.19 | |
| 4 |
IC50 values for 3, 13, and 18 at recombinantly expressed hNav1.8/β1 (Merck Millipore) and at TTX-R in rat and human DRG were determined using manual patch clamp electrophysiology. hNav subtype selectivity for 3 was also measured using manual patch clamp electrophysiology. IC50 values determined using manual patch clamp electrophysiology were determined at the respective V0.5 of inactivation for TTX-R and each channel isoform. For 13 and 18, human sodium channel subtype selectivity was measured using IonWorks Quattro and FRET, respectively.
SHSY5Y cells expressing hNav1.2, 1.3, and 1.7 were also used. The voltage protocol for the IonWorks Quattro and assay methodology for the FRET assay is detailed in the Supporting Information.
In order to improve the lipophilic efficiency (LipE) of 3 above 1.8, the trichloroaryl ring was varied (Table 2).17 In this picolinamide series, Nav1.8 potency was found to be highly dependent on aryl polychlorination with 3 being one of the most potent and LipE efficient compounds synthesized.18
Table 2. Aryl Ring SAR in Picolinamide Seriesa.

| Cmpd | Aryl | Nav1.8 IC50 (μM) | cLogP | LipE |
|---|---|---|---|---|
| 3 | 2,3,5-trichlorophenyl | 2.6 | 3.8 | 1.8 |
| 4 | 2-chlorophenyl | >32 | 2.5 | NA |
| 5 | 3-chlorophenyl | >32 | 2.8 | NA |
| 6 | 4-chlorophenyl | >32 | 2.8 | NA |
| 7 | 2,5-dichlorophenyl | 12 | 3.3 | 1.6 |
| 8 | 3,5-dichlorophenyl | >32 | 3.5 | NA |
