

ABSTRACT
Arenaviruses cause severe hemorrhagic fever diseases in humans, and there are limited preventative and therapeutic measures against these diseases. Previous structural and functional analyses of arenavirus nucleoproteins (NPs) revealed a conserved DEDDH exoribonuclease (RNase) domain that is important for type I interferon (IFN) suppression, but the biological roles of the NP RNase in viral replication and host immune suppression have not been well characterized. Infection of guinea pigs with Pichinde virus (PICV), a prototype arenavirus, can serve as a surrogate small animal model for arenavirus hemorrhagic fevers. In this report, we show that mutation of each of the five RNase catalytic residues of PICV NP diminishes the IFN suppression activity and slightly reduces the viral RNA replication activity. Recombinant PICVs with RNase catalytic mutations can induce high levels of IFNs and barely grow in IFN-competent A549 cells, in sharp contrast to the wild-type (WT) virus, while in IFN-deficient Vero cells, both WT and mutant viruses can replicate at relatively high levels. Upon infection of guinea pigs, the RNase mutant viruses stimulate strong IFN responses, fail to replicate productively, and can become WT revertants. Serial passages of the RNase mutants in vitro can also generate WT revertants. Thus, the NP RNase function is essential for the innate immune suppression that allows the establishment of a productive early viral infection, and it may be partly involved in the process of viral RNA replication.
IMPORTANCE Arenaviruses, such as Lassa, Lujo, and Machupo viruses, can cause severe and deadly hemorrhagic fever diseases in humans, and there are limited preventative and treatment options against these diseases. Development of broad-spectrum antiviral drugs depends on a better mechanistic understanding of the conserved arenavirus proteins in viral infection. The nucleoprotein (NPs) of all arenaviruses carry a unique exoribonuclease (RNase) domain that has been shown to be critical for the suppression of type I interferons. However, the functional roles of the NP RNase in arenavirus replication and host immune suppression have not been characterized systematically. Using a prototype arenavirus, Pichinde virus (PICV), we characterized the viral growth and innate immune suppression of recombinant RNase-defective mutants in both cell culture and guinea pig models. Our study suggests that the NP RNase plays an essential role in the suppression of host innate immunity, and possibly in viral RNA replication, and that it can serve as a novel target for developing antiviral drugs against arenavirus pathogens.
INTRODUCTION
Arenaviruses are bisegmented enveloped RNA viruses that include several hemorrhagic fever (HF)-causing viruses, such as Lassa fever virus (LASV), Junin virus (JUNV), Machupo virus (MACV), and Lujo virus (1, 2). These HF arenaviruses cause severe disease in humans, with mortality rates of up to 30% in hospitalized individuals. With the exception of Junin virus, there is currently no vaccine against these arenaviruses. Ribavirin is the only available antiviral drug, but its use is restricted by the requirement of administration at an early stage of infection and by its strong, undesirable side effects. Due to their virulent natures and the limited preventative and treatment measures against them, these pathogenic arenaviruses are classified as category A biosafety level 4 (BSL-4) pathogens. A nonpathogenic arenavirus, Pichinde virus (PICV), has been used as a prototypic arenavirus, and its infection of guinea pigs can serve as a surrogate model of arenavirus HFs (3–5).
Arenaviruses carry 4 viral genes on 2 genomic RNA segments (S and L), in an ambisense coding strategy (6). The glycoprotein (GPC) mediates receptor binding and virus-cell membrane fusion (7). The nucleoprotein (NP) encapsidates viral genomic RNAs, is essential for viral transcription and replication (8), and also suppresses type I interferon (IFN) production (9–14). The small matrix protein Z is required for virus budding and also regulates viral RNA synthesis (15). The large L protein is the RNA-dependent RNA polymerase protein that mediates viral RNA synthesis (16, 17).
Type I IFNs provide a critical defense against viral infections by mediating antiviral innate immunity and modulating adaptive immune responses. Through autocrine and paracrine pathways, type I IFNs amplify the antiviral and proinflammatory signals by inducing the production of hundreds of IFN-stimulating genes (ISGs) that exert antiviral activities through various mechanisms (18, 19). In addition, type I IFNs affect the proper expansion, differentiation, and survival of virus-specific T cells that are critical for viral clearance (19–21). Therefore, viruses have evolved multiple mechanisms for targeting type I IFN induction and/or signaling, including the arenavirus NP. We previously determined the crystal structure of the LASV NP and revealed a unique DEDDH exoribonuclease (RNase) domain that is important for type I IFN suppression (10). Further structural and functional studies revealed that the LASV NP RNase shows specificity for double-stranded RNA (dsRNA) (12, 14), a known pathogen-associated molecular pattern (PAMP) that triggers IFN production. The NP RNase catalytic residues, D389, E391, D466, D533, and H528, are conserved among all arenaviruses and have been shown to be required for type I IFN suppression (10, 12). Therefore, it appears that NP RNase-dependent IFN suppression is a general mechanism among the arenaviruses. A caveat of the prior studies is that the NPs were generally overexpressed. Carnec and colleagues recently attempted to generate recombinant LASV mutants by targeting the NP RNase domain. Recombinant viruses carrying single mutations at D389 or G392 are either unstable or lethal, highlighting the critical significance of the RNase domain in LASV replication (22). Interestingly, a double mutant carrying both D389A and G392A residue changes can be rescued, is unable to suppress type I IFNs, and replicates less well than the wild-type (WT) virus (22). That study therefore highlights the important role of NP RNase activity in type I IFN suppression during virus infection. However, the nature of the double mutation can complicate the understanding of the NP structure-function analysis, and there is still a lack of direct evidence for the biological role(s) of the NP RNase function in arenavirus infection in vivo.
In this study, we successfully generated recombinant PICVs with individual mutations at the 5 NP RNase catalytic residues by using a reverse genetic system that was previously established in our laboratory (5). These 5 different single catalytic mutants can serve as controls for each other in order to exclude the possible variations in the biological consequences caused by specific changes at different sites, and together they provide a complete and nonbiased conclusion on the functional role of NP RNase activity in vitro and in vivo. Our studies reveal the critical importance of functional NP RNase activity in type I IFN suppression and its role in disease pathogenesis during authentic viral infection both in vitro and in vivo.
MATERIALS AND METHODS
Plasmids and cells.
The pCAGGS-based mammalian expression plasmids for PICV (P2 and P18) NPs were described previously (23). Single alanine substitutions of the conserved RNase catalytic residues, D380, E382, D457, D525, and H520, were generated by either the QuikChange protocol (Stratagene) or overlapped PCRs. The primer sequences used for mutagenesis are listed in Table 1. All the mutations were confirmed by DNA sequencing. For bacterial expression, the full-length cDNAs of the WT and mutant PICV NP genes were subcloned into the pLou3 vector. Sequences were verified by sequence analysis. Human kidney epithelial cells (293T cells), human embryonic lung epithelial cells (A549 cells), baby hamster kidney cells (BHK21 cells), and African green monkey kidney cells (Vero cells) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 50 μg/ml penicillin-streptomycin. BSRT7-5 cells, which stably express the T7 RNA polymerase, were obtained from K.-K. Conzelmann (Ludwig-Maximilians-Universität, Germany) and cultured in minimal essential medium (MEM) supplemented with 10% FBS, 1 mg Geneticin per ml, and 50 mg penicillin-streptomycin per ml.
TABLE 1.
