

ABSTRACT
The extreme stability of the latent HIV-1 reservoir in the CD4+ memory T cell population prevents viral eradication with current antiretroviral therapy. It has been demonstrated that homeostatic T cell proliferation and clonal expansion of latently infected T cells due to viral integration into specific genes contribute to this extraordinary reservoir stability. Nevertheless, given the constant exposure of the memory T cell population to specific antigen or bystander activation, this reservoir stability seems remarkable, unless it is assumed that latent HIV-1 resides exclusively in memory T cells that recognize rare antigens. Another explanation for the stability of the reservoir could be that the latent HIV-1 reservoir is associated with an unresponsive T cell phenotype. We demonstrate here that host cells of latent HIV-1 infection events were functionally altered in ways that are consistent with the idea of an anergic, unresponsive T cell phenotype. Manipulations that induced or mimicked an anergic T cell state promoted latent HIV-1 infection. Kinome analysis data reflected this altered host cell phenotype at a system-wide level and revealed how the stable kinase activity changes networked to stabilize latent HIV-1 infection. Protein-protein interaction networks generated from kinome data could further be used to guide targeted genetic or pharmacological manipulations that alter the stability of latent HIV-1 infection. In summary, our data demonstrate that stable changes to the signal transduction and transcription factor network of latently HIV-1 infected host cells are essential to the ability of HIV-1 to establish and maintain latent HIV-1 infection status.
IMPORTANCE The extreme stability of the latent HIV-1 reservoir allows the infection to persist for the lifetime of a patient, despite completely suppressive antiretroviral therapy. This extreme reservoir stability is somewhat surprising, since the latently HIV-1 infected CD4+ memory T cells that form the structural basis of the viral reservoir should be exposed to cognate antigen over time. Antigen exposure would trigger a recall response and should deplete the reservoir, likely over a relatively short period. Our data demonstrate that stable and system-wide phenotypic changes to host cells are a prerequisite for the establishment and maintenance of latent HIV-1 infection events. The changes observed are consistent with an unresponsive, anergy-like T cell phenotype of latently HIV-1 infected host cells. An anergy-like, unresponsive state of the host cells of latent HIV-1 infection events would explain the stability of the HIV-1 reservoir in the face of continuous antigen exposure.
INTRODUCTION
Despite the importance of latent human immunodeficiency virus type 1 (HIV-1) infection for the ability of the virus to persist even in the face of otherwise successful antiretroviral therapy (ART), our understanding of how latent HIV-1 infection is controlled at the molecular level remains incomplete. As a result, it has proven difficult to develop targeted and efficient therapeutic strategies that trigger HIV-1 reactivation and thus allow for subsequent eradication of HIV-1 infection.
Once antiretroviral therapy is initiated, viral infection is thought to be sustained primarily by a long-lived reservoir associated with the memory CD4+ T-cell population (1–3). This latent HIV-1 reservoir is extremely stable, and natural eradication of a reservoir consisting of only 105 cells could take more than 60 years (4). The fact that to date, latent HIV-1 infection has been described mostly in the memory T cell population seems to justify the extraordinary stability of the viral reservoir. However, the exact functional relationship between lifelong immunological memory and the stability of the latent HIV-1 reservoir has not been defined in detail. While T cell memory can persist for the lifetime of an individual, individual memory T cells have a significantly shorter half-life than the latent HIV-1 reservoir.
Hellerstein et al. determined that the overall half-life of CD4+ or CD8+ T cell populations in healthy subjects was 87 or 77 days, respectively. In untreated HIV-1-seropositive patients, CD4+ or CD8+ T cell populations had significantly reduced half-lives of 24 or 22 days, respectively (5). In subsequent studies, the half-life of individual CD4+ central memory T cells (TCM cells), which are thought to serve as the primary reservoir of latent HIV-1 infection, has been measured at below or around 20 days (6) or as long as 4.8 months (7). The half-life of CD4+ TCM cells seems to be about 50% that of CD8+ TCM cells. While we could not find literature specifically addressing the half-life (τ1/2) of CD4+ TCM cells in HIV-1 patients, a recent study suggested that the CD8+ TCM half-life seems reduced from a τ1/2 of ∼100 days to a τ1/2 of ∼50 days (8). Even if we used a τ1/2 of ∼50 days for latently HIV-1 infected CD4+ TCM cells and ignored results showing that CD4+ TCM cells are generally shorter-lived than CD8+ TCM cells, assuming the presence of 1 × 106 latently HIV-1 infected CD4+ TCM cells at any given time, it would take ∼3 years for the last latently infected TCM cell to disappear. This is obviously not the case.