IC50 data generated in VSP-FRET at hNav1.8 in HEK293 cells (in house cell line) with at least three tests on three different assay runs.16 cLogP was calculated using BioByte program version 4.3.
In a parallel effort, the instability of the acetamide in 2 was also addressed. Through variation of the amide moiety (Table 4) pyrazole 9 and isoxazole 10 were found to be two of the most potent amides identified. Both of these heterocyclic amides retained potency and Nav1.8 subtype selectivity over TTX-S while demonstrating improved amide stability in in vitro systems.18−20 These amides may be chemically stable due to increased conjugation between the carbonyl and heterocycle in comparison to the acetamide group. However, 9 and 10 were poorly soluble (<0.1 μg/mL at pH 7.2), which was attributable to their combination of high cLogP and planar shape.21 In order to reduce lipophilicity, the pyridyl core was replaced by a pyrazine core. Although cLogP was reduced by the pyridine to pyrazine switch (data not shown), the simultaneous potency loss of greater than 10-fold led to Nav1.8 inhibitors that were too weak to permit progression.
Table 4. Aryl Ring and Amide Moiety SAR Observed in the Acetamide Seriesa.
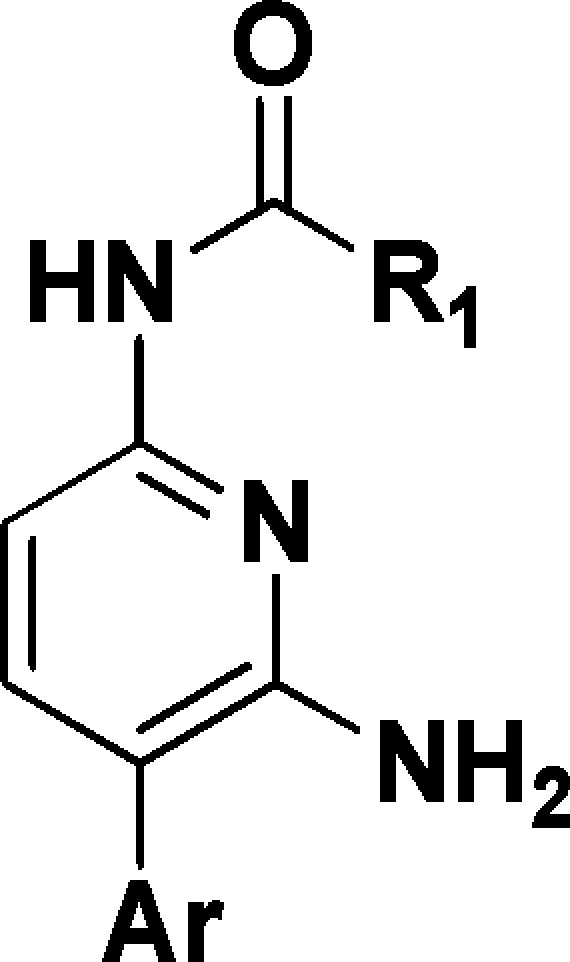
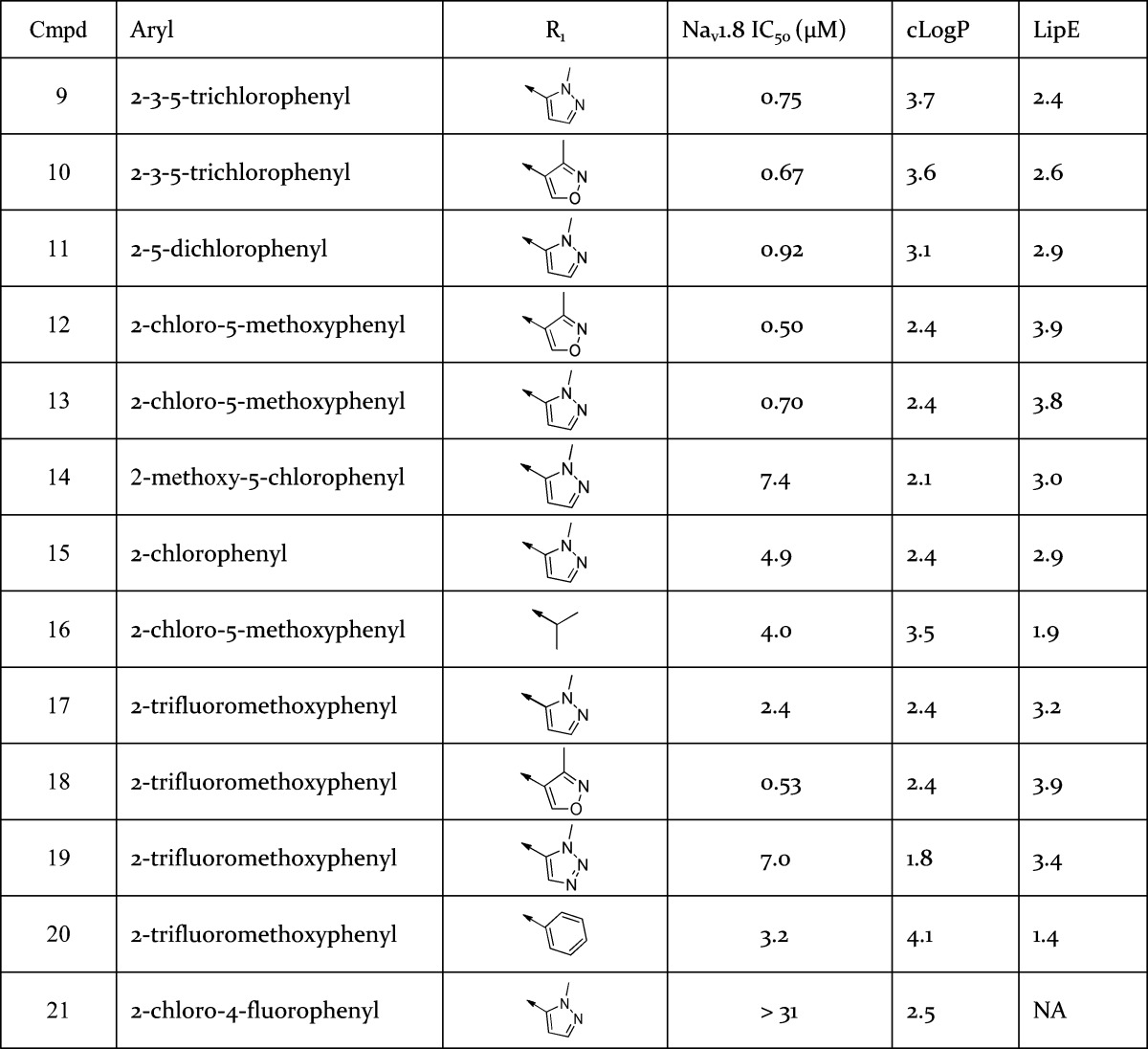
IC50 data generated in VSP-FRET at hNav1.8 in HEK293 cells (in house cell line) with at least three tests on three different assay runs.16 cLogP was calculated using BioByte program version 4.3.
