

Abstract

IRAK4 is a critical upstream kinase in the IL-1R/TLR signaling pathway. Inhibition of IRAK4 is hypothesized to be beneficial in the treatment of autoimmune related disorders. A screening campaign identified a pyrazole class of IRAK4 inhibitors that were determined by X-ray crystallography to exhibit an unusual binding mode. SAR efforts focused on the identification of a potent and selective inhibitor with good aqueous solubility and rodent pharmacokinetics. Pyrazole C-3 piperidines were well tolerated, with N-sulfonyl analogues generally having good rodent oral exposure but poor solubility. N-Alkyl piperidines exhibited excellent solubility and reduced exposure. Pyrazoles possessing N-1 pyridine and fluorophenyl substituents were among the most active. Piperazine 32 was a potent enzyme inhibitor with good cellular activity. Compound 32 reduced the in vivo production of proinflammatory cytokines and was orally efficacious in a mouse antibody induced arthritis disease model of inflammation.
Keywords: Interleukin-1 receptor-associated kinase 4, inflammation, drug discovery, SAR, structure-based drug design
Inflammation, the response by an organism to nonself molecules, pathogens, cellular damage, or cellular debris, is highly controlled in mammalian biology. The dysregulation of this process is the underlying cause of autoimmune disorders, including rheumatoid arthritis and inflammatory bowel disease, which afflict millions worldwide.1,2 Chronic inflammation furthermore plays a role in numerous other disease states including cancer3 and cardiovascular disease.4 While a number of orally administered anti-inflammatory and analgesic agents are available to address the symptoms of inflammation, most of the current medicines that modify the autoimmune disease state itself are biologics.5 These therapies are generally efficacious, but are expensive and require IV administration. The development of an orally available agent to treat autoimmune disorders remains an area of intense investigation.6
The interleukin-1 receptor/Toll-like receptor (IL-1R/TLR) inflammation signaling pathway is an attractive target for therapeutic intervention in autoimmune disease.7 Members of the IL-1R/TLR superfamily of proteins recognize foreign pathogens and endogenous inflammation signaling agents, and are key upstream drivers of inflammation. These proteins all possess a conserved intracellular Toll/interleukin-1R (TIR) domain. Upon ligand binding to the receptor, the TIR domain recruits the scaffolding protein myeloid differentiation primary response gene 88 (MYD88). The resulting complex activates interleukin-1 receptor-associated kinase 4 (IRAK4).8 IRAK4 initiates a signaling cascade terminating in activation of the transcription factors activator protein-1 (AP-1) and nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB), leading to the production of proinflammatory signaling agents including tumor necrosis factor α (TNFα) and interleukin-1 (IL-1).9
The role of IRAK4 in innate immunity is supported by mouse knockout studies.10 Importantly, cells derived from a small human population that lack the IRAK4 protein do not respond to ligands known to activate the TIR pathway.11 The genetic association of IRAK4 with inflammation and the fact that it is the most proximal kinase to the IL-1R/TLR family render it an attractive target for drug discovery. A number of organizations have described IRAK4 inhibitor programs, although the poor kinase selectivity associated with many of these chemotypes limits a full understanding of their in vivo efficacies.12−15 We report herein the discovery of a novel, highly selective IRAK4 inhibitor chemotype and the identification of a compound, which reduces the in vivo production of TLR2-induced cytokines and is orally efficacious in a rodent model of inflammation.
A screening campaign identified pyrazoles 1 and 2 as modest IRAK4 kinase inhibitors (IC50 values = 2.2 and 0.69 μM, respectively, Figure 1). Interest in this series was initially based on the unusual binding mode of 1 as determined by X-ray crystallography. Three H-bonds between 1 and the kinase hinge were observed, with one ring proton from each pyrazole ring functioning as hydrogen bond donors and the amide carbonyl of 1 acting as a hydrogen bond acceptor. An intramolecular hydrogen bond between the inhibitor amide N–H and pyrimidine nitrogen stabilizes the pyrazolopyrimidine moiety into a planar conformation that allows for a favorable slanted face-to-edge π–π interaction with the gatekeeper Tyr262 phenol. The pyrazole phenyl substituent is positioned against the peptide bond between Met192 and Gly193, at the N-terminal end of a glycine rich loop of the protein, and is oriented at about 55° relative to the pyrazole ring. Finally, the pyrazole C-3 methyl group points toward the solvent exposed region.
Figure 1.