As such, latently HIV-1 infected TCM cells must undergo homeostatic proliferation in the absence of HIV-1 reactivation (9). Unlike naïve cells, TCM cells seem to rely on a combination of interleukin 7 (IL-7) and IL-15 for their survival and for occasional cell division without requiring signals stemming from the recognition of cognate antigens presented by major histocompatibility complex (MHC) molecules (10, 11). A combination of IL-7 and IL-15 that would reflect the in vivo situation has never been tested, but it has been shown in vitro that IL-7 can induce limited T cell proliferation, without triggering HIV-1 reactivation in all latently infected cells of a given reservoir (12). In addition, Wagner et al. recently demonstrated that HIV integration into specific genes may promote proliferation and result in clonal expansion of latently HIV infected cells, adding a virus-induced mechanism that would further slow the decay of the viral reservoir during ART (13). Proliferation in the absence of reactivation is certainly a feasible scenario, since latent infection is obviously stable in the presence of cell division, as demonstrated by the existence of many latently HIV-1 infected T cell lines (14–17).
However, despite the existence of cell proliferation mechanisms that stabilize the latent infection pool, it seems most difficult to explain how the latent HIV-1 infection pool remains stable despite likely continuous exposure to antigen. Over time, many latently infected T cells should be exposed to their cognate antigen, which, in turn, would trigger a recall response (18). Given the vigor of an antigen-induced recall response, it is difficult to conceive that a latent HIV-1 infection event would not become reactivated. Under the assumption that latent HIV-1 infection events are established in functional memory T cells, a continuous contraction of the reservoir should be observed, unless it is assumed that latent HIV-1 infection events are established only in memory T cells that recognize extremely rare antigens, or are established exclusively in T cells that recognize HIV-1-specific antigens, which completely disappear after the initiation of ART. In fact, despite the possibility of proliferation as a stabilizing factor for the size of the latent reservoir, some progressive contraction of the reservoir has been reported (19). But that study also suggested the presence of an extremely stable reservoir around a core of less-differentiated memory T cell subsets that no longer contracts.
A logical explanation for the extraordinary stability of this latent HIV-1 reservoir in the face of constant antigen exposure could be that latent HIV-1 infection events are associated with memory T cells that have been altered by the actual infection event to take on an unresponsive or, in some form, anergic phenotype. Such an unresponsive state of the host cells could also explain the recalcitrance of latent HIV-1 infection to previous therapeutic reactivation attempts that aimed at triggering cellular activation mechanisms.
Generally, the term “anergy” defines the inability of an immune cell to mount a complete response against its target; it is a state of lymphocyte unresponsiveness induced by suboptimal stimulation (20). Anergy in T cells affects aspects of cell proliferation, cytokine induction, cell signaling, and cell metabolism (21). Anergy is commonly observed in chronic infections (22). T cell anergy is usually the result of aberrant activation of the T cell receptor and/or lack of costimulation as provided through costimulatory receptors such as CD28 and results in a long-lived but functionally unresponsive state (23, 24). These T cells exit the cell cycle and are arrested in the G0 state (25). Under certain circumstances, T cell anergy is reversible and does not broadly inhibit cell signaling through a particular signal transduction pathway but tends to be selective for particular downstream events (26).
For both simian immunodeficiency virus (SIV) and HIV-1 infection, there are many reports describing a similar loss of T cell functionality for both CD4− and CD8+ T cells. Physical and functional loss of memory CD4+ T cells has been described for SIV infection (27) and for HIV-1 patients (28). In virus controllers, central memory T cell populations seemed to maintain near-normal numbers and to be functionally preserved (29, 30), indicating that exposure to viral antigen plays a key role in the establishment of a dysfunctional, unresponsive memory T cell population. Also, in patients who are placed on therapy very early after infection, and who maintain a high level of immunological functionality, the latent HIV-1 reservoir seems to be smaller than that of patients placed on ART during the chronic stage of infection, after their CD4 counts had significantly dropped (31).
At the single-cell level, lack of function in anergic T cells has been associated with changes in several signal transduction pathways (e.g., NF-κB, extracellular signal-regulated kinase [ERK], Jun N-terminal protein kinase [JNK], cell cycle) (32–34). At the molecular level, T cell anergy is thus the result of stable changes in the kinase activities of several key signal transduction pathways.
