

Abstract

Jumonji AT-rich interactive domain 1A (JARID1A), one of the jumonji C domain-containing histone demethylase (JHDM) family members, plays key roles in cancer cell proliferation and development of drug tolerance. Therefore, selective JARID1A inhibitors are potential anticancer agents. In this study, we searched for cell-active JARID1A inhibitors by screening hydroxamate compounds in our in-house library and the structural optimization based on docking study of the hit-compound to a homology model of JARID1A. As a result, we identified compound 6j, which selectively inhibits JARID1A over three other JHDM family members. Compound 7j, a prodrug form of compound 6j, induced a selective increase in the level of trimethylation of histone H3 lysine 4, a substrate of JARID1A. Furthermore, compound 7j synergistically enhanced A549 human lung cancer cell growth inhibition induced by vorinostat, a histone deacetylase inhibitor. These findings support the idea that JARID1A inhibitors have potential as anticancer agents.
Keywords: Epigenetics, histone demethylase, drug design
Reversible methylation of the ε-amino groups of histone lysine residues is involved in epigenetic gene activation or inactivation independently of DNA sequence.1−3 It occurs at several histone lysine residues, and each methylation site is thought to have a distinct role. Therefore, compounds that can regulate site-specific methylation are of great interest as chemical tools for biological studies. Such compounds are also candidate therapeutic agents for diseases associated with aberrant histone methylation.
Jumonji C domain-containing demethylases (JHDMs) are Fe(II)/α-ketoglutarate-dependent oxygenases that catalyze demethylation of methylated lysines of histones.4,5 JHDM family members identified so far have been categorized into five subfamilies, namely, JHDM1 (also known as KDM2/7), jumonji domain-containing protein 1 (JMJD1, also known as KDM3), JMJD2 (also known as KDM4), jumonji AT-rich interactive domain 1 protein (JARID1, also known as KDM5), and UTX/JMJD3 (also known as KDM6).6 Because each subfamily can distinguish the number of methyl groups and can demethylate lysine in a site-specific manner, JHDMs functionally regulate the state of histone lysine methylation and subsequent gene expression.7 Among the JHDM subfamilies, JARID1s are unique in that their substrates are limited to di- and trimethylated H3K4 (H3K4me2/3), and JARID1s interact with histone deacetylases (HDACs) as polycomb proteins to regulate gene transcription.8−10 In addition, they are involved in cancer cell growth and tumorigenesis.11,12 In particular, JARID1A (also known as KDM5A and RBP2) has been suggested to be associated with cancer cell proliferation13,14 and the development of drug tolerance.15 Therefore, selective inhibitors of JARID1A are of interest not only as biological tools to elucidate the biology of JARID1A and H3K4me2/3 but also as candidate anticancer agents.
To date, several groups, including ours, have reported JHDM inhibitors,7,16−18 such as N-oxalylglycine (NOG, 1),19 4-carboxy-4′-carbomethoxy-2,2′-bipyridine 2,20 NCDM-32a (3),21 NCDM-64 (4),22 and 2-(4-methylphenyl)-1,2-benzisothiazol-3(2H)-one (PBIT, 5)23 (Figure 1). Though some of these inhibitors, such as NCDM-64 (4), selectively inhibit a subfamily of JHDMs, most of them lack selectivity toward subfamily proteins or individual isozymes. It has been reported that compounds 2,245,23 and an inhibitor from EpiTherapeutics12 inhibit JARID1, but few details have been disclosed, and their intracellular activity is unknown. Therefore, there is still a need to develop selective JARID1 inhibitors and to elucidate the structural requirements for JARID1 inhibition. Thus, we set out to identify cell-active JARID1-selective inhibitors. Herein, we describe the discovery, structural optimization, and biological evaluation of JARID1A inhibitors.
Figure 1.

Structures of reported small-molecular JHDM inhibitors.
