

Abstract

Interleukin-1 receptor associated kinase 4 (IRAK4) is an essential signal transducer downstream of the IL-1R and TLR superfamily, and selective inhibition of the kinase activity of the protein represents an attractive target for the treatment of inflammatory diseases. A series of 5-amino-N-(1H-pyrazol-4-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamides was developed via sequential modifications to the 5-position of the pyrazolopyrimidine ring and the 3-position of the pyrazole ring. Replacement of substituents responsible for poor permeability and improvement of physical properties guided by cLogD led to the identification of IRAK4 inhibitors with excellent potency, kinase selectivity, and pharmacokinetic properties suitable for oral dosing.
Keywords: IRAK4; 5-amino-N-(1H-pyrazol-4-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide; SAR; serine-threonine kinase; kinase inhibitor; inflammatory disease
Interleukin-1 receptor associated kinase 4 (IRAK4) is an intracellular serine-threonine kinase that belongs to the IRAK family of kinases (IRAK1, IRAK2, IRAK-M, and IRAK4).1,2 It has been demonstrated that IRAK4 is an essential signal transducer downstream of the interleukin-1 receptor (IL-1R) and toll-like receptor (TLR) superfamily that mediates the innate immune response by upregulating the expression of inflammatory genes in multiple target cells.3 IRAK4–/– mice showed severe defects in cytokine responses and downstream signaling pathways induced by IL-1R and TLRs.4 In addition, cells derived from IRAK4-deficient patients failed to induce downstream cytokines in response to known IL-1R and TLR ligands.5 In view of the generic evidence of the critical role of IRAK4 in IL-1R and TLR signaling, IRAK4 could be an attractive target for the treatment of inflammatory diseases,6 including rheumatoid arthritis (RA),7,8 inflammatory bowel disease (IBD),9,10 asthma,11 and systemic lupus erythematosus (SLE).12 In fact, the discovery of novel IRAK4 inhibitors has been actively pursued by many research groups, and various classes of IRAK4 inhibitors have been reported to date.13−19
In a high-throughput screening (HTS) campaign, N-(1H-pyrazol-5-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamides were identified as promising hits. Further exploration of the scaffold, including hybrids with known IRAK4 inhibitors,20 led to the identification of N-(3-carbamoyl-1-methyl-1H-pyrazol-4-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (1) as a lead. Compound 1 not only displayed attractive lead-like properties such as low molecular weight (285 g/mol), high ligand binding efficiency (LBE) (0.44), and moderate cell permeability (Papp = 17 × 10–6 cm/s) but also offered a good kinome profile (>100-fold selective against 89% of the kinases) (Table 1).
Table 1. Key Properties of Compound 1.
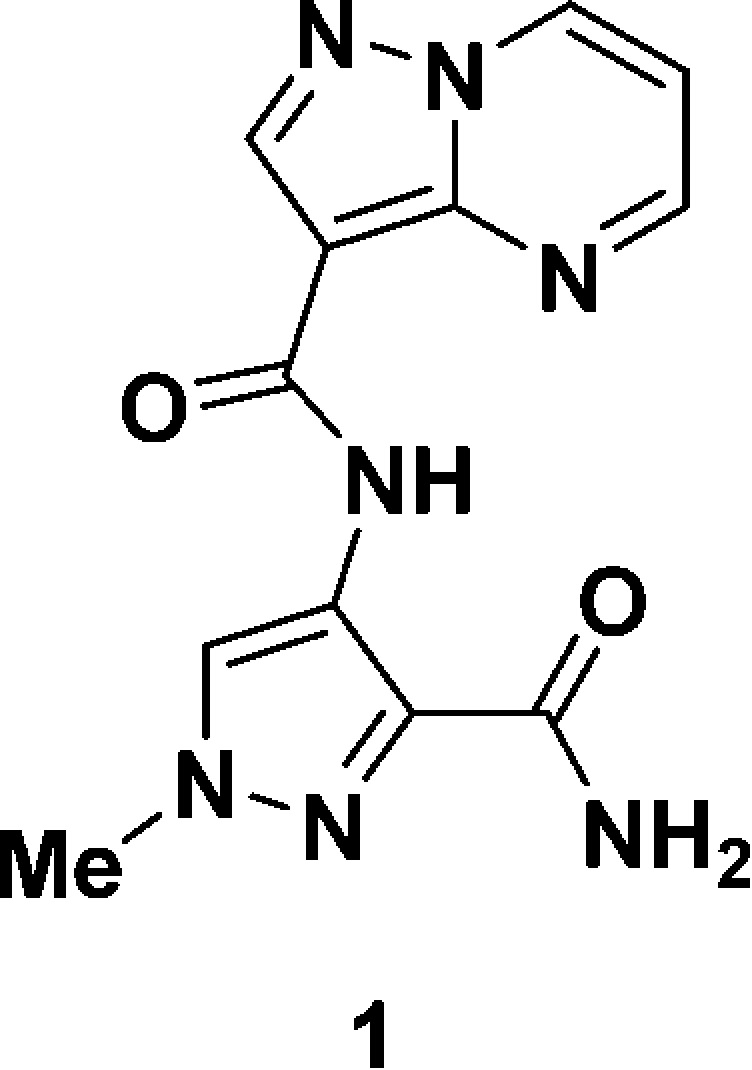
| IRAK4 IC50 (nM) | 110 |
| hPBMC IC50 (nM) | 2300 |
| LBE | 0.44 |
| no. of kinases tested | 101 |
| % of kinases >100× | 89a |
| PSA (Å) | 127 |
| cLogD (pH 7.4) | –1.7b |
| Papp (10–6 cm/s) | 17c |
Fold selectivity against in-house IRAK4 IC50.
From ACD Labs v10.
From MDCK cells.
Herein we report a series of modifications to the 5-position of the pyrazolopyrimidine ring and the 3-position of the pyrazole ring of the N-(1H-pyrazol-4-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide core of 1. The discovery of potent, selective, and bioavailable IRAK4 inhibitors through sequential improvements in cell permeability via cLogD-guided optimization of physical properties is described. In addition, initial results for a highly potent and kinome-selective IRAK4 inhibitor, 14, in a rat pharmacodynamic (PD) model are reported.