Here we demonstrate that latently infected T cells are indeed phenotypically altered in ways that are consistent with the idea of anergy or an unresponsive host cell phenotype. Specifically, we demonstrate that latently HIV-1 infected T cells exhibit an altered NF-κB response and that manipulation of hallmark proteins associated with anergy, such as GRAIL (gene related to anergy in lymphocytes) or ICER (inducible cyclic AMP [cAMP] early repressor), affects the stability of latent HIV-1 infection. Kinome analysis confirms these stable phenotypic changes at the global level, and importantly, we also demonstrate that kinomic data can be used to guide the identification of pharmacological intervention strategies that (i) can alter the level of HIV-1 latency establishment and (ii) can affect the stability of latent HIV-1 infection events.
MATERIALS AND METHODS
Cell culture, plasmids, and reagents.
All T cell lines were maintained in RPMI 1640 medium supplemented with 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% heat-inactivated fetal bovine serum (FBS). FBS was obtained from HyClone (Logan, UT) and was tested on a panel of latently infected cells to ensure that the FBS batch utilized did not spontaneously trigger HIV-1 reactivation (15, 16). The latently infected J89GFP T cells, CA5 T cells, and EF7 T cells have been described previously (15, 17). In CA5 T cells, the virus is integrated into the RMB12/CPNE1 gene in the same-sense orientation relative to the transcriptional direction of the host gene. In EF7 cells, the virus is integrated into the WHSC1 gene in the converse-sense orientation relative to the transcriptional direction of the host gene. The green fluorescent protein (GFP) reporter viruses utilized express GFP as late genes, which means that GFP is expressed only when all viral RNA types are produced and translation of viral RNA is enabled. J2574 reporter T cells have been described previously (35). Briefly, J2574 reporter T cells were generated by lentiviral transduction of Jurkat T cells with an HIV-1 reporter construct (p2574) in which an HIV-1 subtype B long terminal repeat (LTR) controls the expression of GFP. The HIV-1 LTR and the GFP gene are separated by a 2,500-bp spacer element. Following lentiviral transduction of Jurkat cells, all cells that spontaneously expressed GFP were removed by cell sorting. The GFP-negative population was then activated with 13-phorbol-12-myristate acetate (PMA) to identify all cells that would harbor an inducible LTR-GFP-LTR integration event. Cells that turned GFP positive following stimulation were again selected by cell sorting. GFP expression in this population ceased after a few days, leaving a population of GFP-negative reporter cells. The number of founder cells for this population is calculated to represent >100,000 individual integration events (35). Since GFP expression in J2574 cells is driven by an integrated LTR-GFP-LTR vector, in trans provision of HIV Tat protein by the full-length HIV used in the experiments is required. J2574 cells were then infected to generate latently infected T cell clones. The molecular HIV-1 clones utilized have been described previously (36, 37). The LAI subclones utilized are derived from the subtype B LAI molecular clone (38) but have a subtype-specific 3′ LTR from position −147 to +63, stretching from the noncoding part of U3 to the R region, including the complete trans-activation response element (TAR) hairpin. For simplicity, we refer to the viruses as LAI-B for the parental subtype B construct and as LAI-A, LAI-C, and LAI-E for the viruses that have the −147 to −1 LTR sequence of a prototypic HIV-1 subtype A, subtype C, or subtype E (CRF01_AE), respectively. J2574 cells were infected with one of the LAI subclones. On day 2 postinfection, reverse transcriptase (RT) inhibitors were added to prevent ongoing viral replication, which would otherwise interfere with the stability of the established latent reservoir. Once active infection had subsided, T cell clones were established by limiting dilution. Using a high-throughput screening (HTS) flow cytometry setup, we screened each clone generated for baseline GFP expression and for GFP expression 48 h after stimulation with the phorbol ester PMA. Clones that were baseline GFP negative (maximum 5% GFP background) and responded to PMA stimulation with an increase in the percentage of GFP-positive cells to >60% were selected, and the presence of an integrated virus was confirmed by PCR for HIV-1 tat sequences by using genomic cellular DNA from the respective T cell clone as the template.
The phorbol esters PMA and prostratin were purchased from Sigma. Recombinant human tumor necrosis factor alpha (TNF-α) was obtained from R&D. The anticancer compound and protein kinase C (PKC) activator bryostatin was purchased from EMD Millipore. The Dyrk1a inhibitors (1Z)-1-(3-ethyl-5-hydroxy-2(3H)-benzothiazolylidene)-2-propanone (INDY) and proINDY were purchased from Tocris. All antibodies were purchased from BD Pharmingen.
Human subjects.