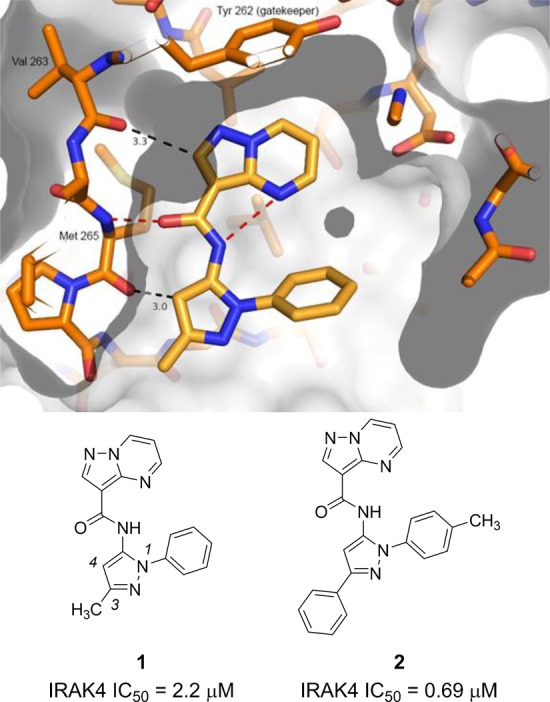
Pyrazoles 1 and 2 and X-ray cocrystal structure of 1 with IRAK4. Not shown on this picture is the interaction with the peptide bond between Met192 and Gly193, which lies above the top clipping plane.
The observation that inhibitor 1 bound to the IRAK4 hinge without a typical kinase binding motif16 such as 2-aminopyrimidine or benzimidazole led us to suspect that analogues in this series may exhibit high selectivity for IRAK4, although a more potent inhibitor would be required in order to accurately interpret selectivity data. With this assumption, an SAR program was undertaken with the goal of identifying an analogue that could be used in a rodent model of inflammation proof-of-concept study. Such a compound should be a potent (<10 nM) enzyme inhibitor with excellent kinase selectivity. Additionally, the analogue sought should possess both oral rodent exposure and good aqueous solubility, which would allow for dosing using various routes of administration.
The general synthesis depicted in Scheme 1, using analogue 6 as an example, was employed for most of the pyrazoles in this Letter.17 α-Ketonitrile 3 was condensed with 4-methylphenylhydrazine to form 4.18 Aminopyrazole 4 was allowed to react with acid chloride 5 to give 6. The analogous reaction between 4 and a carboxylic acid using peptide coupling agents was unsuccessful as only starting materials were recovered even under forcing conditions. In instances where the starting ketonitriles were not commercially available, they were prepared from their corresponding esters and the anion derived from acetonitrile.