Synthetic routes for the target molecules discussed in this communication are described in Scheme 1. Rapid derivatization at the 5-position of the pyrazolo[1,5-a]pyrimidine was realized via SNAr reaction of the 5-chloropyrazolo[1,5-a]pyrimidine intermediates 3 with an array of amines. Compounds 3 were prepared from 5-hydroxypyrazolo[1,5-a]pyrimidine-3-carboxylic acid via chlorination with POCl3 followed by amide formation with various arylamines. Alternatively, amines were introduced to ethyl 5-chloropyrazolo[1,5-a]pyrimidine-3-carboxylate via SNAr reaction. After saponification of esters 5, acids 6 were coupled with various arylamines employing coupling reagents such as HATU for an efficient structure–activity relationship (SAR) study on the arylamides.
Scheme 1. Synthesis of the 5-Aminopyrazolo[1,5-a]pyrimidine-3-carboxamides.

(a) POCl3, Hünig’s base, reflux; (b) ArNH2, Hünig’s base, CH2Cl2; (c) R1R2NH, Hünig’s base, EtOH, reflux; (d) R1R2NH, EtOH, reflux; (e) LiOH, THF/water; (f) ArNH2, HATU, Hünig’s base, CH3CN.
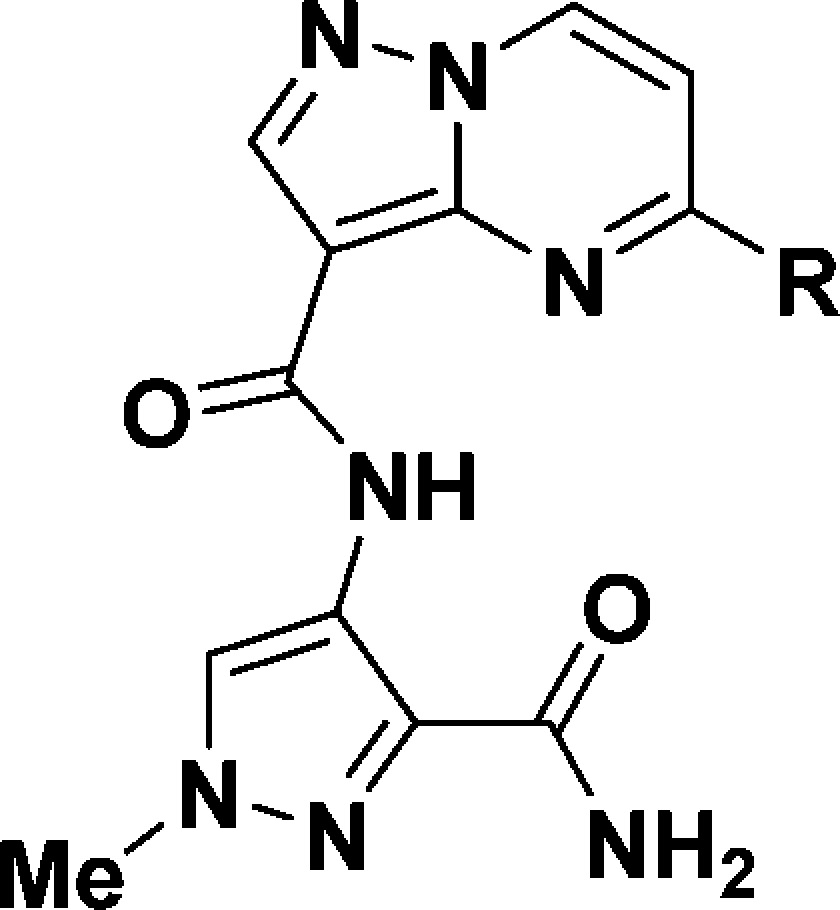
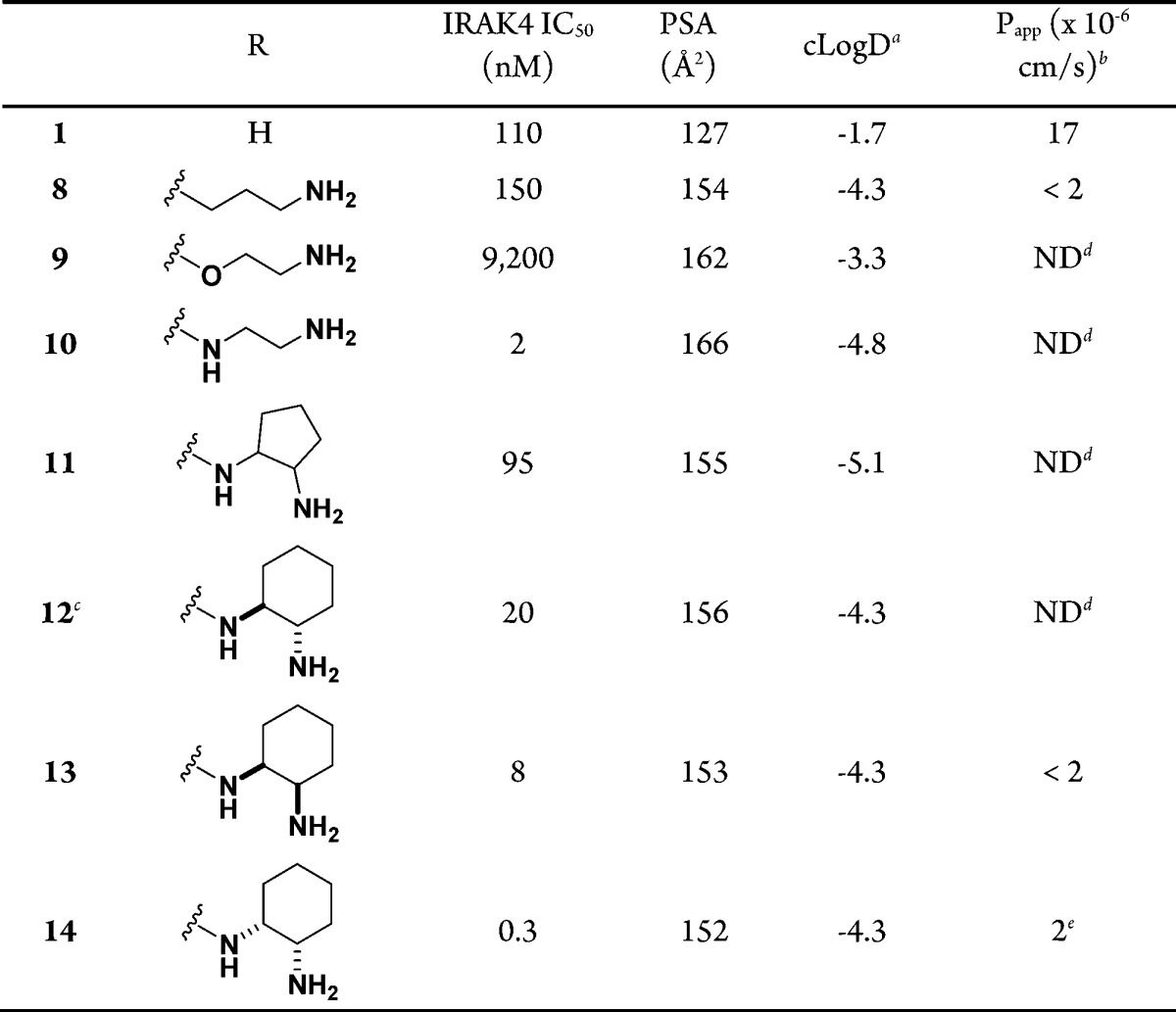
Crystal structures of the IRAK4 kinase domain in the literature21,22 hinted that hydrogen-bond donors growing from the 5-position of the pyrazolopyrimidine ring might have positive interactions with the carboxylate of Asp329 and thereby improve the intrinsic potency. Our initial SAR efforts were directed toward introducing various substituents bearing hydrogen-bond donors at this position to improve the IRAK4 potency. When straight chains were examined, it was found that neither the 3-aminopropyl analogue 8 nor the 2-aminoethoxy analogue 9 was more potent than 1 (Table 2). On the other hand, the 2-aminoethylamino analogue 10 showed a greater than 50-fold potency improvement. The stark difference in potency among 8, 9, and 10 is presumably due to the different trajectories of the substituents in the binding pocket, with the amino linker of 10 positioning the terminal amine toward Asp329 while the methylene linker of 8 and the ether linker of 9 do not.
Table 2. Initial SAR of Amines at the 5-Position of the Pyrazolopyrimidine Ring.
At pH 7.4 from ACD Labs v10.
From MDCK cells.
A racemic mixture.
ND = not determined.
From LLC-PK1 cells.