The aryl unit was also varied (Table 4). Substitution at the 2- and 5-aryl positions with lipophilic groups tended to offer profiles with potency in the submicromolar range, e.g., 11, 12, and 13. In the case of 18, the larger 2-trifluoromethoxy moiety is sufficient to achieve submicromolar potency without the requirement of a 5-aryl substituent. Moreover, the observed SAR exhibited relatively steep activity cliffs, whereby, for instance, addition of a 4-F atom to compound 15 to give 21 decreased Nav1.8 potency from 4.9 to >31 μM.22
The amide group was optimized further (Table 4). Aliphatic amides were less stable to amide hydrolysis in vitro than aromatic amides as exemplified by 16, which readily underwent amide hydrolysis in buffer solutions at pH 7.4 such that in vitro ADME measurements were precluded. As before, this may be attributed to increased conjugation between the carbonyl and aromatic amides in comparison to the aliphatic amides. Furthermore, it appeared that the optimal heterocyclic amide was heavily dependent on the aryl unit it was combined with. For instance, a comparison of compounds 17–20 suggested that the ideal heterocyclic amide to combine with the 2-trifluoromethoxyaryl moiety was 3-methylisoxazole. However, analysis of the 2-chloro, 5-methoxy, and 2,3,5-trichlorophenyl aryl groups present in 9, 10, 12, 13, and 16 indicated very little difference in Nav1.8 potency between the 1-methyl-1H-pyrazole amides and 3-methylisoxazole amides.
SAR development led to the selection of 13 and 18 as compounds of interest to progress based on highest LipE (13 and 18 have a LipE two units greater than 3), Nav1.8 potency and acceptable solubility of ∼10 μg/mL. PK studies in rat with 13 indicated a low CL compound with good bioavailability ∼60% (Table 1). Allometric scaling predicted low CL of 13 in human (CL < 5 mL/min/kg).13 Preclinical oral PK studies of 18 also yielded low human CL estimates. Compounds 13 and 18 were profiled through manual electrophysiology. In a consistent manner to 3, 13 and 18 were selective for hNav1.8 over the other human sodium channel subtypes studied and the hERG channel. Compounds 13 and 18 also inhibited native TTX-R currents in rodent DRG neurons and the IC50 for the inhibition of TTX-R currents in human DRG neurons was determined for 13 (Table 3). The block of both human recombinant Nav1.8 and TTX-R currents from rat DRG neurons by both 13 and 18 were found to be frequency and state dependent.14,15
Compounds 3, 13, and 18 were efficacious in preclinical in vivo models of neuropathic pain: 3 was efficacious in the rat model of spinal nerve injury, while 13 and 18 were efficacious in the tibial nerve transection (TNT) induced mechanical allodynia model in rat (see Table 3 and Figure 2). Analysis of compound concentrations in plasma and cerebrospinal fluid samples suggested that 13 readily crossed the blood–brain barrier in rats (cerebrospinal fluid: unbound plasma concentration ratio 0.75:1). Moreover, an oral dose of 13 (250 mg/kg) achieving unbound exposures up to 0.33 μM in rats had no effect on either horizontal or vertical movements when compared to vehicle control animals indicating little or no effect of 13 on either the peripheral or central nervous system.
Figure 2.
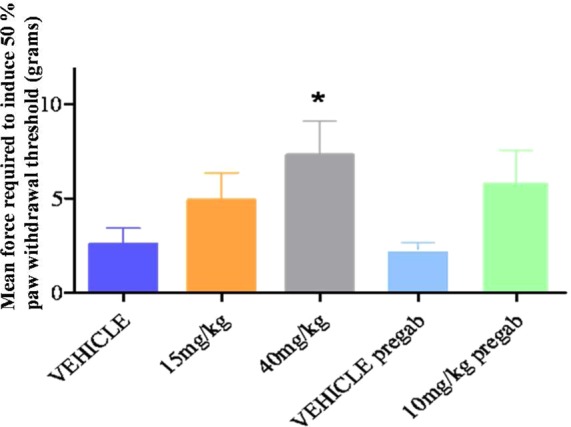
Antiallodynic effects of 13 in the TNT model of neuropathic pain. Error bars represent the SEM. A single dose of 40 mg/kg of 13 equivalent to a free plasma exposure of 0.2 μM, significantly shifted the 50% paw withdrawal threshold in the ipsilateral paw from a baseline of 1.7 ± 0.3 to 7.3 ± 1.8 g, 1.5 h after dosing (*P < 0.05). These effects were comparable to 10 mg/kg of Pregabalin, which shifted the 50% paw withdrawal threshold in the ipsilateral hindpaw from 1.7 ± 0.2 to 5.8 ± 1.7 g, 1.5 h after dosing (P = 0.07).
Based on the favorable Nav1.8 potency, LipE, selectivity and in vivo PK profiles of 3, 13, and 18 these compounds were selected as candidates for further progression.