To find selective inhibitors of JARID1A, we screened analogues of hydroxamates 3 and 4, which we had identified as JMJD2 and JHDM1 inhibitors, respectively.21,22 Our previous docking studies of these inhibitors with JMJD2C (also known as KDM4C and GASC1) and JHDM1F (also known as KDM7B and PHF8) suggested that the hydroxamate group could bidentately coordinate to a catalytic Fe(II) ion in the catalytic site, and the carboxylate group could form hydrogen bonds with amino acid residues such as tyrosine, lysine, or asparagine in the active site of the two enzymes. The positions of the catalytic Fe(II) ion and the above-mentioned amino acid residues appeared to be similar among JARID1A, JMJD2C, and JHDM1F (Figure S1). On the other hand, it was suggested that the pockets around the active site are different among the enzymes.21,22 For example, there is a unique hydrophobic pocket around the active site of JHDM1F, where the alkyl chain of compound 4 is thought to be located.22 We assumed that the pocket around the active site might be unique in each JHDM and could contribute to the substrate specificity, i.e., H3K4 for JARID1A,8 H3K9 and H3K36 for JMJD2C,25 and H3K9 and H3K27 for JHDM1F.26 We also hypothesized that derivatives of compounds 3 and 4 whose alkyl amino group fits the unique pocket of JARID1A could be selective inhibitors of JARID1A. On the basis of this hypothesis, we screened our JHDM inhibitor candidate library, including various alkyl amino group-hydroxamate hybrid compounds. As a result of the screening using previously reported JARID1A, JMJD2C, and JHDM1F assays,21,22 we identified compound 6a, which has an N-methylbutan-1-amino group. Compound 6a efficiently and selectively inhibited JARID1A (JARID1A IC50 = 4.3 μM; JMJD2C IC50 = 55 μM; JMJD1A > 100 μM; JHDM1F IC50 > 100 μM) (Table 1, entry 4), showing much higher selectivity than the hydroxamates 3 and 4. Encouraged by this finding, we next attempted to optimize the structure of compound 6a.
Table 1. In Vitro JARID1A, JMJD2C, JMJD1A, and JHDM1F Inhibitory Activities of Compounds 1–4 and 6a.
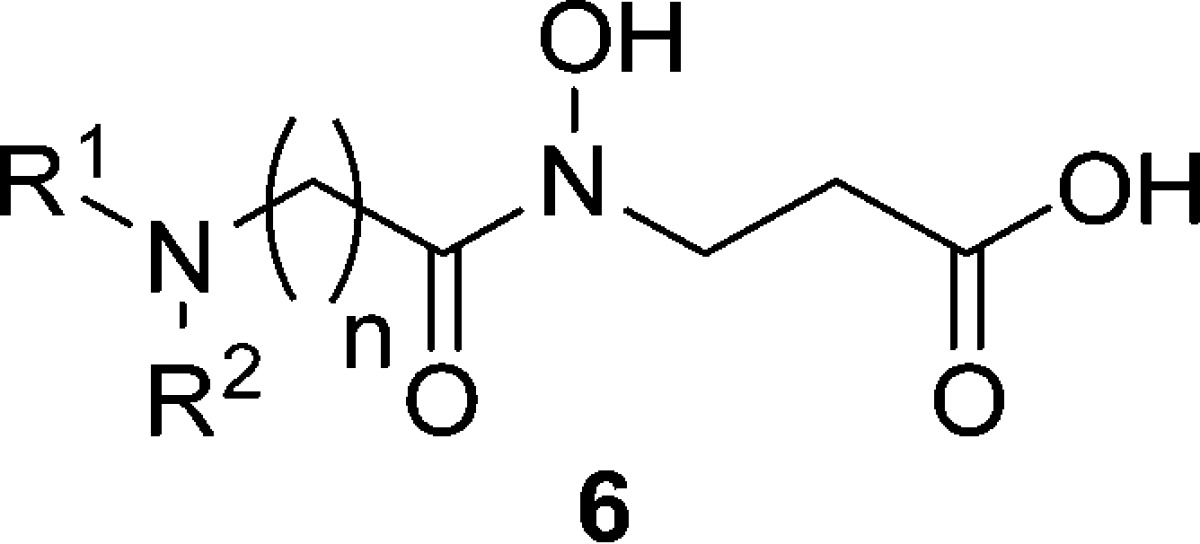
| structure |
IC50 (μM) |
|||||||
|---|---|---|---|---|---|---|---|---|
| entry | compd | R1 | R2 | n | JARID1A | JMJD2C | JMJD1A | JHDM1F |
| 1 | 1 | 250 | 430 | 569 | 640 | |||
| 2 | 3b | 13 | 2.2 | NDc | 19 | |||
| 3 | 4b | 55 | 83 | NDc | 1.2 | |||
| 4 | 6a | n-Bu | Me | 4 | 4.3 ± 0.9 | 55 ± 11 | >100 | >100 |
| 5 | 6b | n-Bu | Me | 2 | 53 ± 8.5 | >100 | NDc | NDc |
| 6 | 6c | n-Bu | Me | 3 | 6.3 ± 1.8 | 63 ± 6.4 | NDc | NDc |
| 7 | 6d | n-Bu | Me | 5 | 4.0 ± 0.6 | 61 ± 1.6 | NDc | 58 ± 16 |
| 8 | 6e | n-Bu | Me | 6 | 1.8 ± 0.1 | 6.5 ± 0.3 | NDc | NDc |
| 9 | 6f | Me | Me | 4 | 18 ± 2.0 | 51 ± 6.3 | NDc | NDc |
| 10 | 6g | Et | Me | 4 | 7.8 ± 1.0 | 62 ± 11 | NDc | NDc |
| 11 | 6h | n-Pr | Me | 4 | 8.9 ± 3.0 | 76 ± 4.7 | NDc | NDc |