Healthy adults (n = 9) and HIV patients (n = 8) provided written informed consent to provide all biological specimens and clinical data used in this study. Research records were kept confidential, meeting specifications for this project approved by the UAB Institutional Review Board for Human Use (IRB).
HIV-1 latency establishment assays.
To determine latency establishment levels under defined experimental conditions, 20 to 30 individual cell cultures are infected over a wide range of multiplicities of infection (MOIs). On day 3 or 4 postinfection, active infection levels are determined by measuring the percentage of GFP-positive cells, since all experiments use either GFP reporter viruses or GFP reporter T cell lines. To determine the size of the latent reservoir formed between days 14 and 21 postinfection (17), a sample of each infection culture is taken and is stimulated with PMA overnight, which will trigger the reactivation of latent HIV-1 infection (GFP-positive cells). By subtracting the percentage of residual active infection in untreated cell cultures from the level of active infection in a sample of each culture in which HIV-1 reactivation has been triggered by PMA, using GFP expression as a surrogate marker of HIV-1 infection, we can calculate the size of the latent reservoir that has been established.
HIV-1 reactivation experiments.
For all HIV-1 reactivation experiments using CA5 cells, EF7 cells (39, 40), or latently infected cells of the J-LAI series (35), GFP is used as a surrogate marker of HIV-1 expression. GFP expression is routinely measured 24 or 48 h postinduction using flow cytometric analysis and is expressed as the percentage of GFP-positive cells.
Expression plasmids.
For the construction of MSCV/puro-IκB, the IκB gene was amplified using an IκB signaling probe (Clontech) as the template and oligonucleotides 5′-BglII-ggccagatctATGTTCCAGGCGGCCGAGCG and 3′-EcoRI-gcgcgaattcTCATAACGTCAGACGCTGGCCTCC (lowercase letters stand for restriction sites), and the product was cloned into the multiple cloning site of pMSCV/puro (Clontech) using BglII and EcoRI. For the construction of pMSCV/puro-ICER1, the ICER gene product was cloned using HUT78 cell-derived cDNA as the template and oligonucleotides 5′-XhoI-tataCTCGAGatggctgtaactggagat and 3′-EcoRI-ctcgGAATTCttactctactttatggca, and the product was cloned into the multiple cloning site of pMSCV/puro (Clontech) using XhoI and EcoRI. For the construction of pMSCV/puro-GRAIL, the GRAIL gene product was cloned using 293T cell-derived cDNA as the template and oligonucleotides 5′-XhoI-aattCTCGAGatggggccgccgcct and 3′-EcoRI-gtccGAATTCttaagatttaatttctcgaacagcag, and the product was cloned into the multiple cloning site of pMSCV/puro (Clontech) using XhoI and EcoRI.
TransAM assays for NF-κB.
NF-κB p50 and p65 activities in nuclear extracts of cells were determined using TransAM assays (Active Motif). All experiments were performed according to the manufacturer's instructions. TransAM assays measure the ability of activated NF-κB to bind to a NF-κB consensus sequence in solution, with a 5- to 10-fold higher sensitivity than gel shift assays.
Flow cytometry.
Infection levels in the cell cultures were monitored by flow cytometric analysis of GFP expression, which serves as a surrogate marker for HIV-1 expression. Flow cytometric analysis was performed on a Guava easyCyte (Guava Technologies, Inc.), FACSCalibur, or LSR II (Becton Dickinson) flow cytometer. Cell sorting experiments were performed using a FACSAria flow cytometer (Becton Dickinson). All antibodies used for immunologically phenotyping primary T cells were obtained from BD Pharmingen. Data were analyzed using CellQuest (Becton Dickinson), FlowJo (Tree Star Inc.), or Guava Express (Guava Technologies, Inc.) software.
Kinomic analysis.
Any kinome array technology is limited to the kinomic space that is covered. However, predictor algorithms that are used to build hypotheses generating protein interaction networks that guide drug target identification or explain the molecular control of various biological processes are more predictive as the quality and quantity of input data increase. To this end, we combined two kinome array methods to generate a comprehensive set of data describing the stable phenotypic changes in kinase expression and activity that characterize latently HIV-1 infected T cells. Kinome data were generated using PamGene kinase substrate peptide-based analysis and Kinex antibody microarray-based analysis.
PamGene kinase substrate peptide-based analysis.