We next explored various cycloalkyl groups in place of the ethylene moiety of the 2-aminoethylamino analogue 10 (Table 2). Compared with 10, the cyclopentane-1,2-diamine analogues showed a greater than 10-fold loss of potency even when the most potent enantiomer of 11 was assumed to exclusively contribute to the potency. It was found that trans-cyclohexane-1,2-diamine 12 and (1S,2R)-cyclohexane-1,2-diamine 13 were 10- and 4-fold less potent, respectively. On the other hand, a 7-fold potency improvement was achieved with 14 bearing the (1R,2S)-cyclohexane-1,2-diamine at the 5-position of the pyrazolopyrimidine ring (IC50 = 0.3 nM).
As envisioned, the IRAK4 potency was greatly improved by introducing optimal diamines at the 5-position of the pyrazolopyrimidine ring, as in 10 and 14. However, the polarity of the molecules was much higher, as indicated by the polar surface area (PSA) and cLogD23,24 values for 10 (PSA = 166 Å2, cLogD = −4.8) and 14 (PSA = 152 Å2, cLogD = −4.3) compared with those of 1 (PSA = 127 Å2, cLogD = −1.7) (Table 2), because of the added high intrinsic polarity of the diamines. As a result of the high polarity, these molecules displayed poor passive permeability (Papp < 5 × 10–6 cm/s).25 More importantly, the oral bioavailability correlated well with the membrane permeability of the molecules in this series in pharmacokinetic (PK) studies in rats.26 Accordingly, none of the molecules with high polarity in this series offered oral bioavailability even though they showed moderate plasma clearance (Clp), which made them unsuitable for oral dosing (see the Supporting Information).
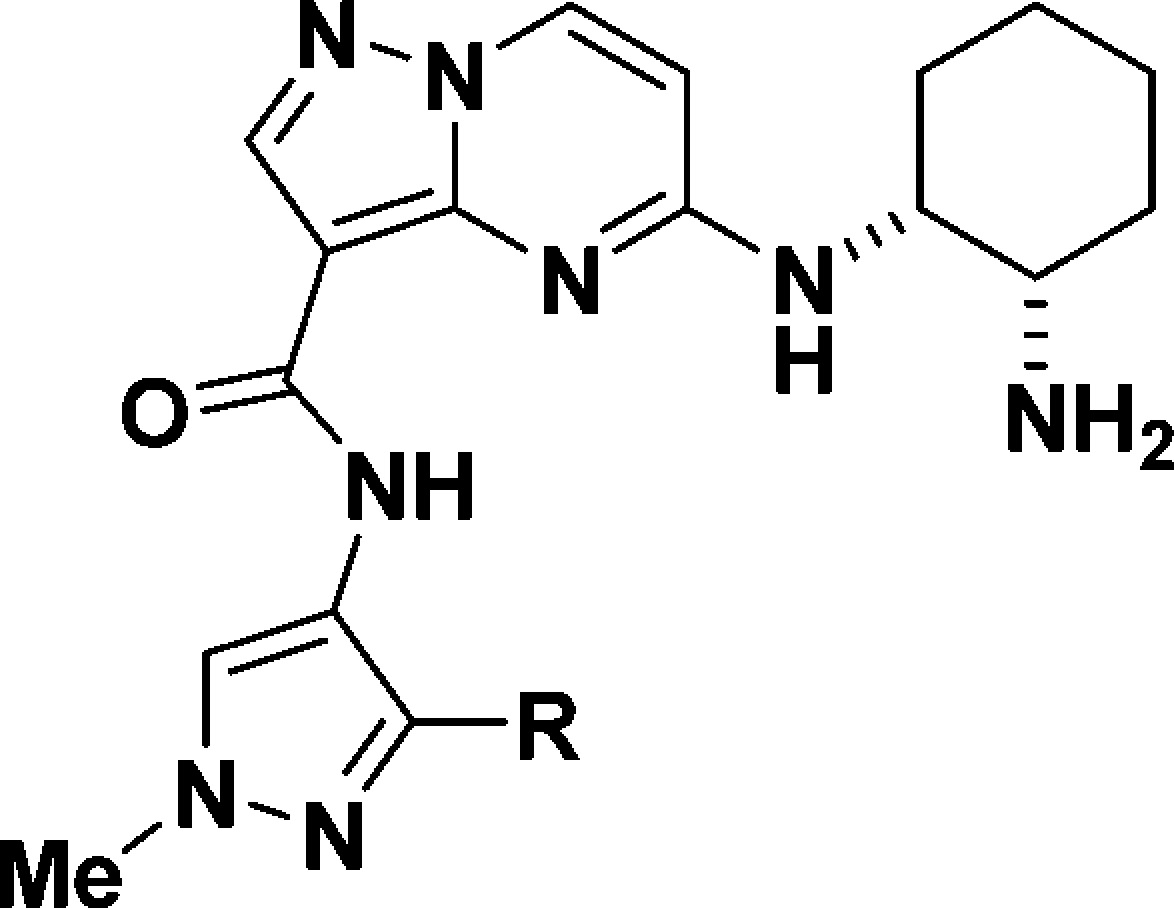
Diamines at the 5-position of the pyrazolopyrimidine ring not only account for high intrinsic potency but also are responsible for poor bioavailability in rats. It was quite challenging to lower polarity of the molecules to obtain high permeability and good oral bioavailability while maintaining high IRAK4 potency. Our strategy was to identify the substituents responsible for the poor membrane permeability and low bioavailability and to find replacements through SAR studies to optimize the polarity as well as the potency. To this end, we examined the company database of historic molecules and found that the carboxamide group was frequently seen in molecules with poor permeability. Next, with (1R,2S)-cyclohexane-1,2-diamine at the 5-position of the pyrazolopyrimidine ring, we explored carboxamide replacements that historically offered improved membrane permeability.
