

Abstract

We describe the synthesis and SAR of 1,2,3-triazolopiperidines as a novel series of potent, brain penetrant P2X7 antagonists. Initial efforts yielded a series of potent human P2X7R antagonists with moderate to weak rodent potency, some CYP inhibition, poor metabolic stability, and low solubility. Further work in this series, which focused on the SAR of the N-linked heterocycle, not only increased the potency at the human P2X7R but also provided compounds with good potency at the rat P2X7R. These efforts eventually delivered a potent rat and human P2X7R antagonist with good physicochemical properties, an excellent pharmacokinetic profile, good partitioning into the CNS, and demonstrated in vivo target engagement after oral dosing.
Keywords: P2X7, neuro-inflammation, depression
Over the past few years, numerous reports have appeared in the literature that implicate cytokines in depression and in particular that cytokines play a role in treatment resistant depression.1,2 As such, several laboratories have reported that reduction of IL-1β levels in mice correlates to symptom improvement in a random stress model of depression.3 Since the activation of the P2X7 receptor (P2X7R) results in the production of IL-1β,4 antagonists of P2X7R, which are known to block IL-1β release, are hypothesized to be useful drugs for the treatment of depression.5
There have been numerous reports of P2X7R antagonists in the literature (Figure 1),6−10 most notably the benzamide class of compounds from Astra-Zeneca (1)11,12 and a structurally distinct benzamide from Pfizer, CE-224,535 (2).13 The Pfizer benzamide was tested in human clinical trials where it was shown to lower peripheral levels of IL-1β in a rheumatoid arthritis trial.14 However, since CE-224,535 is not known to penetrate into the CNS, the effect on reduced CNS levels of IL-1β are not currently known. Herein, we describe a series of potent, brain penetrant P2X7R antagonists that show robust P2X7R target engagement in rodents.
Figure 1.

Previously reported P2X7 antagonists.
Glaxo SmithKline recently disclosed a series of brain penetrant P2X7R antagonists (3, 4)6,7 that had good affinity for the human P2X7R but only modest affinity for the rat P2X7R. We recently disclosed two selective brain penetrant P2X7R antagonists (5, 6) with appreciable affinity for the rat P2X7R and subsequently demonstrated robust P2X7R target engagement in the CNS of the rat as measured by ex vivo autoradiography.15,16
In addition to the various P2X7R chemotypes in Figure 2, we were aware of the 1,2,4-triazolopiperazines disclosed in 2010 (Figure 3).17 In a quest to explore other heterocyclic cores that could serve as competent P2X7R antagonists we decided to embark on a campaign to discover novel heterocycles that were potent P2X7R antagonists, and this work eventually led us to the 1,2,3-triazolopiperidines series disclosed in this report.
Figure 2.

Brain penetrant P2X7R antagonists.
Figure 3.

1,2,4-Triazolopiperazine P2X7R antagonists.
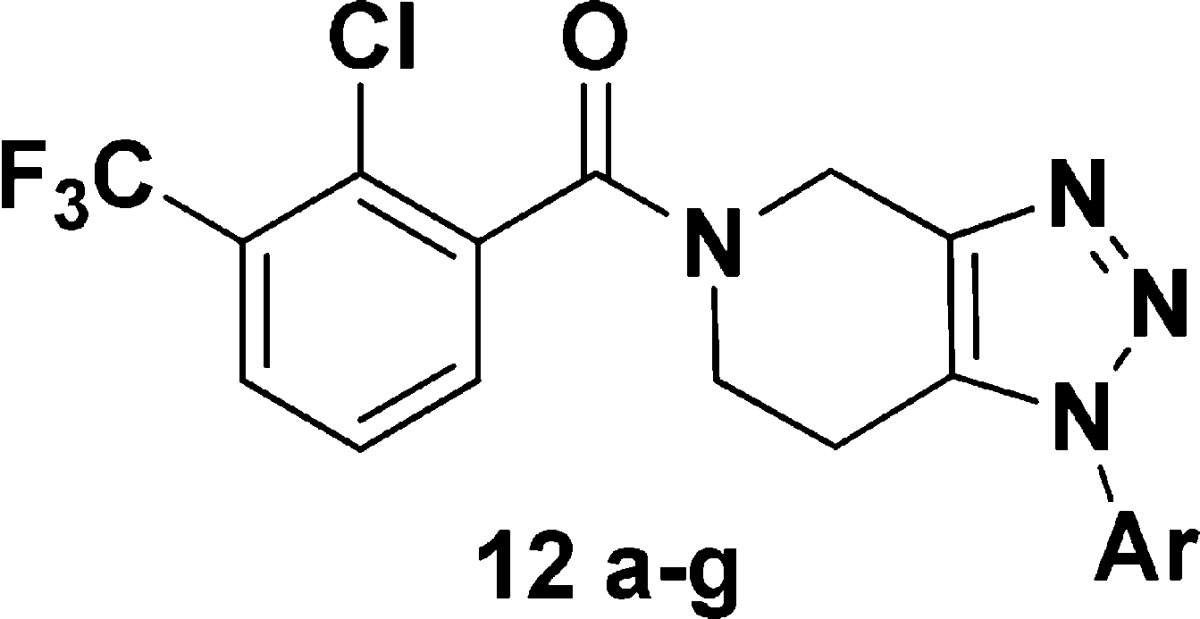
