

Abstract
The hydroxylation and N-dechloroethylation of deuterated ifosfamide (d4IFO) and ifosfamide (IFO) by several human P450s have been determined and compared. d4IFO was synthesized with deuterium at the alpha and alpha′ carbons to decrease the rate of N-dechloroethylation and thereby enhance hydroxylation of the drug at the 4′ position. The purpose was to decrease the toxic and increase the efficacious metabolites of IFO. For all of the P450s tested, hydroxylation of d4IFO was improved and dechloroethylation was reduced as compared with nondeuterated IFO. Although the differences were not statistically significant, the trend favoring the 4′-hydroxylation pathway was noteworthy. CYP3A5 and CYP2C19 were the most efficient enzymes for catalyzing IFO hydroxylation. The importance of these enzymes in IFO metabolism has not been reported previously and warrants further investigation. The catalytic ability of the common polymorphisms of CYP2B6 and CYP2C9 for both reactions were tested with IFO and d4IFO. It was determined that the commonly expressed polymorphisms CYP2B6*4 and CYP2B6*6 had reduced catalytic ability for IFO compared with CYP2B6*1, whereas CYP2B6*7 and CYP2B6*9 had enhanced catalytic ability. As with the wild-type enzymes, d4IFO was more readily hydroxylated by the polymorphic variants than IFO, and d4IFO was not dechloroethylated by any of the polymorphic forms. We also assessed the use of specific inhibitors of P450 to favor hydroxylation in human liver microsomes. We were unable to separate the pathways with these experiments, suggesting that multiple P450s are responsible for catalyzing both metabolic pathways for IFO, which is not observed with the closely related drug cyclophosphamide.
Introduction
Ifosfamide (IFO), a DNA alkylating agent, is used in numerous cancer chemotherapeutic regimens. It is the structural isomer of the more commonly used antineoplastic and immunosuppressive agent cyclophosphamide (CPA) (Zhang et al., 2006). Toxicities associated with IFO treatment limit its use, even though the drug has proven more effective than CPA in a wide range of malignant diseases (Buda et al., 2003; Sorio et al., 2003; Donfrancesco et al., 2004; Pocali et al., 2004; Biagi et al., 2005; Kosmas et al., 2007). IFO-induced toxicities include moderate-to-severe nephrotoxicity, which occurs in ∼30% of the patient population, and neurotoxicities, which occur in ∼20% of the patient population (Loebstein et al., 1999; Klastersky, 2003; McCune et al., 2005). IFO treatment is also associated with life-threatening arrhythmias, heart failure, and severe encephalopathy (Zhang et al., 2006). Since IFO-associated toxicities are believed to be attributable to metabolites of the drug, improving our knowledge of IFO metabolism and its determinants may aid in patient treatment and expand the use of IFO to a broader range of disease states and patient populations.
IFO is a prodrug activated via hydroxylation on the 4′ carbon by cytochrome P450s (P450s; Sladek, 1988). CYP2B6, CYP3A4, and CYP3A5 are the primary P450s responsible for the activation of IFO. However, CYP2C9 and CYP2C19 may also play a role in the activation (Roy et al., 1999; Huang et al., 2000). After hydroxylation, IFO undergoes a series of spontaneous events to produce the active metabolite ifosfamide mustard and equimolar amounts of acrolein (Fig. 1). In our studies, as well as others, acrolein measurement is used to analyze the activation of ifosfamide (Chang et al., 1993; Huang et al., 2000; Chen et al., 2004). In addition, IFO may be N-dechloroethylated by CYP2B6, CYP3A4, and CYP3A5 (Roy et al., 1999; Huang et al., 2000), ultimately leading to the inactivation of IFO and formation of the nephro- and neurotoxic metabolites 2-dechloroethylifosfamide or 3-dechloroethylifosfamide and equimolar amounts of chloroacetaldehyde (CAA). Figure 1 depicts the primary pathways for IFO metabolism. It is estimated that 25–60% of IFO is N-dechloroethylated in humans, whereas only 10% of CPA undergoes this reaction (Bruggemann et al., 1997; Carlson et al., 1998; Borner et al., 2000). It has been suggested that elevated circulating levels of CAA in IFO-treated patients are responsible for the development of IFO-specific nephrotoxicity (Woodland et al., 2000; Dubourg et al., 2001; Aleksa et al., 2004). In vitro studies indicate that the minimum toxic concentration of CAA is 500 μM, which is well above the circulating levels of CAA in IFO-treated patients (0.5–109 μM) (Kurowski and Wagner, 1993; Mohrmann et al., 1993; Springate et al., 1999; Dubourg et al., 2001). Thus, organ-specific toxicities are suggested to be attributable to N-dechloroethylation of IFO at the organ of interest, which would increase local tissue concentrations of CAA to potentially toxic levels (Aleksa et al., 2004; McCune et al., 2005). Tissue expression of the P450s is highly variable; for instance, in the kidney CYP3A5 is highly expressed, whereas CYP3A4 is not (Haehner et al., 1996; Wrighton et al., 2000; Koch et al., 2002). Therefore, it has been suggested that CYP3A5 plays a more prominent role than other P450s in IFO-induced nephrotoxicity. Our studies have focused on characterizing the catalytic abilities of the P450s to ascertain the potential roles of specific P450s in IFO metabolism.
Fig. 1.

The major pathways for the metabolism of IFO. The therapeutic activation pathway is depicted vertically. The hydroxylation of IFO occurs on the 4′ carbon and is catalyzed by multiple P450s as indicated. Once hydroxylated, the drug exists in equilibrium with the open ring form of the compound, aldoifosfamide. Aldoifosfamide undergoes spontaneous β-elimination to produce isophosphoramide mustard and acrolein in equimolar amounts. The inactivation pathway of IFO is depicted horizontally. Numerous P450s can catalyze the N-dechloroethylation of IFO as indicated in the figure. N-dechloroethylation can occur at the 2′ or the 3′ carbons resulting in the formation of dechloroethylifosfamide and chloroacetaldehyde, both of which are toxic.
Several studies have suggested that CAA is primarily responsible for IFO-induced toxicities (Woodland et al., 2000; Dubourg et al., 2001; Aleska et al., 2004). Therefore, suppression of CAA formation by favoring hydroxylation over N-dechloroethylation of IFO may improve efficacy and reduce adverse events. Previously this approach has been referred to as “metabolic switching” (Ludeman et al., 2012). In our research, we examined the ability to initiate metabolic switching of IFO. IFO was synthesized with deuterium at the alpha and alpha′ carbons (Fig. 2) to impede N-dechloroethylation and thereby enhance hydroxylation of the drug. The in vitro metabolism of deuterated IFO (d4IFO) was compared with the metabolism of IFO.
