

ABSTRACT
Virus-like particles (VLPs) built on the Newcastle disease virus (NDV) core proteins, NP and M, and containing two chimeric proteins, F/F and H/G, composed of respiratory syncytial virus (RSV) fusion protein (F) and glycoprotein (G) ectodomains fused to the transmembrane and cytoplasmic domains of the NDV F and HN proteins, respectively, stimulate durable, protective RSV neutralizing antibodies in mice. Here, we report the properties of VLPs constructed to contain mutant RSV F protein ectodomains stabilized in prefusion (pre-F/F) or postfusion (post-F/F) configurations. The structures of the chimeric proteins assembled into VLPs were verified immunologically by their reactivities with a conformationally restricted anti-F protein monoclonal antibody. Following immunization of mice, without adjuvant, pre-F/F-containing VLPs induced significantly higher neutralizing antibody titers than the post-F/F-containing VLPs or the wild-type F/F-containing VLPs after a single immunization but not after prime and boost immunization. The specificities of anti-F IgG induced by the two mutant VLPs were assessed by enzyme-linked immunosorbent assay (ELISA) using soluble forms of the prefusion and postfusion forms of the F protein as targets. While both types of VLPs stimulated similar levels of IgG specific for the soluble postfusion F protein, titers of IgG specific for prefusion F induced by the pre-F/F-containing VLPs were higher than those induced by post-F/F-containing VLPs. Thus, VLPs containing a stabilized prefusion form of the RSV F protein represent a promising RSV vaccine candidate.
IMPORTANCE The development of vaccines for respiratory syncytial virus has been hampered by a lack of understanding of the requirements for eliciting high titers of neutralizing antibodies. The results of this study suggest that particle-associated RSV F protein containing mutations that stabilize the structure in a prefusion conformation may stimulate higher titers of protective antibodies than particles containing F protein in a wild-type or postfusion conformation. These findings indicate that the prefusion F protein assembled into VLPs has the potential to produce a successful RSV vaccine candidate.
INTRODUCTION
Human respiratory syncytial virus (RSV) is the most significant cause of acute viral respiratory disease in infants and young children (1). There are from 34 to 65 million RSV infections resulting in acute lower respiratory disease requiring hospitalization and 160,000 to 199,000 deaths per year worldwide (2). Elderly populations are also at significant risk for serious RSV disease. In the United States, the virus accounts for 10,000 deaths and 14,000 to 60,000 hospitalizations per year among individuals more than 64 years of age (3–5). Indeed, RSV infection of this population is at least as significant as influenza virus infections. RSV infections result in high mortality rates in immunocompromised populations, particularly stem cell transplant recipients (6) and individuals with cardiopulmonary diseases (7). Despite the significance of RSV disease in different populations, there are no vaccines available.
Failure to develop a licensed RSV vaccine is not due to lack of effort as numerous vaccine candidates have been characterized in preclinical and clinical studies spanning 5 decades (summarized in references 8 to 9). While many problems have uniquely hindered RSV vaccine development, a major hurdle has been a lack of understanding of requirements for generation of protective immunity to RSV infection. Many vaccine candidates are protective in animal models and, while stimulating antibody responses in humans, have failed to induce high levels of neutralizing antibodies and protection from virus challenge in human trials (reviewed in references 10 and 11). Although there are likely many reasons for these observations, one important issue has been a lack of clear understanding of the most effective form of the RSV antigens, particularly the F protein, for stimulating potent neutralizing antibodies.
The paramyxovirus F protein is folded into a metastable conformation and upon fusion activation refolds, through a series of conformational intermediates, into the postfusion conformation, which is structurally very different from the prefusion form (12–19). While it is logical to assume that the prefusion form of F protein should be more effective in stimulating optimally neutralizing antibodies, recent structural studies have shown that the postfusion form of the F protein contains at least some epitopes recognized by neutralizing monoclonal antibodies (17, 18). Thus, it has been argued that a postfusion F protein will stimulate protection (20), and this form of F protein is now in clinical trials. In contrast, Magro et al. reported that a significant proportion of neutralizing antibodies in human or rabbit anti-RSV immune serum do not bind to the postfusion F protein (21). These authors have suggested that the majority of effective neutralizing antibody binding sites reside on the prefusion F protein and not on the postfusion form. Extending these studies, McLellan and colleagues solved the crystal structure of a prefusion form of the RSV F protein ectodomain and demonstrated that this form of the protein contained an antigenic site ϕ not present on the postfusion form of the protein (19). Furthermore, monoclonal antibodies specific for this site neutralized RSV at significantly lower concentrations than antibodies specific for sites present on both the pre- and postfusion forms of the protein. In a groundbreaking study, McLellan et al. (22) identified mutations in the F protein ectodomain that stabilized the prefusion form of the protein and reported that soluble forms of stabilized prefusion F protein, in the presence of adjuvant, stimulated significantly higher neutralizing antibody titers in both mice and nonhuman primates than postfusion forms.
Virus-like particles (VLPs) are increasingly recognized as safe, effective vaccines for viral diseases (23). VLPs are virus-sized particles composed of repeating structures on their surfaces and in their cores, structures that mimic those of infectious viruses and contribute to their very potent immunogenicity (23–25). VLPs are formed by the assembly of viral structural proteins and sometimes lipids into particles but without the incorporation of the viral genome. Thus, VLPs are incapable of multiple rounds of infection typical of an infectious virus. The surface glycoproteins of enveloped viruses are folded and inserted into VLP membranes typical of a virus; thus, antigenic sites are retained, and no inactivation is required.
We have recently described a novel, enveloped RSV virus-like particle that stimulates protective immune responses in mice (26, 27). These VLPs were formed with the structural core proteins, nucleocapsid protein (NP) and matrix (M) protein, of Newcastle disease virus (NDV) and the ectodomains of the RSV F and G proteins fused to the transmembrane (TM) and cytoplasmic tail (CT) sequences of the NDV fusion (F) and hemagglutinin-neuraminidase (HN) proteins, respectively. These VLPs are highly immunogenic and stimulated both anti-F protein- and anti-G protein-specific antibodies in the absence of adjuvant (26, 27). We have also reported that immunization of mice with three different versions of these VLPs did not stimulate enhanced respiratory disease upon RSV challenge (26–28) even at late times after immunization, in contrast to some other nonreplicating RSV vaccine candidates (29, 30).
Here, we report the assembly into these VLPs of the ectodomain of the RSV F protein containing mutations reported to stabilize the prefusion or the postfusion form of this protein. We compared in mice and without adjuvant the stimulation of neutralizing antibody responses to the two forms of VLP-associated RSV F proteins. Our results show that the VLP-associated prefusion F protein stimulated significantly higher titers of neutralizing antibodies than the VLP-associated postfusion F protein after a single immunization.
MATERIALS AND METHODS
Cells, virus, and plasmids.
ELL-0 cells (avian fibroblasts), Vero cells, COS-7 cells, and Hep2 cells were obtained from the American Type Culture Collection. ELL-0 cells, Vero cells, COS-7 cells, and Hep2 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with penicillin, streptomycin (Pen-Strep), and 5% (Vero cells) or 10% fetal calf serum. RSV, A2 strain, was obtained from Robert Finberg.
The cDNAs encoding the Newcastle disease virus (NDV) NP and M protein have been previously described (31). The construction, expression, and incorporation of the chimeric protein NDV HN/RSV G (H/G) into VLPs (yielding VLP-H/G constructs) have been previously described (26). The “wild-type” RSV F/NDV F (F/F) chimeric protein was constructed from a Gallus codon-optimized RSV F protein sequence (GeneWiz). The sequence encoding the ectodomain of the RSV F protein (amino acids 1 to 524) was fused to sequences encoding the NDV F protein transmembrane (TM) and cytoplasmic (CT) domains (amino acids 501 to 553) as previously described (27). Using a Stratagene QuikChange II kit, four point mutations were introduced into the RSV sequence (S155C, S190F, V207L, and S290C) to produce the DS-Cav1 F/F mutant.
To generate the pre-F/F chimeric protein (DS-Cav1-foldon-F/F), the RSV F protein sequence encoding amino acids 1 to 520 was fused to a Gallus codon-optimized sequence encoding the T4 fibritin trimerization motif (foldon) (22, 32). RSV sequence encoding amino acids 521 to 524 (the membrane-proximal serine-threonine-rich region common to paramyxovirus F proteins) was fused to the sequences encoding the NDV TM and CT domains and ligated to the RSV F-foldon sequences.