Our initial synthesis began with commercially available 1H-[1,2,3]triazolo[4,5-c]pyridine (9) (Scheme 1). Although arylation of the 1H-[1,2,3]-triazolo-[4,5-c]-pyridine had the potential to give several regioisomers, we anticipated that the electronic effect of the pyridyl nitrogen would favor arylation to the 1-position. In practice, the use of Buchwald’s copper mediated arylation18,19 furnished the desired arylated products in a 5:1 ratio favoring the N-1 aryl regioisomer albeit in low yield and as an inseparable mixture.20 Subsequent hydrogenation of the N-1, N-2 mixture (H-cube, Pt2O, 90 bar, MeOH) did furnish a small amount the 1,2,3-triazolopiperidine; however, this method suffered from incomplete/irreproducible conversion, even after conducting the reaction in continuous flow mode. Regardless, the regioisomers were separated at the 1,2,3-triazolopiperidine stage and then coupled with the corresponding benzoic acids to furnish the desired product(s) (Scheme 1). As the first analogue made (12a) exhibited an hP2X7R IC50 = 2.7 nM, our interest in this series grew.
Scheme 1. First Generation Synthesis of 1,2,3-Triazolopiperidines: Triazole Arylation Route.

The low yield and irreproducibility of the hydrogenation reaction coupled with difficulties encountered with the purification of the regioisomers prompted us to look at alternative methods to more efficiently prepare these compounds. The regiochemical issue was addressed by using 4-chloro-3-nitropyridine as the starting material as shown in Scheme 2. An amino heterocycle displacement of chloro-nitro-pyridine (13) was followed by reduction, which furnished the diamino pyridine (15), and subsequently converted to the 1,2,3-triazolopyridine core (16) after treatment with t-butyl nitrite. Reduction as in Scheme 1 provided 17, which was coupled to the desired carboxylic acid or acid chloride to provide compounds 12.
Scheme 2. Synthetic Route to 1,2,3-Triazolopiperidines.

Alternatively, we were aware that an acylpyridinium21,22 species (18) could enable the synthesis of the final products directly. As such, the acylpyridinium species (18) was formed by the addition of an acid chloride to the triazolopyridine 16 in THF; however, this species (18) was unstable and readily decomposed in the presence of protic solvents such as methanol. Treatment of the intermediate acylpyridinium (18) with sodium or lithium borohydride did not result in reduction to the desired product. However, use of the Hantzsch ester23 cleanly provided the partially reduced derivatives (19); which were subsequently reduced with hydrogen over Pd/C in ethanol to provide final products (12). In some cases the pendant heterocycles were cleaved, and a transfer hydrogenation with ammonium formate and Pd/C was more productive.
The first entry into this series of antagonists was compound 12a (Table 1), which had a potent hP2X7 FLIPR IC50 of 2.7 nM but was significantly less potent at the rat P2X7R with a IC50 = 1900 nM. In addition, compound 12a was rapidly metabolized in vitro and was a CYP 2C19 inhibitor with an IC50 of 0.1 μM. Compound 12a was not cross reactive in a commercial panel of 50 receptors, ion channels, and transporter assays (Eurofins-CEREP, http://www.eurofins.com/) at a screening concentration of 1.0 μM. Since compound 12 had a clogP of ∼4.0, we reasoned that reducing lipophilicity (lower cLogP) could address both the metabolism issues and the CYP profile.