Fig. 2.
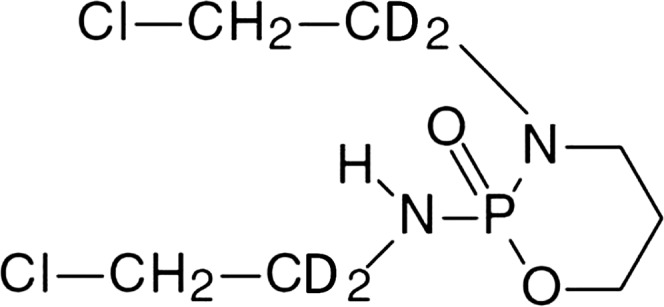
The structure of d4IFO. The structure of d4IFO is shown with the deuteriums located on the alpha and alpha′ carbons.
We report here that deuteration of IFO resulted in enhanced hydroxylation and reduced N-dechloroethylation for all P450s tested (CYP2B6, CYP2C9, CYP2C19, and CYP3A4). Although the differences between d4IFO metabolism and IFO metabolism were moderate, a trend toward metabolic switching is apparent. To more thoroughly investigate IFO and d4IFO metabolism, the polymorphic variants of CYP2B6 (CYP2B6*4, CYP2B6*6–*9) and CYP2C9 (CYP2C9*2 and CYP2C9*3) were assessed for hydroxylation and N-dechloroethylation of IFO and d4IFO. These studies revealed differences in the catalytic rates of the polymorphic variants for both pathways and for both IFO and d4IFO. In brief, CYP2B6*4 and CYP2B6*6 exhibited decreased activation and inactivation for both drugs while CYP2B6*7 and CYP2B6*9 exhibited increased activation and inactivation compared with the wild-type enzymes. d4IFO was more readily activated by the variants than IFO without being inactivated (d4IFO inactivation was only observed with wild-type CYP2B6). The differences observed for the CYP2C9 variants were very small. These findings may relate to patient outcomes with IFO treatment since CYP2B6 is largely responsible for the inactivation of IFO in vivo (Yu and Waxman, 1996).
Materials and Methods
Materials.
NADPH, catalase, IFO, L-α-dilauroyl-phosphocholine, L-α-dioleyl-sn-glycero-3- phosphocholine, L-α-phosphatidylserine, acrolein, CAA, 3-aminophenol, and adenosine were all purchased from Sigma-Aldrich (St. Louis, MO). Pooled human liver microsomes (HLM) were purchased from BD Biosciences (San Jose, CA). d4IFO was prepared as described previously, and NMR analysis confirmed the positions of deuteration (Springer et al., 2007). All other reagents were reagent grade and purchased from commercial sources.
cDNA Expression and Purification of Human P450s, P450 Polymorphic Variants, and Rat Cytochrome P450 Reductase.
Polymorphic variants of CYP2B6 (CYP2B6*4, CYP2B6*6– CYP2B6*9) were overexpressed in Escherichia coli C41 (DE3) cells and purified as described previously (Zhang et al., 2011). All P450s were expressed as their N-terminally truncated forms to increase the expression yields (Jenkins et al., 1998; Scott et al., 2001). Rat cytochrome 450 reductase was expressed and purified as previously described (Hanna et al., 1998). CYP3A5 was a gift from Dr. James Halpert (UCSD), and CYP2C9*2 and CYP2C9*3 were gifts from Dr. Tim Tracy (University of Kentucky) (Domanski et al., 2001; Hustert et al., 2001; Hummel et al., 2005).
Assay for the Metabolism of IFO and d4IFO.
The metabolism of IFO and d4IFO by the human P450s was carried out in a reconstituted system. The reconstituted system consisting of P450 and cytochrome P450 reductase (CPR) in a 1:2 molar ratio was incubated for 30 minutes at 20°C. Dilauroylphosphatidylcholine was not used in the reconstituted systems containing CYP2B6, CYP2C9, or CYP2C19 because it was not required for catalysis (data not shown). For the reactions catalyzed by CYP3A4 and CYP3A5, a mixture of L-α-dilauroyl-phosphocholine, L-α-dioleyl-sn-glycero-3-phosphocholine, and L-α-phosphatidylserine (1:1:1) were included in the reconstituted system for optimal activity. For these studies, 60 μg of the lipid mixture was added to the proteins and the combination was incubated for 45 minutes at 4°C prior to use. The final reaction mixtures contained 1 μM P450, 2 μM CPR, 30 mM potassium phosphate (pH 7.4), 15 units of catalase, and 0–5.0 mM IFO or d4IFO in a total volume of 95 μl. The reaction mixtures were incubated at 37°C for 4 minutes followed by the addition of 5 μl of NADPH (final concentration of 1 mM) to initiate the reaction. After incubation at 37°C for 30 minutes, the reactions were terminated by the addition of 40 μl of ice-cold 5.5% zinc sulfate, 40 μl of ice-cold saturated barium hydroxide, and 20 μl of ice-cold 0.01 M HCl, as described previously (Weber and Waxman, 1993). The samples were centrifuged for 10 minutes at 13,200 rpm and kept on ice prior to derivatization and quantification by high-performance liquid chromatography (HPLC).
IFO Metabolism by Pooled Human Liver Microsomes.
Metabolism of IFO by HLM was determined as described above with the substitution of 0.2 mg/ml of HLM for the reconstituted protein mixture. To inhibit CYP3A4, 5 μM ketoconazole was added to the reaction mixtures prior to initiating the reaction with NADPH. Likewise, to inhibit CYP2B6, 5 μM clopidogrel was added to the reaction mixtures prior to initiating the reaction.
HPLC Analysis of IFO Metabolites.
Acrolein formation was analyzed by HPLC; specifically, 60 μl of the reaction mixture containing the acrolein product was incubated with 40 μl of the fluorescence reagent (30 mg 3-aminophenol and 30 mg hydroxylamine HCl dissolved in 5 ml 1 N HCl) for 25 minutes at 90°C to form the fluorescent product 7-hydroxyquinoline. The derivatized samples were then separated on an Agilent C18 column (Zorbax SB, 3.0 × 150 mm, 5 μm; Agilent Technologies, Santa Clara, CA), using a Shimadzu HPLC system. 7-Hydroxyquinoline was eluted isocratically with 20% methanol in 0.1% phosphoric acid at a flow rate of 0.5 ml/min. Elution of 7-hydroxyquinoline occurred at 3.2 minutes as monitored fluorometrically at 515 nm with excitation of 350 nm (Weber and Waxman, 1993). Calibration curves for acrolein quantification were generated by incubation of acrolein standards in the reaction mixtures containing no substrate or NADPH. The standard curve was linear in the range of 0.5–100 μM acrolein.
