

Abstract
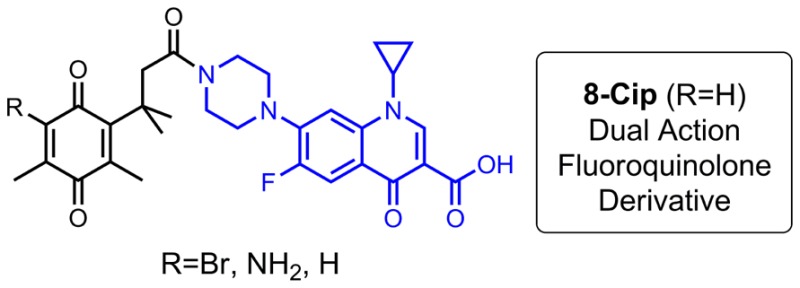
Several N-acyl ciprofloxacin quinone derivatives based on a trimethyl lock structure were synthesized, and their in vitro antibacterial activity against a panel of clinically relevant bacteria was evaluated. A few new analogues displayed enhanced activity against Gram-positive species compared to the parent drug. Additionally, studies of 8-Cip, which was the most potent compound tested, indicate that it may act through a dual-action mechanism.
Keywords: Ciprofloxacin, trimethyl lock, drug release, antibacterial
Since their discovery in 1970s, fluoroquinolone antibiotics have played an important role in treatment of bacterial infections and have saved countless millions of lives. Targeting bacterial DNA gyrase and topoisomerase, fluoroquinolones exhibit their significant bactericidal activity by forming ternary complexes of enzyme–DNA–drug and consequently blocking bacterial replication.1,2 Their notable antimicrobial activity, excellent pharmacokinetic properties, and few side effects have led to the widespread use of fluoroquinolones and, unfortunately, thus also the development of bacterial resistance.3 Therefore, although many advances in the search for new fluoroquinolones have been already made, there is a sense of urgency for new and more effective derivatives.
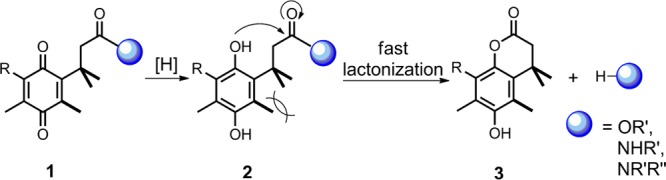
Recently, in our development of linkers to incorporate a drug release function in iron transport-mediated drug delivery, a series of carboxylic acids with a quinone “trimethyl lock” structure and their methyl esters were obtained as synthetic intermediates.4,5 The chemical nature of the trimethyl lock is an o-hydroxylcinnamic acid derivative.6 Reduction of the quinone trimethyl lock system generalized by structure 1 would lead to the corresponding hydroquinones 2 in which severe steric repulsion between three methyl groups promotes rapid lactonization with concomitant release of an alcohol or amine (Scheme 1).7,8 Several quinone trimethyl lock compounds have been investigated for reductase-activated prodrugs such as those based on mustard9 and oxindoles10 for anticancer therapy. In ciprofloxacin, one of the best known fluoroquinolones, it has been demonstrated that chemical modification of the secondary amine in the piperazinyl ring by acylation does not always alter the effect of these molecules toward their bacterial target.11 With the aim of probing the effects of the quinone trimethyl lock on antibacterial activity of fluoroquinonlones, we herein describe the syntheses of several ciprofloxacin derivatives with quinone trimethyl lock precursors as N-acyl substituents and studies of their in vitro antibacterial activity.
Scheme 1. Reaction of a Quinone Trimethyl Lock to Form Lactone via Reduction.
The syntheses of trimethyl locks with different substituents on the quinone are shown in Scheme 2. Both forms 5H and 8H were derived from lactone 4 by oxidative ring opening of the lactone using bromine8 and N-bromosuccinimide,12 respectively. The acids were conveniently converted to their methyl esters 5Me and 8Me via thionyl chloride-mediated or acid catalyzed esterification in methanol. Nucleophilic substitution of 5Me with NaN3 in aqueous methanol provided azide 6 in excellent yield. A triphenylphosphine-mediated reduction of the azide afforded vinylogous amide 7Me in 66% yield, which was subsequently hydrolyzed to furnish acid 7H. Water-soluble carbodiimide-mediated coupling of ciprofloxacin to quinone trimethyl locks 5H, 7H, and 8H provided N-acyl ciprofloxacin derivatives 5-Cip, 7-Cip, and 8-Cip with different substituents (Br-, NH2-, H-) on the quinone moieties in 44–71% yield (Scheme 3).