Although replacements of the carboxamide group with hydrogen (15) or a methoxymethyl group (16) decreased the PSA by 42 and 36 Å2, respectively, both compounds displayed poor membrane permeability (Table 3). However, replacements with nitrile, trifluoromethyl, difluoromethyl, and pentafluoroethyl groups (17–20, respectively) offered moderate membrane permeability, with Papp values in the range of (13–18) × 10–6 cm/s, even though their PSA values are in the same range as those of 15 and 16. Gratifyingly, the intrinsic IRAK4 potency was maintained, especially for 18 and 19 (IC50 = 0.3 and 0.2 nM, respectively). Carboxamide replacements with some heterocyclic groups that historically offered permeability improvements failed to improve the membrane permeability in our case (21–23).
Table 3. Carboxamide Replacements.
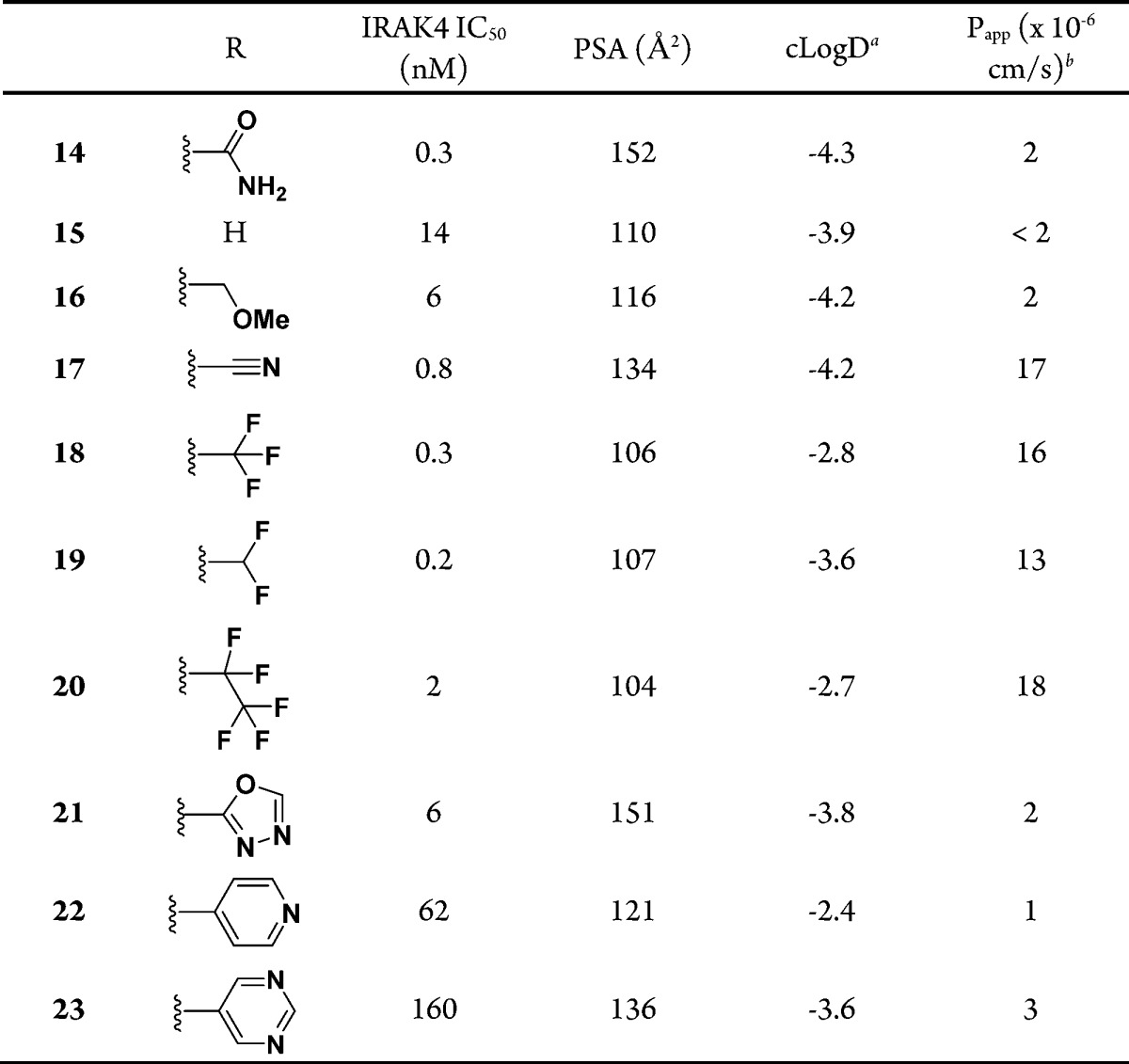
At pH 7.4 from ACD Labs v10.