To detect CAA, 60 μl of the reaction mixture was incubated with 10 μl of 100 mM adenosine in 0.25 N HCl and 10 μl of 2 M sodium acetate (pH 4.5) for 2.5 hours at 80°C. Under these conditions, CAA was derivatized to yield the fluorescent product, 1-N-ethenoadenosine (Weber and Waxman, 1993). The derivatized sample was then resolved on an Agilent C18 column (Zorbax SB, 3.0 × 150 mm, 5 μM, Agilent Technologies) with a mobile phase of 20% methanol in water and it was eluted isocratically with a flow rate of 0.5 ml/min. 1-N-Ethenoadenosine was monitored fluorometrically at 411 nm with excitation at 270 nm. The derivatized product eluted at 12.7 minutes. Calibration curves for CAA quantification were generated by incubation of CAA standards in reaction mixtures containing no substrate or NADPH. The standard curve was linear from 0.5–150 μM CAA.
Data Analysis.
Data were analyzed using GraphPad Prism software (GraphPad Software, Inc., San Diego, CA). The parameters for the enzyme kinetics were determined using nonlinear regression analysis, specifically Michaelis-Menten analysis in GraphPad Prism. Statistical significance was determined using an unpaired t test in GraphPad Prism.
Results
Metabolism of Ifosfamide and Deuterated Ifosfamide by Wild-Type Human Cytochrome P450s.
The ability of the wild-type P450s (CYP2B6, CYP2C9, CYP2C19, CYP3A4, and CYP3A5) to catalyze the activation of IFO and d4IFO was investigated in the purified reconstituted system. Figure 3 shows the kinetic data that were generated using CYP3A4. Similar data were obtained for most of the other human P450s; these data are not shown, but they are summarized in Table 1. Of the P450s tested, CYP2C9 had the least amount of activity for the metabolism of IFO. Kinetic values for CYP2C9-mediated IFO metabolism could not be determined. Previous reports have indicated that CYP3A4 is the most efficient P450 for the activation of IFO (Roy et al., 1999; Huang et al., 2000). However, our results suggest that CYP3A4 has relatively moderate efficiency for this reaction (Vmax/Km = 0.4 minutes–1 mM−1) (Fig. 3 and Table 1). CYP3A5 exhibited the highest catalytic efficiency for the activation of IFO (Vmax/Km = 2.5 minutes–1 mM−1), and the lowest Vmax value (0.1 pmol/min per picomole of P450). CYP2C19 was the next-most efficient P450 for the activation of IFO (Vmax/Km = 0.9 minutes–1 mM−1). CYP2B6 exhibited the largest Vmax (3.8 pmol/min per picomole of P450), which is 38-fold higher than that of CYP3A5. Similar kinetics were observed for d4IFO (Table 1 and Fig. 3). Most notably, the Km for activation by CYP2B6 was lower for d4IFO than IFO, resulting in a more efficient reaction. Likewise, the Km of CYP3A4 for d4IFO activation was reduced by a factor of 3, resulting in a more efficient reaction as well.
Fig. 3.

Time course for the activation of IFO and d4IFO by CYP3A4. IFO (blue) or d4IFO (red) (0.1–5.0 mM) was incubated in the standard reconstituted system containing 1 μM CYP3A4 for 30 minutes at 37°C. The details of the analytical methods are provided in the Materials and Methods. Shown are the nonlinear fits for the activation of the drugs determined using the Michaelis-Menten equation with Prism 6 (GraphPad Software, Inc.). The Km and Vmax values for all of the P450s tested are summarized in Table 1.
TABLE 1.
Kinetic parameters for the metabolism of IFO and d4IFO by several human P450s
The Km and Vmax values for CYP3A4 were obtained by fitting the data shown in Fig. 3 to the Michaelis-Menten equation as described under Materials and Methods. Similar data and fits were produced and the same analyses were performed for each of the P450s indicated in this table. The reported values are averages that were determined from experiments performed in triplicate.
| IFO |
d4IFO |
|||||
|---|---|---|---|---|---|---|
| Vmaxa | Kma | Vmax/Km | Vmaxa | Kma | Vmax/Km | |
| pmol/min/pmol P450 | mM | min–1mM–1 | pmol/min/pmol P450 | mM | min–1mM−1 | |
| Activation | ||||||
| CYP2B6 | 3.8 ± 1 | 4.6 ± 2 | 0.8 | 4.1 ± 0.4 | 2.0 ± 0.4 | 2 |
| CYP2C9 | Trace amounts of acrolein formed | 0 | Trace amounts of acrolein formed | 0 | ||
| CYP2C19 | 1.0 ± 0.2 | 1.1 ± 0.6 | 0.9 | 1.0 ± 0.1 | 1.2 ± 0.4 | 0.8 |
| CYP3A4 | 0.7 ± 0.1 | 1.6 ± 0.6 | 0.4 | 0.4 ± 0.03 | 0.6 ± 0.12 | 0.7 |
| CYP3A5 | 0.1 ± 0.01 | 0.04 ± 0.03 | 2.5 | Not tested | N/A | |
| Inactivation | ||||||
| CYP2B6 | 0.4 ± 0.04 | 2.0 ± 0.4 | 0.2 | 0.2 ± 0.05 | 2.0 ± 0.8 | 0.1 |
| CYP2C9 | Trace amounts of CAA formed | 0 | Trace amounts of CAA formed | 0 | ||
| CYP2C19 | No CAA formed | 0 | No CAA formed | 0 | ||
| CYP3A4 | 0.14 ± 0.03 | 3.5 ± 1.7 | 0.04 | 0.05 ± 0.02 | 1.7 ± 1.2 | 0.03 |
| CYP3A5 | 0.02 ± 0.005 | 0.4 ± 0.3 | 0.05 | Not tested | N/A | |
Mean ± S.E.M.
N/A, CYP3A5 was not assessed for d4IFO metabolism.
The amount of inactivation of IFO and d4IFO by the P450s was investigated in the same samples described above and evaluated for the production of the inactive metabolite CAA (Table 1). CYP2C9 and CYP2C19 did not catalyze the inactivation of either IFO or d4IFO. CYP2B6 was approximately 4-fold more efficient at the inactivation of IFO than any of the other P450s tested, and this held true for d4IFO as well (Table 1). Interestingly, the Vmax values for the inactivation of d4IFO by CYP2B6 and CYP3A4 were reduced compared with IFO, resulting in reduced efficiency for the inactivation of d4IFO compared with IFO for these P450s.