To generate the post-F/F chimeric protein, sequences encoding amino acids 137 to 146 were deleted from F/F using a Stratagene QuikChange II kit.
The soluble pre-F was constructed using the sequences encoding the RSV F ectodomain (amino acids 1 to 513) containing the DS-Cav1 mutations fused to sequences (synthesized by GenWiz) encoding the foldon, the thrombin cleavage site, Strep-Tag II, GSGSG linker, and six copies of a His (6×His) tag (22). The soluble postfusion F was constructed using sequences encoding the RSV F protein ectodomain with the deletion of amino acids 137 to 146 fused to sequences encoding the thrombin cleavage site, Strep-Tag II, GSGSG linker, and 6×His tag (22).
Polyacrylamide gel electrophoresis, silver staining, and Western analysis.
Proteins in extracts, virus, or VLPs were resolved on 8% bis-Tris gels (NuPage; Invitrogen). Silver staining of proteins in the polyacrylamide gels was accomplished as recommended by the manufacturer (Pierce). Quantification of NP, M, different forms of F/F, and H/G proteins in the polyacrylamide gels was accomplished by Western blotting of the proteins as well as protein standards as previously described (27, 33). For Western analysis, proteins in the polyacrylamide gels were transferred to polyvinylidene difluoride (PVDF) membranes using dry transfer (iblot; Invitrogen). Proteins were detected in the blots using antibodies noted on the figures and in the legends.
Antibodies.
RSV F monoclonal antibody (MAb) clone 131-2A (Chemicon) was used in RSV plaque assays. Monoclonal antibody 1112, MAb 1200, MAb 1269, and MAb 1243, generous gifts of J. Beeler (34), and MAb 5C4, a generous gift of B. Graham (19), were used for fluorescence-activated cell sorting (FACS) analysis of transfected cells and enzyme-linked immunosorbent assays (ELISAs) of VLPs and soluble F proteins. Anti-RSV F protein HR2 antibody is a polyclonal antibody specific to the HR2 domain of the RSV F protein (27). Secondary antibodies against goat, mouse, and rabbit IgG were purchased from Sigma.
Detection of cell surface-expressed chimeric F proteins.
Biotinylation of surface-expressed F chimeric proteins was accomplished as previously described (35, 36). Cell surface biotinylated molecules were precipitated from cell lysates using NeutrAvidin-agarose (35, 36), and precipitated F proteins were detected by Western analysis using anti-RSV F HR2 antibody.
Detection of antibody binding to surface-expressed chimeric proteins was accomplished by flow cytometry. Avian cells transfected with pCAGGS, pCAGGS-F/F, pCAGGS-DS-Cav1 F/F, pCAGGS–pre-F/F, or pCAGGS–post-F/F were washed in phosphate-buffered saline (PBS), removed from plates with cell dissociation buffer (Sigma), resuspended, and washed in FACS buffer (Hanks balanced salt solution; Gibco). Cells were then incubated with mouse monoclonal antibody for 30 min on ice, washed twice in FACS buffer, and incubated with goat anti-mouse IgG coupled to Alexa Fluor 488 for 30 min on ice in the dark. After three washes in FACS buffer, cells were resuspended in FACS buffer containing propidium iodide (PI; 10 μg/ml). Flow cytometry was accomplished with a MACSQuant Analyzer flow cytometer (Miltenyi Biotec), and data were analyzed using FlowJo software gating on PI-negative, fluorescein isothiocyanate (FITC)-positive cells. Geometric mean fluorescence intensity (MFI) of these cells was determined using FlowJo software.
VLP preparation, purification, and characterization.
For preparations of VLPs to be used in ELISAs or as immunogens (VLP-H/G+pre-F/F and VLP-H/G+post-F/F), ELL-0 cells growing in T-150 flasks were transfected with cDNAs encoding the NDV M protein, NP, the chimeric protein H/G, and either pre-F/F or post-F/F as previously described (26, 27). At 24 h posttransfection, heparin was added to the cells at a final concentration of 10 μg/ml (26) to inhibit rebinding of released VLPs to cells. At 48, 72, and 96 h posttransfection, cell supernatants were collected, and VLPs were purified by sequential pelleting and sucrose gradient fractionation as previously described (26, 27, 33). Concentrations of proteins in the purified VLPs were determined by silver-stained polyacrylamide gels and by Western analysis using marker proteins for standard curves (27, 33).
Preparation of soluble F proteins.
COS-7 cells transfected with pCAGGS vector or with pCAGGS vector containing sequences encoding the soluble pre-F protein or the soluble post-F protein were grown in DMEM-high glucose, with glutamine, Na pyruvate, Pen-Strep, and insulin, transferrin, and sodium selenite (ITS). At 48 h posttransfection, total cell supernatants were collected, and cell debris was removed by centrifugation. Amounts of soluble F protein in the cell supernatants were determined by Western blotting using anti-HR2 antibodies for detection. Volumes of supernatant containing equivalent amounts of F protein were used to coat microtiter wells. Similar volumes of supernatant from vector-only-transfected cells were used to coat a third set of microtiter wells to provide an indication of nonspecific binding to host cell soluble proteins. F proteins were not purified from the cell supernatants in order to avoid possible alteration of the conformation of the proteins during the purification.
Quantification of soluble F protein and VLP-associated F protein.
Determinations of amounts of F protein in VLPs or in soluble F protein preparations were accomplished by Western blotting using anti-HR2 antibody for detection and comparing the signals obtained with a standard curve of purified F proteins, as previously described (33).
Preparation of RSV, RSV plaque assays, and antibody neutralization.
RSV was grown in Hep2 cells (26, 27), and RSV plaque assays were accomplished on Vero cells as previously described (26, 27).
Antibody neutralization assays in a plaque reduction assay have been previously described (26, 27). Neutralization titer was defined as the dilution of serum that reduced the virus titer by 50%.
Animals, animal immunization, and RSV challenge.
Four-week-old female BALB/c mice from Jackson Laboratories or Taconic laboratories were housed (groups of five) under pathogen-free conditions in microisolator cages at the University of Massachusetts Medical Center animal quarters. Protocols requiring open cages were accomplished in biosafety cabinets. BALB/c mice were immunized by intramuscular (i.m.) inoculation of 30 μg of total VLP protein in 0.05 ml of TNE buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA) containing 10% sucrose. For infection or challenges of mice with RSV, the animals were lightly anesthetized with isoflurane and then infected by intranasal (i.n.) inoculation of RSV (titers are indicated in the figure legends). All animal procedures and infections were performed in accordance with the University of Massachusetts Medical School IACUC- and IBC-approved protocols.
Detection of virus in lung tissue.
Four days after RSV challenge (37), mice were sacrificed by CO2 asphyxiation. Lungs were removed aseptically, and each lobe was placed separately in 0.5 ml of 30% sucrose in PBS and frozen on dry ice. Lungs were stored at −80°C. Upon thawing, lungs were weighed and then homogenized in the storage buffer using a Dounce homogenizer (Kontes). The homogenate was centrifuged at 12,000 rpm for 15 min, and the virus titer in the supernatant was determined by plaque assay as described previously (26, 27).
ELISA protocols.
For determination of anti-F protein serum antibody titers, blood was obtained from immunized animals by tail vein nicks and centrifuged in BD Microtainer serum separator tubes to remove blood cells. For ELISAs, wells of microtiter plates (Costar) were coated with cell supernatant containing either soluble prefusion F protein or soluble postfusion F protein. Volumes containing equivalent amounts of the two forms of soluble F protein were determined by reactivity to anti-HR2 antibody measured in Western blots. Negative controls were equivalent volumes of supernatants from cells transfected with empty vector.
For antibody binding to VLPs, microtiter wells were coated with anti-RSV antisera in 50 μl of PBS for 24 to 30 h at 4°C. Wells were then incubated in 100 μl of PBS–1% bovine serum albumin (BSA) for 16 h at 4°C. Equivalent amounts of pre-F/F- or post-F/F-containing VLPs were then added to the wells. Alternatively, equivalent amounts of VLPs in PBS were added directly to the microtiter wells and incubated for 24 to 30 h at 4°C. Wells were then incubated in PBS–1% BSA for 16 h. After wells were washed three times with PBS, different concentrations of selected antibodies were added to each well, and wells were incubated for 2 h at room temperature. After six washes in PBS, sheep anti-mouse antibody coupled to horseradish peroxidase (HRP) or goat anti-rabbit antibody coupled to HRP was added in 50 μl of PBS–1% BSA and incubated for 1.5 h at room temperature. Bound HRP was detected by the addition of 50 μl of TMB (3,3′5,5′-tetramethylbenzidine; Sigma) and incubation for 5 to 20 min at room temperature until the development of blue pigment in the wells. The reaction was stopped with 50 μl of 1N sulfuric acid. Color was read in a SpectraMax Plus plate reader (Molecular Devices) using SoftMax Pro software.