From MDCK cells.
An X-ray cocrystal structure of 18 bound to the kinase domain of IRAK4 revealed the expected mode of binding (Figure 1). The (2S)-amino group is in the axial position of the cyclohexane, and it interacts with the side-chain amide oxygen of Asn316 and, with the (1R)-amino group, donates a hydrogen-bonding interaction to the side-chain carboxylate of Asp329. The pyrazolopyrimidine core interacts at an angle with the phenol side-chain of the gatekeeper residue, Tyr262. There is a single polar hinge interaction provided by the amide oxygen with the backbone NH of Met265. The CF3 group stacks intramolecularly with the cyclohexyl ring on the other side of the compound.
Figure 1.
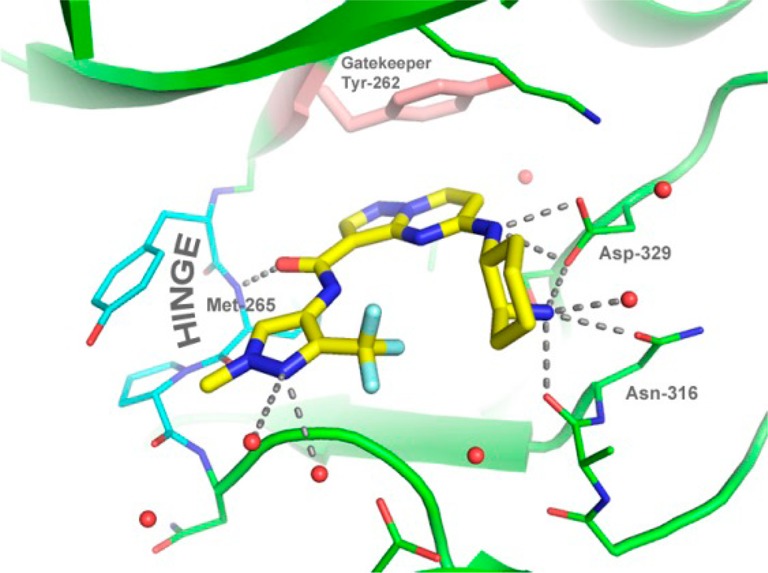
X-ray crystal structure of 18 bound to IRAK4.
Encouraged by the permeability improvements with some carboxamide replacements such as CF3 and CHF2 groups, we next looked for calculated values for the molecules that offer a good correlation with measured membrane permeability so that we could prioritize target molecules on the basis of the calculated values and efficiently drive our SAR efforts. To this end, we plotted all of the Papp values of compounds in this series versus calculated values such as PSA, cLogD, and LogP (see the Supporting Information). It was found that Papp correlated reasonably well with cLogD23 with a minimal number of outliers. Accordingly, we focused our SAR efforts on improving the cLogD value while maintaining high IRAK4 potency.
As previously discussed, diamines at the 5-position of the pyrazolopyrimidine ring account for the high polarity and poor membrane permeability of the molecules. Having identified promising carboxamide replacements, we next explored an array of diamines at the 5-position of the pyrazolopyrimidine ring with either CF3 or CHF2 at the 3-position of the pyrazole ring to further improve the membrane permeability (Table 4). As expected, removal of the primary amine offered much higher cLogD and Papp values (24, cLogD = 1.2, Papp = 23 × 10–6 cm/s). However, 24 suffered from greater than 1000-fold loss of potency compared with 18, confirming that a hydrogen-bond donor is required for high IRAK4 potency. The (1R,2S)-2-hydroxycyclohexylamine analogues 25 and 26 also displayed much higher cLogD and Papp values, but they were intrinsically less potent than 18 and 19 by 70-fold and 25-fold, respectively. A piperazine group was also pursued, since piperazine is less basic than cyclohexane-1,2-diamine (ACD Labs pKa = 9.8 and 8.5 for 18 and 27, respectively), thereby increasing the cLogD value. As expected, piperazines 27 and 28 displayed higher cLogD values by 1.5 units and high membrane permeabilities (Papp = 29 × 10–6 and 24 × 10–6 cm/s for 27 and 28, respectively). Potency loss was also observed for 27 and 28 compared with 18 and 19, albeit to a lesser extent. It was generally observed that the CHF2 analogues offered higher intrinsic IRAK4 potency and comparable membrane permeability compared with the corresponding CF3 analogues, as seen with (1R,2S)-cyclohexane-1,2-diamine, (1R,2S)-2-hydroxycyclohexylamine, and piperazine groups on the pyrazolopyrimidine ring (18, 19, 25, 26, 27, and 28 in Table 4).
Table 4. SAR of Diamines at the 5-Position of the Pyrazolopyrimidine Ring.