Primer sequences used for mutagenesis, qRT-PCR, and nested PCRs
| Primer name and use | Primer sequence |
|---|---|
| Primers for mutagenesis | |
| P18 NP-m1 D380A | GCTACCACATGGATTGCCATTGAAGGAACACCAAATG |
| CATTTGGTGTTCCTTCAATGGCAATCCATGTGGTAGC | |
| P18 NP-m2 E382A | CCACATGGATTGACATTGCAGGAACACCAAATGATCC |
| GGATCATTTGGTGTTCCTGCAATGTCAATCCATGTGG | |
| P18 NP-m18 D525A | CACTGTGCATTGCTTGCTACCATCATGTTTGATGCAAC |
| GTTGCATCAAACATGATGGTAGCAAGCAATGCACAGTG | |
| P18 NP-m19 H520A | CAAACCACTGCGAATCCTGCCTGTGCATTGCTTGATACC |
| GGTATCAAGCAATGCACAGGCAGGATTCGCAGTGGTTTG | |
| P18 NP-m22 D457A | CTGCTCAAGGTTCAGATGCTATAATCAGTTTGTTCGAAATGC |
| GCATTTCGAACAAACTGATTATAGCATCTGAACCTTGAGCG | |
| Primers for qRT-PCR | |
| GAPDH | ACAGTGACAGCCATTCTTCC |
| AGCCGAACTCATTGTCATACC | |
| IFN-α1 | CAGACTGGCTGTGAGGAAATAC |
| GCATCTGACGATTTCTGCTCT | |
| IFN-β1 | AGGGATACCATGACCACTCT |
| GACTACTGTCCAGGCACAAAT | |
| ISG15 | ACAGTGAAGATGCTGGATGG |
| CTGGAAGGCAGGTACTTTAGTC | |
| IRF7 | GCTGTGCCGGACATACTT |
| GTCATCATAGAGACTGCTGGTG | |
| RIG-I | TTGCTACTTCCGTTGCTGATG |
| CTTTTGCTCTTCCTCTGCCTC | |
| MDA5 | CCCATCATCCATAACTTTTCTCTG |
| GCAGTGCTTTGTTTTCTCTTGTAAC | |
| Primers for nested RT-PCR | |
| Outer primers for first-round PCR | |
| Forward primer | GAGATCAATCCAGAGGGACAATG |
| Reverse primer | GAGGTTGATCAGATCTGTGTTG |
| Inner primers for second-round PCR | |
| Primers for detection of WT revertants in D380A mutant | |
| Forward primer | GCTACCACATGGATCGA |
| Reverse primer | TTGCATCAAACATGATGGTAT |
| Primers for detection of D380A mutant | |
| Forward primer | GCTACCACATGGATTGC |
| Reverse primer | TTGCATCAAACATGATGGTAT |
| Primers for detection of WT revertants in E382A mutant | |
| Forward primer | ACATGGATTGACATCGA |
| Reverse primer | TTGCATCAAACATGATGGTAT |
| Primers for detection of E382A mutant | |
| Forward primer | ACATGGATTGACATTGC |
| Reverse primer | TTGCATCAAACATGATGGTAT |
| Primers for detection of WT revertants in D525A mutant | |
| Forward primer | GCTACCACATGGATTGA |
| Reverse primer | ATCAAACATGATGGCAT |
| Primers for detection of D525A mutant | |
| Forward primer | GCTACCACATGGATTGA |
| Reverse primer | ATCAAACATGATGGTAG |
| Primers for detection of WT revertants in H520A mutant | |
| Forward primer | GCTACCACATGGATTGA |
| Reverse primer | CAAGCAATGCACAATG |
| Primers for detection of H520A mutant | |
| Forward primer | GCTACCACATGGATTGA |
| Reverse primer | CAAGCAATGCACAGGC |
| Primers for detection of WT revertants in D457A mutant | |
| Forward primer | GCTCAAGGTTCAGACGA |
| Reverse primer | TTGCATCAAACATGATGGTAT |
| Primers for detection of D457A mutant | |
| Forward primer | GCTCAAGGTTCAGATGC |
| Reverse primer | TTGCATCAAACATGATGGTAT |
Protein expression and purification from bacteria.
As previously described (10), the pLou3-based plasmids expressing the full-length PICV NP and NP mutants were transformed into Rosetta cells (Novagen). After IPTG (isopropyl-β-d-thiogalactopyranoside) induction at a final concentration of 0.03 mM overnight at 20°C, the cells were lysed and the proteins purified through a nickel column followed by a gel filtration column.
In vitro 3′-5′ exoribonuclease assay.
The in vitro RNase assay was conducted as described previously (12). In brief, a 15-nucleotide (nt) RNA oligonucleotide was 5′-end labeled with 32P in a polynucleotide kinase reaction mixture (Promega), purified through an Illustra MicroSpin G25 column (GE Healthcare Life Sciences), and annealed with a 5-fold excess of unlabeled complementary oligonucleotides to form dsRNA substrates. Each exoribonuclease reaction mixture contained 1 pmol of radiolabeled RNA substrates and purified NP in an RNase buffer (25 mM Tris-HCl, pH 7.5, 10 mM NaCl, 15 mM KCl, 1 mM MnCl2, and 0.1 mg/ml bovine serum albumin) and was incubated at 37°C for 60 min. EDTA was added to a final concentration of 10 mM to terminate the reaction. Radiolabeled RNA products were heated at 95°C for 5 min, rapidly cooled on ice, separated in a 17% urea-polyacrylamide gel, and detected by exposure on an X-ray film (Kodak).
Generation of recombinant PICV mutants.
The respective NP RNase mutations (D380A, E382A, D525A, H520A, and D457A) were subcloned from the pCAGGS-based vectors carrying the individual mutations into a plasmid encoding the full-length antisense S segment of the P18 virus and confirmed by sequencing. Recombinant PICVs were generated by use of the reverse genetic system developed in our lab (5). Briefly, a plasmid encoding either a WT or mutant P18 S segment, together with a plasmid encoding the P18 L segment, was transfected into BSRT7-5 cells that stably expressed T7 RNA polymerase. The medium was replaced at 4 h posttransfection, and supernatants were collected at 48 and 72 h. Viruses recovered in supernatants were plaque purified in Vero cells. The NP sequence was verified by sequencing of the reverse transcription-PCR (RT-PCR) products. The recombinant viruses were amplified once in BHK-21 cells, and viral titers were determined by plaque assay.
Determination of the purity of recombinant virus stocks.
In order to determine the purity of the virus stocks, we carried out nested RT-PCR analysis of cDNA products generated from RNAs isolated from the virus stocks by using primers with sequences at the 3′ distal ends that had sequence mismatches with either the WT or the RNase mutations in such a way that the WT NP sequence should not have been amplified from the mutant RNA virus stocks if no WT virus was present in these virus stocks but only the mutated NP RNase sequences should have been amplified. The primer sequences used for the nested PCRs are listed in Table 1. Briefly, viral RNAs were extracted from the WT and NP RNase mutant viruses (D380A, E382A, D525A, H520A, and D457A) by use of a Qiagen viral RNA minikit. The cDNAs were generated with Moloney murine leukemia virus (M-MLV) reverse transcriptase according to the manufacturer's instructions (Promega). The PCR step was carried out using Taq polymerase according to the manufacturer's protocol (New England BioLabs). The first round of PCR amplification, using the outer primer pairs (Table 1), included 1 cycle at 95°C for 30 s followed by 10 cycles of 95°C for 30 s, 58°C for 30 s, and 68°C for 45 s and 1 cycle of 68°C for 5 min. The second round of PCR amplifications included primers with sequences specific to the individual NP RNase mutant sequences (Table 1). The PCR amplification steps were as follows: 1 cycle of 95°C for 30 s, 32 cycles of 95°C for 30 s, 58°C for 30 s, and 68°C for 45 s, and 1 cycle of 68°C for 5 min. The PCR products were separated in a 1% agarose gel.
Growth curve analysis.