| 12 | 6i | n-pentyl | Me | 4 | 2.3 ± 0.9 | 37 ± 26 | >100 | >100 |
| 13 | 6j | n-hexyl | Me | 4 | 3.3 ± 1.3 | 43 ± 11 | >100 | 73 ± 9 |
| 14 | 6k | n-Bu | Et | 4 | 14 ± 5.0 | 65 ± 13 | NDc | NDc |
| 15 | 6l | n-Bu | n-Bu | 4 | 19 ± 6.2 | >100 | NDc | NDc |
| 16 | 6m | n-Bu | n-hexyl | 4 | 4.5 ± 0.9 | >100 | >100 | 29 ± 13 |
| 17 | 6n | n-Bu | n-octyl | 4 | 3.1 ± 0.3 | 83 ± 17 | >100 | 24 ± 8 |
| 18 | 7i | 63 ± 16 | NDc | NDc | NDc | |||
| 19 | 7j | 21 ± 3.7 | NDc | NDc | NDc | |||
Value are means of at least three experiments.
Taken from the literature (ref (22)).
ND = no data available.
To guide optimization of the inhibitor structure, we initially performed a binding simulation of compound 6a with JARID1A and JMJD2C. The binding model of compound 6a to JARID1A (Figure 2A,B) showed three characteristic features: (i) the NH group forms a hydrogen bond with Asp 412, which is positioned near Ser 464 and Tyr 472; (ii) the n-butyl group is positioned near a small hydrophobic pocket formed by Pro 449, Val 468, and Trp 470; (iii) the methyl group is located in a large space at the entrance to the catalytic site of JARID1A. However, , in the binding model of JMJD2C (Figure 2C,D), (i) the NH group forms a hydrogen bond with Asn 292, (ii) the n-butyl group is located in a spacious tunnel-like pocket of the catalytic site where no interaction is observed between the n-butyl group and the amino acid residues of JMJD2C, and (iii) the methyl group is located near the backbone amide between Ser 290 and Thr 291. On the basis of these simulation results, we designed compounds 6b–n and evaluated their JARID1A- and JMJD2C inhibitory activity.
Figure 2.

(A) View of the conformation of compound 6a (ball and stick) docked into the JARID1A active site. (B) Schematic diagram of binding of compound 6a to JARID1A (a homology model based on the crystal structure of JMJD2A). (C) View of the conformation of compound 6a (ball and stick) docked into the JMJD2C active site (PDB code 2MXL). (D) Schematic diagram of binding of compound 6a to JMJD2C.
Compounds 6b–e with various linker lengths were expected to form a hydrogen bond with Ser 464 or Tyr 472 of JARID1A (Figure 2A,B), but should not form a hydrogen bond with JMJD2C because of loss of the interaction between the NH group and Asn 292 of JMJD2C. As shown in Table 1, JARID1A inhibitory activity and selectivity were distinctly dependent on linker length (Table 1, entries 5–8). Among compounds 6b–e, compound 6e (n = 6) showed the most potent JARID1A inhibitory activity with an IC50 of 1.8 μM. However, compound 6e also potently inhibited JMJD2C (IC50 = 6.5 μM), which resulted in low JARID1A selectivity as compared with compound 6a. With respect to the selectivity for JARID1A over JMJD2C, compounds 6a (n = 4) and 6d (n = 5) were superior to the other compounds (Table 1, entries 4 and 7).