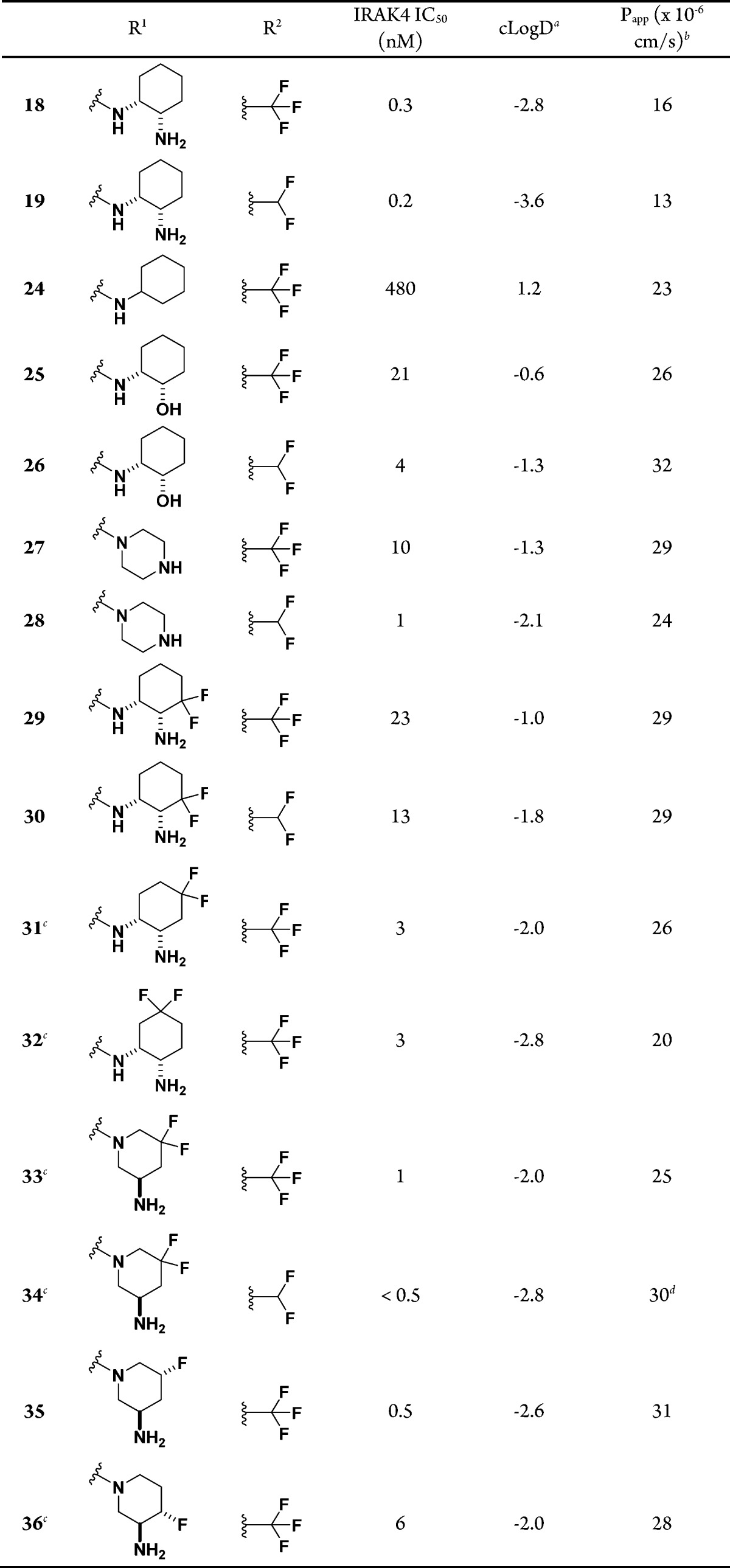
At pH 7.4 from ACD Labs v10.
From MDCK cells.
Absolute stereochemistry unknown.
From LLC-PK1 cells.
As fluorine atoms β, γ, or δ to a basic nitrogen would reduce the basicity of the nitrogen, increase the cLogD value, and potentially improve the metabolic stability of the molecule, fluorine atoms at various positions of the promising amines on the pyrazolopyrimidine ring were explored (Table 4). Compared with 18 and 19, the 3,3-, 4,4-, and 5,5-difluoro analogues (29–32) showed improved high membrane permeability (Papp ≥ 20 × 10–6 cm/s). It was observed that the IRAK4 potency deteriorated as the carbon atoms bearing fluorine atoms were closer to the basic amine. The (3R)-3-aminopiperidine analogues with fluorine atoms at the 5,5-, (5R)-, and (4S)-positions (33–36) displayed not only high membrane permeability (Papp ≥ 25 × 10–6 cm/s) but also high IRAK4 intrinsic potency, especially in the case of 33, 34, and 35 (IC50 ≤ 1 nM).
Throughout the SAR studies, IRAK4 inhibitors with high intrinsic potency were evaluated in both human peripheral blood mononuclear cell (hPBMC) assay and rat whole-blood (rWB) assay to assess their cell activities. The kinome selectivity profile was also closely monitored against a broad panel of kinases (101 or 264 kinases) for key IRAK4 inhibitors in order to assess potential in vivo adverse events caused by inhibition of off-target kinases. Key inhibitors with high passive permeability in Table 4 were dosed to rats in standard iv/po dosing protocols to evaluate their PK properties.
Compounds 14 and 18, which displayed low to moderate passive permeability (Papp = 2 × 10–6 and 16 × 10–6 cm/s, respectively), were poorly bioavailable in rats (F = 0 and 5%, respectively; Table 5). On the other hand, the highly permeable compounds 25, 26, 29, 30, and 34 (Papp = (26–32) × 10–6 cm/s) offered significantly improved bioavailability in the range of 24–49% in rats. The (1R,2S)-2-hydroxycyclohexylamine analogues 25 and 26, however, showed a moderate erosion in kinase selectivity compared with 14 and 18, and 25 suffered from a loss of rat whole-blood potency. Balanced properties in cell potency, kinase selectivity, and PK properties were observed with the (1R,2R)-2-amino-3,3-difluorocyclohexylamine analogues 29 and 30 and the (3R)-3-amino-5,5-difluoropiperidine analogue 34. They offered good rat whole-blood potency, excellent kinase selectivity, and moderate Clp and good bioavailability in rats suitable for oral in vivo studies.
Table 5. Cell Potencies, Kinase Selectivities, and Pharmacokinetic Profiles of Selected IRAK4 Inhibitors.
| compd | hPBMC IC50 (nM) | rWB IC50 (nM) | no. of kinases tested | % of kinases >100×a | rat Clp (mL min–1 kg–1)b | rat %Fc |
|---|---|---|---|---|---|---|
| 14 | 31 | 300 | 264 | 99 | 56 | 0 |
| 18 | 9 | 89 | 101 | 96 | 98 | 5 |
| 25 | 130 | 5500 | 101 | 91 | 41 | 26 |
| 26 | 53 | 500 | 101 | 87 | 45 | 49 |
| 29 | 310 | 590 | 101 | 100 | 48 | 24 |
| 30 | 320 | 890 | 101 | 94 | 29 | 46 |
| 34 | 12 | 81 | 101 | >95 | 46 | 42 |
Fold selectivity against in-house IRAK4 IC50.
Dosed iv at 0.5 mg/kg as a solution in DMSO/PEG400/H2O (20:40:20 v/v/v).
Dosed po at 1 mg/kg as a solution in DMSO/PEG400/H2O (20:40:20 v/v/v).