Statistical analysis.
Statistical analyses (Student t test) of data were accomplished using GraphPad Prism, version 7, software.
RESULTS
Expression and antigenicity of mutant F proteins.
To construct NDV VLPs containing a stabilized prefusion form of the RSV F protein and VLPs containing a stabilized postfusion F protein, we modified our previously characterized RSV F/NDV F chimeric protein (F/F) (27) in three ways. First, to produce a chimeric protein containing the stabilized prefusion RSV F protein ectodomain, we introduced four point mutations in the ectodomain (S190F, V207L, S155C, and S290C), mutations previously identified by McLellan et al. (22) as stabilizing the secreted form of the prefusion RSV F protein. McLellan and colleagues designated this mutant protein DS-Cav1; thus, our mutant chimeric protein was designated DS-Cav1 F/F (Fig. 1A). In a second construction, we added to the DS-Cav1 F/F mutant a foldon sequence at the carboxyl terminus of the RSV F protein ectodomain, a sequence found necessary for the stabilization of the secreted form of the prefusion F protein trimer by McLellan et al. (19, 22) In this construction, this sequence was placed in between the RSV F protein ectodomain sequence and the NDV F protein transmembrane domain sequence (Fig. 1A). The resulting chimeric protein is designated pre-F/F. We characterized the expression of both the DS-Cav1 F/F and the pre-F/F in order to compare the effect of the addition of the foldon sequence on expression of the chimeric proteins.
FIG 1.
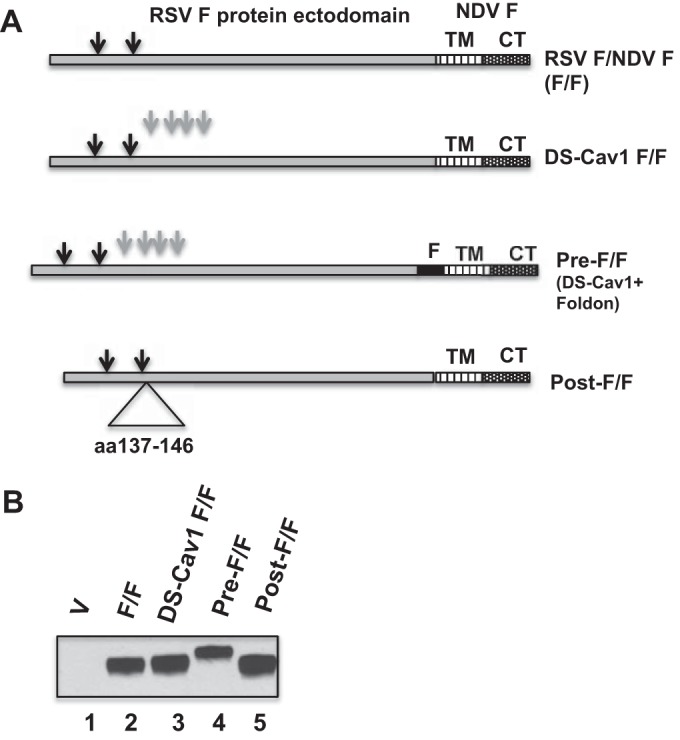
Expression of wild-type and mutant chimeric proteins. (A) Diagram of the wild-type chimeric protein (F/F) and three chimeric proteins with mutations in the RSV F protein ectodomain. DS-Cav1 F/F contains four point mutations, S155C, S190F, V207L, and S290C (gray arrows). Pre-F/F contains the mutations in DS-Cav1 F/F as well as the foldon (F) sequence inserted between the RSV F protein ectodomain and the NDV TM domain. Post-F/F contains a deletion of sequences encoding amino acids (aa) 137 to 146. TM, NDV F protein transmembrane domain; CT, NDV F protein cytoplasmic domain. Black arrows indicate RSV F protein cleavage sites. (B) Surface expression of chimeric F proteins. Avian cells (1 × 105 cells) transfected with pCAGGS vector (V) or pCAGGS containing the gene encoding F/F, DS-Cav1 F/F, pre-F/F, or post-F/F were biotinylated and then lysed. Biotinylated molecules were precipitated and then denatured and reduced, and chimeric proteins in the precipitate were detected by Western blotting using anti-RSV HR2 antibody, which detects the F1 protein. The figure shows the results of one of three comparable experiments.
To compare immune responses to the prefusion F protein with responses to a postfusion form of the F protein, we constructed an F/F chimeric protein containing a stabilized postfusion form of the RSV F protein ectodomain. McLellan and colleagues have reported that deletion of amino acids 137 to 146 results in a stable postfusion RSV F protein (38). Thus, we deleted this sequence from wild-type F/F to create a post-F/F chimeric protein.
Incorporation of the mutant chimeric proteins into VLPs requires their efficient expression on cell plasma membranes. To determine if the alterations in the F/F chimeric protein affected the cell surface expression of the chimeric proteins, surfaces of cells expressing the wild-type and mutant chimeric proteins were biotinylated, and after cell lysis, biotinylated molecules were precipitated with NeutrAvidin. The levels of biotinylated wild-type and mutant F/F proteins in the precipitates were determined in Western blots using an anti-RSV F protein polyclonal antibody for detection. Representative results of multiple experiments are shown in Fig. 1B. Consistently, the levels of F/F, DS-Cav1 F/F, pre-F/F, and post-F/F chimeric proteins were very similar. As expected, the pre-F/F migrates on polyacrylamide gels slightly more slowly than the F/F due to the presence of the foldon sequence, which adds 27 amino acids to the protein. The post-F/F protein migrates slightly faster than F/F or the DS-Cav1 F/F due to the deletion of amino acids 137 to 146. We have previously shown that the F/F protein is proteolytically cleaved. Thus, the migration of the DS-Cav1 F/F, the pre-F/F, and the post-F/F proteins on polyacrylamide gels is consistent with proteolytic cleavage of these mutant proteins.
McLellan et al. have identified an antigenic site, termed site ϕ, which is unique to the prefusion form of the RSV F protein (19, 22). This site, which is present on the prefusion soluble F protein but not the postfusion soluble F protein, has been defined by two human anti-RSV F monoclonal antibodies and one murine anti-F monoclonal antibody, 5C4 (19, 22). To verify that chimeric proteins DS-Cav1 F/F and pre-F/F contain this site and that the post-F/F chimeric protein does not, the binding of anti-F 5C4 antibody to cells transfected with cDNAs encoding the three mutant chimeric proteins as well as with the cDNA encoding the wild-type F/F protein was determined by flow cytometry. Figure 2A to D show representative profiles obtained. Figure 3A shows the geometric mean fluorescence intensity after multiple separate determinations. Clearly the DS-Cav1 F/F and the pre-F/F chimeric proteins contain site ϕ (Fig. 2B and C and 3A), whereas the post-F/F protein does not contain significant levels of this site (Fig. 2D and 3A). The wild-type F/F also contains site ϕ (Fig. 2A) but at a much reduced level relative to DS-Cav1 F/F and pre-F/F. 5C4 binding to DS-Cav1 F/F was comparable to that seen with pre-F/F (Fig. 2B and C), suggesting that the foldon sequence has little influence on expression of site ϕ on cell surfaces in these chimeric proteins.
FIG 2.
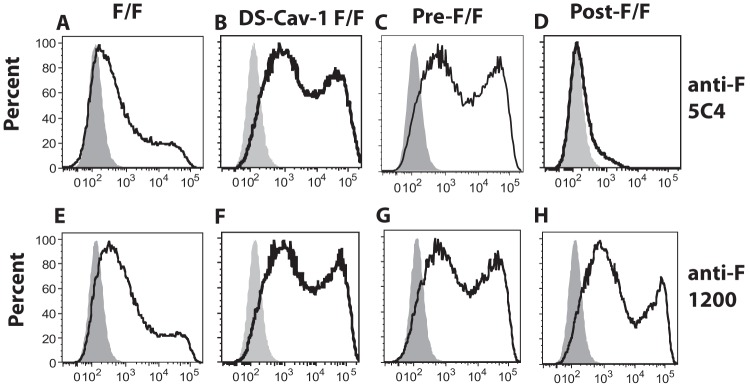
Detection of monoclonal antibody binding to avian cells expressing chimeric F proteins. Representative monoclonal antibody binding to chimeric F protein-expressing cells was detected by flow cytometry. Avian cells transfected with empty vector pCAGGS or pCAGGS containing the gene encoding the F/F, the DS-Cav1 F/F, the pre-F/F, or the post-F/F chimeric protein were incubated with MAb anti-F 5C4 antibody or MAb anti-F 1200. (A to D) Binding to anti-5C4 antibody. (E to H) Binding to MAb anti-F 1200. Antibody binding to cells transfected with empty vector is shown in the gray peaks.