Vero or A549 cells were seeded in six-well plates at 90% to 100% confluence and infected (in triplicate) with viruses at a multiplicity of infection (MOI) of 0.01 for 1 h at 37°C. After the cells were washed with phosphate-buffered saline (PBS), a fresh aliquot of medium was added to the culture. At different time points postinfection, aliquots of the supernatant were harvested for plaque assay on Vero cells. To determine the viral growth in the presence of IFN-β, cells were infected with the respective viruses at an MOI of 0.01 for 1 h, washed three times with PBS, and replaced with fresh medium containing 100 IU of human IFN-β (R&D Systems, Inc.). Viral titers in the supernatants at various time points postinfection were quantified by plaque assay on Vero cells.
PICV plaque assay.
Vero cells were seeded into six-well plates at 90% to 100% confluence and infected with 0.5 ml of serial 10-fold dilutions of viruses in MEM for 1 h at 37°C. After removal of the medium, the cells were incubated in fresh MEM supplemented with 0.5% agar and 10% FBS and cultured for 4 days at 37°C. Plaques were stained overnight with diluted neutral red solution (1:50) in 0.5% agar–MEM–10% FBS.
In vitro passaging of mutant viruses.
Recombinant PICV mutants were serially passaged in Vero or A549 cells, with each infection performed at an MOI of 0.01. At 48 h postinfection (hpi), the supernatants were collected for the subsequent infection and for viral titer determination by plaque assay, in which the large plaques were counted and analyzed by sequencing.
rNDV-GFP-based biological assay of type I IFNs.
The IFN biological assay was conducted as described previously (24). In brief, supernatants from mock- or virus-infected cells were treated with UV light for 30 min to inactivate the viruses. Serial dilutions of the inactivated supernatants were added to Vero cells in 24-well plates for 24 h, followed by infection with rNDV-GFP. Green fluorescent protein (GFP) expression at 24 hpi was observed by fluorescence microscopy and measured with a Biotek Synergy 2 plate reader with the fluorescence option.
ELISA.
The levels of IFN-β in the supernatants of the virus-infected cells were quantified using a human IFN-β enzyme-linked immunosorbent assay (ELISA) kit (Thermo Scientific) following the manufacturer's instructions.
Guinea pig experiments.
The experimental procedures for guinea pigs followed the approved protocol of the Institutional Animal Care and Use Committee (IACUC) of the University of Minnesota. Briefly, healthy 350- to 400-g male outbred Hartley guinea pigs were housed for 3 days for acclimatization, after which they were randomly divided into different groups and injected intraperitoneally with 10,000 PFU of each of the virus strains or with PBS as a mock infection. The viruses used in these experiments were recombinant viruses generated from the PICV reverse genetic system that were plaque purified in Vero cells and amplified once in BHK-21 cells. Rectal temperature and body weight were measured daily for 18 days. Blood was drawn from the saphenous vein at different days postinfection (dpi) and used for virus titer determination by plaque assay. Guinea pigs were declared moribund and were euthanized if their body weight decreased 30% compared to that of the control group or if the rectal temperature fell below 38°C in addition to body weight loss.
Quantitative RT-PCR analysis of guinea pig type I IFNs and ISGs.
Guinea pigs were infected intraperitoneally with the respective viruses at 10,000 PFU. At 1 and 3 dpi, three animals from each group were euthanized. Peritoneal cavity cells, livers, lymph nodes, and spleens were collected for total RNA extraction by use of TRIzol reagent (Life Technologies). Single-stranded cDNAs were generated by use of an oligo(dT) primer and Superscript III reverse transcriptase following the manufacturer's protocol (Life Technologies). Quantitative PCR was conducted with specific primers designed to detect guinea pig genes by using Perfecta SYBR green supermix (Quanta) on a Bio-Rad CFX quantitative PCR machine. The PCR primer sequences are provided in Table 1. The PCR mixtures were incubated at 95°C for 3 min, followed by 40 cycles of amplification at 95°C for 10 s and 55°C for 30 s. The RNA level of each gene from virus-infected cells was first normalized to that of the housekeeping glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene and then compared to that in mock-treated cells and shown as the 2−ΔΔCT value in the graph. Results shown are the averages for three independent experiments.
Coimmunoprecipitation assay.
293T cells were grown overnight to 90% confluence in 10-cm2 plates in complete DMEM supplemented with 1% penicillin-streptomycin and 10% FBS (Invitrogen). Using a standard calcium phosphate transfection method, cells were transfected with plasmids expressing the wild-type PICV or LASV NP or the individual PICV NP RNase catalytic mutants (D525A, H520A, and D457A) along with the FLAG-tagged IKKε plasmid. Forty-eight hours after transfection, cells were lysed in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, and protein inhibitor cocktail [Roche]). The cell lysates were incubated with anti-FLAG antibody or with mouse IgG (Genescript) (as a control) at 4°C for 2 to 4 h, with rotation. Protein A/G beads (Santa Cruz) were added, and the samples were incubated with constant rotation at 4°C overnight. The protein complexes were washed 5 times with lysis buffer, standard 2× Laemmli sample buffer was added, and the samples were boiled for 5 min and then loaded onto an SDS-PAGE gel for Western blotting using a mouse anti-FLAG monoclonal antibody (MAb) (Sigma) to detect IKKε and anti-NP polyclonal Abs present in the serum of a rabbit that was immunized with a conserved NP peptide sequence (LDPNAKTWMDIEGRP).
RESULTS
Roles of PICV NP RNase activity in IFN suppression and viral RNA synthesis.
Sequence analysis of PICV NP reveals a conserved 3′-5′ DEDDH exoribonuclease (RNase) domain, with the catalytic residues D380, E382, D457, D525, and H520 absolutely conserved, in all known arenaviral NPs. To demonstrate that PICV NP indeed contains functional RNase activity, we expressed and purified the full-length PICV P18 NP and the five catalytic mutants from bacteria and analyzed their 3′-5′ RNase activity against dsRNA substrates by using an established in vitro RNase assay (12). As shown in Fig. 1A, wild-type PICV NP (P18 NP) degraded the dsRNA substrates with an efficiency comparable to that of LASV NP, while alanine substitutions at each of the five catalytic residues completely abolished the RNase activity in vitro. We then asked whether the PICV NP RNase was involved in IFN suppression, as expected based on previous studies of the LASV and Tacaribe virus (TCRV) NPs (10, 12). We showed that each of the PICV NP RNase mutants was expressed at levels similar to those of the WT NPs of the P2 and P18 PICV strains, as determined by Western blotting of the transfected 293T cell lysates (Fig. 1B). The NPs of the avirulent P2 and virulent P18 PICV strains differ at only a single residue (R374K) (23) and do not differ in their RNase domains. Both the P2 and P18 NPs strongly suppressed Sendai virus-induced IFN-β activation, whereas all five RNase mutants had lost the IFN-suppressive activity (Fig. 1B), demonstrating that, similar to that of other arenaviral NPs (12), the RNase activity of the PICV NP is critical for IFN-β suppression. As NP is required for viral RNA replication and transcription, we wondered whether the RNase activity also affects its function in viral RNA synthesis. We therefore quantified the levels of viral RNA synthesis in an established minigenome (MG) replicon assay (25) in which the L polymerase protein, NP, and a luciferase (LUC)-encoding viral RNA template were reconstituted in 293T cells. WT PICV L and NP efficiently transcribed the MG RNA template, leading to 5,000- to 9,000-fold increases of LUC expression over that of a negative control (Fig. 1C). Consistent with our previous report (25), P2 L and NP produced lower LUC levels than did P18 L and NP, which we previously attributed mainly to the difference in the L polymerases between these two PICV strains. Each of the RNase mutations in the P18 NP backbone could still support significant levels of MG RNA synthesis, i.e., ∼5,000-fold increases in LUC expression, which were nonetheless about 2-fold lower than the level catalyzed by the parental WT P18 proteins (Fig. 1C), suggesting that although the NP RNase function is not absolutely required for mediating PICV RNA synthesis, it may contribute to optimal viral RNA transcription. We also examined whether the RNase mutations affected the recently described interaction between NP and IKKε (26) by coimmunoprecipitation. Both LASV and PICV NPs interacted with IKKε (Fig. 1D), while three of the PICV NP RNase mutants tested (D525A, H520A, and D457A) showed various degrees of interaction with IKKε, indicating that an individual catalytic residue(s), rather than the RNase activity itself, may be involved in the IKKε interaction.