Scheme 1.

Reagents and conditions: (a) p-tolylhydrazine, montmorillonite K-10, i-PrOH, Δ, 75%; (b) pyrazolo[1,5-a]pyrimidine-3-carbonyl chloride (5), DIPEA, CH2Cl2, 28%.
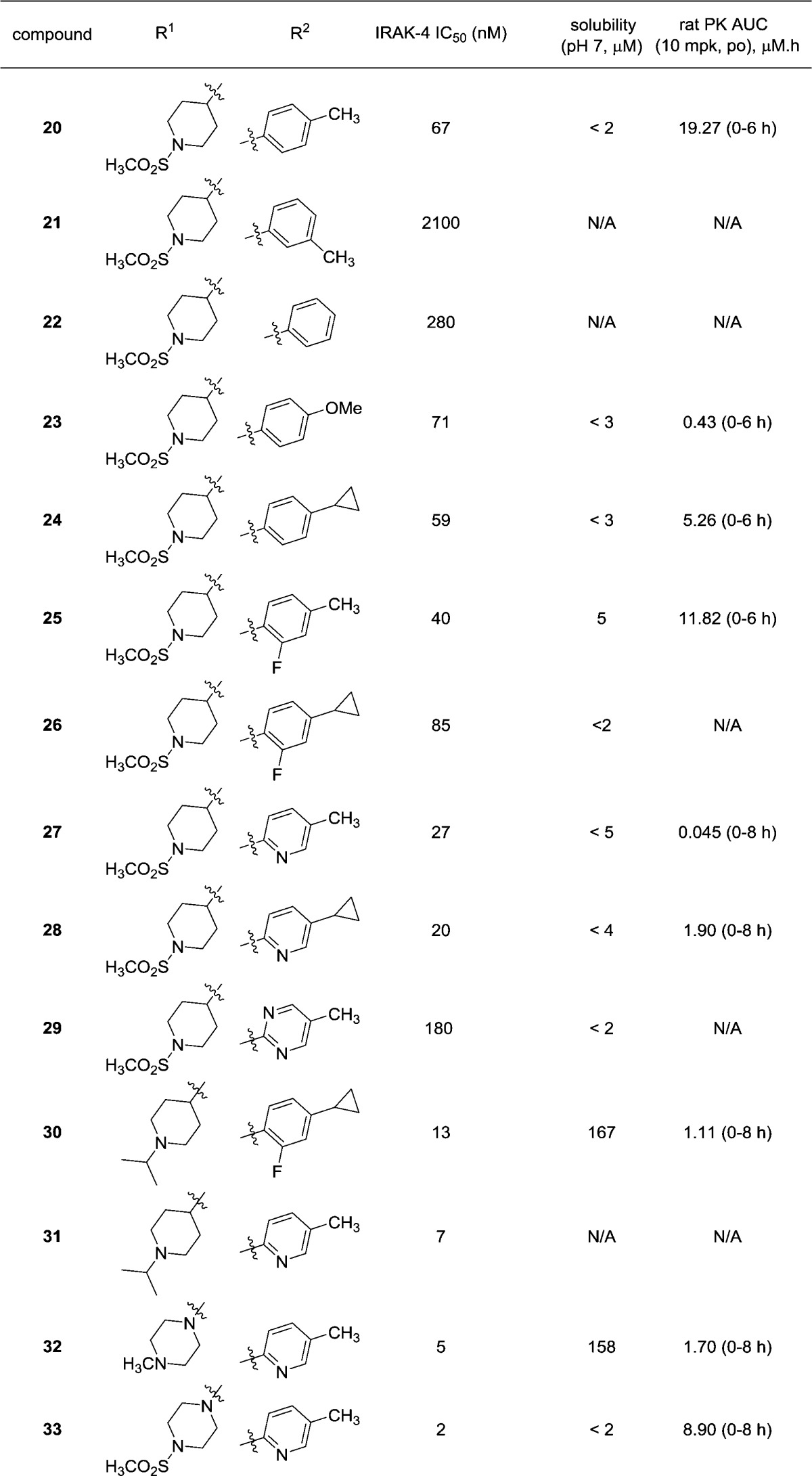
Initial SAR studies focused on the pyrazole C-3 position (Table 1). In addition to IRAK4 inhibition, kinetic aqueous solubility at pH 7 was measured for most analogues. Rat oral exposure was determined in cassette mode for select inhibitors. In these studies,19 Sprague–Dawley rats were dosed p.o. at 10 mg/kg, in 20% HPbCD, with drug plasma concentrations measured at multiple time points and AUC values from 0 to 6 h calculated. The pyrazole C-3 phenyl analogue 2 was chosen as a starting point for SAR development. Since the C-3 substituent points toward solvent in the X-ray structure, it was hypothesized that the incorporation of polar groups on the phenyl ring may improve enzyme activity through desolvation. This was generally found to be true. The addition of a para-methoxyphenyl substituent (6) resulted in an approximately 4-fold increase in potency relative to 2. Inhibitor 6 was also assessed for rat pharmacokinetics (PK) and kinase selectivity. Although 6 exhibited no oral exposure, it was found to be very selective: against a panel of 108 kinases, >80% inhibition was observed for only 2 kinases (IRAK4 and MINK1) at 10 μM. This confirmed the expectation that analogues in this series are highly selective.
Table 1. SAR Studies at Pyrazole C-3 Substituent.