Metabolism of Ifosfamide and Deuterated Ifosfamide by the Polymorphic Variants of CYP2B6 and CYP2C9.
The polymorphic variants of CYP2B6 and CYP2C9 were examined for their ability to catalyze the activation of IFO and d4IFO. The polymorphic variants exhibited variable activation of IFO compared with the wild-type forms (Fig. 4A). CYP2B6*7 and CYP2B6*9 produced nearly 3-fold more acrolein than CYP2B6*1 at 1 mM IFO. However, the common polymorphic variants CYP2B6*4 and CYP2B6*6 produced approximately half of the acrolein that wild-type CYP2B6 produced. With regard to the activation of d4IFO, the polymorphic variants followed a trend similar to that observed for the activation of IFO but consistently produced more of the activated metabolite from d4IFO than from IFO (Table 2). For CYP2B6*1 and CYP2B6*7 this enhancement was statistically significant. The variants of CYP2C9 exhibited small variations in catalyzing IFO activation. As with the CYP2B6 variants, the activation of d4IFO by CYP2C9*2 and CYP2C9*3 followed the same pattern of variation as for IFO but with significantly higher levels of activated metabolite produced (Table 2).
Fig. 4.

Metabolism of IFO and d4IFO by the polymorphic variants of CYP2B6 and CYP2C9. IFO (blue) or d4IFO (red) (1 mM) was incubated in the reconstituted system with 1 μM P450 for 30 minutes at 37°C. Half of each sample was analyzed for acrolein to determine the rate of activation of IFO for each of the polymorphic variants (A). The other half of each sample was analyzed for CAA to determine the rate of inactivation of IFO for each of the polymorphic variants (B). The details of the analytical methods are provided in Materials and Methods. The average for each experimental condition that was performed in triplicate is shown. Statistical significance was determined using an unpaired t test in Prism 6 (GraphPad Software, Inc.). †P < 0.05, ††P < 0.01, †††P < 0.005 for comparisons within polymorphic variants; **P < 0.01, ***P < 0.005 for comparisons between IFO and d4IFO. The data are summarized in Table 2.
TABLE 2.
Metabolism of IFO and d4IFO by the polymorphic variants of CYP2B6 and CYP2C9
The data shown in Fig. 4 are summarized here. Data are expressed as metabolite formation in pmol/min per picomole of P450, and the metabolite production compared with wild-type metabolite production in terms of fold change. Also, the percent increase or decrease of the metabolite produced from d4IFO compared with IFO is given. The data were determined in triplicate. Statistical significance was determined using the unpaired t test in Prism 6 (GraphPad Inc.).
| IFO |
d4IFO |
Increase in Active Metabolite over IFO | |||
|---|---|---|---|---|---|
| Acrolein Produceda | Fold Change over Wild-Type | Acrolein Produceda | Fold Change over Wild Type | ||
| pmol/min/pmol P450 | pmol/min/pmol P450 | % | |||
| Activation | |||||
| CYP2B6*1 | 0.5 ± 0.06 | 1.0 | 1.1 ± 0.08* | 1.0 | 120.0 |
| CYP2B6*4 | 0.1 ± 0.02† | 0.2 | 0.2 ± 0.02†† | 0.2 | 100.0 |
| CYP2B6*6 | 0.3 ± 0.1 | 0.6 | 0.3 ± 0.08†† | 0.3 | 0.0 |
| CYP2B6*7 | 1.7 ± 0.05††† | 3.4 | 3.6 ± 0.04†††** | 3.3 | 111.8 |
| CYP2B6*9 | 1.6 ± 0.3††† | 3.2 | 2.5 ± 0.3†† | 2.3 | 56.3 |
| CYP2C9*1 | 0.05 ± 0.01 | 1.0 | 0.05 ± 0.02 | 1.0 | 0.0 |
| CYP2C9*2 | 0.2 ± 0.1 | 4.0 | 0.5 ± 0.05†† | 10.0 | 150.0 |
| CYP2C9*3 | 0 ± 0 | 0.0 | 0.2 ± 0.04 | 4.0 | N/A |
| IFO | d4IFO | Decrease in Inactive Metabolite over IFO | |||
| CAA Produceda | Fold Change over Wild Type | CAA Produceda | Fold Change over Wild Type | ||
| pmol/min/pmol P450 | pmol/min/pmol P450 | % | |||
| Inactivation | |||||
| CYP2B6*1 | 0.1 ± 0.02 | 1.0 | 0.06 ± 0.02 | 1.0 | 40.0 |
| CYP2B6*4 | 0.04 ± 0.001 | 0.4 | 0 ± 0* | 0.0 | 100.0 |
| CYP2B6*6 | 0.02 ± 0.01 | 0.2 | 0 ± 0 | 0.0 | 100.0 |
| CYP2B6*7 | 0 ± 0† | 0.0 | 0 ± 0 | 0.0 | N/A |
| CYP2B6*9 | 0.02 ± 0.01 | 0.2 | 0 ± 0 | 0.0 | 100.0 |
| CYP2C9*1 | 0.01 ± 0.003 | 1.0 | 0 ± 0 | 1.0 | 100.0 |
| CYP2C9*2 | 0 ± 0 | 0.0 | 0 ± 0 | 0.0 | N/A |
| CYP2C9*3 | 0 ± 0 | 0.0 | 0 ± 0 | 0.0 | N/A |
P < 0.05, ††P < 0.01, †††P, 0.005 for comparisons within polymorphic variants
P < 0.01, **P < 0.005 for comparisons between IFO and d4IFO
Mean ± S.E.M.
N/A no metabolite was detected with IFO, therefore percent change could not be calculated.
The inactivation of IFO and d4IFO by these polymorphic variants was also assessed using the same samples described above. All of the polymorphic variants of CYP2B6 tested produced less of the inactivated metabolite than wild-type CYP2B6 (Fig. 4B). In fact, the pathway leading to the inactivation of d4IFO by the CYP2B6 variants was essentially abolished. CYP2C9 catalyzed the inactivation IFO to a small extent, but its variants also failed to produce the inactive metabolites for both IFO and d4IFO (Table 2).
Metabolism of Ifosfamide by Human Liver Microsomes.