FIG 3.

Quantification of monoclonal antibody binding to cells expressing chimeric proteins. Quantifications of the geometric mean fluorescence intensity in multiple experiments (three to five) was accomplished by analysis of flow cytometry profiles, as shown in Fig. 2, using FlowJo software. Antibodies and sites are as indicated on the figure.
To determine the influence of these mutations on other previously defined RSV F protein antigenic sites, the binding of representative anti-RSV F monoclonal antibodies, specific to sites I, II, and IV (34), to the mutant and wild-type chimeric proteins expressed on cell surfaces was characterized. Figure 2E to H show representative flow cytometry profiles for anti-F 1200, a site II antibody. Additional determinations using this antibody as well as antibodies from sites I, II, and IV are shown as the average geometric mean fluorescence intensity (MFI) in Fig. 3B to E. With the exception of anti-F 1112, there was no statistical difference in the binding of monoclonal antibodies to sites I, II, and IV to cells expressing F/F, pre-F/F, or post-F/F chimeric proteins. These results are consistent with the previous conclusion that both the post- and prefusion forms of the RSV F protein contain antigenic sites I, II, and IV (18, 19).
Characterization of virus-like particles containing pre-F/F and post-F/F chimeric proteins.
To prepare VLPs containing the prefusion or postfusion forms of the RSV F protein ectodomain, ELL-0 cells were transfected with plasmids encoding NDV M protein, NDV NP, the H/G chimeric protein (26), and either the pre-F/F or the post-F/F chimeric protein. VLPs were harvested and purified from the supernatants of these transfected cells as we have previously described (26, 27, 39). Because the DS-Cav1 F/F and the pre-F/F proteins contained similar levels of site ϕ and because the two proteins were expressed on cell surfaces at similar levels, we focused on producing VLPs with the pre-F/F because addition of the foldon sequence had the potential to increase the stability of the prefusion form of the RSV F protein ectodomain during preparation and purification of VLPs as well as immunization.
Figure 4 shows the characterization of the protein content of the resulting VLPs. Panel A shows a Western blot detecting RSV F protein content in equivalent amounts (lanes 3 and 4) of total VLP protein using an anti-RSV F protein polyclonal peptide antibody for detection. To compare the efficiencies of chimeric protein incorporation into the VLPs, the NDV NP protein content of each VLP preparation was determined by Western blotting using anti-NDV antibody (Fig. 4B). Clearly, the VLPs contained similar levels of NP (lanes 3 and 4), and the ratios of RSV chimeric F proteins to NDV NP protein in each VLP preparation were similar, indicating that the efficiencies of pre-F/F and post-F/F chimeric protein incorporation into the VLPs were similar. As shown in Fig. 4C, there was no differential effect of the mutant F proteins on H/G chimeric protein incorporation into VLPs. Figure 4D shows the silver stain of a polyacrylamide gel containing total proteins in the VLP preparations. The levels of NDV M protein, as well as NP, in the VLPs were similar (lanes 3 and 4), supporting the conclusion that the efficiencies of assembly of the pre-F and the post-F proteins into VLPs were similar. In addition, there is little detectable host protein content in these VLPs.
FIG 4.
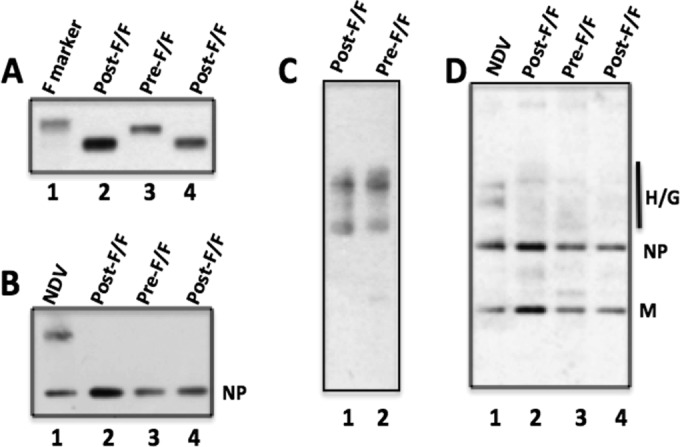
Proteins in VLPs. VLPs harvested from cells expressing the NDV NP, M, and H/G chimeric proteins as well as the pre-F/F or the post-F/F protein were purified, and proteins in the VLPs were characterized by Western blotting (A to C) and silver staining (D). Labels at the top of panels indicate the chimeric F protein in the VLPs shown. Lanes 2 in panels A, B, and D contained twice the VLP F protein content of lanes 3 and 4, determined in a preliminary Western blot, in order to demonstrate that differences in protein content could be detected. (A) RSV F protein content detected with anti-RSV HR2 peptide antibody. Lane 1, purified RSV F (Novavax, Inc.). (B) NDV NP protein content detected with polyclonal anti-NDV. (C) RSV H/G detected with polyclonal anti-RSV antibody (which does not detect F sequences [27]). Lanes 1 and 2 contained equivalent amounts of VLPs as determined by F protein content. (D) Silver stain of proteins in VLPs or NDV. NP, NDV nucleocapsid protein; M, NDV membrane protein; H/G, chimeric protein containing RSV G protein ectodomain. All samples were boiled and reduced in sample buffer containing β-mercaptoethanol prior to electrophoresis.
Antibody binding to purified VLPs.
To determine if assembly of the chimeric F proteins into VLPs influences the F protein conformation and, therefore, the antigenic sites on the pre-F/F or the post-F/F chimeric protein, binding of representative monoclonal antibodies to the VLPs was characterized. In an ELISA, the binding of increasing amounts of representative monoclonal antibodies to VLPs bound to microtiter plates was determined. Amounts of VLPs containing equivalent levels of F protein, determined by Western blotting as shown in Fig. 4, were bound to microtiter wells. Figure 5A shows the binding of the VLPs to a polyclonal antibody raised against the HR2 (HRB) domain of the RSV F protein, a domain present in both chimeric proteins. Clearly the levels of binding were similar, indicating that amounts of VLP loaded on the plates were equivalent. Fig. 5B shows the binding to the two VLPs of anti-F 5C4, the antibody specific to the prefusion site ϕ. VLPs containing the pre-F/F bound this antibody while the VLPs containing the post-F/F chimeric protein did not, indicating that the VLP-associated pre-F/F chimeric protein retained site ϕ during assembly and purification of the VLPs and that the post-F/F chimeric protein did not acquire this site.
FIG 5.

Monoclonal antibody binding to purified VLPs. Pre-F/F- and post-F/F-containing VLPs containing equivalent amounts of F protein, as determined by Western blotting as described in the legend of Fig. 4, were bound to microtiter plates as described in Materials and Methods. Increasing amounts of representative monoclonal antibodies were bound to the plates, and binding was detected using anti-mouse monoclonal antibody, as described in Materials and Methods. That virtually the same amounts of F protein-containing VLPs were bound to the plates is shown in panel A, in which RSV F protein sequences were detected with anti-RSV HR2 antibody. Antibodies (Abs) and sites are as indicated on the figure. Results are representative of four separate determinations.
We also measured binding of representative antibodies to sites I, II, and IV to both types of VLPs (Fig. 5C to G). Consistently, we found that none of the monoclonal antibodies bound to VLPs containing the pre-F/F at levels comparable to binding to VLPs containing the post-F/F chimeric protein. Binding of the antibodies to the pre-F/F- or post-F/F-containing VLPs was saturated at similar antibody concentrations, but the total binding at saturation to the pre-F/F-containing VLPs was lower. This result was obtained in at least four separate experiments. In the experiment shown, the VLPs were bound to plates previously coated with anti-RSV polyclonal antibody. Experiments in which the VLPs were directly bound to microtiter wells were also conducted (data not shown). Virtually identical results were obtained using both protocols.