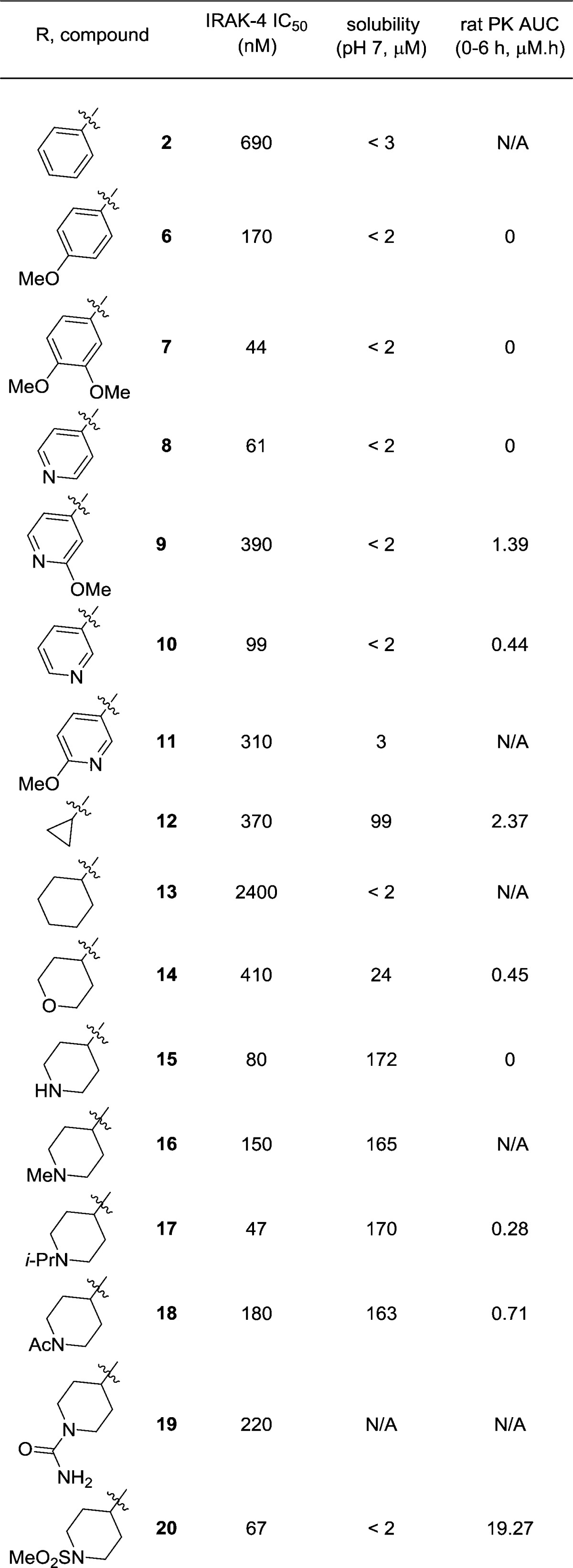
The dimethoxyphenyl analogue 7 exhibited excellent IRAK4 inhibition (IC50 = 44 nM). As with 6, compound 7 had no measurable plasma exposure in rat. The poor PK was investigated using metabolite identification studies, whereby analogue 7 was incubated with rat hepatocytes. Monooxidation was found to be the dominant route of metabolism, with the oxidation occurring on the tolyl, pyrazole, or dimethoxyphenyl ring. Although this study did not conclusively identify the site of metabolism, it was suspected that the oxidation may be occurring at the electron rich dimethoxyphenyl ring.
Replacement of the methoxyphenyl ring with more electron deficient aromatic systems was explored next. The 4-pyridyl analogue 8 maintained good IRAK4 activity and selectivity (6/108 kinases, >80% inhibition at 10 μM). However, no improvement in plasma exposure was observed.
Analogue 9 was designed with the expectation that the addition of the methoxy group would both improve IRAK4 activity, in a similar manner as seen with the phenyl analogues, and reduce any oxidative metabolism of the pyridine nitrogen and increase rat PK. Although some loss in activity was observed, it was significant that 9 became the first compound in this chemotype to exhibit any rat exposure. The regioisomeric pyridines 10 and 11 displayed similar IRAK4 IC50 values relative to their 4-pyridyl counterparts. It is noteworthy that the unsubstituted 3-pyridine 10 also exhibited measurable but poor rodent exposure.
Collectively the above findings demonstrated that modification of the pyrazole C-3 substituent was a viable strategy to improve both IRAK4 potency and rodent PK in this series. It was hypothesized that the poor solubilities of 2 and 6–11 were due to the expected planar nature of the compounds.20 In order to address this, alicyclic C-3 substituents were explored next. Replacement of the methyl group in the original hit 1 (IRAK4 IC50 = 2.2 μM) with a cyclopropyl ring (12) gave an approximately 6-fold improvement in IRAK4 potency. It was encouraging that 12 also exhibited better solubility and oral exposure than analogues in the (hetero)aryl series. However, the more lipophilic cyclohexyl analogue 13 was only equipotent to 1 and was poorly soluble.
In a similar manner as described above, heteroatom containing substituents were prepared with the goals of improving IRAK4 potency and decreasing lipophilicity and thus improving solubility relative to 13. The THP species 14 was a modest inhibitor of IRAK4, but showed improved solubility and measurable exposure. Encouraged by this result, piperidine analogues were explored next. The unsubstituted 4-piperidine 15 was a relatively potent enzyme inhibitor with excellent solubility, likely due to the charged nature of this species at pH 7. N-Alkylated analogues 16 and 17 displayed similar activities and maintained excellent solubilities relative to N-H piperidine 15. Disappointingly, exposure was below the limit of detection for 15 and minimal for N-iso-propyl analogue 17.