Table 1. SAR for N-1-Aryl 1,2,3-Triazolopiperidines.
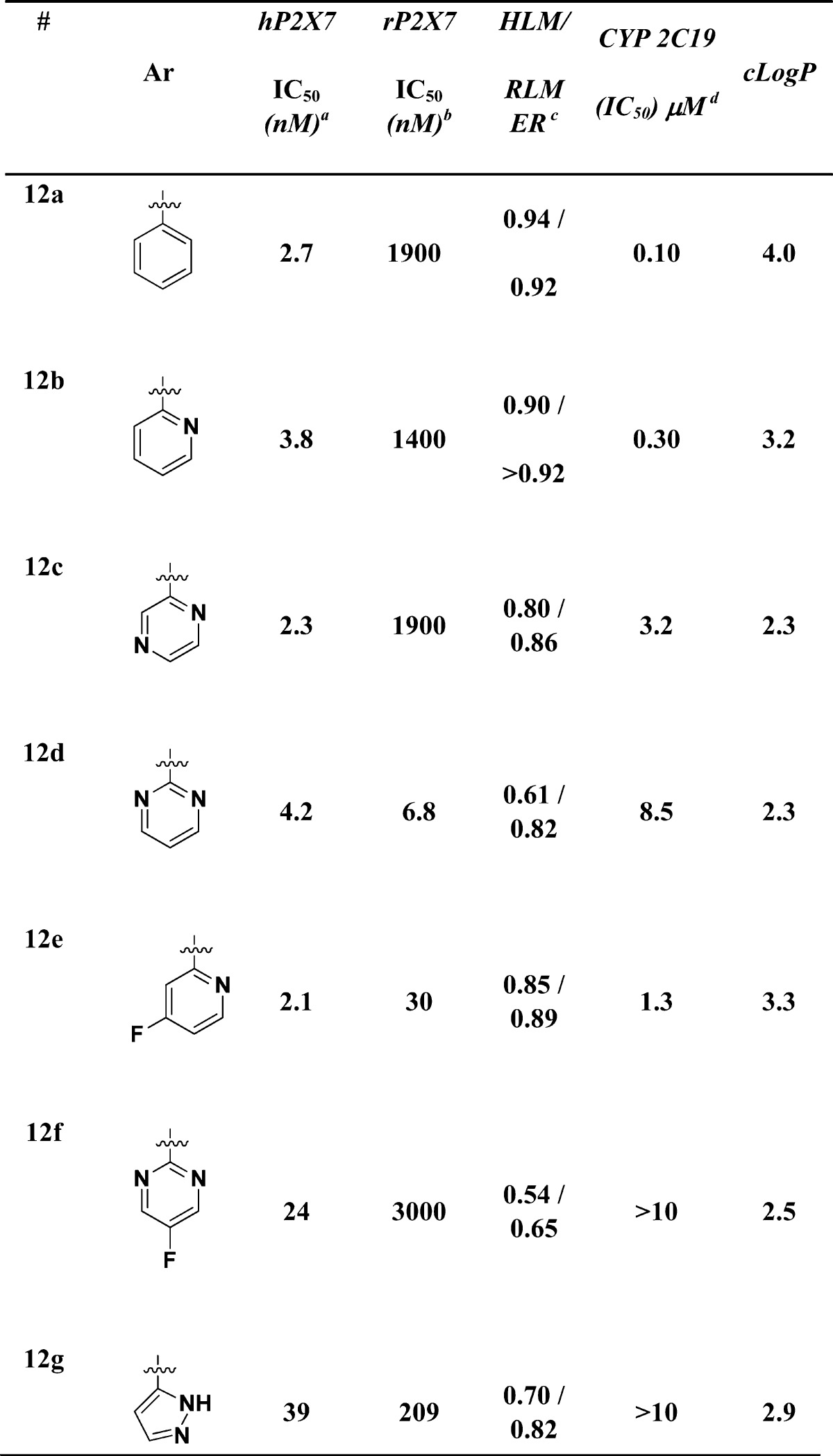
Human FLIPR pIC50 measured in a Ca2+ flux assay.
Rat FLIPR pIC50 measured in a Ca2+ flux assay; all data are the result of at least three assays run in triplicate with the mean value reported. The standard deviation is all cases was less than 2-fold.