Because the levels of binding of the antibodies to sites I, II, and IV to surfaces of cells transfected with pre-F/F or post-F/F were virtually the same but binding to VLPs was not, we considered the possibility that the presence of the H/G chimeric protein in the VLPs affected the binding of antibodies to the RSV F protein sequences. To test this possibility, we determined binding of antibodies to surfaces of cells transfected with cDNAs encoding the H/G chimeric protein and either the pre-F/F or post-F/F protein. Figure 6 shows that the coexpression of the H/G chimeric protein in cells with either chimeric RSV F protein had little effect on the binding of antibodies to site I or II. Thus, expression of the H/G chimeric protein did not affect monoclonal antibody binding to either chimeric F protein expressed on cell surfaces.
FIG 6.
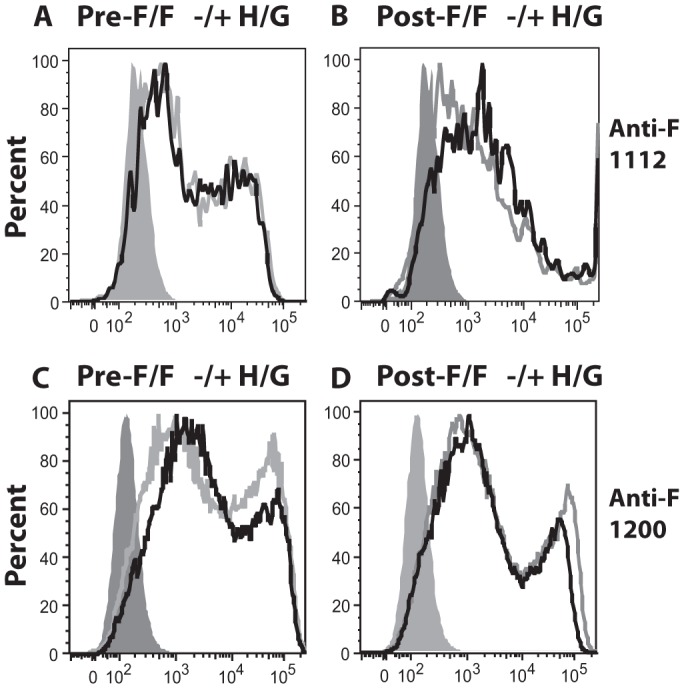
Effect of H/G protein expression on anti-F protein monoclonal antibody binding to avian cells expressing chimeric F proteins. Avian cells transfected with empty vector pCAGGS or pCAGGS–pre-F/F in the presence or absence of pCAGGS-H/G (A and C) or with pCAGGS–post-F/F in the presence or absence of pCAGGS-H/G (B and D) were incubated with MAb anti-1112 (A and B) or MAb anti-1200 (C and D). Binding of anti-F monoclonal antibodies was determined by flow cytometry. Gray peak, binding of antibody to cells transfected with empty vector; black line, chimeric F-expressing cells in the presence of H/G; gray line, chimeric F proteins expressed alone.
Anti-F protein IgG immune responses to the VLPs.
We next compared the ability of the two different VLPs to stimulate anti-RSV F protein antibody responses. Groups of five BALB/c mice were immunized with either VLP-H/G+pre-F/F or VLP-H/G+post-F/F or sham immunized with buffer, and sera were harvested by tail vein bleeding at different times after the prime immunization. Another group was infected intranasally (i.n.) with RSV. At 118 or 109 days post-prime immunization, respectively, the animals were boosted, and sera were harvested after 2 weeks. Sera obtained from the five mice at each time point were pooled for determination of anti-F protein IgG responses. To measure specificity of these responses induced by the prefusion and postfusion forms of the F protein, we prepared soluble versions of both forms to use as targets in ELISAs as previously described (18, 22). To eliminate any conformational changes that may occur during purification, we used unpurified supernatants of cells secreting the soluble F proteins. To verify the conformation of these soluble forms of F protein, the binding of representative anti-F monoclonal antibodies to these secreted proteins was characterized by ELISA. Equivalent amounts of the two forms of soluble RSV F protein, as determined by Western blotting, were bound to microtiter plates, and, indeed, there were similar levels of binding of anti-RSV F protein HR2 antibody to both forms of soluble protein, verifying that equivalent amounts of F protein bound to the plates (Fig. 7A). That these soluble F proteins retained antigenic properties reported for the soluble pre- and postfusion forms of F protein is shown by the binding of soluble pre-F to anti-F 5C4 and the failure of soluble post-F to bind this antibody (Fig. 7B). Thus, the preparation of soluble forms of the RSV F protein did not impair the conformation of the pre-F protein, nor did the preparation of post-F protein result in the acquisition of this site. In addition, and as reported previously (18, 19, 38), the soluble pre-F protein contained antigenic sites I, II, and IV as did the soluble post-F protein (Fig. 7C to F). However, antibodies to sites I, II, and IV did not bind to the soluble pre-F protein at levels typical of the soluble post-F protein, a result similar but not identical to the differential binding to VLP-associated pre-F and post-F proteins.
FIG 7.

Monoclonal antibody binding to soluble pre-F and post-F proteins. Soluble forms of the prefusion RSV F protein and the postfusion RSV F proteins as well as cell supernatants from empty vector-transfected cells, prepared as described in Materials and Methods, were bound to microtiter plates and then incubated with increasing amounts of representative monoclonal antibodies. Binding of antibodies was detected using anti-mouse IgG (or anti-rabbit IgG for panel A). Antibodies and sites are as indicated on the figure. Results are representative of two separate determinations. OD, optical density.
To measure specificity of antibodies in murine serum after VLP immunization or RSV infection, the soluble F proteins were used as targets in ELISAs (Fig. 8). After the prime immunization, the levels of post-F-specific IgG in sera from mice immunized with VLP-H/G+pre-F/F were similar to levels stimulated by VLP-H/G+post-F/F and levels stimulated by RSV infection (Fig. 8A and C). After the boost immunization, the VLP-H/G+post-F/F sera contained slightly higher levels of post-F protein-specific IgG than sera from VLP-H/G+pre-F/F-immunized or RSV-infected mice, but these differences were not statistically significant (Fig. 8C). In contrast, after both the prime and boost immunizations, there were significantly higher titers of pre-F-specific anti-F IgG in sera obtained from VLP-H/G+pre-F/F-immunized mice than in sera obtained from VLP-H/G+post-F/F-immunized mice (Fig. 8B and D).
FIG 8.

IgG Immune responses to the pre-F/F- and post-F/F-containing VLPs. BALB/c mice, in groups of five, were immunized i.m. with 30 μg of VLP protein/animal (3.8 μg of F protein) with either pre-F/F- or post-F/F-containing VLPs. Another group received buffer only, while a fourth group was infected i.n. with RSV (1 × 106 PFU/mouse). Serum was harvested by tail vein bleed at various times after the prime immunization. At day 118 (VLP-H/G+pre-F/F) or 109 (VLP-H/G+post-F/F), mice received a boost immunization (10 μg of VLP protein/mouse containing 1.25 μg of F protein or 1 × 106 RSV PFU/mouse). Two weeks after boost, serum was harvested. At 18 days after boost, animals were challenged with RSV, and 4 days after challenge they were sacrificed for serum and lung harvest. (A and B) At each time point, sera from each group were pooled, and amounts of total IgG that bound to the soluble pre-F and of total IgG that bound to soluble post-F were determined as described in Materials and Methods. (C and D) Total anti-F protein IgG in serum of individual mice was determined for day 90, for 2 weeks postboost, and for terminal bleeds. Graphs show the average anti-F IgG amount in serum from individual mice, with variations indicated. For results using soluble post-F as a target, differences between the results for three groups at day 90 and differences between the results for the groups at 2 weeks postboost or in terminal bleeds are not statistically significant. Using soluble pre-F as a target, differences between the results for all three groups at day 90 are statistically significant. Differences between the results for pre- and post-F/F VLP sera and differences between the results for pre-F/F VLP sera and RSV sera at 2 weeks postboost are significant while the differences between the results for post-F/F VLP sera and RSV sera are not. Arrows indicate time of boost immunizations.
RSV infection induced levels of antibodies that bound to the soluble prefusion F protein at levels similar to those induced by the post-F/F-containing VLPs. However, sera from all three groups of immunized mice contained overall lower levels of anti-F IgG antibodies that would bind to the prefusion soluble F protein than levels that would bind the postfusion soluble F protein.
Neutralizing antibody responses stimulated by the VLPs.