Replacement of the piperidine N-alkyl substituents with other functional groups was explored with the hope that this may improve rat PK. N-Acetamide 18 had a similar profile to the parent piperidine 15, although improved pharmacokinetics. This result led to the design of piperidines with other nitrogen functional groups. The urea 19 was somewhat less potent than 15. Importantly, methanesulfonamide 20 was identified as having reasonable IRAK4 potency and high plasma exposure in rat. Furthermore, the excellent kinase selectivity observed with earlier analogues was preserved, as 20 inhibits only 2/108 kinases with >80% inhibition at 10 μM (IRAK4, TRKB).
Although the enzyme potency of 20 was below that of our desired criteria, the excellent selectivity and rat oral exposure made this compound suitable for the development of an in vivo biomarker assay. In this experiment, female Lewis rats (n = 9) were dosed by oral gavage at 100 mg/kg with 20. One hour later, the rats were injected intraperitoneally with the TLR2 agonist PAM2CSK4.21 Three hours postinjection, plasma was collected and levels of TLR2-derived cytokines were measured relative to the control group that was dosed with only the vehicle. Modest but not statistically significant reductions of TNFα (21%) and IL-1β (28%) were observed, with no decrease seen of either IFNγ or IL-6. The average exposure of 20 in this study was 2.66 μM at 3 h postinjection.
The lack of a robust decrease in cytokine levels observed with 20 was attributed to its modest IRAK4 enzyme activity. Although permeability22 for 20 was good (Papp = 31 × 10–6 cm/s), we were concerned that the poor solubility of 20 (<2 μM) conflated an understanding of its cellular activity (IC50 = 2.4 μM). We thus undertook SAR investigations into the pyrazole N-1 position. For these studies, the C-3 substituents were restricted to either the piperidine sulfonamide (due to the high exposure of 20) or N-iso-propyl piperidine (given the excellent potency and solubility of 17). The findings are summarized in Table 2. Rat AUC values were measured over 6 or 8 h as indicated.
Table 2. SAR Studies at Pyrazole C-3 and N-1 Substituents.
Movement of the para-methyl group to the meta position (21) led to marked decrease in IRAK4 inhibition compared to 20, while the removal of this substituent altogether (22) gave a 4-fold loss in activity. Subsequent studies thus focused on analogues with para-substituted aromatic rings at the N-1 position. Replacement of the methyl group with a methoxy functionality had no effect on potency but did result in a dramatic loss in rat exposure, possibly due to metabolism of 23 via demethylation of the ether. Replacement of the methyl group with a cyclopropyl substituent gave analogue 24 that had similar enzyme potency to 20 and approximately 4-fold less plasma exposure. This was curious since it was expected that the cyclopropyl group would confer improved metabolic stability relative to its methyl counterpart.
None of Table 2 analogues thus far (21–24) exhibited improved solubility or potency relative to 20. A strategy involving the addition of an ortho substituent was designed. This was expected to improve solubility by forcing the pyrazole and phenyl rings to adopt a noncoplanar arrangement in solution. It was also known from the X-ray structure of 1 that these groups are oriented at a 55° angle relative to one another in the enzyme active site. It was thus believed that the ortho substituent would have the additional benefit of preorganizing the inhibitor into its biologically active conformation, as the predicted dihedral angle is 5° larger for an analogue with an ortho-fluoro substituent.
ortho-Fluorophenyl analogue 25 had IRAK4 IC50 = 40 nM. Although solubility remained poor, 25 was the first compound in the sulfonamide series for which a measurable value (5 μM) could be obtained. Replacement of the para-methyl substituent with a cyclopropyl group (26) resulted in a slight decrease in potency and a loss of whatever solubility had been gained with 25.
It was believed that replacing the fluorophenyl rings of 25 and 26 with their bioisosteric pyridines may lead to an improvement in solubility. Although this was not observed with pyridines 27 and 28, these compounds were slightly more potent than their phenyl or fluorophenyl counterparts. Further, it was found that 27 was highly selective: in a panel of 108 kinases, the only kinase with >80% inhibition at 10 μM was IRAK4. We attempted to further capitalize on this finding by using pyrimidine 29, but this led to a loss in IRAK4 activity relative to 27.
Having been unsuccessful in the quest to use N-1 substituents to improve solubility, C-3 N-alkyl piperidines were revisited. Analogue 30 was designed with a fully basic nitrogen for solubility and an ortho-fluorophenyl pyrazole for improved potency. Compound 30 was a potent enzyme inhibitor (IC50 = 13 nM) with excellent solubility and modest exposure in rat. An X-ray cocrystal structure shows that 30 retained the same interactions with the protein as compound 1 (Figure 2). The pyrazolopyrimidine and Tyr262 are oriented at approximately 40° to one another (slanted face-to-edge interaction). The pyrazole and fluorophenyl rings are as expected perpendicular to one another. The fluorine atom engages in a hydrogen bond with the N–H of Ser269. Replacement of the fluorophenyl with a pyridine gave analogue 31, which was the first inhibitor in this series to possess IRAK4 IC50 < 10 nM. We suspect that the additional interactions between the fluorine atom and, by extended reasoning, the pyridine nitrogen are responsible for the improved potencies of 30 and 31.