FIG 1.

Roles of PICV NP RNase activity in IFN suppression and viral RNA synthesis. (A) In vitro RNase activities of WT PICV NP and catalytic mutants of PICV NP. Purified PICV (P2 and P18) NPs and the RNase catalytic mutants were incubated with a 5′-32P-labeled dsRNA oligonucleotide in in vitro RNase reaction mixtures. (B) Alanine substitution mutations at each of the 5 catalytic residues abolished the ability of PICV NP to suppress the Sendai virus (SeV)-induced IFN-β activation (10). 293T cells were transfected with an IFN-β promoter-directed LUC plasmid and a β-galactosidase (β-Gal) plasmid, together with an empty vector or the respective NP plasmids, followed by Sendai virus infection. LUC activity was measured and normalized for transfection efficiency by using the β-Gal activity. The results shown are the averages for three independent experiments. Expression of the WT (rP2 and rP18) and mutant PICV NPs was detected by Western blotting using an anti-NP serum antibody raised in a rabbit. β-Actin was detected as a loading control. Results shown are the averages for three independent experiments. Each sample was compared to the vector control by statistical analysis using the Student t test. ***, P < 0.001; ns, no statistical significance. (C) PICV NP RNase mutants are slightly attenuated in supporting viral RNA synthesis in a minigenome assay (25). 293T cells were transfected with expression plasmids encoding PICV L and NP (WT or mutants) and a viral RNA template carrying the LUC reporter gene. Results shown are the averages for three independent experiments. Statistical significance was determined by the Student t test. ***, P < 0.001; ns, no statistical significance. (D) Interactions of NP with IKKε. 293T cells were transfected with plasmids containing the wild-type PICV or LASV NP or the individual PICV NP RNase catalytic mutations (D525A, H520A, and D457A) along with the FLAG-tagged IKKε plasmid. Immunoprecipitation (IP) was conducted with an anti-FLAG antibody. Western blotting (WB) was done to detect the NP by using a rabbit serum reactive to a conserved NP peptide sequence or anti-FLAG antibody to detect IKKε.
Recombinant PICV NP RNase mutants induce strong IFN responses in vitro.
Using the PICV reverse genetic system developed in our laboratory (5), we successfully generated recombinant P18 PICVs carrying individual RNase mutations, which were confirmed by sequencing. These RNase mutant viruses were used individually to infect human airway epithelial A549 cells at an MOI of 1. Type I IFN production at 24 hpi and 48 hpi was quantified using the established rNDV-GFP biological assay (24). Both of the WT recombinant (rP2 and rP18) viruses produced low levels of IFNs, similar to mock infection, as evidenced by the high levels of GFP expression viewed by fluorescence microscopy (Fig. 2A) and as quantified by use of a fluorescence plate reader (Fig. 2B). This was not surprising, as the NPs of both the rP2 and rP18 strains carried a functional RNase domain and did not seem to differ in the ability to suppress IFN production (Fig. 1B) (23). In contrast, the RNase mutant viruses produced significantly more IFNs, as evidenced by the greatly reduced levels of GFP expression at both 24 hpi and 48 hpi (Fig. 2A and B). We next used ELISA to directly quantify the levels of IFN-β present in the cell culture supernatants. As expected, rP2 or rP18 infection as well as mock infection did not lead to any appreciable levels of IFN-β production, whereas each of the RNase mutants produced significantly larger amounts of IFN-β than those in mock infection at both time points tested (Fig. 2C). Thus, we have demonstrated that the NP RNase activity is required for effective inhibition of type I IFNs in virus-infected cells.
FIG 2.

Recombinant PICV NP RNase mutants induce strong IFN responses in vitro. A549 cells were mock infected or infected with the respective recombinant PICVs at an MOI of 1. At 24 and 48 hpi, the supernatants were analyzed by the rNDV-GFP-based biological assay to measure the levels of type I IFNs (A and B) and by ELISA to quantify the IFN-β levels (C). Representative GFP images (A) and the average GFP values (i.e., GFP fluorescence intensities) and standard deviations for three independent experiments (B) are shown. (C) Results shown are the averages for three independent experiments. Each sample was compared to the rP18 group by statistical analysis using the Student t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, no statistical significance.
The PICV NP RNase function is essential for virus replication in immunocompetent cells.
We observed that the RNase mutant viruses produced plaques of sizes similar to those of the avirulent rP2 virus but slightly smaller than those of the parental rP18 virus (Fig. 3A). For quantitative comparison, viral growth kinetics were examined in Vero cells and A549 cells at an MOI of 0.01. All RNase mutant viruses replicated reasonably well in the IFN-deficient Vero cells, reaching levels above 105 PFU/ml at 48 hpi, similar to the level of the avirulent rP2 virus but ∼0.5 to 1 log lower than that of the virulent rP18 virus (Fig. 3B, left panel), consistent with the observed plaque sizes (Fig. 3A). In sharp contrast, these RNase mutant viruses barely grew in IFN-competent A549 cells, while both rP2 and rP18 grew to relatively high titers (Fig. 3B, right panel). We next asked whether addition of exogenous IFNs could inhibit PICV growth. Similar viral growth kinetic analyses were conducted in Vero and A549 cells in the presence of exogenously added human IFN-β (Fig. 3C). In Vero cells, IFN-β addition did not completely abolish viral replication but did reduce the growth of both the WT and mutant viruses, by similar degrees (∼2 log) (Fig. 3C, left panel). The fact that the RNase mutants grew to levels ∼0.5 log lower than that of the rP18 virus in Vero cells, with or without exogenous IFN-β, suggests that the NP RNase activity may be involved in mediating optimal virus replication. This is consistent with the finding that the RNase mutations led to a slight decrease in viral RNA synthesis as monitored by the viral replicon assay (Fig. 1C). In A549 cells, exogenous addition of IFN-β almost completely suppressed the growth of rP2 and rP18 and completely suppressed the growth of the RNase mutants (Fig. 3C, right panel), suggesting that inhibition of IFN production by NP RNase activity is essential for viral replication in IFN-competent A549 cells. Taken together, our results suggest that the NP RNase activity is not absolutely required for the basic virus life cycle but is essential for productive virus replication in IFN-competent cells by effectively inhibiting IFN production, consistent with a recent analysis of an LASV double mutant (D389A/G392A) (22).
FIG 3.

Viral growth kinetics of recombinant PICV NP RNase mutants in vitro. (A) Plaques formed in Vero cells by the respective viruses. (B) Viral growth kinetics in IFN-defective Vero and IFN-competent A549 cells. Cells were infected with the respective viruses at an MOI of 0.01. At different times postinfection, virus titers in the supernatants were quantified by plaque assay. The results shown are the averages for three independent experiments. (C) Viral growth kinetics in Vero and A549 cells in the presence of exogenously added IFN-β. Cells were infected with the respective viruses at an MOI of 0.01 for 1 h. After a PBS wash, fresh medium containing 100 IU of human IFN-β was added. Viral titers in the supernatants at different times postinfection were quantified by plaque assay. Each sample was compared to the rP18 group by statistical analysis using the Student t test. *, P < 0.05; **, P < 0.01.
In vivo infection by recombinant PICV NP RNase mutants.