To determine the neutralizing antibody titer of sera obtained in relation to time after immunization with VLP-H/G+pre-F/F or VLP-H/G+post-F/F or after RSV infection, sera from each group of animals were pooled and used in an in vitro plaque reduction assay. Figure 9 shows that VLP-H/G+pre-F/F stimulated significantly higher titers of neutralizing antibody than VLP-H/G+post-F/F after a single dose of VLPs (prime immunization). At 75 days postprime, the neutralization titers stimulated by the pre-F/F- and post-F/F-containing VLPs were approximately 312 and 65 (log2 8.3 and 6, respectively), respectively. The boost significantly increased the neutralizing antibody titer in mice immunized with VLP-H/G+pre-F/F. The titer obtained at this time point was approximately 4,000 (approximately 12 log2). The boost with the post-F/F-containing VLPs also increased titers significantly to approximately 2,700 (approximately 11.3 log2). Titers after a single or double RSV infection were 74 and 465, respectively.
FIG 9.

Neutralization titers in sera from immunized mice. Sera from each group of immunized mice at each time point were pooled. The neutralization titers were determined in a standard plaque reduction assay as described in Materials and Methods. Titer was defined as the reciprocal of the dilution that inhibited RSV plaque formation by 50% (22). Results are the averages of two to five separate determinations. Differences between the results for pre-F/F-containing VLP sera and post-F/F-containing VLP sera at days 60, 75, and 90 are significant (P < 0.05). Differences between the results at 2 weeks postboost and in the terminal bleed are not significant.
Protection of immunized mice from RSV challenge.
To determine the protection from RSV replication afforded by VLP immunization, the virus titers in lungs of immunized mice after RSV challenge were determined by plaque assay (Fig. 10). Clearly immunization with both VLP-H/G+pre-F/F and VLP-H/G+post-F/F protected mice from RSV replication upon challenge, as did RSV infection. This result was expected since we have previously shown that the VLP-H/G+F/F immunization protected mice from RSV replication upon challenge (27).
FIG 10.

Protection from RSV replication in lungs by immunization. The graph shows virus titers in lungs of mice at 4 days postchallenge with infectious RSV. Mice were immunized as described in legend to Fig. 8. Three weeks post-boost immunization, mice were challenged with RSV (1.5 × 106 PFU/mouse). At 4 days postchallenge, lungs were harvested, and virus titers were determined by plaque assay. Data from nonimmunized (TNE) mice were statistically significantly different from data obtained from all groups of immunized mice.
DISCUSSION
Recent studies of the RSV F protein have made it clear that most previously tested RSV vaccine candidates contained predominantly the postfusion form of the F protein. It is also clear from the seminal work of McLellan et al., (19, 22) and Magro et al. (21) that the postfusion form of the RSV F protein is missing epitopes that stimulate the most effective neutralizing antibodies in experimental animals. McLellan et al. (19, 22) defined one such epitope, which they called ϕ, and showed that point mutations in the RSV F protein ectodomain along with a trimerization domain (foldon) derived from phage T4 fibritin (22, 40) resulted in the stabilization of the soluble form of the prefusion form of the RSV F protein.
We have previously shown that the RSV F protein and G protein ectodomain sequences, fused to the cytoplasmic and transmembrane domain sequences of the NDV F and HN proteins, respectively, could be incorporated into VLPs formed with the NDV NP and M protein and that these VLPs stimulated, in murine systems, protective, neutralizing antibody responses (27). Furthermore, we also reported that the chimeric F protein contained antigenic site ϕ (28). However, the proportion of VLP-associated chimeric F proteins containing this epitope was unclear as was the stability of prefusion F protein in these VLPs. It is conceivable that injection of VLPs into an animal may result in the conversion of any VLP-associated prefusion forms to a postfusion form and, thus, in a diminished stimulation of the more effective neutralizing antibody responses. Thus, to stabilize the prefusion form of the chimeric F protein in the ND VLPs, we introduced point mutations in the RSV F protein ectodomain sequence, mutations previously defined as stabilizing site ϕ (22). In addition, as was done in the preparation of the soluble form of the prefusion F protein (22), we added the foldon trimerization domain in between the RSV F and the NDV F sequences to further stabilize the structure. In order to directly compare immune responses to VLPs containing the prefusion F protein with responses to VLP-associated postfusion F protein, VLPs containing a stabilized postfusion F protein were also prepared by introducing a deletion mutation in the fusion peptide and HR1 (HRA) domain, a deletion that favors the formation of the postfusion form of the RSV F (18).
The two forms of the mutant chimeric proteins were incorporated with similar efficiencies with respect to NP and M protein content into ND VLPs, and the two chimeric proteins had no differential effect on incorporation of the H/G chimeric protein. Furthermore, the VLP-associated chimeric proteins differentially bound an antibody specific for site ϕ, verifying their conformation. Surprisingly, however, antibodies specific to sites I, II, and IV bound the two VLPs differently. In all cases, binding of antibody at saturation was lower for the pre-F/F-containing VLPs although saturation was reached at similar antibody concentrations for the two VLPs. We considered the possibility that these results could be due to the presence of the H/G chimeric protein, but no difference in binding of anti-F monoclonal antibodies to surfaces of cells expressing the F chimeric proteins in the presence or absence of the H/G chimeric protein was observed. However, the packing of the molecules on cell surfaces and on VLP surfaces may be very different. Close packing of the pre-F/F with H/G protein in the VLPs may well inhibit binding of antibodies to sites I, II, or IV. The post-F/F protein may extend beyond the H/G protein molecule on VLP surfaces, making the sites more accessible to antibody binding. Indeed, studies of the structure of the G protein of human metapneumovirus (hMPV), a closely related paramyxovirus, indicated that the MPV G protein extends from the virion surface somewhat beyond that predicted for the prefusion F protein while the postfusion F protein would extend beyond the G protein (41). Thus, it is possible that in VLPs the H/G chimeric protein limits the accessibility of the prefusion F/F to antibody while having less effect on site accessibility in the postfusion F/F protein. However, it is also possible that the affinity of the monoclonal antibodies to the prefusion F protein is lower than affinity to the postfusion F protein.
In order to compare immune responses induced by the particle-associated pre-F/F and the post-F/F chimeric proteins, mice were immunized, without adjuvant, with the two VLPs, and neutralizing antibody titers, anti-F protein serum IgG responses, and protection from challenge were assessed. The key results presented here are the neutralizing antibody titers stimulated by the two VLPs. It has been estimated that the protective threshold is a titer of 1/100 (dilution resulting in 50% inhibition) (22) or a titer of 6 log2 (42). Significantly, after a single or prime immunization, without adjuvant, the pre-F/F-containing VLPs stimulated neutralizing antibody titers of approximately 312 and 324 (log2 of 8.3) by days 75 and 90, respectively, while the titers stimulated by the post-F/F-containing VLPs were significantly lower (65 and 150, respectively) and very similar to neutralization titers induced after a single immunization with VLPs containing the wild-type F/F chimeric protein (27). Thus, VLPs containing a stabilized prefusion F protein are more effective in inducing neutralizing antibodies after a single immunization than VLPs that contain chimeric protein in primarily a post-F conformation.
A prime followed by a boost immunization with the pre-F/F-containing VLPs resulted in titers of approximately 4,000 (12 log2). Interestingly, serum titers after a prime and boost immunization with the post-F/F-containing VLPs were also very high, approximately 2,700 (11 log2). The neutralization titers after a prime and boost with the post-F/F-containing VLPs can be due, in part, to antibodies to the RSV G protein. In addition, it has also been shown that monoclonal antibodies specific to sites I, II, and IV on the F protein are neutralizing but at a much higher concentration than antibodies to site ϕ (22). Therefore, after a prime and boost with VLP-H/G+post-F/F, the antibody titers to these sites may well be high enough to neutralize virus effectively. Indeed, a humanized monoclonal antibody, palivizumab, which is specific for site II, is an effective prophylactic for infants at risk for RSV disease. Thus, immunization with pre-F/F-containing VLPs resulted in the highest neutralizing antibody titers although the post-F/F-containing VLPs after a boost were also effective in stimulating high titers of neutralizing antibodies. A prime and boost of RSV resulted in titers at 2 weeks postboost of approximately 465 (8.9 log2) and 1,200 after the subsequent RSV challenge, titers significantly lower than those stimulated by either VLP type at most time points. However, the route of immunization of RSV was different, which may account for lower serum neutralizing antibody titers.