HLM and RLM refer to human and rat liver microsomal preparations with data reported as the extraction ratio (ER).
CYP2C19 data was obtained from human liver microsomes. Details of all assay conditions are provided in the Supporting Information.
Indeed, alternative heterocycles proved to be a fruitful expedition (Table 1). Changing from a phenyl (12a) to a pyridyl (12b) maintained affinity at the human receptor (IC50 = 3.8 nM) and provided a slight improvement in the CYP 2C19 inhibition (IC50 = 0.3 μM), though the rat affinity and metabolic stability were not improved. The addition of a second nitrogen, as in the pyrimidine (12c), also maintained affinity for the human receptor (IC50 = 2.3 nM), showed a further improvement in the CYP 2C19 inhibition (IC50 = 3.2 μM), and showed a modest improvement in the human and rat microsomal stability. The pyrimidine compound (12d) was especially gratifying as it was potent at both the human (IC50 = 4.2 nM) and rat (IC50 = 6.8 nM) P2X7R in addition to further improving the CYP inhibition (IC50 = 8.5 μM) and the microsomal stability. This was one of the first compounds in this series that showed substantial affinity for the rat P2X7R. The 4-fluoropyridine (12e) also had significantly improved potency at the rat receptor (IC50 = 30 nM), relative to the pyridyl parent (12b, IC50 = 1400 nM), and reduced CYP inhibition (IC50 = 1.3 μM) but was still rapidly metabolized. The 5-fluoro pyrimidine (12f) had good human potency (IC50 = 24 nM), improved CYP inhibition (IC50 = >10 μM), and microsomal stability when compared to the parent pyrimidine (12d) though the rat affinity (IC50 = 3000 nM) was reduced. Finally, the pyrazole (12g) had good human affinity (IC50 = 39 nM), reduced rat affinity (IC50 = 209 nM), moderate microsomal stability and no CYP inhibition (IC50 = >10 μM).
After optimizing the N-1-aryl portion of the molecule, we prepared a limited number of benzamides while keeping the aryl group as a pyrimidine (Table 2). The 2-chloro-3-trifluoromethyl (12d) had an IC50 = 4.2 nM. Replacing the chloro (12c) with a fluoro (12h) provided a compound with reduced potency at the human (IC50 = 22 nM) and rat (IC50 = 258 nM) receptor, though human microsomal stability and CYP 2C19 liability were slightly