On the basis of its excellent rat whole-blood potency (IC50 = 300 nM) and kinase selectivity profile (>100-fold selective against 99% of the 264 kinases tested) (Table 5), compound 14 was chosen for in vivo proof-of-mechanism studies before orally active compounds were available. In this in vivo model, female Lewis rats were dosed with either vehicle or compound 14 subcutaneously at 3, 10, 30, and 50 mg/kg 1 h prior to stimulation with R848 (5 mg/kg, ip), a TLR7 agonist. At 1.5 h post R848 stimulation, blood samples were obtained from the animals and cytokine levels were measured (Figure 2). The IL-6 level increased markedly in vehicle-treated animals in response to stimulation with R848. Rats dosed with 14, however, showed inhibition of IL-6 secretion in a dose-dependent manner compared with vehicle-treated animals. It is noteworthy that the percent inhibition of IL-6 and terminal exposure of 14 in this in vivo study correlated well with the rat whole-blood potency of 14 (Figure 2).
Figure 2.
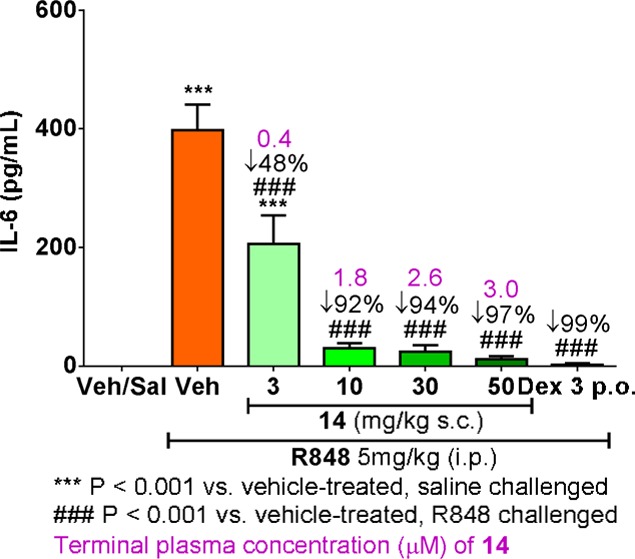
Effect of compound 14 on the IL-6 level in an R848-induced rat PD model.
In summary, the development of a series of 5-amino-N-(1H-pyrazol-4-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamides as IRAK4 inhibitors was achieved via sequential modifications to the 5-position of the pyrazolopyrimidine ring and the 3-position of the pyrazole ring. While the intrinsic potency was markedly improved when diamines were introduced at the 5-position of the pyrazolopyrimidine ring, the passive permeability and bioavailability of this series were initially poor. Replacement of the substituents responsible for the poor permeability and improvement of the physical properties guided by cLogD led to the identification of IRAK4 inhibitors with excellent potency, kinase selectivity, and PK properties suitable for oral dosing. Robust PK/PD response in the R848-induced rat model was observed with compound 14 in a dose-dependent manner.
Acknowledgments
We thank William McElroy and Ginny Ho for following up the HTS hits and Ziping Liu and Tongqian Chen at Pharmaron and Chandra Korapala and Senthilkumar S P at Syngene for preparation of key compounds.
Supporting Information Available
Synthetic procedures and analytical data of selected compounds, conditions for all of the biological assays, PK profiles of 13 and 14, X-ray statistics for 18, and Papp plots. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00107.
Accession Codes
The PDB code for 18 is 4Y73.
The authors declare no competing financial interest.
Supplementary Material
References
- Li S.; Strelow A.; Fontana E. J.; Wesche H. IRAK-4: A Novel Member of the IRAK Family with the Properties of an IRAK Kinase. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 5567–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens S.; Beyaert R. Functional Diversity and Regulation of Different Interleukin-1 Receptor-Associated Kinase (IRAK) Family Members. Mol. Cell 2003, 11, 293–302. [DOI] [PubMed] [Google Scholar]
- Suzuki N.; Suzuki S.; Yeh W.-C. IRAK-4 as the Central TIR Signaling Mediator in Innate Immunity. Trends Immunol. 2002, 23, 503–506. [DOI] [PubMed] [Google Scholar]
- Suzuki N.; Suzuki S.; Duncan G. S.; Millar D. G.; Wada T.; Mirtsos C.; Takada H.; Wakeham A.; Itie A.; Li S.; Penninger J. M.; Wesche H.; Ohashi P. S.; Mak T. W.; Yeh W.-C. Severe Impairment of Interleukin-1 and Toll-Like Receptor Signaling in Mice Lacking IRAK-4. Nature 2002, 416, 750–756. [DOI] [PubMed] [Google Scholar]
- Picard C.; Puel A.; Bonnet M.; Ku C. L.; Bustamante J.; Yang K.; Soudais C.; Dupuis S.; Feinberg J.; Fieschi C.; Elbim C.; Hitchcock R.; Lammas D.; Davies G.; Al-Ghonaium A.; Al-Rayes H.; Al-Jumaah S.; Al-Hajjar S.; Al-Mohsen I. Z.; Frayha H. H.; Rucker R.; Hawn T. R.; Aderem A.; Tufenkeji H.; Haraguchi S.; Day N. K.; Good R. A.; Gougerot-Pocidalo M.-A.; Ozinsky A.; Casanova J.-L. Pyogenic Bacterial Infections in Humans with IRAK-4 Deficiency. Science 2003, 299, 2076–2079. [DOI] [PubMed] [Google Scholar]
- Li X. IRAK4 in TLR/IL-1R Signaling: Possible Clinical Applications. Eur. J. Immunol. 2008, 38, 614–618. [DOI] [PubMed] [Google Scholar]
- Huang Q.; Ma Y.; Adebayo A.; Pope R. M. Increased Macrophage Activation Mediated through Toll-like Receptors in Rheumatoid Arthritis. Arthritis Rheum. 2007, 56, 2192–2201. [DOI] [PubMed] [Google Scholar]
- Joosten L. A. B.; Netea M. G. Interleukin-1 Receptor-Associated Kinase 4 Links Innate Immunity to the Pathogenesis of Rheumatoid Arthritis. Arthritis Rheum. 2009, 60, 1571–1574. [DOI] [PubMed] [Google Scholar]