IFO activation by HLM was determined at concentrations of 1.0 and 10.0 mM IFO (Fig. 5A). These concentrations of IFO were chosen to ensure metabolism by the respective P450s on the basis of Km values shown in Table 1. Acrolein production increased with the increased concentration of IFO, as expected. The inhibition of CYP3A4 by ketoconazole reduced IFO activation by approximately 85% at both IFO concentrations. Inhibition of CYP2B6 by clopidogrel inhibited the activation of IFO by 67% and 47% at 1 mM and 10 mM IFO, respectively.
Fig. 5.

Inhibition of the metabolism of IFO by HLMs with the addition of P450 inhibitors. IFO (1.0 or 10 mM) was incubated with HLM (0.2 mg/ml). A short preincubation with 5 μM ketoconazole (red) or 5 μM clopidogrel (green) was used to inhibit CYP3A4 or CYP2B6, respectively. The total amount of acrolein formed was measured to determine the effect of the P450 inhibitors on IFO activation (A). Similarly, the total amount of CAA formed was measured to determine the effect of inhibitors of CYP3A4 or CYP2B6 on IFO inactivation (B). The analytical methods are described in Materials and Methods. The experiments were performed twice in triplicate and the statistical significance was determined using an unpaired t test in Prism 6 (GraphPad Software, Inc.). *P < 0.05, **P < 0.01.
IFO inactivation by HLM was also monitored. Incubation with ketoconazole resulted in a decrease of almost 90% in CAA production at both IFO concentrations (Fig. 5B). Inhibition of CYP2B6 by clopidogrel decreased the inactivation of IFO by approximately 40% at both concentrations.
Discussion
We have used the purified reconstituted system to assess the contributions to the hydroxylation and the N-dechloroethylation of IFO by CYP2B6, CYP2C9, CYP2C19, CYP3A4, CYP3A5, and several commonly occurring polymorphic variants of CYP2B6 and CYP2C9. We also investigated deuterium isotope effects on the metabolism of IFO. Michaelis-Menten kinetic values have been determined for all wild-type P450s in the reconstituted system, allowing for accurate comparisons between the efficiencies of the enzymes for the activation and inactivation of both IFO and d4IFO. This is the first time that CYP2C9, CYP2C19, and CYP3A5 have been evaluated for both reactions in this manner. Additionally, this is the first investigation of the metabolism of IFO by the polymorphic variants of CYP2B6 and CYP2C9.
The Km values for the CYP2B6- and CYP3A4-mediated activation of IFO are similar to those previously reported (Table1; Roy et al., 1999; Huang et al., 2000). In our hands, CYP2B6 yielded a larger Vmax than did CYP3A4, resulting in a higher efficiency for this reaction (Vmax/Km for CYP2B6 = 0.8 minutes–1mM−1 versus 0.4 minutes–1mM−1 for CYP3A4). This observation is in contrast with previous studies that analyzed the activation of IFO using Supersomes, where it was found that CYP3A4 was the most efficient enzyme for this reaction (Roy et al., 1999; Huang et al., 2000). This discrepancy is likely a result of the different expression systems used in the two studies and the potential effect of b5, which was present in previous studies but not in our experiments. During preliminary optimization experiments we examined the effect of b5 by adding it to the reconstitution mixture at a range of ratios (0.5:1.0:0.25 to 0.5:1.0:2.0, P450/CPR/b5). We observed little enhancement in the activation of IFO by any of the P450s, with the exception of CYP3A4, for which absolutely no change was detected (data not shown). Therefore, b5 was not included in our optimized studies. Of the P450s tested, CYP3A5 had the highest efficiency for the activation of IFO (2.5 minutes–1mM−1). Furthermore, in patients the plasma concentrations of IFO are well above our determined Km (0.04 mM and up to ∼1 mM) (Boddy et al., 1996; Highley et al., 1997). Given the low Km, CYP3A5 may be important inside tumors where IFO concentrations are considered lower than in plasma owing to the low cell membrane permeability of IFO. In addition, expression of CYP3A5 varies as much as 10% in certain races, which may implicate CYP3A5 as more relevant in populations with relatively high expression of CYP3A5 (Hustert et al., 2001). Also, CYP3A5 is more ubiquitously expressed than CYP3A4 and therefore may be more relevant for tissue-specific activation of IFO (Haehner et al., 1996; Wrighton et al., 2000; Aleksa et al., 2004).
Involvement of CYP2C19 in the metabolism of IFO may have been previously overlooked. Previous work in Supersomes expressing CYP2C19 observed a small amount of activation but did not determine the steady state kinetics of the enzyme (Huang et al., 2000). We determined CYP2C19 was the second-most efficient enzyme for IFO activation and did not catalyze the inactivation of IFO, rendering it an ideal P450 for the metabolism of IFO.
With respect to the inactivation of IFO, only CYP2B6, CYP3A4, and CYP3A5 are involved. The efficiency of inactivation of IFO by CYP3A4 and CYP3A5 was similar, supporting previous results (McCune et al., 2005). We found the inactivation of IFO by CYP2B6 to be approximately 4-fold more efficient than CYP3A4 and CYP3A5. The inclusion of b5 (at ratios in the range of 0.5:1:0.25 to 0.5:1:2, P450/CPR/b5) into the recombinant mixture somewhat enhanced the inactivation of IFO for most of the P450s, with the exception of CYP3A4, which showed no enhancement (data not shown), supporting previous observations that CYP3A5-mediated IFO inactivation was enhanced by the addition of purified b5, but that the CYP3A4-mediated inactivation of IFO was not (McCune et al., 2005).
Deuteration of IFO enhanced the activation of IFO by CYP2B6 and CYP3A4. This is probably attributable to the inhibition of N-dechloroethylation caused by the presence of deuterium on the chloroethyl groups of IFO (Figs. 1 and 2). This is supported by the determined Vmax values for the inactivation of d4IFO, which were lower than the Vmax values for the inactivation of IFO for all of the P450s tested. An overall enhancement in the metabolism of IFO is best observed with CYP2B6, whose activation of d4IFO (Vmax/Km = 2 minutes–1mM−1) was ∼2.5-fold more efficient than IFO (Vmax/Km = 0.8 minutes–1mM−1), and the inactivation of d4IFO (Vmax/Km = 0.1 minutes–1mM−1) was approximately 50% less efficient than for IFO (Vmax/Km = 0.2 minutes–1mM−1). Thus, for the CYP2B6-mediated metabolism of d4IFO we observe an overall 5-fold change in the ratio of activation to inactivation that results in a significant enhancement of IFO metabolism. Similar changes were observed with CYP3A4 whose activation of d4IFO (Vmax/Km = 0.7 minutes–1mM−1) was ∼2-fold more efficient than IFO (Vmax/Km = 0.4 minutes–1mM−1), and the inactivation of d4IFO (Vmax/Km = 0.03 minutes–1mM−1) was approximately 25% less efficient than for IFO (Vmax/Km = 0.04 minutes–1mM−1). Thus, a >2-fold change in the ratio of activation to inactivation of IFO by CYP3A4 was observed. The deuterium isotope effects reported may be a viable tool to enhance the therapeutic efficacy and reduce the adverse effects of IFO. Metabolization of IFO to the inactivated and toxic metabolites dechloroethylifosfamide and chloroacetaldehyde was 25–60%. Utilizing d4IFO may inhibit the formation of the toxic metabolites (Bruggemann et al., 1997; Carlson et al., 1998; Borner et al., 2000). Additional studies with d4IFO to address the effect of the deuterium isotopes on the DNA alkylating properties of the phosphoramide mustard of IFO merit further investigation.