To compare the levels of viral virulence in vivo, we infected 6 Hartley outbred guinea pigs intraperitoneally with 1 × 104 PFU of each virus, as previously described (5, 25, 27). The IACUC at the University of Minnesota reviewed and approved all procedures. Animals were monitored daily for body weight and disease signs for up to 18 dpi and were euthanized when they reached the predetermined terminal points (were moribund), such as a weight loss of >30% compared to a nomogram or a rectal temperature below 38°C in combination with continuing weight loss. As expected, all rP2-infected animals survived and cleared the virus, while rP18-infected animals developed an early onset of fever, showed greatly reduced body weights starting at 7 dpi, and reached the terminal points by 13 dpi (Fig. 4A). The RNase mutant viruses caused various disease outcomes (Fig. 4B and Table 2). All E382A mutant virus-infected animals survived without any evidence of body weight loss, while animals infected with each of the other three mutant viruses (D525A, H520A, and D457A) showed various degrees of viral attenuation (Fig. 4B). Most D380A mutant-infected animals, however, succumbed to the infection, albeit at a later time point than that for animals infected with rP18.
FIG 4.
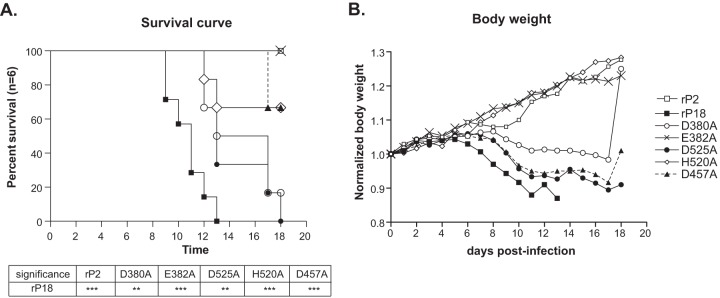
In vivo infection by recombinant PICV NP RNase mutants. Guinea pigs (n = 6) were infected intraperitoneally with the respective viruses at 1,000 PFU. Survival rates (A) and normalized body weights (B) are shown. Statistical analyses of the survival curves were performed with the log rank (Mantel-Cox) χ2 test, using GraphPad prism 5 software. ***, P < 0.001; **, P < 0.01. The body weight of each animal was monitored daily and normalized to that at day 0. The results shown are the averages of the normalized body weights for each group. Increases in the average body weights of some groups of animals are due to euthanization of some moribund animals and/or recovered animals.
TABLE 2.
Disease characteristics in guinea pigs infected with recombinant PICVs
| Recombinant PICV strain | Disease outcome | Viremiaa (PFU/ml) | Viral sequenceb |
|---|---|---|---|
| rP2c | Survived | ND | NA |
| rP18 | Moribund | 2.00E+06 | WT |
| Moribund | 5.80E+05 | WT | |
| Moribund | 2.80E+05 | WT | |
| Moribund | 1.80E+06 | WT | |
| Moribund | 2.30E+05 | WT | |
| Moribund | 8.00E+05 | WT | |
| D380A mutant | Moribund | 5.00E+04 | WT reversion |
| Moribund | 3.10E+05 | WT reversion | |
| Moribund | 3.30E+06 | WT reversion | |
| Moribund | 1.60E+04 | WT reversion | |
| Moribund | 5.00E+04 | WT reversion | |
| Survived | ND | NA | |
| E382A mutant | Survived | ND | NA |
| Survived | 2.13E+03 | WT reversion | |
| Survived | ND | NA | |
| Survived | 9.00E+02 | WT reversion | |
| Survived | ND | NA | |
| Survived | ND | NA | |
| D525A mutant | Moribund | 7.50E+04 | WT reversion |
| Moribund | 6.25E+04 | WT reversion | |
| Moribund | 1.75E+05 | WT reversion | |
| Moribund | 1.40E+04 | WT reversion | |
| Moribund | 2.40E+04 | WT reversion | |
| Moribund | 2.10E+05 | WT reversion | |
| H520A mutant | Moribund | 5.75E+05 | WT reversion |
| Survived | ND | NA | |
| Moribund | 7.50E+05 | WT reversion | |
| Survived | ND | NA | |
| Survived | ND | NA | |
| Survived | ND | NA | |
| D457A mutant | Survived | ND | NA |
| Survived | ND | NA | |
| Survived | ND | NA | |
| Survived | 6.20E+02 | WT reversion | |
| Moribund | 5.50E+04 | WT reversion | |
| Moribund | 1.10E+05 | WT reversion |
Virus titers in blood collected at the time of euthanization when animals reached terminal points or at day 18 postinfection. ND, not detectable by plaque assay.
Sequences of viruses isolated from animals at the time of euthanization when animals reached terminal points or at day 18 postinfection. NA, not applicable.
All 6 animals in the rP2-infected group survived, with no detectable viremia at day 18 postinfection.
To determine whether WT revertants occurred, we measured the viremia levels at the endpoint and sequenced the viruses (Table 2). Moribund animals were associated with high viremia, while all animals that survived the infections had very low to undetectable levels of virus. This is consistent with what we previously observed in this PICV-guinea pig model (5, 25, 27). Viruses isolated from some mutant-infected animals had reverted back to the WT NP sequence (Table 2). This was unlikely to be due to PCR contamination or sequencing error, as the viruses isolated from other mutant virus-infected animals at the same time still contained the expected mutations (data not shown). To determine that the mutant virus stocks did not contain WT virus contaminants, we sequenced the NP gene after RT-PCR amplification of the genomic RNAs isolated from the virions and found that all mutant viruses contained the intended NP mutant sequences (data not shown). As an additional measure of integrity of the virus stocks, we carried out nested RT-PCR analysis of cDNA products generated from RNAs isolated from the virus stocks by using DNA oligonucleotides with sequences at the 3′ distal ends that had sequence mismatches that would amplify only the WT NP sequence. Indeed, no PCR amplifications of the WT NP sequence resulted from all 5 mutant virus stocks (D380, E382, D457, D525, and H520), and only the respective RNase mutant sequences were amplified (Fig. 5), again demonstrating that all mutant virus stocks were pure. Taking these data together, we believe that the disease phenotype observed for each of the mutant viruses (Fig. 4) was in fact caused by the WT revertants and that the degrees of illness among the animals infected with the mutant viruses were determined by how fast the reversion occurred in vivo. For example, two of the E832A mutant-infected animals and one of the D457A mutant-infected animals carried very low levels of the WT revertant (2 × 103 PFU/ml or less) at day 18 postinfection, suggesting a delay in appearance of the WT reversion, and thus did not show any signs of disease throughout the course of the infection.
FIG 5.
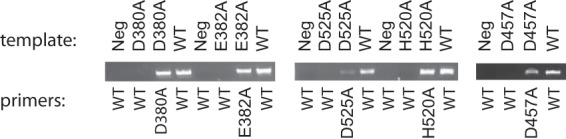
Determination of the purity of the recombinant virus stocks. Nested RT-PCR analysis was performed on cDNA products (templates) generated from RNAs isolated from the virus stocks (WT, D380A, E382A, D525A, H520A, and D457A) by using primers with sequences at the 3′ distal ends that had sequence mismatches with either the WT or the RNase mutations in such a way that the WT NP sequence should not have been amplified from the mutant RNA virus templates if no WT virus was present in these virus stocks but only the mutant NP RNase sequences should have been amplified. On the other hand, the WT NP sequence was amplified using the cDNA template generated from the WT virus stock and the primer pairs with sequences specific to the WT sequence. Neg, negative controls in which no cDNA templates were included in the reaction mixtures. The PCR products were separated and visualized in an ethidium bromide-stained 1% agarose gel.
In vitro passaging of recombinant PICV NP RNase mutants.