McLellan et al. reported that a prime and boost immunization of mice with soluble forms of the prefusion form of RSV F resulted in neutralization titers of 4,000 (22). This result was obtained using an adjuvant and significantly more F antigen (10 μg/animal versus 3.8 μg/animal) than we used in our VLPs. Titers after a single immunization with soluble protein were not reported. Thus, at lower antigen levels without adjuvant, the pre-F/F-containing VLPs induced similar levels of neutralizing antibodies as those reported for soluble forms of the prefusion form of the protein with adjuvant after both a prime and boost. Interestingly, titers after a prime-boost immunization with soluble postfusion F protein were reported to be 600 (22) or 200 (20), considerably less than those we observed with VLP-associated post-F/F protein. This difference may be due to the presence of the G protein sequences in the VLPs. However, such differences in responses to soluble and particulate forms of an antigen have been repeatedly observed in many systems. As discussed by Bachmann and colleagues (23, 25, 43), presentation of an antigen in a particulate form and in an ordered array typical of a virus particle results in a more effective immune response than one to soluble proteins, a response that does not require the complications of adjuvants.
To assess total anti-F protein IgG responses specific for the prefusion and postfusion F proteins, we first prepared the soluble forms of pre-F and post-F proteins as targets in ELISAs, and their differential binding to antibody specific to site ϕ was verified. Surprisingly, the reactivities of the soluble prefusion F protein to antibodies to sites I, II, and IV were significantly different from reactivities to comparable amounts of the soluble postfusion F protein, results somewhat similar but not identical to those obtained with VLP-associated chimeric proteins. In contrast to results of antibody binding to VLP-associated chimeric F proteins, antibody binding to the soluble pre-F protein was saturated at higher concentrations than binding to the soluble post-F protein. These results suggest that the accessibility of the soluble prefusion F protein to antibodies to sites I, II, and IV is less than that of the soluble postfusion F protein. Alternatively, the conformation of an antigenic site in the two differently folded forms of F is somewhat different, resulting in a different affinity of an antibody to that site. It will be important for future improvements of vaccine design to clarify the reasons for reduced binding of antibodies to the prefusion F protein compared to binding to the postfusion F.
Immunization of mice with both types of VLPs induced similar levels of IgG specific to the postfusion forms of F protein, indicating that both forms are similarly antigenic. After a prime and boost, the levels of post-F-specific IgG stimulated by pre-F/F-containing VLPs were slightly lower than those stimulated by comparable amounts of post-F/F-containing VLPs, but the differences were not statistically significant. Surprisingly, neither VLP type induced pre-F-specific IgG at levels comparable to post-F protein-specific IgG levels. This result, coupled with the results of our antibody binding to VLP-associated pre-F/F and soluble pre-F protein, suggests that antibody binding sites on the pre-F protein are less accessible to antibody or, alternatively, that antibodies have lower avidity to pre-F protein. The pre-F/F-containing VLPs did stimulate significantly higher levels of antibody reactive to the soluble prefusion F protein than the post-F/F-containing VLPs, a result consistent with stimulation of antibody to sites not present on the postfusion forms. However, it is also possible that this difference is due, at least in part, to antibodies induced by the foldon sequences present in the pre-F/F-containing VLPs and the soluble pre-F protein target but not in the post-F/F-containing VLPs or soluble post-F target.
RSV infection stimulated responses to the two forms of soluble F, but the levels induced to both the pre- and postfusion targets were similar to those induced by the VLPs containing the postfusion F protein. This result is consistent with the report that purified RSV contains predominantly postfusion forms of F protein (44).
All three immunizations resulted in protection of mice from RSV replication upon RSV challenge. This result is not surprising since we have shown that VLPs that stimulate considerably lower titers of neutralizing antibody provide statistically significant protection in mice upon challenge (27). In addition, protection may also be due in part to responses to the H/G chimeric protein present in both VLPs.
Results presented here show that a prefusion F antigen associated with VLPs will stimulate high titers of neutralizing antibodies and antibodies specific for the prefusion form of the RSV F protein. This form of the RSV F protein will also induce antibodies specific for sites present on the postfusion form of the protein. A humanized monoclonal antibody, palivizumab, which is specific for site II (45, 46) has proven to be a useful prophylactic for RSV in infants. Thus, inclusion of the prefusion form of the F protein in virus-like particle RSV vaccine candidates should stimulate antibodies to all sites associated with protective responses and enhance protective responses without the necessity of added adjuvants.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants R41AI109926 (to J. Blanco, Sigmovir, Inc., and T.G.M) and R01AI084800 (to M.R.S. and R.T.W.).
We thank Barney Graham for the anti-F 5C4 monoclonal antibody and Judith Beeler for anti-F monoclonal antibodies 1112, 1269, 1200, 1237, and 1243.
REFERENCES
- 1.Karron RA. 2008. Respiratory syncytial virus and parainfluenza virus vaccines, 5th ed Saunders-Elsevier, Philadelphia, PA. [Google Scholar]
- 2.Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, O'Brien KL, Roca A, Wright PF, Bruce N, Chandran A, Theodoratou E, Sutanto A, Sedyaningsih ER, Ngama M, Munywoki PK, Kartasasmita C, Simoes EAF, Rudan I, Weber MW, Campbell H. 2010. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 375:1545–1555. doi: 10.1016/S0140-6736(10)60206-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Han LL, Alexander JP, Anderson LJ. 1999. Respiratory syncytial virus pneumonia among the elderly: an assessment of disease burden. J Infect Dis 179:25–30. doi: 10.1086/314567. [DOI] [PubMed] [Google Scholar]
- 4.Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. 2005. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med 352:1749–1759. doi: 10.1056/NEJMoa043951. [DOI] [PubMed] [Google Scholar]
- 5.Falsey AR, Walsh EE. 2000. Respiratory syncytial virus infection in adults. Clin Microbiol Rev 13:371–384. doi: 10.1128/CMR.13.3.371-384.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raboni SM, Nogueira MB, Tsuchiya LR, Takahashi GA, Pereira LA, Pasquini R, Siqueira MM. 2003. Respiratory tract viral infections in bone marrow transplant patients. Transplantation 76:142–146. doi: 10.1097/01.TP.0000072012.26176.58. [DOI] [PubMed] [Google Scholar]
- 7.Walsh EE, Falsey AR, Hennessey PA. 1999. Respiratory syncytial and other virus infections in persons with chronic cardiopulmonary disease. Am J Respir Crit Care Med 160:791–795. doi: 10.1164/ajrccm.160.3.9901004. [DOI] [PubMed] [Google Scholar]
- 8.Graham BS. 2011. Biological challenges and technological opportunities for respiratory syncytial virus vaccine development. Immunol Rev 239:149–166. doi: 10.1111/j.1600-065X.2010.00972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karron RA, Buchholz UJ, Colline PL. 2013. Live-attenuated respiratory syncytial virus vaccines, p 259–284. In Anderson LJ, Graham BS (ed), Challenges and opportunities for respiratory syncytial virus vaccines. Springer, Heidelberg, Germany. [Google Scholar]
- 10.Morrison TG, Walsh EE. 2013. Subunit and virus-like particle vaccine approaches for respiratory syncytial virus, p 285–306. In Anderson LJ, Graham BS (ed), Challenges and opportunities for respiratory syncytial virus vaccines. Springer, Heidelberg, Germany. [DOI] [PubMed] [Google Scholar]
- 11.Power UF. 2008. Respiratory syncytial virus (RSV) vaccines–two steps back for one leap forward. J Clin Virol 41:38–44. doi: 10.1016/j.jcv.2007.10.024. [DOI] [PubMed] [Google Scholar]
- 12.Jardetzky TS, Lamb RA. 2004. A class act. Nature 427:307–308. doi: 10.1038/427307a. [DOI] [PubMed] [Google Scholar]