Scheme 2. Synthesis of Quinone Trimethyl Locks 5H, 7H, and 8H and Their Methyl Esters.

Reagents and conditions: (a) Br2, AcOH, rt, 80%; (b) SOCl2, MeOH, reflux, 77%; (c) NaN3, MeOH–H2O, rt, 91%; (d) 1. PPh3, CH2Cl2, rt; 2. AcOH, THF–H2O, reflux, 66% for 2 steps; (e) LiOH, MeOH–H2O, rt, 91%; (f) NBS, CH3CN–H2O–acetone, rt, 79%; (g) H2SO4 (cat.), MeOH, reflux, 95%.
Scheme 3. Synthesis of N-Acyl Ciprofloxacin Derivatives.

N-Acyl ciprofloxacin quinone derivatives 5-Cip, 7-Cip, and 8-Cip were initially screened for antibiotic activity against representative Gram-positive and Gram-negative bacteria as well as Mycobacterium vaccae using agar diffusion assays (Table 1). All three ciprofloxacin derivatives displayed strong antibacterial activities when tested at 0.1 mM, as indicated by the large zones of inhibition they induced. The trimethyl lock cores, as represented by methyl esters 5Me, 7Me, and 8Me, were generally inactive. Moreover, all three ciprofloxacin derivatives showed moderate to good activity against Mycobacterium vaccae, a nonpathogenic model organism for Mycobacterium tuberculosis that causes tuberculosis, suggesting their potential in antituberculosis agent development.
Table 1. Diameter of Growth Inhibition Zones (mm) in the Agar Diffusion Antibacterial Susceptibility Assaya.
| 5-Cip | 5Me | 7-Cip | 7Me | 8-Cip | 8Me | Cip | ||
|---|---|---|---|---|---|---|---|---|
| 1 | B.subtilis ATCC 6633 | 35 | 0 | 36 | 0 | 39 | 0 | 20/24Pc |
| 2 | S. aureus SG511 | 25 | 0 | 31 | 0 | 33 | 0 | 21b |
| 3 | M. luteus ATCC10240 | 19 | 0 | 17V | 0 | 28 | 0 | 0b |
| 4 | M. vaccae IMET10670 | 37 | 0 | 24/30P | 0 | 32/42P | 0 | 23b |
| 5 | A. baumannii ATCC17961 | 18/23P | 0 | 24/30P | 0 | 26/32P | 0 | 18/21Pb |
| 6 | P. aeruginosa K799/wt | 29/35P | 0 | 22/32P | 0 | 41 | 0 | 15c |
| 7 | P. aeruginosa K799/61 | 29/38P | 0 | 31/35P | 0 | 35/42P | 0 | 19/24Pc |
| 8 | E. coli X580 | 33/40P | 0 | 31/37P | 0 | 32/40P | 0 | 23d |
| 9 | E. coli DC0 | 17/21P | 0 | 16/23P | 0 | 21/29P | 0 | 18b |
| 10 | E. coli DC2 | 24 | 0 | 23 | 0 | 27 | 0 | 22b |
P, unclear inhibition zone/many colonies in the inhibition zone; V, very unclear inhibition zone. Exactly 50 μL of a 0.1 mM solution of each compound dissolved in 1:9 DMSO/MeOH was added to 9 mm wells in agar media (Standard I Nutrient Agar, Serva, or Mueller Hinton II Agar, Becton, Dickinson and Company). Inhibition zones were read after incubation at 37 °C for 24 h. M. vaccae was incubated at 37 °C for 44 h.
Ciprofloxacin was tested at 5 μg/mL in H2O.
Ciprofloxacin was tested at 1.66 μg/mL in H2O.
Ciprofloxacin was tested at 0.33 μg/mL in H2O.
All ciprofloxacin derivatives were subjected to further assays to determine their minimum inhibitory concentrations (MIC) (Table 2). Two additional N-acyl derivatives 9 and 10 that have similar trimethyl substituted side chain structures but without the quinone fragment were also synthesized and included in the assay. In general, the trimethyl lock ciprofloxacin quinone derivatives possessed equal or enhanced in vitro activity against Gram-positive species compared to the parent drug. Among the three derivatives, compound 8-Cip was the most active with MIC values less than 0.05 and 0.2 μM against S. aureus and M. luteus, respectively, against which ciprofloxacin is less active (MIC in 0.47 and 8 μM, respectively). However, 9 and 10 were less active than the trimethyl lock ciprofloxacin derivatives, indicating that the quinone structure plays a key role for the activity enhancement observed. In fact, trimethyl lock compound 8Me has very weak antibacterial activity (MIC = 50–80 μM) against the Gram-positive species tested except S. aureus, suggesting that the trimethyl lock ciprofloxacin derivatives may be dual-action antibiotics.