Schemes 1 and 2 demonstrate a typical synthetic route for both the picolinamide and acetamide series, in this case illustrated by the synthesis of analogues 3 and 13. Further synthesis details are available in the Supporting Information.23
Scheme 1. Preparation of Compound 3.

Scheme 2. Preparation of Compound 13.

In conclusion, optimization of a biaryl lead has led to highly selective Nav1.8 series. Three key compounds 3, 13, and 18 have also demonstrated good pharmacokinetics in preclinical species leading to low human CL projections. Moreover, these compounds are efficacious in preclinical studies of neuropathic and inflammatory pain. Further data on the progression of 3, 13, and 18 will be reported in due course.
Acknowledgments
We thank Chan W. Huh and Wendy B. Wang for the generation of compound characterization data. Compound 13 (PF-04531083) is commercially available via Sigma-Aldrich (catalog # PZ0273). Compound 3 (PF-01247324) is commercially available via Sigma-Aldrich (catalog # PZ0274).
Glossary
ABBREVIATIONS
- Cmpd
compound
- PK
pharmacokinetics
- ADME
absorption distribution, metabolism, excretion
- CL
clearance
- PK
pharmacokinetics
- DRG
dorsal root ganglion neuron
- TNT
tibial nerve transection
- TM
transmembrane
- TTX-S
tetrodotoxin-sensitive
- TTX-R
tetrodotoxin-resistant
- LipE
lipophilic efficiency
- SNL
spinal nerve injury
- CNS
central nervous system
- L/kg
liters per kilogram
- μg/mL
microgram per milliliter
- h
hour
- i.v.
intravenous
- p.o.
pharmacokinetic study with oral administration
- T1/2
pharmacokinetic half-life
- Tmax
time of maximum concentration in vivo
- Vd
volume of distribution
- oral F
oral bioavaibility
Supporting Information Available
Experimental procedures and analytical data for the preparation of compounds 2–21, additional biological data, PK and efficacy study information, and experimental details for the in vitro assays. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00059.
The authors declare no competing financial interest.
Dedication
This publication is dedicated to the memory of Bill Million.
Supplementary Material
References
- Lampert A.; Eberhardt M.; Waxman Stephen G. Altered sodium channel gating as molecular basis for pain: contribution of activation, inactivation, and resurgent currents. Handb. Exp. Pharmacol. 2014, 221, 91–110. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj S. D.; Cummins T. R.; Black J. A.; Waxman S. G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 2010, 33, 325–347. [DOI] [PubMed] [Google Scholar]
- Amaya F.; Decosterd I.; Samad T. A.; Plumpton C.; Tate S.; Mannion R. J.; Costigan M.; Woolf C. J. Diversity of expression of the sensory neuron-specific TTX-resistant voltage-gated sodium ion channels SNS and SNS2. Mol. Cell. Neurosci. 2000, 15, 331–342. [DOI] [PubMed] [Google Scholar]
- Shields S. D.; Ahn H.-S.; Yang Y.; Han C.; Seal R. P.; Wood J. N.; Waxman S. G.; Dib-Hajj S. D. Nav1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain 2012, 153, 2017–2030. [DOI] [PubMed] [Google Scholar]
- England S.; Bevan S.; Docherty R. J. PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurons via the cyclic AMP-protein kinase A cascade. J. Physiol. 1996, 495, 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj S. D.; Fjell J.; Cummins T. R.; Zheng Z.; Fried K.; LaMotte R.; Black J. A.; Waxman S. G. Plasticity of sodium channel expression in DRG neurons in the chronic constriction injury model of neuropathic pain. Pain 1999, 83, 591–600. [DOI] [PubMed] [Google Scholar]
- Akopian A. N.; Souslova V.; England S.; Okuse K.; Ogata N.; Ure J.; Smith A.; Kerr B. J.; McMahon S. B.; Boyce S.; Hill R.; Stanfa L. C.; Dickenson A. H.; Wood J. N. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat. Neurosci. 1999, 2, 541–548. [DOI] [PubMed] [Google Scholar]
- Kerr B. J.; Souslova V.; McMahon S. B.; Wood J. N. A role for the TTX-resistant sodium channel Nav 1.8 in NGF-induced hyperalgesia, but not neuropathic pain. NeuroReport 2001, 12, 3077–3080. [DOI] [PubMed] [Google Scholar]