Next, we examined the activity of compounds 6f–j, in which the n-butyl group of compound 6a is replaced with various alkyl groups (Table 1, entries 9–13). The longer alkyl chains of compounds 6i and 6j were expected to be located at the hydrophobic pocket formed by Pro 449, Val 468, and Trp 470 of JARID1A (Figure 2A,B) without interacting with any amino acid residues of JMJD2C (Figure 2C and 2D). Compounds 6i and 6j with a longer alkyl group (Table 1, entries 12 and 13) were slightly superior JARID1A inhibitors to compound 6a with the n-butyl group (Table 1, entry 4), although compounds 6f–h with a shorter alkyl group (Table 1, entries 9–11) were inferior. As we had expected, compounds 6f–j showed moderate activity against JMJD2C (IC50 = 37–76 μM) (Table 1, entries 9–13), similar to compound 6a (IC50 = 55 μM) (Table 1, entry 4). Compound 6i showed the highest selectivity among compounds 6f–j.
Next, we focused on changing the methyl group of compound 6a to various alkyl groups (6k–n). We expected that the R2 group of compounds 6k–n would be accommodated in the large space at the entrance of JARID1A, whereas there could be steric repulsion between the alkyl group and the backbone amides of Ser 290 and Thr 291 of JMJD2C (Figure 2). As we had expected, the longer alkyl group of compounds 6m and 6n was effective for JARID1A inhibition (Table 1, entries 16 and 17), but the shorter alkyl group of compounds 6k and 6l was not (Table 1, entries 14 and 15). In addition, compounds 6m and 6n with a longer alkyl group showed only weak activity against JMJD2C (Table 1, entries 16 and 17).
We next investigated the JMJD1A (also known as KDM3A) and/or JHDM1F inhibitory activity of compounds 6d, 6i, 6j, 6m, and 6n (Table 1, entries 7, 12, 13, 16, and 17). These compounds showed moderate JHDM1F inhibitory activities (IC50 > 24 μM) and no JMJD1A inhibitory activities (IC50 > 100 μM). As a result, they showed high selectivity for JARID1A over JMJD1A and JHDM1F as compared with compounds 3 and 4. Among them, compound 6i and 6j displayed relatively high JARID1A inhibitory activity and selectivity (Table 1, entry 12; Table S1).
The structure–activity and structure–selectivity relationships may be summarized as follows. With regard to the length between the amino group and the hydroxamate, n > 4 is better for JARID1A inhibition, and n = 4 or n = 5 is suitable for JARID1A-selective inhibition. As for the R1 and R2 groups, the combination of a methyl group and a long alkyl group, such as n-butyl, n-pentyl, and n-hexyl, is preferred for both JARID1A inhibition and JARID1A selectivity. These structure–activity and structure–selectivity relationships are consistent with the calculation results of the binding mode of compounds 6a, 6e, 6i, and 6j (Figures 2 and S2–S4 and Table S2).
Next, we investigated whether the identified inhibitors behave as JARID1 inhibitors in cell-based assays by using Western blot analysis to evaluate accumulation of H3K4me3, a physiological substrate of JARID1A.8 We applied JARID1A inhibitors 6i and 6j to human lung cancer A549 cells, which overexpress JARID1A,27 and analyzed the cell extracts after 24 h treatment. However, compounds 6i and 6j had little effect on the level of H3K4me3 (Figure 3A). On the basis of the idea that this might be due to poor membrane permeability, we next examined the methyl ester prodrugs 7i and 7j (Figure 3A), which were expected to permeate the cell membrane more efficiently than the parent compound and to be converted to 6i and 6j, respectively, by enzymatic hydrolysis within the cell.20 Though compounds 7i and 7j did not inhibit JARID1A strongly in enzyme assays (Table 1, entries 18 and 19), they increased the accumulated amount of H3K4me3 in a dose-dependent matter in cell-based assays (Figure 3B). The activity of compound 7j was a little higher than that of compound 7i. Then, to examine the selectivity in cells, we evaluated the effect of compound 7j on H3K9me3 and H3K27me2, substrates of JMJD2s and JHDM1s,25,26 respectively. Treatment with 100 μM compound 7j had little effect on the level of H3K9me3 and H3K27me2 (Figure 3C). The Western blot analysis data suggest that compound 7j inhibits JARID1 selectively over JMJD2 and JHDM1 in the cells.