Kinome profiling using substrate chips was performed on the PamStation 12 platform (PamGene, Hertogenbosch, The Netherlands) as described previously (41). Utilizing high-throughput peptide microarrays spotted with either 144 individual tyrosine-phosphorylatable peptides on the PTK array or 144 serine- and/or threonine-phosphorylatable peptides on the STK array, the phosphorylation activity of the kinase composition in different cell lysates was measured. Briefly, all peptides are composed of 12 to 15 amino acids that are imprinted onto an aluminum oxide matrix allowing exposure to kinases in order to measure activity in lysates that are pumped through these peptide-rich matrices. Phosphospecific fluorescein isothiocyanate (FITC)-conjugated antibodies were used to detect peptide phosphorylation. Images of FITC-dependent fluorescent signals were captured via a computer-controlled charge-coupled device (CCD) camera with kinetic image capture over time and over multiple exposures.
The kinomic profiles of different samples were compared by using BioNavigator software, version 5 (PamGene), to identify significantly different phosphopeptides (P, <0.05 by t test). Upstream kinases were identified by scoring potential kinases based on their prevalences in the top five kinase scoring lists for each phosphopeptide as mapped in the Kinexus upstream kinase database (www.phosphonet.ca).
Kinex antibody microarray-based analysis.
To perform the Kinex analysis, 50 μg of lysate protein (∼5 × 106 cells) from each sample was covalently labeled with a proprietary fluorescent dye according to the manufacturer's instructions (Kinexus, Canada). After the completion of the labeling reaction, free dye was removed by gel filtration. After blocking of nonspecific binding sites on the array, an incubation chamber was mounted onto the microarray to permit the loading of one control sample and one latently HIV-1 infected sample side by side on the same chip. Following sample incubation, unbound proteins were washed away. The KAM-850 chips utilized are spotted in duplicate with >850 antibodies. Five hundred ten panspecific antibodies used in the chip provide for the detection of 189 protein kinases, 31 protein phosphatases, and 142 regulatory subunits of these enzymes and other cell signaling proteins. By this means, the microarrays provide information about the expression levels of these target proteins. Three hundred forty phosphosite-specific antibodies track the nonredundant phosphorylation of 128 sites in protein kinases, 4 sites in protein phosphatases, and 155 sites in other cell signaling proteins. The microarrays provide information about the activation states of the various kinases, which are usually linked to their phosphorylation states.
Each array produced a pair of 16-bit images, which were captured with a Perkin-Elmer ScanArray Reader laser array scanner (Waltham, MA). Signal quantification was performed with ImaGene, version 8.0, from BioDiscovery (El Segundo, CA) with predetermined settings for spot segmentation and background correction. The background-corrected raw intensity data were logarithmically transformed with base 2. Since Z normalization in general displays greater stability as a result of examining where each signal falls in the overall distribution of values within a given sample, as opposed to adjusting all of the signals in a sample by a single common value, Z scores are calculated by subtracting the overall average intensity of all spots within a sample from the raw intensity for each spot, and dividing by the standard deviations (SD) of all of the intensities measured within each sample (42). Z ratios were further calculated by taking the difference between the averages of the observed protein Z scores and dividing by the SD of all of the differences for that particular comparison. Calculated Z ratios have the advantage that they can be used in multiple comparisons without further reference to the individual conditional SD by which they were derived.
Data analysis.
MetaCore software (Thomson Reuters) was used to generate the shortest-pathway interaction networks that are presented. Pathway-specific Gene Ontology (GO) filters or tissue-specific filters were used to prioritize nodes and edges. MetaCore was further used to identify the pathway associations of the input kinases identified.
Statistical analysis.
For the immune phenotyping study of HIV patients, statistical analyses using unpaired t tests were carried out across biomarkers of interest using GraphPad Prism, version 5.0d (GraphPad Software, La Jolla, CA, USA).
RESULTS
Functional correlation between anergy induction and the establishment of HIV-1 latency.