improved. The 2-methyl (12h) analogue was slightly more potent at the human receptor (IC50 = 2.2 nM), though significantly less potent at the rat receptor (349 nM). Human and rat microsomal stability and CYP 2C19 inhibition were also improved. The 2,3-dichloro derivative (12j) was potent at the human receptor (IC50 = 2.0 nM), less potent at rat receptor (IC50 = 53 nM), and had improved microsomal stability, but slightly worse in the CYP 2C19 assay. Finally, both of the 2,3-bis halo derivatives (12k,l) were less active.
Table 2. SAR for Benzamide of 1,2,3-Triazolopiperidines.

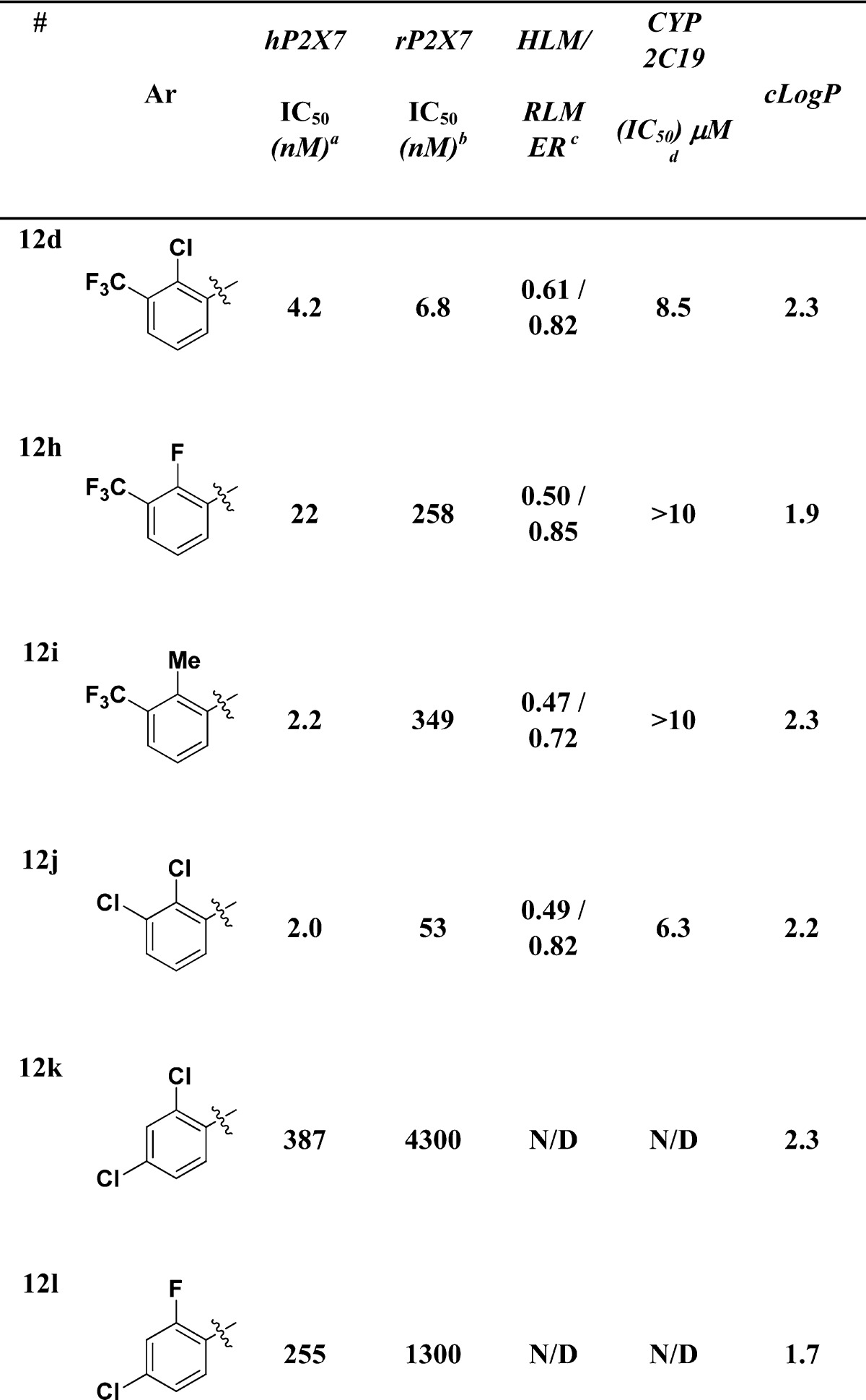
Human FLIPR pIC50 measured in a Ca2+ flux assay.
Rat FLIPR pIC50 measured in a Ca2+ flux assay; all data are the result of at least three assays run in triplicate with the mean value reported. The standard deviation is all cases was less than 2-fold.
HLM and RLM refer to human and rat liver microsomal preparations with data reported as the extraction ratio (ER).
CYP2C19 data was obtained from human liver microsomes. Details of all assay conditions are provided in the Supporting Information.
Since we had now generated potent P2X7 compounds in this series, we were interested to see how these compounds behaved in vivo, and in particular if the compounds would distribute efficiently into the CNS. Although we were especially interested in compound (12d), as it was active at both the human and the rat P2X7R, we obtained additional in vitro ADME characterization of three of the more promising compounds (12c, 12d, and 12f). The profiles of those three compounds are shown in Tables 3 and 4. In general all three compounds have reasonable physical properties, including solubility, permeability, and protein binding to warrant in vivo testing.
Table 3. In Vitro Pharmacology for Compounds 12c, 12d, and 12f.
| human FLIPR IC50a | rat FLIPR IC50b | mouse FLIPR IC50c | human Ki | rat Ki | hERGd IC50 (μM) | human WB IC50e | |
|---|---|---|---|---|---|---|---|
| 12c | 2.3 nM | 1.9 μM | 15 nM | 23 | 5.0 | >10 | 316 nM |
| 12d | 4.2 nM | 6.8 nM | 4.0 nM | 16 | 2.8 | >10 | 79 nM |
| 12f | 24 nM | 3.0 μM | 588 nM | 32 | 20 | >10 | 251 nM |
Human FLIPR pIC50 measured in a Ca2+ flux assay.
Rat FLIPR pIC50 measured in a Ca2+ flux assay.