- Asquith M.; Powrie F. An Innately Dangerous Balancing Act: Intestinal Homeostasis, Inflammation, and Colitis-Associated Cancer. J. Exp. Med. 2010, 207, 1573–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivakumar P. V.; Westrich G. M.; Kanaly S.; Garka K.; Born T. L.; Derry J. M.; Viney J. L. Interleukin 18 Is a Primary Mediator of the Inflammation Associated with Dextran Sulphate Sodium Induced Colitis: Blocking Interleukin 18 Attenuates Intestinal Damage. Gut 2002, 50, 812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasamy A.; Kuokkanen M.; Vedantam S.; Gajdos Z. K.; Alves A. C.; Lyon H. N.; Ferreira M. A. R.; Strachan D. P.; Zhao J. H.; Abramson M. J.; Brown M. A.; Coin L.; Dharmage S. C.; Duffy D. L.; Haahtela T.; Heath A. C.; Janson C.; Kahonen M.; Khaw K.-T.; Laitinen J.; Le Souef P.; Lehtimaki T.; Madden P. A. F.; Marks G. B.; Martin N. G.; Matheson M. C.; Palmer C. D.; Palotie A.; Pouta A.; Robertson C. F.; Viikari J.; Widen E.; Wjst M.; Jarvis D. L.; Montgomery G. W.; Thompson P. J.; Wareham N.; Eriksson J.; Jousilahti P.; Laitinen T.; Pekkanen J.; Raitakari O. T.; O’Connor G. T.; Salomaa V.; Jarvelin M.-R.; Hirschhorn J. N. Genome-Wide Association Studies of Asthma in Population-Based Cohorts Confirm Known and Suggested Loci and Identify an Additional Association Near HLA. PLoS One 2002, 7, e44008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrat F. J.; Meeker T.; Chan J. H.; Guiducci C.; Coffman R. L. Treatment of Lupus-Prone Mice with a Dual Inhibitor of TLR7 and TLR9 Leads to Reduction of Autoantibody Production and Amelioration of Disease Symptoms. Eur. J. Immunol. 2007, 37, 3582–3586. [DOI] [PubMed] [Google Scholar]
- Powers J. P.; Li S.; Jaen J. C.; Liu J.; Walker N. P. C.; Wang Z.; Wesche H. Discovery and Initial SAR of Inhibitors of Interleukin-1 Receptor-Associated Kinase-4. Bioorg. Med. Chem. Lett. 2006, 16, 2842–2845. [DOI] [PubMed] [Google Scholar]
- Buckley G. M.; Gowers L.; Higueruelo A. P.; Jenkins K.; Mack S. R.; Morgan T.; Parry D. M.; Pitt W. R.; Rausch O.; Richard M. D.; Sabin V.; Fraser J. L. IRAK-4 Inhibitors. Part I: A Series of Amides. Bioorg. Med. Chem. Lett. 2008, 18, 3211–3214. [DOI] [PubMed] [Google Scholar]
- Buckley G. M.; Ceska T. A.; Fraser J. L.; Gowers L.; Groom C. R.; Higueruelo A. P.; Jenkins K.; Mack S. R.; Morgan T.; Parry D. M.; Pitt W. R.; Rausch O.; Richard M. D.; Sabin V. IRAK-4 Inhibitors. Part II: A Structure-Based Assessment of Imidazo[1,2-a]pyridine Binding. Bioorg. Med. Chem. Lett. 2008, 18, 3291–3295. [DOI] [PubMed] [Google Scholar]
- Buckley G. M.; Fosbeary R.; Fraser J. L.; Gowers L.; Higueruelo A. P.; James L. A.; Jenkins K.; Mack S. R.; Morgan T.; Parry D. M.; Pitt W. R.; Rausch O.; Richard M. D.; Sabin V. IRAK-4 Inhibitors. Part III: A Series of Imidazo[1,2-a]pyridines. Bioorg. Med. Chem. Lett. 2008, 18, 3656–3660. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Wesche H.; Stevens T.; Walker N.; Yeh W.-C. IRAK-4 Inhibitors for Inflammation. Curr. Top. Med. Chem. 2009, 9, 724–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumey L. N.; Boschelli D. H.; Bhagirath N.; Shim J.; Murphy E. A.; Goodwin D.; Bennett E. M.; Wang M.; Lin L.-L.; Press B.; Shen M.; Frisbie R. K.; Morgan P.; Mohan S.; Shin J.; Rao V. R. Identification and Optimization of Indolo[2,3-c]quinoline Inhibitors of IRAK4. Bioorg. Med. Chem. Lett. 2014, 24, 2066–2072. [DOI] [PubMed] [Google Scholar]
- Chaudhary D.; Robinson S.; Romero D. L. Recent Advances in the Discovery of Small Molecule Inhibitors of Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) as a Therapeutic Target for Inflammation and Oncology Disorders. J. Med. Chem. 2015, 58, 96–110. [DOI] [PubMed] [Google Scholar]
- Inami H.; Mizutani T.; Maeda J.; Usuda H.; Nagashima S.; Ito T.; Aoyama N.; Kontani T.; Hayashida H.; Terasawa T.; Seo R.; Akamatsu M.; Ishikawa T.; Hayashi K.. Oxazole Compound. WO 2011/043371, 2011.
- Wang Z.; Liu J.; Sudom A.; Ayres M.; Li S.; Wesche H.; Powers J. P.; Walker N. P. C. Crystal Structures of IRAK-4 Kinase in Complex with Inhibitors: A Serine/Threonine Kinase with Tyrosine as a Gatekeeper. Structure 2006, 14, 1835–1844. [DOI] [PubMed] [Google Scholar]
- Kuglstatter A.; Villasenor A. G.; Shaw D.; Lee S. W.; Tsing S.; Niu L.; Song K. W.; Barnett J. W.; Browner M. F. Cutting Edge: IL-1 Receptor-Associated Kinase 4 Structures Reveal Novel Features and Multiple Conformations. J. Immunol. 2007, 178, 2641–2645. [DOI] [PubMed] [Google Scholar]
- Log D Suite, version 10; Advanced Chemistry Development, Inc.: Toronto, ON, Canada; http://www.acdlabs.com/.
- Hill A. P.; Young R. J. Getting Physical in Drug Discovery: A Contemporary Perspective on Solubility and Hydrophobicity. Drug Discovery Today 2010, 15, 648–655. [DOI] [PubMed] [Google Scholar]
- Irvine J. D.; Takahashi L.; Lockhart K.; Cheong J.; Tolan J. W.; Selick H. E.; Grove J. R. MDCK (Madin–Darby Canine Kidney) Cells: A Tool for Membrane Permeability Screening. J. Pharm. Sci. 1999, 88, 28–33. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H. Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.