- Minett M. S.; Falk S.; Santana-Varela S.; Bogdanov Y. D.; Nassar M. A.; Heegaard A.-M.; Wood J. N. Pain without nociceptors? Nav1.7-independent pain mechanisms. Cell Rep. 2014, 6, 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar M. A.; Levato A.; Stirling L. C.; Wood J. N. Neuropathic pain develops normally in mice lacking both Nav1.7 and Nav1.8. Mol. Pain 2005, 1, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber C. G.; Lauria G.; Merkies I. S. J.; Cheng X.; Han C.; Ahn H.-S.; Persson A.-K.; Hoeijmakers J. G. J.; Gerrits M. M.; Pierro T.; Lombardi R.; Kapetis D.; Dib-Hajj S. D.; Waxman S. G. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 19444–19449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis M. F.; Honore P.; Shieh C.-C.; Chapman M.; Joshi S.; Zhang X.-F.; Kort M.; Carroll W.; Marron B.; Atkinson R.; Thomas J.; Liu D.; Krambis M.; Liu Y.; McGaraughty S.; Chu K.; Roeloffs R.; Zhong C.; Mikusa J. P.; Hernandez G.; Gauvin D.; Wade C.; Zhu C.; Pai M.; Scanio M.; Shi L.; Drizin I.; Gregg R.; Matulenko M.; Hakeem A.; Gross M.; Johnson M.; Marsh K.; Wagoner P. K.; Sullivan J. P.; Faltynek C. R.; Krafte D. S. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 8520–8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell G. W.; Masucci J. A.; Yan Z.; Hageman W. Allometric scaling of pharmacokinetic parameters in drug discovery: Can human CL, Vss and t1/2 be predicted from in-vivo rat data?. Eur. J. Drug Metab. Pharmacokinet. 2004, 29, 133–143. [DOI] [PubMed] [Google Scholar]
- Payne C. E. Brown A. R.; Theile J. W.; Mahoney J. H.; Loucif A. J. C.; Alexandrou A. J.; Fuller M. D.; Antonio B. M.;. Gerlach A. C.; Prime R. L.; Stockbridge G.; Kirkup A. J.; Bagal S. K.; Bannon A. W.; England S.; Chapman M. L.; Roeloffs R.; Bungay P.; Anand U.; Anand P.; Kemp M.; Butt R. P.; Stevens E. B.. A novel oral NaV 1.8 blocker PF-01247324 attenuates nociception and neuronal excitability. Br. J. Pharmacol. 2015, 172 ( (10), ), 2654–2670; DOI: 10.1111/bph.13092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagal S. K.; Brown A. D.; Cox P. J.; Omoto K.; Owen R. M.; Pryde D. C.; Sidders B.; Skerratt S. E.; Stevens E. B.; Storer R. I.; Swain N. A. Ion channels as therapeutic targets: a drug discovery perspective. J. Med. Chem. 2013, 56 (3), 593–624. [DOI] [PubMed] [Google Scholar]
- hNav1.8 data generated in VSP-FRET hNav1.8 in HEK293 cells with at least three tests on three different assay runs. TTX-S data generated in VSP-FRET in SHSY5Y neuroblastoma cell line expressing hNav1.2, hNav1.3, and hNav1.7.
- Freeman-Cook K. D.; Hoffman R. L.; Johnson T. W. Lipophilic efficiency: the most important efficiency metric in medicinal chemistry. Future Med. Chem. 2013, 5 (2), 113–115. [DOI] [PubMed] [Google Scholar]
- Lane C. A. L.; Maw G. N.; Rawson D. J.; Thompson L. R.. Pyridine derivatives. WO2006011050, 2006.
- Bagal S. K.; Gibson K. R.; Kemp M. I.; Poinsard C.; Stammen B. L.; Denton S. M.; Glossop M. S.. Pyridine derivatives. WO2008135826, 2008.
- Kemp M. I.2,6-Diaminopyrazine-amide derivatives as NaV1.8 channel modulators and their preparation, pharmaceutical compositions and use in the treatment of pain. WO2008135830, 2008.
- Ran Y.; Jain N.; Yalkowsky S. H. Prediction of aqueous solubility of organic compounds by the general solubility equation (GSE). J. Chem. Inf. Comput. Sci. 2001, 41, 1208–1217. [DOI] [PubMed] [Google Scholar]
- Hu X.; Hu Y.; Vogt M.; Stumpfe D.; Bajorath J. MMP-Cliffs: systematic identification of activity cliffs on the basis of matched molecular pairs. J. Chem. Inf. Model. 2012, 52, 1138–45. [DOI] [PubMed] [Google Scholar]
- Fray M. J.; Gillmore A. T.; Glossop M. S.; McManus D. J.; Moses I. B.; Praquin C. F. B.; Reeves K. A.; Thompson L. R. Optimization of permanganate oxidation and Suzuki-Miyaura coupling steps in the synthesis of a Nav1.8 sodium channel modulator. Org. Process Res. Dev. 2010, 14, 263–271. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.