Table 2. MIC Values of the N-Acyl Ciprofloxacin Derivatives against Selected Bacterial Strainsa.
| 5-Cip | 7-Cip | 8-Cip | 9 | 10 | 8Me | Cip | |
|---|---|---|---|---|---|---|---|
| B.subtilis ATCC 6633 | <0.05 | <0.05 | <0.05 | <0.05 | <0.05 | 50 | 0.03 |
| S. aureus SG511 | 0.1 | <0.05 | <0.05 | 0.4 | 0.4 | >100 | 0.47 |
| M. luteus ATCC10240 | 1.56 | 12.5 | 0.2 | >100 | 12.5 | 50 | 8b |
| M. vaccae IMET10670 | 2 | 2 | 0.5 | 2 | 2 | 80 | 0.47 |
| A. baumannii ATCC17961 | 0.78 | 0.6 | 0.39 | >100 | 25 | >100 | 0.24 |
| P. aeruginosa K799/wt | 5 | 2.5 | 1.25 | >100 | >100 | >100 | 0.47 |
| P. aeruginosa K799/61 | 2.5 | 5 | 2.5 | >100 | >100 | >100 | 0.24 |
| E. coli DC0 | 12.5 | 25 | 12.5 | >100 | >100 | >100 | 0.47 |
| E. coli DC2 | 8.33 | 12.5 | 6.25 | >100 | 100 | >100 | 0.24 |
The trimethyl lock ciprofloxacin derivatives displayed decreased activity (2–50-fold) against mycobacteria and Gram-negative bacteria compared to the parent drug, presumably because of the loss of zwitterionic structure of ciprofloxacin, which favors membrane penetration.15 However, 9 and 10 were virtually inactive (MIC > 100 μM) against Gram-negative bacteria, suggesting that the quinone fragment partially compensates for the loss of activity caused by the nonzwitterionic structure.
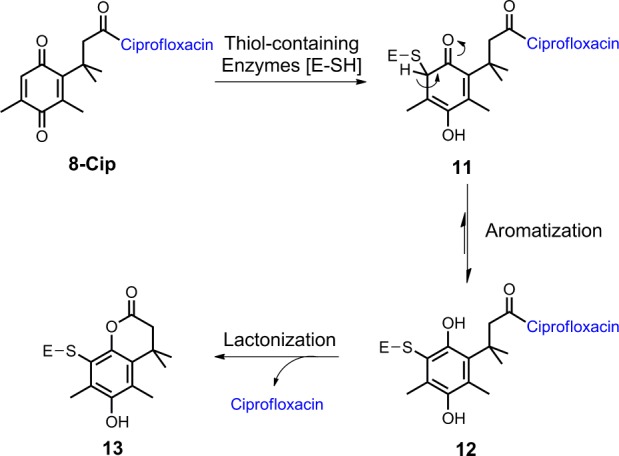
Based on results from antibacterial assays and the chemical nature of the trimethyl lock system, we have proposed modes of action of the trimethyl lock ciprofloxacin derivatives. While all three derivatives could serve as prodrugs, which are activated by potential reductases as depicted in Scheme 1, compound 8-Cip may have an additional dual-action mechanism by serving as a prodrug and as a covalent enzyme inhibitor, which is consistent with its superior antibacterial activity. Conjugate addition of sulfur nucleophiles to quinones is well-known and has been exploited in design of certain enzyme inhibitors.16 Similarly, the activation of compound 8-Cip could be initiated by the reductive alkylation with certain thiol-containing enzymes (Scheme 4). The resulting thiol-quinone adduct 11 would be expected to aromatize to the corresponding hydroquinone 12. The rapid lactonization of 12 induced by the trimethyl lock could release ciprofloxacin and meanwhile result in a covalent inactivated enzyme 13. In contrast, such a pathway with an aromatization step could not be established for compounds 5-Cip and 7-Cip.
Scheme 4. Proposed Modes of Action of Compound 8-Cip.