The polymorphic variants of CYP2B6 and CYP2C9 exhibit altered catalytic activities for both the activation and inactivation of IFO and d4IFO compared with the wild-type P450s. Lamba et al. (2003) identified differences between genders and ethnic groups in CYP2B6 activity with 3.6- and 5.0-fold higher levels in Hispanic females than in Caucasian or African-American females. The polymorphic variants CYP2B6*4 (K262R) and CYP2B6*6 (Q172H/K262R) had reduced activation and inactivation of IFO compared with CYP2B6*1. Other polymorphic variants, CYP2B6*7 (Q172H/K262R/R487C) and CYP2B6*9 (Q172H), produced more of the activated metabolite of IFO and less of the inactivated metabolite of IFO than did the wild-type enzyme. This is interesting in light of studies performed with the structural analog CPA, where Ariyoshi et al. (2011) demonstrated that CYP2B6*4 had reduced activation and conversely CYP2B6*6 had enhanced activation of CPA. The authors concluded that the Q172R variant is capable of reinstating the activity that was lost by the introduction of the K262R mutation. With IFO this was not the case, but the addition of a third single-nucleotide polymorphism (R487C in CYP2B6*7) did fully reverse the effects of the first polymorphisms. This is the first evidence that a third point mutation in CYP2B6 can reverse the direction of the effect and restore activity that was lost by the Q172H and K262R mutations. The polymorphic variant CYP2B6*8 was also investigated and determined to be catalytically inactive with IFO, as expected (data not shown) (Zhang et al., 2011). The polymorphic variants for CYP2C9 exhibited relatively small variations in catalytic efficiency compared with CYP2C9*1.
In following with our observations of the wild-type enzymes, the activation of d4IFO was improved and its inactivation was reduced or completely abolished compared with IFO for each of the polymorphic variants. At the time of this publication, two deuterated drugs (Auspex Pharmaceuticals’ SD-809 and Concert Pharmaceuticals’ CTP-499) were in clinical trials and moving toward FDA approval. This is encouraging and supports the development of d4IFO as a potential therapeutic, as the incorporation of deuterium does not appear to introduce any unexpected perturbations during metabolism by the wild-type P450s as well as the polymorphic variants tested.
To determine the ability to favor activation over inactivation in cell culture, we measured IFO metabolism by pooled HLMs and used several specific P450 inhibitors. The inhibition of CYP2B6 in HLMs by clopidogrel inhibited both the activation and inactivation of IFO by ∼40–70%. This indicates a greater contribution of CYP2B6 to the metabolism of IFO than was previously reported by CYP2B6 inhibition using the monoclonal antibody inhibitor Mab2B6. Under those conditions, only an ∼20% loss of activation and a 40% loss of inactivation were observed (Huang et al., 2000). With CYP2B6-genotyped HLMs, the inactivation observed with CYP2B6 inhibition was highly variable (30–90%) (McCune et al., 2005). The large variations in percentage inhibition between these experiments probably result from differences in the expression of P450s in the HLM samples and from the polymorphic nature of the P450s. Similar results were obtained with CYP3A4 inhibition, where we observed significant inhibition of both activation and inactivation of IFO (∼90% for both reactions). Studies focusing only on the inactivation of IFO reported similar results with ketoconazole treatment, whereas others only observed ∼50% loss of both activation and inactivation in pooled HLM inhibited with the CYP3A4 inhibitor troleandomycin (Huang et al., 2000; McCune et al., 2005). Typically CYP3A4 comprises 30–50% of all hepatic P450 enzymes, which may explain the ability of CYP3A4 to significantly contribute to the inactivation of IFO in HLM in spite of its lower catalytic efficiency for this reaction (Shimada et al., 1994). Thus, we were unable to easily separate the activation and inactivation pathways with our experimental setup.
In summary, we determined that the P450s CYP3A5 and CYP2C19 play significant roles in the metabolism of IFO. Furthermore, the polymorphic variants of CYP2B6 and CYP2C9 exhibited significant changes in IFO metabolism, indicating that interpatient metabolite levels may be greatly altered by the polymorphic nature of these enzymes. Our studies with d4IFO indicate that deuteration of IFO results in a significantly enhanced activation of IFO over inactivation for all of the P450s tested and their respective polymorphic variants, suggesting that deuterium labeling of IFO can induce metabolic switching and may be of significant value in improving the therapeutic index of this widely used chemotherapeutic drug.
Acknowledgments
The authors thank Dr. James Halpert (UCSD) for his contribution of purified CYP3A4, as well as Dr. Tim Tracy (University of Kentucky) for the gift of purified CYP2C9*2 and CYP2C9*3.
Abbreviations
- CAA
chloroacetaldehyde
- CPA
cyclophosphamide
- CPR
cytochrome P450 reductase
- d4IFO
deuterated ifosfamide
- HLM
human liver microsomes
- HPLC
high-performance liquid chromatography
- IFO
ifosfamide
- P450
cytochrome P450
Authorship Contributions
Participated in research design: Calinski, Ludeman, Dolan, Zhang, Hollenberg.
Conducted experiments: Calinski.
Wrote or contributed to the writing of the manuscript: Calinski, Zhang, Hollenberg.
Footnotes
This work was supported in part by the National Institutes of Health National Cancer Institute [Grant CA16954].