Figure 3.

(A) Structures of compounds 7i and 7j. (B) Western blot detection of methylated histone levels in A549 cells treated with JARID1 inhibitors. H3K4me3 levels after 24 h incubation with 6i, 6j, 7i, and 7j. Values of H3K4me3/H3 ratio determined by optical density measurement of the blots are shown. (C) Western blot detection of methylated histone levels in A549 cells treated with 7j. H3K4me3, H3K9me3, and H3K27me2 levels after 24 h incubation with 7j. Values of H3K4me3/H3, H3K9me3/H3, and H3K27me2/H3 ratio determined by optical density measurement of the blots are shown. (D) Cell growth inhibition of A549 cells after 72 h incubation with combination of 7j and vorinostat, an HDAC inhibitor. Error bars represent the mean standard deviation (SD) of at least three samples. Combination with 100 μM compound 7j. *P < 0.05, **P < 0.01 (Student t test).
Finally, we evaluated the effects of compound 7j on proliferation of A549 cells. Contrary to our expectation, compound 7j was found to be inactive at concentrations up to 300 μM (Figure S5). We next tested whether compound 7j could act synergistically with vorinostat, a clinically used HDAC inhibitor, in growth inhibition assays using A549 cells. We expected that the combination of compound 7j and vorinostat would cause synergistic inhibition of A549 cell growth because it has been reported that HDACs are associated with A549 cell proliferation,28,29 and JARID1A is a participant in polycomb repressive complexes that down-regulate gene transcription cooperatively with HDACs.8 Combined treatment of A549 cells with vorinostat and 100 μM compound 7j induced more potent cell growth inhibition than that obtained with vorinostat alone (Figure 3D). However, JMJD2 inhibitor NCDM-32b (8), a prodrug form of NCDM-32a (3), did not enhance the antiproliferative activity of vorinostat (Figure S6). In addition, JARID1A inhibitors 6i and 6j and prodrug 7j did not inhibit HDAC1 (Figure S7). These results suggest that JARID1 inhibition, but not JMJD2 inhibition or HDAC inhibition, by compound 7j is involved in the synergistic inhibition of A549 cell growth with vorinostat. These results of cell-based assays suggest that JARID1A inhibitors could have potential as anticancer agents in combination with vorinostat.
In conclusion, we identified compound 6j, which selectively inhibits JARID1A over three other JHDM family members. In cell-based assays, compound 7j, a prodrug of compound 6j, induced selective accumulation of H3K4me3. Furthermore, the combination of compound 7j with vorinostat, a clinically used HDAC inhibitor, caused synergistic inhibition of A549 lung cancer cell growth. We believe that the compounds described here will be useful tools for probing the biology of JARID1A and are also candidate agents, or at least lead compounds, for cancer treatment.
Acknowledgments
We thank Ms. Mie Tsuchida, Dr. Yasunao Hattori, and Prof. Kenichi Akaji for their technical support.
Glossary
ABBREVIATIONS
- JARID
jumonji AT-rich interactive domain
- JHDM
jumonji C domain-containing demethylases
- JMJD
jumonji domain-containing protein
- HDAC
histone deacetylase
- NOG
N-oxalyl glycine
Supporting Information Available
Figures S1–S7, Table S1 and S2, and experimental procedures. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00083.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported in part by CREST, JST (to T.S.), Takeda Science Foundation (to T.S.), Mochida Memorial Foundation for Medical and Pharmaceutical Research (to T.S., and Y.I.), and Kyoto Prefectural Public University Corporation for Young Researcher Grant (to Y.I.).
The authors declare no competing financial interest.