The idea that anergy-related mechanisms at the molecular level could contribute to the stability of latent HIV-1 infection in T cells can be experimentally probed at several levels, most importantly by directly testing the effect of anergy induction on HIV-1 latency establishment. Stimulation with ionomycin, a calcium ionophore, has been demonstrated to trigger T cell anergy. This effect has been reported for primary T cells as well as for Jurkat T cells (43, 44). We thus tested whether ionomycin treatment in our model system would lead to increased establishment of latent HIV-1 infection at the population level. Over a 7-day period, Jurkat T cell cultures were treated with three doses of ionomycin. Four days following the last ionomycin treatment, the cells were infected with a GFP reporter virus, and for each infection culture, active infection levels were initially determined on day 3 postinfection using flow cytometry. The results generated for 30 individual paired infection cultures by infecting control and ionomycin-treated cells with increasing amounts of virus are shown in Fig. 1. Active infection levels determined on day 4 postinfection were similar (Fig. 1A), but the pool of silent HIV-1 infection events was already found to be increased in ionomycin-treated T cells (Fig. 1B). On day 14 postinfection, we determined the size of the stable latent HIV-1 reservoir in each infection culture (Fig. 1C). The results show that ionomycin treatment altered the T cells to allow for an increased level of HIV-1 latency establishment. Independently of the initial infection levels, the established latent HIV-1 infection event pool in ionomycin-treated cells was found to be about twice as large as the latent infection pool in untreated control T cells, suggesting that anergy induction provides an intracellular host cell environment that is conducive to HIV-1 latency establishment.
FIG 1.
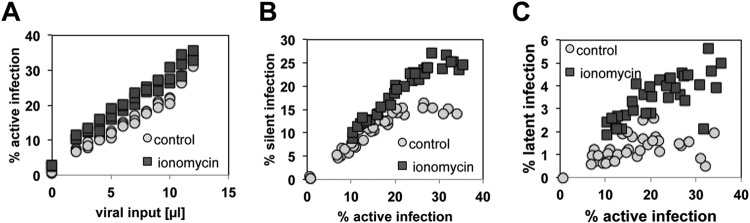
Anergy induction results in a higher level of HIV-1 latency establishment. The calcium ionophore ionomycin induces T cell anergy. Following three applications of ionomycin to Jurkat cells over a period of 7 days and a resting period of 4 days, 30 individual infection cultures each of untreated control Jurkat cells and ionomycin-treated Jurkat cells were initiated over a wide range of MOIs. (A) Active initial infection levels were determined on day 4 postinfection by using PMA-induced reactivation, with GFP as a surrogate marker for HIV-1 infection. (B) The size of the silent HIV-1 reservoir in each culture was plotted against the level of initially active infection as determined on day 3 postinfection. (C) The size of the latent HIV-1 reservoir in each culture as determined on day 14 postinfection was plotted against the level of initially active infection.
CDK2 inhibition promotes the establishment of HIV-1 latency.
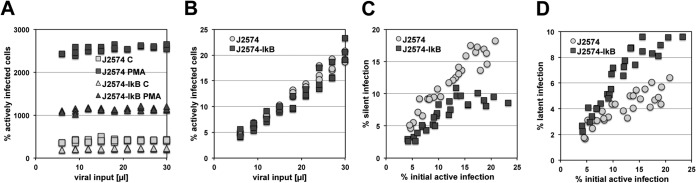
Chunder et al. provided genetic evidence that in the absence of cyclin-dependent kinase 2 (CDK2) activity, CD4+ T cells enter a hyporesponsive state even when stimulated by signals that normally allow T cells to escape anergy (45). By extension, we proposed that CDK2 inhibitors would at least transiently create a similar cell phenotype and thereby affect the establishment of HIV-1 latency. To test this idea, we treated Jurkat T cells with roscovitine, a clinically relevant CDK2 inhibitor (46, 47). In independent infection experiments, control and roscovitine-treated cells were then infected over a wide range of MOIs, resulting in initial infection levels ranging from 15 to 60% in control cells and slightly lower in the paired roscovitine-treated infection cultures, capping maximal achievable infection in these cell populations at ∼40% (Fig. 2A). An important consideration here is a possible inhibitory side effect of roscovitine on CDK9, a key component of the p-TEFb complex, which, in turn, is essential for HIV-1 transcription and is thought to be involved in latency control (48–51). However, as seen in Fig. 2B, maximal HIV-1 expression, measured by using GFP mean channel fluorescence (MCF) as a direct surrogate marker, was only marginally reduced in roscovitine-treated infected cells. These data suggest that in this system, roscovitine has only minor inhibitory effects on CDK9 and that the lower infection levels observed are thus likely based on roscovitine-induced changes in the overall intracellular environment and are not limited to a roscovitine effect that specifically affects transcriptional initiation at the integrated HIV-1 LTRs. As determined on day 4 postinfection, the silent infection reservoir in roscovitine-treated cells was larger than that in control cells (Fig. 2C). This difference increased further on day 14, when stable latent HIV-1 infection reservoirs had formed in the various infection cultures. In agreement with data we have generated previously (17), the size of the stable latent HIV-1 reservoir in control cells decreased during the transition period from silent infection (day 4) to stable latent infection (day 14). However, the size of the silent reservoir in roscovitine-treated cells remained almost stable during this phase of transition to stable latency. Since roscovitine was added only at the beginning of the experiment, this would suggest that roscovitine had long-term effects on the intracellular environment of the host cells that provided for greater stability of the integrated latent infection events.