To determine whether 8-Cip could be activated in the presence of thiol-containing enzymes as proposed, model reactions were examined for the reaction of thiols with N-acyl phenylpiperazine 8-PP, a more soluble alternative to 8-Cip, and the reaction was monitored by HPLC (Scheme 5). The thiol methyl mercaptoacetate was chosen for the model studies principally because its pKa (7.9) was similar to that of the thiol in cysteine (pKa = 8), which is ubiquitous in biological systems. We were pleased to find that in the presence of the thiol, 8-PP (retention time = 11.1 min) was largely converted to lactone 14 (retention time = 9.2 min) with concomitant release of the phenylpiperazine (retention time = 8.5 min) in buffered aqueous acetonitrile after 3 h. In sharp contrast, no reaction was observed for compound 7-PP, a representative system lacking the aromatization pathway, under the same conditions that led to rapid lactonization for 8-PP. These data provide evidence for 8-Cip being a dual-action prodrug that might utilize a unique thiol-activated pathway.
Scheme 5.
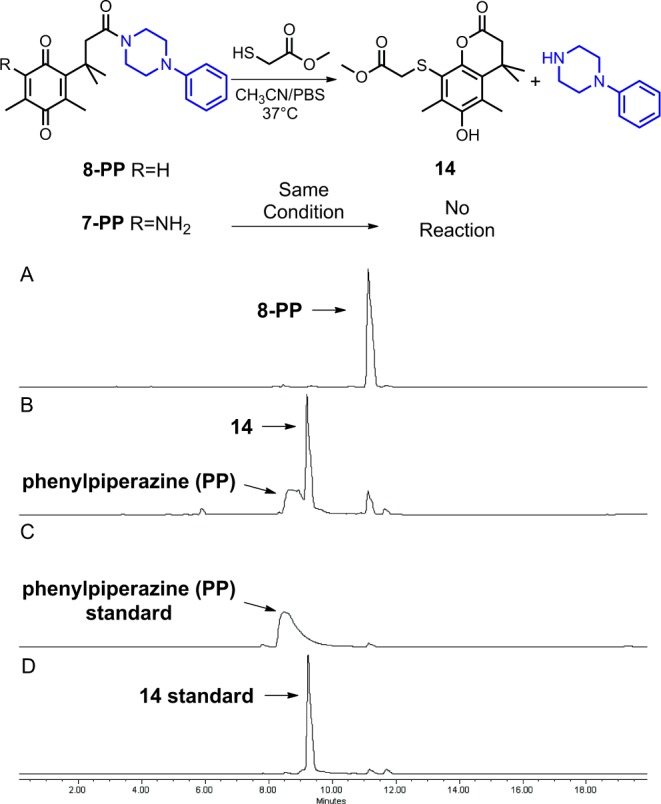
Top: model reaction for enzyme activation of trimethyl lock derivatives. Bottom: HPLC studies of the activation of 8-PP. Panel A, 8-PP standard; panel B, 8-PP after treatment with methyl mercaptoacetate in CH3CN/PBS solution at 37 °C for 3 h; panel C, phenylpiperazine (PP) standard; panel D, 14 standard.
In summary, we have described the syntheses of a series of ciprofloxacin derivatives with a quinone trimethyl lock as the N-acyl substituent and their antibacterial activity. Among the derivatives, compound 8-Cip displayed enhanced activity against several Gram-positive species compared to the parent drug. The superior activity of 8-Cip suggests that it may act through a dual-action mechanism by serving as a prodrug and as a covalent thiol-containing enzyme inhibitor. This dual-action mechanism also has great potential for use in enzyme-activated prodrug strategies for site-selective drug delivery. Future work will focus on syntheses of other drug derivatives based on 8H and examine the release of drug in appropriate enzyme assays.
Acknowledgments
We gratefully acknowledge the use of the NMR facilities provided by the Lizzadro Magnetic Resonance Research Center at the University of Notre Dame (UND) under the direction of Dr. Jaroslav Zajicek and the mass spectrometry services provided by the UND Mass Spectrometry & Proteomics Facility (Mrs. N. Sevova, Dr. W. Boggess, and Dr. M. V. Joyce; supported by the National Science Foundation under CHE-0741793).
Glossary
ABBREVIATIONS
- DNA
DNA
- Cip
ciprofloxacin
- MIC
minimum inhibitory concentration required to inhibit the growth of 90% of organisms
- HPLC
high-performance liquid chromatography
- PP
phenylpiperazine
Supporting Information Available
Experimental procedures, copies of spectral data, and characterization data. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00146.