Supplementary Material
References
- Kubicek S.; Jenuwein T. A crack in histone lysine methylation. Cell 2004, 119, 903–906. [DOI] [PubMed] [Google Scholar]
- Lee J. S.; Smith E.; Shilatifard A. The language of histone crosstalk. Cell 2010, 142, 682–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A.; Cuddapah S.; Cui K.; Roh T. Y.; Schones D. E.; Wang Z.; Wei G.; Chepelev I.; Zhao K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [DOI] [PubMed] [Google Scholar]
- Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat. Rev. Genet. 2007, 8, 829–833. [DOI] [PubMed] [Google Scholar]
- Kooistra S. M.; Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 2012, 13, 297–311. [DOI] [PubMed] [Google Scholar]
- Rose N. R.; Woon E. C.; Tumber A.; Walport L. J.; Chowdhury R.; Li X. S.; King O. N.; Lejeune C.; Ng S. S.; Krojer T.; Chan M. C.; Rydzik A. M.; Hopkinson R. J.; Che K. H.; Daniel M.; Strain-Damerell C.; Gileadi C.; Kochan G.; Leung I. K.; Dunford J.; Yeoh K. K.; Ratcliffe P. J.; Burgess-Brown N.; von Delft F.; Muller S.; Marsden B.; Brennan P. E.; McDonough M. A.; Oppermann U.; Klose R. J.; Schofield C. J.; Kawamura A. Plant growth regulator daminozide is a selective inhibitor of human KDM2/7 histone demethylases. J. Med. Chem. 2012, 55, 6639–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T.; Miyata N. Lysine demethylases inhibitors. J. Med. Chem. 2011, 54, 8236–8250. [DOI] [PubMed] [Google Scholar]
- Pasini D.; Hansen K. H.; Christensen J.; Agger K.; Cloos P. A.; Helin K. Coordinated regulation of transcriptional repression by the RBP2 H3K4 demethylase and Polycomb-Repressive Complex 2. Genes Dev. 2008, 22, 1345–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose R. J.; Yan Q.; Tothova Z.; Yamane K.; Erdjument-Bromage H.; Tempst P.; Gilliland D. G.; Zhang Y.; Kaelin W. G. Jr. The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell 2007, 128, 889–900. [DOI] [PubMed] [Google Scholar]
- Christensen J.; Agger K.; Cloos P. A.; Pasini D.; Rose S.; Sennels L.; Rappsilber J.; Hansen K. H.; Salcini A. E.; Helin K. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell 2007, 128, 1063–1076. [DOI] [PubMed] [Google Scholar]
- Teng Y. C.; Lee C. F.; Li Y. S.; Chen Y. R.; Hsiao P. W.; Chan M. Y.; Lin F. M.; Huang H. D.; Chen Y. T.; Jeng Y. M.; Hsu C. H.; Yan Q.; Tsai M. D.; Juan L. J. Histone demethylase RBP2 promotes lung tumorigenesis and cancer metastasis. Cancer Res. 2013, 73, 4711–4721. [DOI] [PubMed] [Google Scholar]
- Rasmussen P. B.; Staller P. The KDM5 family of histone demethylases as targets in oncology drug discovery. Epigenomics 2014, 6, 277–286. [DOI] [PubMed] [Google Scholar]
- Liang X.; Zeng J.; Wang L.; Fang M.; Wang Q.; Zhao M.; Xu X.; Liu Z.; Li W.; Liu S.; Yu H.; Jia J.; Chen C. Histone demethylase retinoblastoma binding protein 2 is overexpressed in hepatocellular carcinoma and negatively regulated by hsa-miR-212. PLoS One 2013, 8, e69784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J.; Ge Z.; Wang L.; Li Q.; Wang N.; Björkholm M.; Jia J.; Xu D. The histone demethylase RBP2 is overexpressed in gastric cancer and its inhibition triggers senescence of cancer cells. Gastroenterology 2010, 138, 981–992. [DOI] [PubMed] [Google Scholar]
- Hou J.; Wu J.; Dombkowski A.; Zhang K.; Holowatyj A.; Boerner J. L.; Yang Z. Q. Genomic amplification and a role in drug-resistance for the KDM5A histone demethylase in breast cancer. Am. J. Transl. Res. 2012, 4, 247–256. [PMC free article] [PubMed] [Google Scholar]
- Itoh Y.; Suzuki T.; Miyata N. Small-molecular modulators of cancer-associated epigenetic mechanisms. Mol. Biosyst. 2013, 9, 873–896. [DOI] [PubMed] [Google Scholar]