References
- Aleksa K, Ito S, Koren G. (2004) Renal-tubule metabolism of ifosfamide to the nephrotoxic chloroacetaldehyde: pharmacokinetic modeling for estimation of intracellular levels. J Lab Clin Med 143:159–162. [DOI] [PubMed] [Google Scholar]
- Ariyoshi N, Ohara M, Kaneko M, Afuso S, Kumamoto T, Nakamura H, Ishii I, Ishikawa T, Kitada M. (2011) Q172H replacement overcomes effects on the metabolism of cyclophosphamide and efavirenz caused by CYP2B6 variant with Arg262. Drug Metab Dispos 39:2045–2048. [DOI] [PubMed] [Google Scholar]
- Biagi JJ, Herbert KE, Smith C, Abdi E, Leahy M, Falkson C, Wolf M, Januszewicz H, Seymour JF, Richards K, et al. (2005) A phase II study of dexamethasone, ifosfamide, cisplatin and etoposide (DICE) as salvage chemotherapy for patients with relapsed and refractory lymphoma. Leuk Lymphoma 46:197–206. [DOI] [PubMed] [Google Scholar]
- Boddy AV, Yule SM, Wyllie R, Price L, Pearson AD, Idle JR. (1996) Intrasubject variation in children of ifosfamide pharmacokinetics and metabolism during repeated administration. Cancer Chemother Pharmacol 38:147–154. [DOI] [PubMed] [Google Scholar]
- Börner K, Kisro J, Brüggemann SK, Hagenah W, Peters SO, Wagner T. (2000) Metabolism of ifosfamide to chloroacetaldehyde contributes to antitumor activity in vivo. Drug Metab Dispos 28:573–576. [PubMed] [Google Scholar]
- Brüggemann SK, Kisro J, Wagner T. (1997) Ifosfamide cytotoxicity on human tumor and renal cells: role of chloroacetaldehyde in comparison to 4-hydroxyifosfamide. Cancer Res 57:2676–2680. [PubMed] [Google Scholar]
- Buda A, Dell’Anna T, Signorelli M, Mangioni C. (2003) Role of ifosfamide in cervical cancer: an overview. Oncology 65 (Suppl 2):63–66. [DOI] [PubMed] [Google Scholar]
- Carlson L, Goren MP, Bush DA, Griener JC, Quigley R, Tkaczewski I, Kamen BA, Weitman SD. (1998) Toxicity, pharmacokinetics, and in vitro hemodialysis clearance of ifosfamide and metabolites in an anephric pediatric patient with Wilms’ tumor. Cancer Chemother Pharmacol 41:140–146. [DOI] [PubMed] [Google Scholar]
- Chang TK, Weber GF, Crespi CL, Waxman DJ. (1993) Differential activation of cyclophosphamide and ifosphamide by cytochromes P-450 2B and 3A in human liver microsomes. Cancer Res 53:5629–5637. [PubMed] [Google Scholar]
- Chen CS, Lin JT, Goss KA, He YA, Halpert JR, Waxman DJ. (2004) Activation of the anticancer prodrugs cyclophosphamide and ifosfamide: identification of cytochrome P450 2B enzymes and site-specific mutants with improved enzyme kinetics. Mol Pharmacol 65:1278–1285. [DOI] [PubMed] [Google Scholar]
- Domanski TL, Finta C, Halpert JR, Zaphiropoulos PG. (2001) cDNA cloning and initial characterization of CYP3A43, a novel human cytochrome P450. Mol Pharmacol 59:386–392. [DOI] [PubMed] [Google Scholar]
- Donfrancesco A, Jenkner A, Castellano A, Ilari I, Milano GM, De Sio L, Cozza R, Fidani P, Deb G, De Laurentis C, et al. (2004) Ifosfamide/carboplatin/etoposide (ICE) as front-line, topotecan/cyclophosphamide as second-line and oral temozolomide as third-line treatment for advanced neuroblastoma over one year of age. Acta Paediatr Suppl 93:6–11. [DOI] [PubMed] [Google Scholar]
- Dubourg L, Michoudet C, Cochat P, Baverel G. (2001) Human kidney tubules detoxify chloroacetaldehyde, a presumed nephrotoxic metabolite of ifosfamide. J Am Soc Nephrol 12:1615–1623. [DOI] [PubMed] [Google Scholar]
- Haehner BD, Gorski JC, Vandenbranden M, Wrighton SA, Janardan SK, Watkins PB, Hall SD. (1996) Bimodal distribution of renal cytochrome P450 3A activity in humans. Mol Pharmacol 50:52–59. [PubMed] [Google Scholar]
- Hanna IH, Roberts ES, Hollenberg PF. (1998) Molecular basis for the differences in lidocaine binding and regioselectivity of oxidation by cytochromes P450 2B1 and 2B2. Biochemistry 37:311–318. [DOI] [PubMed] [Google Scholar]
- Highley MS, Schrijvers D, Van Oosterom AT, Harper PG, Momerency G, Van Cauwenberghe K, Maes RA, De Bruijn EA, Edelstein MB. (1997) Activated oxazaphosphorines are transported predominantly by erythrocytes. Ann Oncol 8:1139–1144. [DOI] [PubMed] [Google Scholar]
- Huang Z, Roy P, Waxman DJ. (2000) Role of human liver microsomal CYP3A4 and CYP2B6 in catalyzing N-dechloroethylation of cyclophosphamide and ifosfamide. Biochem Pharmacol 59:961–972. [DOI] [PubMed] [Google Scholar]
- Hummel MA, Locuson CW, Gannett PM, Rock DA, Mosher CM, Rettie AE, Tracy TS. (2005) CYP2C9 genotype-dependent effects on in vitro drug-drug interactions: switching of benzbromarone effect from inhibition to activation in the CYP2C9.3 variant. Mol Pharmacol 68:644–651. [DOI] [PubMed] [Google Scholar]
- Hustert E, Haberl M, Burk O, Wolbold R, He YQ, Klein K, Nuessler AC, Neuhaus P, Klattig J, Eiselt R, et al. (2001) The genetic determinants of the CYP3A5 polymorphism. Pharmacogenetics 11:773–779. [DOI] [PubMed] [Google Scholar]
- Jenkins CM, Pikuleva I, Waterman MR. (1998) Expression of eukaryotic cytochromes P450 in E. coli. Methods Mol Biol 107:181–193. [DOI] [PubMed] [Google Scholar]
- Klastersky J. (2003) Side effects of ifosfamide. Oncology 65 (Suppl 2):7–10. [DOI] [PubMed] [Google Scholar]
- Koch I, Weil R, Wolbold R, Brockmöller J, Hustert E, Burk O, Nuessler A, Neuhaus P, Eichelbaum M, Zanger U, et al. (2002) Interindividual variability and tissue-specificity in the expression of cytochrome P450 3A mRNA. Drug Metab Dispos 30:1108–1114. [DOI] [PubMed] [Google Scholar]