This research was supported by grant 2R01AI054193 from the National Institutes of Health (NIH). C.J. also acknowledges a fellowship from the Notre Dame CBBI training grant (NIH T32 GM075762).
The authors declare no competing financial interest.
Supplementary Material
References
- Mitscher L. A. Bacterial topolsomerase inhibitors: quinolone and pyridone antibacterial agents. Chem. Rev. 2005, 105, 559–592. [DOI] [PubMed] [Google Scholar]
- Drlica K.; Hiasa H.; Kerns R.; Malik M.; Mustaev A.; Zhao X. L. Quinolones: action and resistance updated. Curr. Top. Med. Chem. 2009, 9, 981–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drlica K.; Zhao X.; Malik M.; Salz T.; Kerns R. Fluoroquinolone resistance: mechanisms, restrictive dosing, and anti-mutant screening strategies for new compounds. Antibiot. Discovery Dev. 2012, 485–514. [Google Scholar]
- Ji C.; Miller M. J. Chemical syntheses and in vitro antibacterial activity of two desferrioxamine B-ciprofloxacin conjugates with potential esterase and phosphatase triggered drug release linkers. Bioorg. Med. Chem. 2012, 20, 3828–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C.; Miller M. J. Siderophore-fluoroquinolone conjugates containing potential reduction-triggered linkers for drug release: synthesis and antibacterial activity. BioMetals 2015, 28, 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M. N.; Raines R. T. Trimethyl lock: a trigger for molecular release in chemistry, biology, and pharmacology. Chem. Sci. 2012, 3, 2412–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B. H.; Liu S. M.; Borchardt R. T. Development of a novel redox-sensitive protecting group for amines which utilizes a facilitated lactonization reaction. J. Org. Chem. 1995, 60, 539–543. [Google Scholar]
- Carpino L. A.; Triolo S. A.; Berglund R. A. Reductive lactonization of strategically methylated quinone propionic-acid esters and amides. J. Org. Chem. 1989, 54, 3303–3310. [Google Scholar]
- Volpato M.; Abou-Zeid N.; Tanner R. W.; Glassbrook L. T.; Taylor J.; Stratford I.; Loadman P. M.; Jaffar M.; Phillips R. M. Chemical synthesis and biological evaluation of a NAD(P)H: quinone oxidoreductase-1-targeted tripartite quinone drug delivery system. Mol. Cancer Ther. 2007, 6, 3122–3130. [DOI] [PubMed] [Google Scholar]
- Maskell L.; Blanche E. A.; Colucci M. A.; Whatmore J. L.; Moody C. J. Synthesis and evaluation of prodrugs for anti-angiogenic pyrrolylmethylidenyl oxindoles. Bioorg. Med. Chem. Lett. 2007, 17, 1575–1578. [DOI] [PubMed] [Google Scholar]
- Cormier R.; Burda W. N.; Harrington L.; Edlinger J.; Kodigepalli K. M.; Thomas J.; Kapolka R.; Roma G.; Anderson B. E.; Turos E.; Shaw L. N. Studies on the antimicrobial properties of N-acylated ciprofloxacins. Bioorg. Med. Chem. Lett. 2012, 22, 6513–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng A. L.; Shan D. X.; Shi X. L.; Wang B. H. A novel resin linker for solid-phase peptide synthesis which call be cleaved using two sequential mild reactions. J. Org. Chem. 1999, 64, 7459–7466. [Google Scholar]
- Wencewicz T. A.; Yang B.; Rudloff J. R.; Oliver A. G.; Miller M. J. N-O chemistry for antibiotics: discovery of N-alkyl-N-(pyridin-2-yl)hydroxylamine scaffolds as selective antibacterial agents using nitroso Diels-Alder and ene chemistry. J. Med. Chem. 2011, 54, 6843–6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, 8th ed; Clinical and Laboratory Standards Institute (CLSI): Villanova, PS, 2009; approved standard document M07-A7. [Google Scholar]
- Cramariuc O.; Rog T.; Javanainen M.; Monticelli L.; Polishchuk A. V.; Vattulainen I. Mechanism for translocation of fluoroquinolones across lipid membranes. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 2563–2571. [DOI] [PubMed] [Google Scholar]
- Vadnere M. K.; Maggiora L.; Mertes M. P. Thiol addition to quinones-model reactions for the inactivation of thymidylate synthase by 5-para-benzoquinonyl-2′-deoxyuridine 5′-phosphate. J. Med. Chem. 1986, 29, 1714–1720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.