Figure 2.
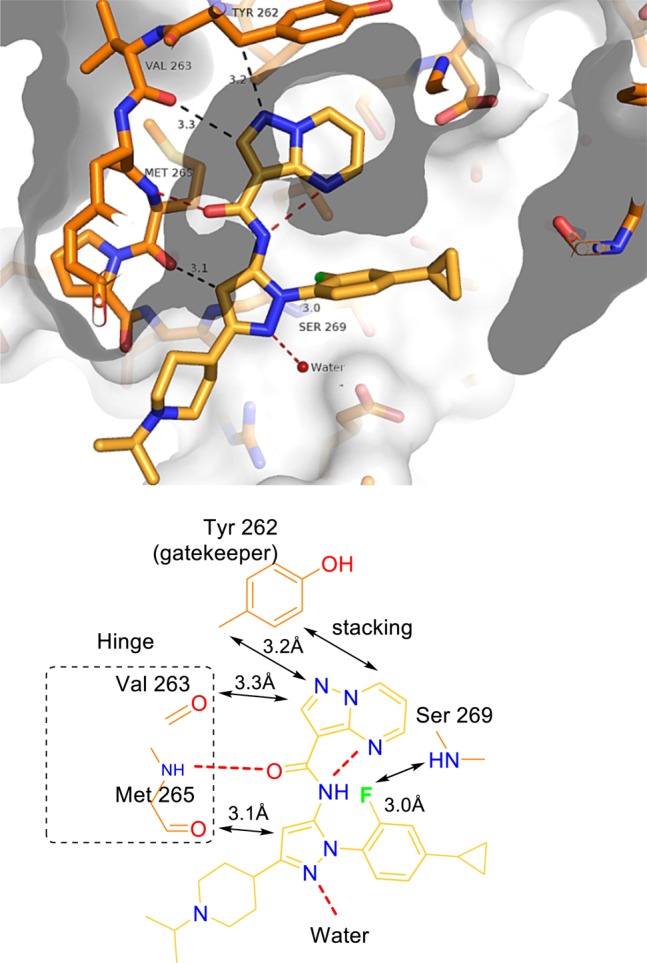
X-ray cocrystal of 30 with IRAK4.
A key finding was the discovery that replacement of the pyrazole C-3 piperidine ring with a piperazine provided analogues displaying small but significant improvements in both enzyme inhibition and exposure. Thus, N-methyl piperazine 32 had IRAK4 IC50 = 5 nM, excellent aqueous solubility, and rat AUC = 1.7 μM·h (0–8 h). As expected, replacement of the N-methyl group with a methanesulfonamide (33) improved plasma exposure but decreased solubility.
Piperazine 32 was selected for further profiling (Figure 3). In order to determine its cellular activity, THP1-XBlue cells were preincubated with inhibitor 32. Inflammatory signaling was then stimulated with either the TLR4 agonist LPS, which signals through IRAK4, or TNFα, which acts through IRAK4 independent pathways. Activation of the downstream transcription factor NF-κB was then determined. Analogue 32 exhibits an IC50 = 83 nM in cells stimulated by LPS, and no measurable inhibition at concentrations up to 31.6 μM in cells stimulated by TNFα. Compound 32 was highly selective, as the only kinase with >80% inhibition at 1 μM in a panel of 108 is IRAK4. Bioavailability when dosed at 10 mg/kg was 28%, with 21% unbound in rat plasma. This analogue was assessed for its ability to reduce proinflammatory cytokine production in vivo, using a similar experiment as performed with 20 (vide supra). Good reduction in levels of TNFα (32%), IL-1β (28%, p < 0.05), IL-6 (85%, p < 0.005), IL-8 (34%), and IFNγ (30%, p < 0.005) were observed upon dosing at 100 mg/kg.
Figure 3.

Profile of analogue 32 and paw size reduction in mouse ABIA study.
Analogue 32 was assessed in the antibody induced arthritis model of inflammation in mice.23 In this study, BALB/c female mice were immunized with an arthritogenic monoclonal antibody (CIA-MAB-50, 2.5 mg) on day 0. On day 3, the mice were given LPS (25 μg i.p.) as a booster. On days 3–10, mice were dosed q.d. by oral gavage with 100 mg/kg of compound 32 or dexamethasone (2 mg/kg) as a positive control. Paw size was measured on days 0, 3, 6, 8, and 10. On day 10 the study was terminated. The results of the paw measure on day 10 are shown in Figure 3. Compound 32 gave a robust decrease in paw size relative to the control vehicle group (dosed with methylcellulose, MC) and displayed similar efficacy to dexamethasone.