- Kosmas C, Tsavaris N, Mylonakis N, Tsakonas G, Gassiamis A, Skopelitis H, Polyzos A, Malamos N, Karabelis A. (2007) Docetaxel-ifosfamide combination in patients with advanced breast cancer failing prior anthracycline-based regimens: results of a phase I-II study. J Chemother 19:322–331. [DOI] [PubMed] [Google Scholar]
- Kurowski V, Wagner T. (1993) Comparative pharmacokinetics of ifosfamide, 4-hydroxyifosfamide, chloroacetaldehyde, and 2- and 3-dechloroethylifosfamide in patients on fractionated intravenous ifosfamide therapy. Cancer Chemother Pharmacol 33:36–42. [DOI] [PubMed] [Google Scholar]
- Lamba V, Lamba J, Yasuda K, Strom S, Davila J, Hancock ML, Fackenthal JD, Rogan PK, Ring B, Wrighton SA, et al. (2003) Hepatic CYP2B6 expression: gender and ethnic differences and relationship to CYP2B6 genotype and CAR (constitutive androstane receptor) expression. J Pharmacol Exp Ther 307:906–922. [DOI] [PubMed] [Google Scholar]
- Loebstein R, Atanackovic G, Bishai R, Wolpin J, Khattak S, Hashemi G, Gobrial M, Baruchel S, Ito S, Koren G. (1999) Risk factors for long-term outcome of ifosfamide-induced nephrotoxicity in children. J Clin Pharmacol 39:454–461. [PubMed] [Google Scholar]
- Ludeman SM, D’Alessandro MA, Huang RS, Spasojevic I, and Dolan ME (2012) The use of deuterium isotope effects to shift the partitioning of cyclophosphamide and ifosfamide among competing, P450-dependent, oxidative pathways, in Proceedings American Association for Cancer Research 103rd Annual Meeting, 2012 Mar 31–Apr 4; Chicago, IL. American Association for Cancer Research, Philadelphia, PA. [Google Scholar]
- McCune JS, Risler LJ, Phillips BR, Thummel KE, Blough D, Shen DD. (2005) Contribution of CYP3A5 to hepatic and renal ifosfamide N-dechloroethylation. Drug Metab Dispos 33:1074–1081. [DOI] [PubMed] [Google Scholar]
- Mohrmann M, Pauli A, Walkenhorst H, Schönfeld B, Brandis M. (1993) Effect of ifosfamide metabolites on sodium-dependent phosphate transport in a model of proximal tubular cells (LLC-PK1) in culture. Ren Physiol Biochem 16:285–298. [DOI] [PubMed] [Google Scholar]
- Pocali B, De Simone M, Annunziata M, Palmieri S, D’Amico MR, Copia C, Viola A, Mele G, Schiavone EM, Ferrara F. (2004) Ifosfamide, epirubicin and etoposide (IEV) regimen as salvage and mobilization therapy for refractory or early relapsing patients with aggressive non-Hodgkin’s lymphoma. Leuk Lymphoma 45:1605–1609. [DOI] [PubMed] [Google Scholar]
- Roy P, Yu LJ, Crespi CL, Waxman DJ. (1999) Development of a substrate-activity based approach to identify the major human liver P-450 catalysts of cyclophosphamide and ifosfamide activation based on cDNA-expressed activities and liver microsomal P-450 profiles. Drug Metab Dispos 27:655–666. [PubMed] [Google Scholar]
- Scott EE, Spatzenegger M, Halpert JR. (2001) A truncation of 2B subfamily cytochromes P450 yields increased expression levels, increased solubility, and decreased aggregation while retaining function. Arch Biochem Biophys 395:57–68. [DOI] [PubMed] [Google Scholar]
- Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. (1994) Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther 270:414–423. [PubMed] [Google Scholar]
- Sladek NE. (1988) Metabolism of oxazaphosphorines. Pharmacol Ther 37:301–355. [DOI] [PubMed] [Google Scholar]
- Sorio R, Lombardi D, Spazzapan S, La Mura N, Tabaro G, Veronesi A. (2003) Ifosfamide in advanced/disseminated breast cancer. Oncology 65 (Suppl 2):55–58. [DOI] [PubMed] [Google Scholar]
- Springate J, Chan K, Lu H, Davies S, Taub M. (1999) Toxicity of ifosfamide and its metabolite chloroacetaldehyde in cultured renal tubule cells. In Vitro Cell Dev Biol Anim 35:314–317. [DOI] [PubMed] [Google Scholar]
- Springer JB, Colvin OM, Ludeman SM. (2007) Labeled oxazaphosphorines for applications in MS studies. Synthesis of deuterium labeled cyclophosamides and ifosfamides. J Labelled Compounds and Radiopharmaceuticals 50:115–122. [DOI] [PubMed] [Google Scholar]
- Weber GF, Waxman DJ. (1993) Activation of the anti-cancer drug ifosphamide by rat liver microsomal P450 enzymes. Biochem Pharmacol 45:1685–1694. [DOI] [PubMed] [Google Scholar]
- Woodland C, Ito S, Granvil CP, Wainer IW, Klein J, Koren G. (2000) Evidence of renal metabolism of ifosfamide to nephrotoxic metabolites. Life Sci 68:109–117. [DOI] [PubMed] [Google Scholar]
- Wrighton SA, Schuetz EG, Thummel KE, Shen DD, Korzekwa KR, Watkins PB. (2000) The human CYP3A subfamily: practical considerations. Drug Metab Rev 32:339–361. [DOI] [PubMed] [Google Scholar]
- Yu L, Waxman DJ. (1996) Role of cytochrome P450 in oxazaphosphorine metabolism. Deactivation via N-dechloroethylation and activation via 4-hydroxylation catalyzed by distinct subsets of rat liver cytochromes P450. Drug Metab Dispos 24:1254–1262. [PubMed] [Google Scholar]
- Zhang H, Sridar C, Kenaan C, Amunugama H, Ballou DP, Hollenberg PF. (2011) Polymorphic variants of cytochrome P450 2B6 (CYP2B6.4-CYP2B6.9) exhibit altered rates of metabolism for bupropion and efavirenz: a charge-reversal mutation in the K139E variant (CYP2B6.8) impairs formation of a functional cytochrome p450-reductase complex. J Pharmacol Exp Ther 338:803–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Tian Q, Zhou SF. (2006) Clinical pharmacology of cyclophosphamide and ifosfamide. Curr Drug Therapy 1:55–84. [Google Scholar]