We decided to also monitor the appearance of potential WT revertants in vitro by passaging each of the mutant viruses in Vero and A549 cells at an MOI of 0.01. We conducted a plaque assay for each viral passage and picked a number of plaques for sequence analysis. Viral titers after each passage in Vero cells increased greatly for the D380A, E382A, D525A, and D457A viruses after the 2nd passage (P2), but only slightly for the H520A virus (Fig. 6A). Indeed, large plaques were observed for the D380A, E382A, D525A, and D457A viruses at P3, and sequence analysis of these plaques confirmed that they were indeed WT revertants (Table 3), implicating a role for the NP RNase in mediating optimal virus replication (Fig. 1C and 3A). When viruses were passaged in A549 cells, viral titers increased sharply for the D380A, E382A, D525A, and D457A viruses after P2, and slightly for the H520A virus (Fig. 6B). WT reversions were detected at P2 for the D525A and D457A viruses and, not surprisingly, at P3 for all mutants but the H520A mutant (Table 3). The rate of WT reversions in vitro seems to be determined by multiple factors. One such factor is the number of nucleotide changes required. For example, the H520A mutant contained a 2-nt change (CA → GC) and thus showed a much lower rate of reversion in both Vero and A549 cells than the other mutants, which had a 1-nt change (A → C). The selection pressure is another driving factor, as revertants generally arose at a slower pace in the IFN-deficient Vero cells (detected at P3) than in the IFN-competent A549 cells (detected at P2). On the other hand, WT reversion in vivo (Table 2) is more difficult to predict, due partly to the natural complexity of an animal system and to the relatively small animal sample size (n = 6 for each group).
FIG 6.
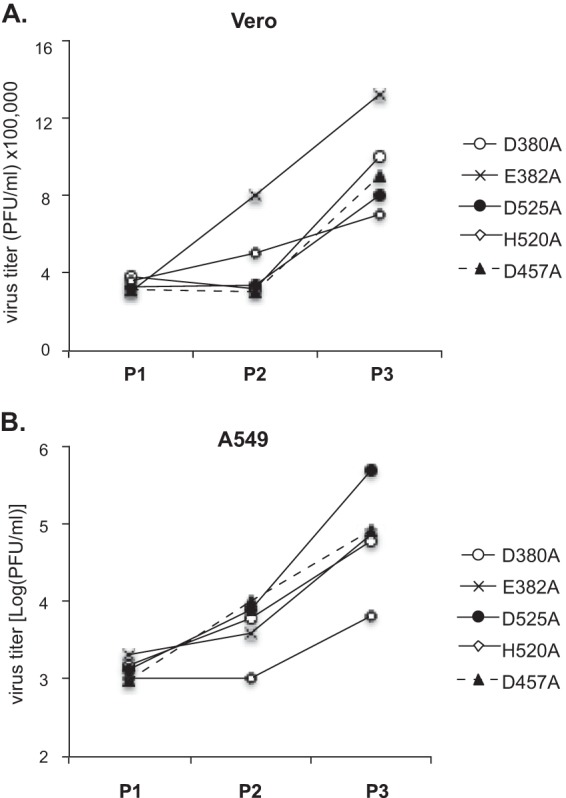
In vitro passaging of recombinant PICV NP RNase mutants. Each mutant virus was passaged successively in Vero (A) and A549 (B) cells at an MOI of 0.01. The viral titer at each passage was determined by plaque assay.
TABLE 3.
In vitro passage characteristics of PICV RNase mutants
| PICV mutant | P0 |
Vero cells |
A549 cells |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 |
P2 |
P3 |
P1 |
P2 |
P3 |
|||||||||
| % large plaquesa | Detection of WT reversionb | % large plaquesa | Detection of WT reversionb | % large plaquesa | Detection of WT reversionb | % large plaquesa | Detection of WT reversionb | % large plaquesa | Detection of WT reversionb | % large plaquesa | Detection of WT reversionb | % large plaquesa | Detection of WT reversionb | |
| D380A | <1 | − | <1 | − | <1 | − | 2 | + | <1 | − | <1 | − | 6.7 | + |
| E382A | <1 | − | <1 | − | <1 | − | 3 | + | <1 | − | <1 | − | 5.7 | + |
| D525A | <1 | − | <1 | − | <1 | − | 5 | + | <1 | − | 2.5 | + | 12 | + |
| H520A | <1 | − | <1 | − | <1 | − | 3 | − | <1 | − | <1 | − | 3 | − |
| D457A | <1 | − | <1 | − | <1 | − | 7 | + | <1 | − | 2 | + | 7.5 | + |
Percentage of large plaques among the total plaques.
Identification of WT revertants by sequencing. −, no revertants identified; +, revertants were identified.
Recombinant PICV RNase mutants induce strong IFN responses early in infection in vivo.
To examine the role(s) of RNase in early viral infection and in innate immune responses in infected animals, we monitored virus spread and quantified the levels of expression of some representative innate antiviral genes at 1 dpi and 3 dpi. At day 1, viruses were detected at low levels in the livers and spleens of the rP18-infected animals and in the livers of some animals infected with the rP2 or RNase mutant viruses (Fig. 7A). At day 3, both rP18 and rP2 replicated to relatively high levels in both the livers and spleens. In contrast, RNase mutant viruses appeared to be cleared from the livers and were detected in only a few spleens, at very low levels (Fig. 7A). We quantified the levels of representative ISGs, i.e., IFN-α1, IFN-β1, ISG15, IRF7, RIG-I, and MDA5, in peritoneal cavity cells on day 1 by quantitative RT-PCR (qRT-PCR) (Fig. 7B). These genes were highly activated in animals infected by the RNase mutant viruses but not by the WT viruses (rP2 and rP18), demonstrating that NP RNase is required for arenavirus-induced innate immune suppression in vivo.
FIG 7.

The PICV NP RNase mutants induced early and strong IFN responses in vivo. Guinea pigs were infected intraperitoneally with 1 × 104 PFU of the respective recombinant viruses (n = 3 animals per virus group) for 1 and 3 days. (A) Viral titers in livers and spleens at 1 and 3 dpi. (B) The levels of type I IFNs and selected ISGs in peritoneal cavity cells at day 1 were measured by qRT-PCR. Primer sequences will be provided upon request. Each dot represents a single animal. Statistical analyses were conducted by Student's t test.
DISCUSSION
In this study, we provide direct evidence for the biological roles of NP RNase activity in IFN suppression and arenavirus infection in vitro and in vivo. Structural and functional analyses of arenavirus NP revealed a unique immune suppression mechanism mediated by its C-terminal RNase activity (9–14). A previous study using the LASV NP D389A/G392A double mutant showed that the mutant virus was severely altered in the ability to suppress type I IFNs in macrophages and dendritic cells (DCs) (22). By introducing a single alanine substitution at each of the five catalytic residues in the PICV NP, we demonstrated in the current study that the NP RNase activity indeed plays a critical role in type I IFN suppression during authentic arenavirus infection in human epithelial A549 cells (Fig. 2). Exogenous addition of IFN-β almost completely suppressed the rPICV growth in A549 cells and significantly reduced the rPICV growth (∼2 log) in Vero cells (Fig. 3C), highlighting the importance of IFN suppression in arenavirus replication. Our data are consistent with previous studies showing that type I IFNs inhibit LASV replication in vitro (28, 29). The fact that exogenously applied IFN-β shows differential effects on PICV replication in A549 and Vero cells may be explained by the cell type- and/or species-specific antiviral activity of IFNs. Taken together, our data suggest that NP RNase-mediated IFN suppression may be critical for arenaviruses to establish productive infections in permissive cell types.