In summary, a series of potent, selective, and orally bioavailable pyrazole IRAK4 inhibitors have been described. A screening campaign identified pyrazoles 1 and 2 as promising hits, and an X-ray structure of 1 indicated that the pyrazole C-3 substituent, pointing toward the solvent exposed portion of the protein, and N-1 substituent offered opportunities to improve IRAK4 activity. SAR studies at the C-3 position indicated that piperidine and piperazine substituents were well tolerated, and the identity of the nitrogen substituent could be used to modulate potency, solubility, and rat oral exposure. Although sulfonamides conferred excellent rat PK, they were universally poorly soluble. In contrast, alkyl substituted piperidines and a piperazine were highly soluble and very potent. Limited SAR studies at the pyrazole N-1 position indicated that ortho-fluoro and pyridine substituents provided the best levels of potency. These studies ultimately led to the discovery of analogue 32, which reduced the production of TLR4-mediated cytokines and was efficacious in a mouse antibody induced arthritis model.
Acknowledgments
The authors thank the Merck departments of preclinical development and pharmacokinetics, pharmacodynamics, and drug metabolism for providing solubility and rat PK analyses, respectively.
Glossary
ABBREVIATIONS
- IRAK4
interleukin-1 receptor-associated kinase 4
- SAR
structure–activity relationship
- IL-1R/TLR
interleukin-1 receptor/Toll-like receptor
- TIR
Toll/interleukin-1R
- MYD88
myeloid differentiation primary response gene 88
- AP-1
activator protein-1
- NF-κB
nuclear factor κ-light-chain-enhancer of activated B cells
- TNFα
tumor necrosis factor α
- IL-1
interleukin-1
- TLR2
Toll-like receptor 2
- i-PrOH
iso-propanol
- DIPEA
diisopropylethylamine
- N/A
not available
- HPbCD
(2-hydroxypropyl)-β-cyclodextrin
- AUC
area under curve
- PK
pharmacokinetic
- MINK1
misshapen-like kinase 1
- THP
tetrahydropyran
- TRKB
tyrosine receptor kinase B
- PAM2CSK4
palmitoyl-2-cysteine-serine-lysine
- IL-1β
interleukin-1 β
- IL-6
interleukin-6
- IFNγ
Interferon γ
- TLR4
Toll-like receptor 4
- LPS
lipopolysaccharide
- IL-8
interleukin-8
- ABIA
antibody induced arthritis
- MC
methylcellulose
Supporting Information Available
Compound synthesis and characterization, and assay protocols. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00106.
Accession Codes
X-ray coordinates for compounds 1 and 30 in the Protein Data Bank are 4YO6 and 4YP8, respectively.
The authors declare no competing financial interest.
Supplementary Material
References
- Pawelec G.; Goldeck D.; Derhovanessian E. Inflammation, ageing and chronic disease. Curr. Opin. Immunol. 2014, 29, 23–28. [DOI] [PubMed] [Google Scholar]
- Jacobson D. L.; Gange S. J.; Rose N. R.; Graham N. M. H. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin. Immunol. Immunopathol. 1997, 84, 223–243. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. Cancer and Inflammation: An old intuition with rapidly evolving new concepts. Annu. Rev. Immunol. 2012, 30, 677–706. [DOI] [PubMed] [Google Scholar]
- Libby P. Inflammation and cardiovascular disease mechanisms. Am. J. Clin. Nutr. 2006, 83, 456s–460s. [DOI] [PubMed] [Google Scholar]
- Choy E. H.; Kavanaugh A. F.; Jones S. A. The problem of choice: current biologic agents and future prospects in RA. Nat. Rev. Rheumatol. 2013, 9, 154–163. [DOI] [PubMed] [Google Scholar]
- Ghoreschi K.; Gadina M. Jakpot! New small molecules in autoimmune and inflammatory diseases. Exp. Dermatol. 2014, 23, 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. H.; Smith C.; Yin H. Targeting Toll-like receptors with small molecule agents. Chem. Soc. Rev. 2013, 42, 4859–4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. Y.; Strelow A.; Fontana E. J.; Wesche H. IRAK-4: A novel member of the IRAK family with the properties of an IRAK-kinase. Proc. Natl. Acad. Sci. USA 2002, 99, 5567–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill L. A. J. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol. Rev. 2008, 226, 10–18. [DOI] [PubMed] [Google Scholar]