Mouse FLIPR pIC50 measured in a Ca2+ flux assay.
hERH IC50 determined in a tritiated dofetilide binding assay.
Human blood was primed with lipopolysaccharide (LPS; 30 ng/mL) followed by addition of the test antagonist or vehicle with the final P2X7 stimulus of Bz-ATP. Test compounds were added and incubated for an additional 30 min. The P2X7 agonist, Bz-ATP (1 mM), was finally added and incubated for 1.5 h at 37 °C. Postincubation, the plates were centrifuged (low-speed spin) and the supernatant collected for IL-1β ELISA analysis as per manufacturer’s protocol (human IL-1β: Thermo Scientific; catalogue # EH2IL1B5). All data are the result of at least three assays run in triplicate with the mean value reported.
Table 4. Physical Properties and In Vitro DMPK Parameters for Compounds 12c, 12d, and 12f.
| MW | clog P | human ERa | rat ERa | mouse ERa | human/rat ppbb | Caco-2 A to B/B to Ac | brain pbd | solubility pH 2/pH 7e | |
|---|---|---|---|---|---|---|---|---|---|
| 12c | 408.7 | 2.3 | 0.80 | 0.86 | 0.84 | 89.6/91.6 | 81.2/38.4 | 94.0 | 304/235 |
| 12d | 408.8 | 2.3 | 0.61 | 0.82 | 0.89 | 84.3/88.6 | 71.4/45.4 | 90.9 | 48/50 |
| 12f | 426.8 | 2.5 | 0.54 | 0.65 | 0.87 | 83.4/83.9 | 75.5/55.4 | 92.5 | 49/202 |
Human, rat, or mouse extraction ratio as measured in a microsomal preparation.
Human or rat protein binding reported as % bound.
Papp reported in units of cm/sec × 10–6.
Rat brain protein binding reported as % bound.
Reported in μM.
The rat pharmacokinetic data for all three compounds is shown in Figure 4. The pyrazine (12c) had bioavailability of 33% and reached a Cmax of 1.0 μM. The half-life of 0.7 h is consistent with the relatively high microsomal extraction ratio (0.82) as clearance of 12c was near the rat hepatic blood flow. Disappointingly, compound 12d, which had the best rat P2X7R affinity (4.2 nM), had the lowest oral exposure of all three compounds with a bioavailability of 1%. Compound 12f proved to have the best PK with measured bioavailability greater than 100% and a half-life of 2.3 h, which is slightly longer than predicted by the rat extraction ratio (0.65). Although we do not know the reason for the low bioavailability of compound 12d, it is interesting to note that 12d has the lowest solubility of the three compounds.
Figure 4.

Rat PK data for compounds 12c, 12d, and 12f.
Compound 12f was also tested in a rat ex vivo autoradiography (ARG) experiment to assess the brain P2X7 receptor occupancy (Figure 5). When 12f was dosed at 10 mg/kg PO in the rat, it achieved a maximum occupancy of 45% at 30 min. The occupancy decreased over time in conjunction with decreasing concentrations of 12f in the brain and plasma, which were measured in the same experiment. Therefore, compound 12f demonstrated proof of concept that the 1,2,3-triazolopiperidines are a new class of brain penetrant P2X7R antagonists that are capable of occupying the receptor when dosed orally.
Figure 5.

Rat P2X7 receptor occupancy data for 12f.
We describe the synthesis and SAR of 1,2,3-triazolopiperidines as a new series of potent and brain penetrant P2X7 antagonists. The prototype N-1-phenyl triazole (12a) was potent at the human P2X7R, but had poor affinity for the rat P2X7R, had poor microsomal stability, was a strong inhibitor of CYP 2C19, and had a high cLogP. By replacing the phenyl group with various heterocycles we discovered compound 12f, a potent human and rat P2X7R ligand with sufficiently good physicochemical properties, to enable oral dosing in rats and provide high levels of the compound in the plasma and the brain, and demonstrated target engagement as measured by the rat ARG. Additional studies of this novel class of P2X7R antagonists will be the subject of future publications.
Acknowledgments
The authors would like to thank Professor David MacMillan for useful discussions toward optimizing the synthetic strategy, and especially the use of the Hantzsch ester reduction of the acylpyridinium species.
Supporting Information Available
Synthesis, characterization, and assay conditions. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00089.