FIG 2.

CDK2 inhibition boosts HIV-1 latency establishment. Reduced CDK2 activity has been associated with an unresponsive T cell phenotype. The data presented thus test the effect of the CDK2 inhibitor roscovitine on the establishment of latent infection. (A) Effect of the CDK2 inhibitor roscovitine (10 μM) on HIV-1 susceptibility or active de novo HIV-1 infection levels on day 4 postinfection. (B) Effect of the CDK2 inhibitor roscovitine (10 μM) on maximum HIV-1 expression levels on day 4 postinfection, plotted as a function of the active infection level. (C) Establishment of reservoirs of silent infection events (day 4 p.i.) in the presence or absence of roscovitine, plotted as a function of initial active infection levels achieved in each individual culture. (D) Establishment of latent HIV-1 infection reservoirs in control and roscovitine-treated cell cultures as determined on day 14 postinfection. Each data set represents a summary of 30 paired infection cultures generated at different MOIs.
We next tested the ability of a series of other CDK2 inhibitors to stabilize latent HIV-1 infection. According to an outstanding study performed by Anastassiadis et al. that compared the inhibitory effects of 178 known kinase inhibitors on 300 kinases, the most potent and specific CDK2 inhibitors are purvalanol, roscovitine, CDK2 inhibitor 3, and kenpaullone (52).
We tested the concentration-dependent effect of each of those inhibitors on PMA-induced HIV-1 reactivation in two different latently HIV-1 infected T cell lines. In agreement with the idea that CDK2 activity levels are a crucial component of the ability of the virus to remain in a latent state, all inhibitors efficiently suppressed HIV-1 reactivation. The data for latently infected CA5 T cells are shown in Fig. 3. CA5 T cells have one integrated copy of an NL4-3-based GFP reporter virus, which is found integrated in the rbm12 gene. The virus and the host gene are expressed in the same transcriptional direction. Following stimulation with PMA, prostratin, or TNF-α, HIV-1 reactivation is triggered in ∼90% of the CA5 T cell population. In the experiments with CA5 T cells, all CDK2 inhibitors efficiently inhibited PMA-induced HIV-1 reactivation without affecting viability. The results of experiments using EF7 T cells, in which the latent HIV-1 infection event is integrated in the converse sense orientation relative to the transcriptional orientation of the whsc1 host gene, confirm these results (see Fig. S1 in the supplemental material).
FIG 3.

CDK2 inhibition affects activation-induced HIV-1 reactivation. If a reduction in CDK2 activity renders T cells unresponsive, we would expect CDK2 inhibitors to reduce the ability of otherwise potent agents, such as PMA, to trigger HIV-1 reactivation. We thus tested the concentration-dependent effects of a series of specific CDK2 inhibitors in preventing PMA-induced HIV-1 reactivation in CA5 T cells. The CDK2 inhibitors used were roscovitine (A), purvalanol (B), CDK2 inhibitor III (CDK2i III) (C), and kenpaullone (D). Cells were pretreated for 2 h with the indicated concentration of the respective CDK2 inhibitor, followed by stimulation with a high dose of PMA (10 nM). HIV-1 reactivation levels were determined as the percentage of GFP-positive cells 48 h post-PMA addition. Data for at least 3 independent experiments are presented.
In agreement with published data showing that lack of CDK2 activity produces a host cell state that is unresponsive to relevant activating stimuli (45), these data demonstrate that cellular regulation of CDK2 activity is central to latency control and that low-level CDK2 activity produces a host cell environment that stabilizes latent infection.
Effect of altered GRAIL or ICER expression on the stability of latent HIV-1 infection.