It has been reported that, besides NPs, the Z proteins of New World (NW) pathogenic arenaviruses, i.e., MACV, JUNV, Sabia virus (SABV), and Guanarito virus (GTOV), but not those of Old World (OW) pathogenic ones, i.e., LASV and lymphocytic choriomeningitis virus (LCMV), can inhibit IFN production (30). We recently reported that the Z proteins of all known human-pathogenic arenaviruses, but not those of nonpathogens, inhibit type I IFN expression by binding to RIG-I and MDA5, thereby disrupting their interactions with the downstream MAVS factor (31). The determinant for RIG-I and MDA5 binding and inhibition maps to the N-terminal domain (NTD) of the pathogenic Z proteins. Swapping of a pathogenic Z NTD with the sequence in a nonpathogenic PICV genome did not affect viral growth in Vero cells but significantly inhibited type I IFN responses and increased viral replication in human primary macrophages. Therefore, it appears that at least two IFN antagonists (i.e., NP and pathogenic Z) are encoded in the arenaviral genome. While arenavirus NP can strongly inhibit IFN production via an RNase domain that is conserved among all arenaviruses (9–14; the current study), the pathogenic Z protein inhibits IFN expression by binding directly to RIG-I and MDA5 and thereby disrupting their interactions with MAVS (31). Further studies are required to evaluate the relative contributions of pathogenic Z and NP to type I IFN suppression in different permissive cell types and at different stages of viral infection.
It is worth noting that although all arenaviruses encode a conserved NP RNase domain to suppress IFN induction (10, 12), variations in the ability to induce IFN expression and in the viral sensitivity to IFN-mediated antiviral effects have been noted for different arenaviruses and in different cell types. For example, we showed in this study that PICV barely induces IFN production and is very sensitive to IFNs in A549 cells. In contrast, virulent JUNV has been shown to induce type I IFNs in A549 cells (32) but not in human macrophages and monocytes (33). Since the JUNV NP RNase mutant was not tested in A549 cells (32), it is unclear whether the JUNV NP RNase can function to inhibit IFN induction to certain degrees in JUNV-infected A549 cells. Nevertheless, the differential degrees of IFN inhibition in PICV- and JUNV-infected A549 cells and in JUNV-infected A549 cells and macrophages suggest that the IFN induction is regulated at both virus- and cell type-specific levels.
Studies of arenavirus animal models and clinical samples have also suggested that IFNs have both protective and pathogenic roles in arenavirus-induced diseases (34–36). A functional IFN system has been shown to be required for arenavirus clearance in several mouse models (37–41). We showed in this study that the NP RNase-defective rPICVs induced early and strong IFN responses in guinea pigs that limited virus replication in various organs (Fig. 7A), demonstrating a strong correlation between early IFN induction and arenavirus clearance in vivo. Our data are consistent with clinical studies and animal models of LASV infections in that early and strong immune responses are associated with the effective control of LASV replication (42, 43). On the other hand, high levels of IFN-α have been detected in JUNV patient sera and correlate with disease severity (44). The source of the high levels of IFN-α has not been identified for JUNV-infected patients but is unlikely to be macrophages or monocytes (33). We speculate that the conserved NP RNase activity is used by arenaviruses to suppress type I IFN induction in early target cells (e.g., macrophages and dendritic cells), where the IFN responses function to control virus infection in vivo, whereas a sustained elevation of type I IFNs through virus- and/or cell type-specific mechanisms (e.g., in JUNV-infected patients) may contribute to overt disease pathogenesis.
In addition to the IFN suppression activity, whether NP RNase is involved in another step(s) of the viral life cycle requires further investigation. Our previous mutational analysis of LASV NP in a minigenome replicon assay did not reveal any defects in viral RNA transcription (10). In the current study, we showed that although the PICV NP RNase mutant proteins were still highly effective at supporting transcription of the viral MG RNA, they nonetheless showed a 50% reduction in LUC reporter expression (Fig. 1C), suggesting that the NP RNase domain may be required for optimal RNA synthesis. This may partly explain the observation that the PICV NP RNase catalytically inactive mutant viruses grew to levels ∼0.5 log lower than those of the rP18 WT virus in Vero cells in the presence or absence of exogenous IFN-β (Fig. 3B and C). Further studies are thus required to determine whether NP RNase activity has a biological function(s) besides mediating type I IFN suppression and whether this other function(s) is virus and/or cell type specific.
In summary, our studies with recombinant PICVs carrying NP RNase catalytic mutations have not only confirmed the important role of the NP RNase function in mediating type I IFN suppression and viral replication in vitro but also provided unequivocal evidence for its essential role in early innate immune suppression to allow the establishment of a productive infection in vivo. Given that the NP RNase-dependent IFN suppression mechanism is conserved among many arenaviruses (12), our results can be extrapolated to other arenaviral pathogens and implicate the NP RNase function as an ideal target for antiviral development.
ACKNOWLEDGMENTS
We thank J. F. Aronson (University of Texas Medical Branch) for providing the P2 and P18 stock viruses, K.-K. Conzelmann (Ludwig-Maximilians-Universität, Germany) for the BSRT7-5 cells, and A. García-Sastre (Icahn School of Medicine at Mount Sinai) for the rNDV-GFP virus.
This work was supported by NIH grants R01 AI093580 (to H.L.) and R01 AI083409 (to Y.L.).
REFERENCES
- 1.McLay L, Ansari A, Liang Y, Ly H. 2013. Targeting virulence mechanisms for the prevention and therapy of arenaviral hemorrhagic fever. Antiviral Res 97:81–92. doi: 10.1016/j.antiviral.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McLay L, Liang Y, Ly H. 2014. Comparative analysis of disease pathogenesis and molecular mechanisms of New World and Old World arenavirus infections. J Gen Virol 95:1–15. doi: 10.1099/vir.0.057000-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jahrling PB, Hesse RA, Rhoderick JB, Elwell MA, Moe JB. 1981. Pathogenesis of a Pichinde virus strain adapted to produce lethal infections in guinea pigs. Infect Immun 32:872–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aronson JF, Herzog NK, Jerrells TR. 1994. Pathological and virological features of arenavirus disease in guinea pigs. Comparison of two Pichinde virus strains. Am J Pathol 145:228–235. [PMC free article] [PubMed] [Google Scholar]
- 5.Lan S, McLay Schelde L, Wang J, Kumar N, Ly H, Liang Y. 2009. Development of infectious clones for virulent and avirulent Pichinde viruses: a model virus to study arenavirus-induced hemorrhagic fevers. J Virol 83:6357–6362. doi: 10.1128/JVI.00019-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buchmeier MJ, De La Torre JC, Peters CJ. 2007. Arenaviridae: the viruses and their replication, p 1791–1827. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 7.Burri DJ, da Palma JR, Kunz S, Pasquato A. 2012. Envelope glycoprotein of arenaviruses. Viruses 4:2162–2181. doi: 10.3390/v4102162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pinschewer DD, Perez M, de la Torre JC. 2003. Role of the virus nucleoprotein in the regulation of lymphocytic choriomeningitis virus transcription and RNA replication. J Virol 77:3882–3887. doi: 10.1128/JVI.77.6.3882-3887.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez-Sobrido L, Zuniga EI, Rosario D, Garcia-Sastre A, de la Torre JC. 2006. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol 80:9192–9199. doi: 10.1128/JVI.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qi X, Lan S, Wang W, Schelde LM, Dong H, Wallat GD, Ly H, Liang Y, Dong C. 2010. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 468:779–783. doi: 10.1038/nature09605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hastie KM, Liu T, Li S, King LB, Ngo N, Zandonatti MA, Woods VL Jr, de la Torre JC, Saphire EO. 2011. Crystal structure of the Lassa virus nucleoprotein-RNA complex reveals a gating mechanism for RNA binding. Proc Natl Acad Sci U S A 108:19365–19370. doi: 10.1073/pnas.1108515108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang X, Huang Q, Wang W, Dong H, Ly H, Liang Y, Dong C. 2013. Structures of arenaviral nucleoproteins with triphosphate dsRNA reveal a unique mechanism of immune suppression. J Biol Chem 288:16949–16959. doi: 10.1074/jbc.M112.420521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hastie KM, King LB, Zandonatti MA, Saphire EO. 2012. Structural basis for the dsRNA specificity of the Lassa virus NP exonuclease. PLoS One 7:e44211. doi: 10.1371/journal.pone.0044211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hastie KM, Kimberlin CR, Zandonatti MA, Macrae IJ, Saphire EO. 2011. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. Proc Natl Acad Sci U S A 108:2396–2401. doi: 10.1073/pnas.1016404108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fehling SK, Lennartz F, Strecker T. 2012. Multifunctional nature of the arenavirus RING finger protein Z. Viruses 4:2973–3011. doi: 10.3390/v4112973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salvato M, Shimomaye E, Oldstone MB. 1989. The primary structure of the lymphocytic choriomeningitis virus L gene encodes a putative RNA polymerase. Virology 169:377–384. doi: 10.1016/0042-6822(89)90163-3. [DOI] [PubMed] [Google Scholar]
- 17.Sanchez AB, de la Torre JC. 2005. Genetic and biochemical evidence for an oligomeric structure of the functional L polymerase of the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol 79:7262–7268. doi: 10.1128/JVI.79.11.7262-7268.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takeuchi O, Akira S. 2009. Innate immunity to virus infection. Immunol Rev 227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stetson DB, Medzhitov R. 2006. Type I interferons in host defense. Immunity 25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 20.Ng CT, Snell LM, Brooks DG, Oldstone MB. 2013. Networking at the level of host immunity: immune cell interactions during persistent viral infections. Cell Host Microbe 13:652–664. doi: 10.1016/j.chom.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huber JP, Farrar JD. 2011. Regulation of effector and memory T-cell functions by type I interferon. Immunology 132:466–474. doi: 10.1111/j.1365-2567.2011.03412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carnec X, Baize S, Reynard S, Diancourt L, Caro V, Tordo N, Bouloy M. 2011. Lassa virus nucleoprotein mutants generated by reverse genetics induce a robust type I interferon response in human dendritic cells and macrophages. J Virol 85:12093–12097. doi: 10.1128/JVI.00429-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lan S, McLay L, Aronson J, Ly H, Liang Y. 2008. Genome comparison of virulent and avirulent strains of the Pichinde arenavirus. Arch Virol 153:1241–1250. doi: 10.1007/s00705-008-0101-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park MS, Shaw ML, Munoz-Jordan J, Cros JF, Nakaya T, Bouvier N, Palese P, Garcia-Sastre A, Basler CF. 2003. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J Virol 77:1501–1511. doi: 10.1128/JVI.77.2.1501-1511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McLay L, Lan S, Ansari A, Liang Y, Ly H. 2013. Identification of virulence determinants within the L genomic segment of the Pichinde arenavirus. J Virol 87:6635–6643. doi: 10.1128/JVI.00044-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pythoud C, Rodrigo WW, Pasqual G, Rothenberger S, Martinez-Sobrido L, de la Torre JC, Kunz S. 2012. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase IKKepsilon. J Virol 86:7728–7738. doi: 10.1128/JVI.00187-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar N, Wang J, Lan S, Danzy S, McLay Schelde L, Seladi-Schulman J, Ly H, Liang Y. 2012. Characterization of virulence-associated determinants in the envelope glycoprotein of Pichinde virus. Virology 433:97–103. doi: 10.1016/j.virol.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asper M, Sternsdorf T, Hass M, Drosten C, Rhode A, Schmitz H, Gunther S. 2004. Inhibition of different Lassa virus strains by alpha and gamma interferons and comparison with a less pathogenic arenavirus. J Virol 78:3162–3169. doi: 10.1128/JVI.78.6.3162-3169.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baize S, Pannetier D, Faure C, Marianneau P, Marendat I, Georges-Courbot MC, Deubel V. 2006. Role of interferons in the control of Lassa virus replication in human dendritic cells and macrophages. Microbes Infect 8:1194–1202. doi: 10.1016/j.micinf.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Fan L, Briese T, Lipkin WI. 2010. Z proteins of New World arenaviruses bind RIG-I and interfere with type I interferon induction. J Virol 84:1785–1791. doi: 10.1128/JVI.01362-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xing J, Ly H, Liang Y. 2015. The Z proteins of pathogenic but not nonpathogenic arenaviruses inhibit the RIG-I-like receptor-dependent interferon production. J Virol 89:2944–2955. doi: 10.1128/JVI.03349-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang C, Kolokoltsova OA, Yun NE, Seregin AV, Poussard AL, Walker AG, Brasier AR, Zhao Y, Tian B, de la Torre JC, Paessler S. 2012. Junin virus infection activates the type I interferon pathway in a RIG-I-dependent manner. PLoS Negl Trop Dis 6:e1659. doi: 10.1371/journal.pntd.0001659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Groseth A, Hoenen T, Weber M, Wolff S, Herwig A, Kaufmann A, Becker S. 2011. Tacaribe virus but not Junin virus infection induces cytokine release from primary human monocytes and macrophages. PLoS Negl Trop Dis 5:e1137. doi: 10.1371/journal.pntd.0001137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trinchieri G. 2010. Type I interferon: friend or foe? J Exp Med 207:2053–2063. doi: 10.1084/jem.20101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, Schreiber RD, de la Torre JC, Oldstone MB. 2013. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340:207–211. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, Brooks DG. 2013. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340:202–207. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yun NE, Poussard AL, Seregin AV, Walker AG, Smith JK, Aronson JF, Smith JN, Soong L, Paessler S. 2012. Functional interferon system is required for clearance of Lassa virus. J Virol 86:3389–3392. doi: 10.1128/JVI.06284-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yun NE, Seregin AV, Walker DH, Popov VL, Walker AG, Smith JN, Miller M, de la Torre JC, Smith JK, Borisevich V, Fair JN, Wauquier N, Grant DS, Bockarie B, Bente D, Paessler S. 2013. Mice lacking functional STAT1 are highly susceptible to lethal infection with Lassa virus. J Virol 87:10908–10911. doi: 10.1128/JVI.01433-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kolokoltsova OA, Yun NE, Poussard AL, Smith JK, Smith JN, Salazar M, Walker A, Tseng CT, Aronson JF, Paessler S. 2010. Mice lacking alpha/beta and gamma interferon receptors are susceptible to Junin virus infection. J Virol 84:13063–13067. doi: 10.1128/JVI.01389-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bradfute SB, Stuthman KS, Shurtleff AC, Bavari S. 2011. A STAT-1 knockout mouse model for Machupo virus pathogenesis. Virol J 8:300. doi: 10.1186/1743-422X-8-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patterson M, Seregin A, Huang C, Kolokoltsova O, Smith J, Miller M, Smith J, Yun N, Poussard A, Grant A, Tigabu B, Walker A, Paessler S. 2014. Rescue of a recombinant Machupo virus from cloned cDNAs and in vivo characterization in interferon (αβ/γ) receptor double knockout mice. J Virol 88:1914–1923. doi: 10.1128/JVI.02925-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baize S, Marianneau P, Loth P, Reynard S, Journeaux A, Chevallier M, Tordo N, Deubel V, Contamin H. 2009. Early and strong immune responses are associated with control of viral replication and recovery in Lassa virus-infected cynomolgus monkeys. J Virol 83:5890–5903. doi: 10.1128/JVI.01948-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mahanty S, Bausch DG, Thomas RL, Goba A, Bah A, Peters CJ, Rollin PE. 2001. Low levels of interleukin-8 and interferon-inducible protein-10 in serum are associated with fatal infections in acute Lassa fever. J Infect Dis 183:1713–1721. doi: 10.1086/320722. [DOI] [PubMed] [Google Scholar]
- 44.Grant A, Seregin A, Huang C, Kolokoltsova O, Brasier A, Peters C, Paessler S. 2012. Junin virus pathogenesis and virus replication. Viruses 4:2317–2339. doi: 10.3390/v4102317. [DOI] [PMC free article] [PubMed] [Google Scholar]