The authors declare no competing financial interest.
Supplementary Material
References
- Raison C. L.; Capuron L.; Miller A. H. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006, 27, 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowlati Y.; Herrmann N.; Swardfager W.; Liu H.; Sham L.; Reim E. K.; Lanctot K. L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–57. [DOI] [PubMed] [Google Scholar]
- Koo J. W.; Duman R. S. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solle M.; Labasi J.; Perregaux D. G.; Stam E.; Petrushova N.; Koller B. H.; Griffiths R. J.; Gabel C. A. Altered cytokine production in mice lacking P2X(7) receptors. J. Biol. Chem. 2001, 276, 125–32. [DOI] [PubMed] [Google Scholar]
- Chrovian C. C.; Rech J. C.; Bhattacharya A.; Letavic M. A. P2X7 antagonists as potential therapeutic agents for the treatment of CNS disorders. Prog. Med. Chem. 2014, 53, 65–100. [DOI] [PubMed] [Google Scholar]
- Abberley L.; Bebius A.; Beswick P. J.; Billinton A.; Collis K. L.; Dean D. K.; Fonfria E.; Gleave R. J.; Medhurst S. J.; Michel A. D.; Moses A. P.; Patel S.; Roman S. A.; Scoccitti T.; Smith B.; Steadman J. G.; Walter D. S. Identification of 2-oxo-N-(phenylmethyl)-4-imidazolidinecarboxamide antagonists of the P2X(7) receptor. Bioorg. Med. Chem. Lett. 2010, 20, 6370–4. [DOI] [PubMed] [Google Scholar]
- Abdi M. H.; Beswick P. J.; Billinton A.; Chambers L. J.; Charlton A.; Collins S. D.; Collis K. L.; Dean D. K.; Fonfria E.; Gleave R. J.; Lejeune C. L.; Livermore D. G.; Medhurst S. J.; Michel A. D.; Moses A. P.; Page L.; Patel S.; Roman S. A.; Senger S.; Slingsby B.; Steadman J. G.; Stevens A. J.; Walter D. S. Discovery and structure-activity relationships of a series of pyroglutamic acid amide antagonists of the P2X7 receptor. Bioorg. Med. Chem. Lett. 2010, 20, 5080–4. [DOI] [PubMed] [Google Scholar]
- Beswick P. J.; Billinton A.; Chambers L. J.; Dean D. K.; Fonfria E.; Gleave R. J.; Medhurst S. J.; Michel A. D.; Moses A. P.; Patel S.; Roman S. A.; Roomans S.; Senger S.; Stevens A. J.; Walter D. S. Structure-activity relationships and in vivo activity of (1H-pyrazol-4-yl)acetamide antagonists of the P2X7 receptor. Bioorg. Med. Chem. Lett. 2010, 20, 4653–4656. [DOI] [PubMed] [Google Scholar]
- Gleave R. J.; Walter D. S.; Beswick P. J.; Fonfria E.; Michel A. D.; Roman S. A.; Tang S.-P. Synthesis and biological activity of a series of tetrasubstituted-imidazoles as P2X7 antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 4951–4954. [DOI] [PubMed] [Google Scholar]
- Guile S. D.; Alcaraz L.; Birkinshaw T. N.; Bowers K. C.; Ebden M. R.; Furber M.; Stocks M. J. Antagonists of the P2X(7) receptor. From lead identification to drug development. J. Med. Chem. 2009, 52, 3123–41. [DOI] [PubMed] [Google Scholar]
- Baxter A.; Bent J.; Bowers K.; Braddock M.; Brough S.; Fagura M.; Lawson M.; McInally T.; Mortimore M.; Robertson M.; Weaver R.; Webborn P. Hit-to-Lead studies: the discovery of potent adamantane amide P2X7 receptor antagonists. Bioorg. Med. Chem. Lett. 2003, 13, 4047–50. [DOI] [PubMed] [Google Scholar]
- Furber M.; Alcaraz L.; Bent J. E.; Beyerbach A.; Bowers K.; Braddock M.; Caffrey M. V.; Cladingboel D.; Collington J.; Donald D. K.; Fagura M.; Ince F.; Kinchin E. C.; Laurent C.; Lawson M.; Luker T. J.; Mortimore M. M. P.; Pimm A. D.; Riley R. J.; Roberts N.; Robertson M.; Theaker J.; Thorne P. V.; Weaver R.; Webborn P.; Willis P. Discovery of potent and selective adamantane-based small-molecule P2X7 receptor antagonists/interleukin-1β inhibitors. J. Med. Chem. 2007, 50, 5882–5885. [DOI] [PubMed] [Google Scholar]