- Suzuki N.; Saito T. IRAK-4 - a shared NF-kappa B activator in innate and acquired immunity. Trends Immunol. 2006, 27, 566–572. [DOI] [PubMed] [Google Scholar]
- Picard C.; Puel A.; Bonnet M.; Ku C. L.; Bustamante J.; Yang K.; Soudais C.; Dupuis S.; Feinberg J.; Fieschi C.; Elbim C.; Hitchcock R.; Lammas D.; Davies G.; Al-Ghonaium A.; Al-Rayes H.; Al-Jumaah S.; Al-Hajjar S.; Al-Mohsen I. Z.; Frayha H. H.; Rucker R.; Hawn T. R.; Aderem A.; Tufenkeji H.; Haraguchi S.; Day N. K.; Good R. A.; Gougerot-Pocidalo M. A.; Ozinsky A.; Casanova J. L. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science 2003, 299, 2076–2079. [DOI] [PubMed] [Google Scholar]
- Dou H.; Song Y.; Liu X.; Yang L.; Jiang N.; Chen D.; Li E.; Tan R.; Hou Y. A novel benzenediamine derivate rescued mice from experimental sepsis by attenuating proinflammatory mediators via IRAK4. Am. J. Respir. Cell Mol. 2014, 51, 191–200. [DOI] [PubMed] [Google Scholar]
- Tumey L. N.; Boschelli D. H.; Bhagirath N.; Shim J.; Murphy E. A.; Goodwin D.; Bennett E. M.; Wang M. M.; Lin L. L.; Press B.; Shen M.; Frisbie R. K.; Morgan P.; Mohan S.; Shin J.; Rao V. R. Identification and optimization of indolo[2,3-c]quinoline inhibitors of IRAK4. Bioorg. Med. Chem. Lett. 2014, 24, 2066–2072. [DOI] [PubMed] [Google Scholar]
- For a recent review, seeChaudhary D.; Robinson S.; Romero D. L. Recent advances in the discovery of small molecule inhibitors of interleukin-1 receptor-associated kinase 4 (IRAK4) as a therapeutic target for inflammation and oncology disorders. J. Med. Chem. 2015, 58, 96–110. [DOI] [PubMed] [Google Scholar]
- For a recent review, seeHynes J.; Nair S. K. Advances in the discovery of small-molecule IRAK4 inhibitors. Annu. Rep. Med. Chem. 2014, 49, 117–133. [Google Scholar]
- Toledo L. M.; Lydon N. B.; Elbaum D. The structure-based design of ATP-site directed protein kinase inhibitors. Curr. Med. Chem. 1999, 6, 775–805. [PubMed] [Google Scholar]
- See Supporting Information for complete description of compound synthesis.
- Reddy G. J.; Latha D.; Rao K. S. A clean and rapid synthesis of 5-amino and 5-alkoxycarbonylpyrazoles using montmorillonite under acid free conditions. Org. Prep. Proced. Int. 2004, 36, 494–498. [Google Scholar]
- Korfmacher W. A.; Cox K. A.; Ng K. J.; Veals J.; Hsieh Y. S.; Wainhaus S.; Broske L.; Prelusky D.; Nomeir A.; White R. E. Cassette-accelerated rapid rat screen: a systematic procedure for the dosing and liquid chromatography/atmospheric pressure ionization tandem mass spectrometric analysis of new chemical entities as part of new drug discovery. Rapid Commun. Mass Spectrom. 2001, 15, 335–340. [DOI] [PubMed] [Google Scholar]
- Ritchie T. J.; Macdonald S. J. F. The impact of aromatic ring count on compound developability - are too many aromatic rings a liability in drug design?. Drug Discovery Today 2009, 14, 1011–1020. [DOI] [PubMed] [Google Scholar]
- Palsson-McDermott E. M.; O’Neill L. A. J. The potential of targeting Toll-like receptor 2 in autoimmune and inflammatory diseases. Irish J. Med. Sci. 2007, 176, 253–260. [DOI] [PubMed] [Google Scholar]
- He H.; Lyons K. A.; Shen X.; Yao Z.; Bleasby K.; Chan G.; Hafey M.; Salituro G. M.; Cohen L. H.; Tang W. Utility of unbound plasma drug levels and P-glycoprotein transport data in prediction of central nervous system exposure. Xenobiotica 2009, 39, 687–693. [DOI] [PubMed] [Google Scholar]
- Asquith D. L.; Miller A. M.; McInnes I. B.; Liew F. Y. Animal models of rheumatoid arthritis. Eur. J. Immunol. 2009, 39, 2040–2044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.