- Pachaiyappan B.; Woster P. M. Design of small molecule epigenetic modulators. Bioorg. Med. Chem. Lett. 2014, 24, 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X.; Liu Y.; Kubicek S.; Myllyharju J.; Tumber A.; Ng S.; Che K. H.; Podoll J.; Heightman T. D.; Oppermann U.; Schreiber S. L.; Wang X. A selective inhibitor and probe of the cellular functions of Jumonji C domain-containing histone demethylases. J. Am. Chem. Soc. 2011, 133, 9451–9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloos P. A.; Christensen J.; Agger K.; Maiolica A.; Rappsilber J.; Antal T.; Hansen K. H.; Helin K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 2006, 442, 307–311. [DOI] [PubMed] [Google Scholar]
- Rose N. R.; Ng S. S.; Mecinovic J.; Lienard B. M. R.; Bello S. H.; Sun Z.; McDonough M. A.; Opperman U.; Schofield C. J. Inhibitor scaffolds for 2-oxoglutarate-dependent histone lysine demethylases. J. Med. Chem. 2008, 51, 7053–7056. [DOI] [PubMed] [Google Scholar]
- Hamada S.; Suzuki T.; Mino K.; Koseki K.; Oehme F.; Flamme I.; Ozasa H.; Itoh Y.; Ogasawara D.; Komaarashi H.; Kato A.; Tsumoto H.; Nakagawa H.; Hasegawa M.; Sasaki R.; Mizukami T.; Miyata N. Design, synthesis, enzyme-inhibitory activity, and effect on human cancer cells of a novel series of Jumonji domain-containing protein 2 histone demethylase inhibitors. J. Med. Chem. 2010, 53, 5629–5638. [DOI] [PubMed] [Google Scholar]
- Suzuki T.; Ozasa H.; Itoh Y.; Zhan P.; Sawada H.; Mino K.; Walport L.; Ohkubo R.; Kawamura A.; Yonezawa M.; Tsukada Y.; Tumber A.; Nakagawa H.; Hasegawa M.; Sasaki R.; Mizukami T.; Schofield C. J.; Miyata N. Identification of the KDM2/7 histone lysine demethylase subfamily inhibitor and its antiproliferative activity. J. Med. Chem. 2013, 56, 7222–7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayegh J.; Cao J.; Zou M. R.; Morales A.; Blair L. P.; Norcia M.; Hoyer D.; Tackett A. J.; Merkel J. S.; Yan Q. Identification of small molecule inhibitors of Jumonji AT-rich interactive domain 1B (JARID1B) histone demethylase by a sensitive high throughput screen. J. Biol. Chem. 2013, 288, 9408–9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotili D.; Tomassi S.; Conte M.; Benedetti R.; Tortorici M.; Ciossani G.; Valente S.; Marrocco B.; Labella D.; Novellino E.; Mattevi A.; Altucci L.; Tumber A.; Yapp C.; King O. N.; Hopkinson R. J.; Kawamura A.; Schofield C. J.; Mai A. Pan-histone demethylase inhibitors simultaneously targeting Jumonji C and lysine-specific demethylases display high anticancer activities. J. Med. Chem. 2014, 57, 42–55. [DOI] [PubMed] [Google Scholar]
- Hillringhaus L.; Yue W. W.; Rose N. R.; Ng S. S.; Gileadi C.; Loenarz C.; Bello S. H.; Bray J. E.; Schofield C. J.; Oppermann U. Structural and evolutionary basis for the dual substrate selectivity of human KDM4 histone demethylase family. J. Biol. Chem. 2011, 286, 41616–41625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada Y.; Ishitani T.; Nakayama K. I. KDM7 is a dual demethylase for histone H3 Lys 9 and Lys 27 and functions in brain development. Genes Dev. 2010, 24, 432–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y. C.; Lee C. F.; Li Y. S.; Chen Y. R.; Hsiao P. W.; Chan M. Y.; Lin F. M.; Huang H. D.; Chen Y. T.; Jeng Y. M.; Hsu C. H.; Yan Q.; Tsai M. D.; Juan L. Histone demethylase RBP2 promotes lung tumorigenesis and cancer metastasis. Cancer Res. 2013, 73, 4711–4721. [DOI] [PubMed] [Google Scholar]
- Han S.; Fukazawa T.; Yamatsuji T.; Matsuoka J.; Miyachi H.; Maeda Y.; Durbin M.; Naomoto Y. Anti-tumor effect in human lung cancer by a combination treatment of novel histone deacetylase inhibitors: SL142 or SL325 and retinoic acids. PLoS One 2010, 5, e13834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundall B. K.; Denlinger C. E.; Jones D. R. Combined histone deacetylase and NF-κB inhibition sensitizes non-small cell lung cancer to cell death. Surgery 2004, 136, 416–425. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.