- Duplantier A. J.; Dombroski M. A.; Subramanyam C.; Beaulieu A. M.; Chang S. P.; Gabel C. A.; Jordan C.; Kalgutkar A. S.; Kraus K. G.; Labasi J. M.; Mussari C.; Perregaux D. G.; Shepard R.; Taylor T. J.; Trevena K. A.; Whitney-Pickett C.; Yoon K. Optimization of the physicochemical and pharmacokinetic attributes in a 6-azauracil series of P2X7 receptor antagonists leading to the discovery of the clinical candidate CE-224,535. Bioorg. Med. Chem. Lett. 2011, 21, 3708–11. [DOI] [PubMed] [Google Scholar]
- Stock T. C.; Bloom B. J.; Wei N.; Ishaq S.; Park W.; Wang X.; Gupta P.; Mebus C. A. Efficacy and safety of CE-224,535, an antagonist of P2X7 receptor, in treatment of patients with rheumatoid arthritis inadequately controlled by methotrexate. J. Rheumatol. 2012, 39, 720–7. [DOI] [PubMed] [Google Scholar]
- Letavic M. A.; Lord B.; Bischoff F.; Hawryluk N. A.; Pieters S.; Rech J. C.; Sales Z.; Velter A. I.; Ao H.; Bonaventure P.; Contreras V.; Jiang X.; Morton K. L.; Scott B.; Wang Q.; Wickenden A. D.; Carruthers N. I.; Bhattacharya A. Synthesis and pharmacological characterization of two novel, brain penetrating P2X7 antagonists. ACS Med. Chem. Lett. 2013, 4, 419–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A.; Wang Q.; Ao H.; Shoblock J. R.; Lord B.; Aluisio L.; Fraser I.; Nepomuceno D.; Neff R. A.; Welty N.; Lovenberg T. W.; Bonaventure P.; Wickenden A. D.; Letavic M. A. Pharmacological characterization of a novel centrally permeable P2X7 receptor antagonist: JNJ-47965567. Br. J. Pharmacol. 2013, 170, 624–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean D. K.; Munoz-Muriedas J.; Sime M.; Steadman J. G. A.; Thewlis R. E. A.; Trani G.; Walter D. S.. Preparation of tetrahydro[1,2,4]triazolo[4,3-a]pyrazine derivatives for use as P2X7 modulators. WO 2008124153, April 2009.
- Altman R. A.; Koval E. D.; Buchwald S. L. Copper-catalyzed N-arylation of imidazoles and benzimidazoles. J. Org. Chem. 2007, 72, 6190–6199. [DOI] [PubMed] [Google Scholar]
- Antilla J. C.; Baskin J. M.; Barder T. E.; Buchwald S. L. Copper–diamine-catalyzed N-arylation of pyrroles, pyrazoles, indazoles, imidazoles, and triazoles. J. Org. Chem. 2004, 69, 5578–5587. [DOI] [PubMed] [Google Scholar]
- Note that the N-1 arylated product was observed in only trace amounts and was not isolated in pure form due to difficulties with purification either by normal or reverse phase chromatography.
- Khanna I. K.; Weier R. M. Regiospecific addition of Grignard reagents to the 4-position of activated imidazo[4,5-c]pyridine. A convenient method for the synthesis of 4-alkylarylimidazopyrazines. Tetrahedron Lett. 1993, 34, 1885–1888. [Google Scholar]
- Chen P.; Caldwell C. G.; Mathvink R. J.; Leiting B.; Marsillo F.; Patel R. A.; Wu J. K.; He H.; Lyons K. A.; Thornberry N. A.; Weber A. E. Imidazopiperidine amides as dipeptidyl peptidase IV inhibitors for the treatment of diabetes. Bio. Org. Med. Chem. Lett. 2007, 17, 5853–5857. [DOI] [PubMed] [Google Scholar]
- Ouellet S. J.; Tuttle J. B.; MacMillan D. W. C. Enantioselective organocatalytic hydride reduction. J. Am. Chem. Soc. 2005, 127, 32–33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.