- 13.Smith EC, Popa A, Chang A, Masante C, Dutch RE. 2009. Viral entry mechanisms: the increasing diversity of paramyxovirus entry. FEBS J 276:7217–7227. doi: 10.1111/j.1742-4658.2009.07401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamb RA, Parks GD. 2007. Paramyxoviridae: the viruses and their replication, p 1450–1496. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 15.Yin H-S, Paterson RG, Wen X, Lamb RA, Jardetzky TS. 2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc Natl Acad Sci U S A 102:9288–9293. doi: 10.1073/pnas.0503989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin H-S, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44. doi: 10.1038/nature04322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swanson KA, Settembre EC, Shaw CA, Dey AK, Rappuoli R, Mandl CW, Dormitzer PR, Carfi A. 2011. Structural basis for immunization with postfusion respiratory syncytial virus fusion F glycoprotein (RSV F) to elicit high neutralizing antibody titers. Proc Natl Acad Sci U S A 108:9619–9624. doi: 10.1073/pnas.1106536108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLellan JS, Yang Y, Graham BS, Kwong PD. 2011. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J Virol 85:7788–7796. doi: 10.1128/JVI.00555-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McLellan JS, Chen M, Leung S, Graepel KW, Du X, Yang Y, Zhou T, Baxa U, Yasuda E, Beaumont T, Kumar A, Modjarrad K, Zheng Z, Zhao M, Xia N, Kwong PD, Graham BS. 2013. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 340:1113–1117. doi: 10.1126/science.1234914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith G, Raghunandan R, Wu Y, Liu Y, Massare M, Nathan M, Zhou B, Lu H, Boddapati S, Li J, Flyer D, Glenn G. 2012. Respiratory syncytial virus fusion glycoprotein expressed in insect cells form protein nanoparticles that induce protective immunity in cotton rats. PLoS One 7:e50852. doi: 10.1371/journal.pone.0050852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Magro M, Mas V, Chappell K, Vazquez M, Cano O, Luque D, Terron MC, Melero JA, Palomo C. 2012. Neutralizing antibodies against the preactive form of respiratory syncytial virus fusion protein offer unique possibilities for clinical intervention. Proc Natl Acad Sci U S A 109:3089–3094. doi: 10.1073/pnas.1115941109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McLellan JS, Chen M, Joyce MG, Sastry M, Stewart-Jones GBE, Yang Y, Zhang B, Chen L, Srivatsan S, Zheng A, Zhou T, Graepel KW, Kumar A, Moin S, Boyington JC, Chuang G-Y, Soto C, Baxa U, Bakker AQ, Spits H, Beaumont T, Zheng Z, Xia N, Ko S-Y, Todd J-P, Rao S, Graham BS, Kwong PD. 2013. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 342:592–598. doi: 10.1126/science.1243283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jennings GT, Bachmann MF. 2008. The coming of age of virus-like particle vaccines. Biol Chem 389:521–536. doi: 10.1515/BC.2008.064. [DOI] [PubMed] [Google Scholar]
- 24.Noad R, Roy P. 2003. Virus-like particles as immunogens. Trends Microbiol 11:438–444. doi: 10.1016/S0966-842X(03)00208-7. [DOI] [PubMed] [Google Scholar]
- 25.Bachmann MF, Jennings GT. 2010. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol 10:787–796. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- 26.Murawski MR, McGinnes LW, Finberg RW, Kurt-Jones EA, Massare M, Smith G, Heaton PM, Fraire A, Morrison TG. 2010. Newcastle disease virus-like particles containing respiratory syncytial virus G protein induced protection in BALB/c mice with no evidence of immunopathology. J Virol 84:1110–1123. doi: 10.1128/JVI.01709-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGinnes LW, Gravel KA, Finberg RW, Kurt-Jones EA, Massare MJ, Smith G, Schmidt MR, Morrison TG. 2011. Assembly and immunological properties of Newcastle disease virus-like particles containing the respiratory syncytial virus F and G proteins. J Virol 85:366–377. doi: 10.1128/JVI.01861-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt MR, McGinnes-Cullen LW, Kenward SA, Willems KN, Woodland RT, Morrison TG. 2014. Modification of the respiratory syncytial virus F protein in virus-like particles impacts generation of B cell memory. J Virol 88:10165–10176. doi: 10.1128/JVI.01250-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Connors M, Collins PL, Firestone C-Y, Sotnikov AV, Waitze A, Davis AR, Hung PP, Chanock RM, Murphy BR. 1992. Cotton rats previously immunized with a chimeric RSV FG glycoprotein develop enhanced pulmonary pathology when infected with RSV, a phenomenon not encountered following immunization with vaccinia-RSV recombinants or RSV. Vaccine 10:475–484. doi: 10.1016/0264-410X(92)90397-3. [DOI] [PubMed] [Google Scholar]
- 30.Delgado MF, Coviello S, Monsalvo AC, Melendi GA, Hernandez JZ, Batalle JP, Diaz L, Trento A, Chang H-Y, Mitzner W, Ravetch J, Melero JA, Irusta PM, Polack FP. 2009. Lack of antibody affinity maturation due to poor Toll-like receptor stimulation leads to enhanced respiratory syncytial virus disease. Nat Med 15:34–41. doi: 10.1038/nm.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pantua HD, McGinnes LW, Peeples ME, Morrison TG. 2006. Requirements for the assembly and release of Newcastle disease virus-like particles. J Virol 80:11062–11073. doi: 10.1128/JVI.00726-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frank S, Kammerer RA, Mechling D, Schulthess T, Landwehr R, Bann J, Guo Y, Lustig A, Bachinger HP, Engel J. 2001. Stabilization of short collagen-like triple helices by protein engineering. J Mol Biol 308:1081–1089. doi: 10.1006/jmbi.2001.4644. [DOI] [PubMed] [Google Scholar]
- 33.McGinnes LW, Morrison TG. 2013. Newcastle disease virus-like particles: preparation, purification, quantification, and incorporation of foreign glycoproteins, current protocols in microbiology. John Wiley and Sons, Inc., Hoboken, NJ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beeler JA, van Wyke Coelingh K. 1989. Neutralization epitopes of the F glycoprotein of respiratory syncytial virus: effect of mutation upon fusion function. J Virol 63:2941–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jain S, McGinnes LW, Morrison TG. 2007. Thiol/disulfide exchange is required for membrane fusion directed by the Newcastle disease virus fusion protein. J Virol 81:2328–2339. doi: 10.1128/JVI.01940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jain S, McGinnes LW, Morrison TG. 2008. Overexpression of thiol/disulfide isomerases enhances membrane fusion directed by Newcastle disease virus fusion protein. J Virol 82:12039–12048. doi: 10.1128/JVI.01406-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murawski MR, Bowen GN, Cerny AM, Anderson LJ, Haynes LM, Tripp RA, Kurt-Jones EA, Finberg RW. 2009. Respiratory syncytial virus activates innate immunity through Toll-like receptor 2. J Virol 83:1492–1500. doi: 10.1128/JVI.00671-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McLellan JS, Chen M, Kim A, Yang Y, Graham BS, Kwong PD. 2010. Structural basis of respiratory syncytial virus neutralization by motavizumab. Nat Struct Mol Biol 17:248–250. doi: 10.1038/nsmb.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGinnes LW, Pantua H, Laliberte JP, Gravel KA, Jain S, Morrison TG. 2010. Assembly and biological and immunological properties of Newcastle disease virus-like particles. J Virol 84:4513–4523. doi: 10.1128/JVI.01931-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miroshnikov KA, Marusich EI, Cerritelli ME, Cheng N, Hyde CC, Steven AC, Mesyanzhinov VV. 1998. Engineering trimeric fibrous proteins based on bacteriophage T4 adhesins. Protein Eng 11:329–332. doi: 10.1093/protein/11.4.329. [DOI] [PubMed] [Google Scholar]
- 41.Leyrat C, Paesen GC, Charleston J, Renner M, Grimes JM. 2014. Structural insights into the human metapneumovirus glycoprotein ectodomain. J Virol 88:11611–11616. doi: 10.1128/JVI.01726-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Piedra PA, Cron SG, Jewell A, Hamblett N, McBride R, Palacio MA, Ginsberg R, Oermann CM, Hiatt PW. 2003. Immunogenicity of a new purified fusion protein vaccine to respiratory syncytial virus: a multi-center trial in children with cystic fibrosis. Vaccine 21:2448–2460. doi: 10.1016/S0264-410X(03)00098-7. [DOI] [PubMed] [Google Scholar]
- 43.Jennings GT, Bachmann MF. 2007. Designing recombinant vaccines with viral properties: a rational approach to more effective vaccines. Curr Mol Med 7:143–155. doi: 10.2174/156652407780059140. [DOI] [PubMed] [Google Scholar]
- 44.Liljeroos L, Krzyzaniak MA, Helenius A, Butcher SJ. 2013. Architecture of respiratory syncytial virus revealed by electron cryotomography. Proc Natl Acad Sci U S A 110:11133–11138. doi: 10.1073/pnas.1309070110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cardenas S, Auais A, Piedimonte G. 2005. Palivizumab in the prophylaxis of respiratory syncytial virus infection. Expert Rev Anti Infect Ther 3:719–726. doi: 10.1586/14787210.3.5.719. [DOI] [PubMed] [Google Scholar]
- 46.Simoes EA, Groothuis JR, Carbonell-Estrany X, Rieger CH, Mitchell I, Fredrick LM, LKJ. 2007. Palivizumab prophylaxis, respiratory syncytial virus, and subsequent recurrent wheezing. J Pediatr 151:34–42. doi: 10.1016/j.jpeds.2007.02.032. [DOI] [PubMed] [Google Scholar]