Overexpression of GRAIL (gene related to anergy in lymphocytes) or ICER (inducible cAMP early repressor) is a reported hallmark of T cell anergy (53, 54). GRAIL, or RNF128, is a type I transmembrane zinc RING finger protein in the endocytic pathway that functions as an E3 ubiquitin ligase that is upregulated in anergic T cells. ICER, also called the cAMP response element modulator (CREM), is a protein that in anergic T cells has been reported to form complexes with CREB and bind to the −180 site of the IL-2 promoter (55). Further, it has been shown that ICER specifically binds to several NFAT/AP-1 (nuclear factor of activated T cells/activating protein-1) composite DNA sites essential for the activation of the IL-2 promoter. The similarities of the IL-2 promoter and the HIV-1 LTR are apparent, and we also recently produced evidence that the AP-1 site adjacent to the NF-κB/NFAT binding sites in the LTR is essential for latency control (35). As seen in Fig. 4, overexpression of either GRAIL or ICER by means of retroviral transduction, without enrichment of transduced cells by puromycin selection, indeed resulted in lower levels of HIV-1 reactivation following stimulation with PMA in two latently HIV-1 infected T cell lines, CA5 and EF7 cells. These results demonstrate that expression of two hallmark T cell anergy genes stabilized latent HIV-1 infection by rendering cells unresponsive to a T cell stimulus.
FIG 4.
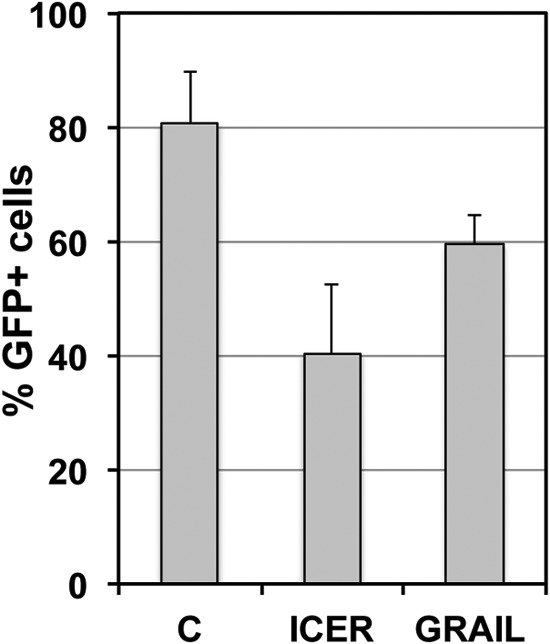
Effects of GRAIL (gene related to anergy in lymphocytes) and ICER (inducible cAMP early repressor) expression on activation-induced HIV-1 reactivation. High levels of GRAIL and ICER are hallmarks of T cell anergy. Latently HIV-1 infected CA5 T cells were retrovirally transduced to express increased levels of ICER or GRAIL. Two days postransduction, the parental cells and the retrovirally transduced cell populations were stimulated with PMA (10 ng/ml), and the level of HIV-1 reactivation was measured by flow cytometry using GFP as a surrogate marker of HIV-1 expression. The cells were not selected with puromycin. For many transgenic cell lines, we observe relatively fast adaptation to a new phenotype or reversion to the parental phenotype within days, which no longer represents the actual initial transgenic effect. The estimated transduction efficacy was 40 to 60%.
Latently HIV-1 infected T cells exhibit an altered kinetic NF-κB response.
Another reported hallmark of T cell anergy is an altered NF-κB response, likely resulting from changes in NF-κB protein availability and an altered kinase composition upstream of NF-κB (33, 56). As an extension of the idea that latently HIV-1 infected host cells would exhibit an anergy-like, unresponsive phenotype, we compared the kinetic NF-κB responses of several latently HIV-1 infected clonal T cell lines with the kinetic NF-κB response in Jurkat T cells. As seen in Fig. 5A, following PMA stimulation, Jurkat cells exhibited the typical sinus wave-shaped kinetic NF-κB p50 and NF-κB p65 responses (57). In stark contrast, latently HIV infected T cells (here CA5 T cells) would respond to stimulation with a single NF-κB activity spike with massively increased amplitude, which was not further sustained. This aberrant NF-κB response was found in all latently HIV-1 infected T cell clones we tested. Of particular interest is the fact that the altered kinetic NF-κB response profile was also found in four GFP reporter T cell lines infected with several different HIV-1 LAI mutants. The LAI mutants have LTR sequences derived from prototypic LTR enhancer/core sequences for the HIV-1 subtypes A, B, C1, and E (LAI-A, LAI-B, LAI-C, and LAI-E) (36), and we had demonstrated previously that these viruses exhibit different HIV-1 latency establishment behaviors (35). However, independently of the frequency of latent infection events that can be established by these viruses, all latently HIV-1 infected host T cells exhibited the same change in the kinetic NF-κB response (Fig. 5B to F), providing further evidence that phenotypic changes of the host cells affecting cell signaling pathways and specific transcription factor regulation are essential to the stability of latent HIV-1 infection. Since altered NF-κB functionality is a major hallmark of T cell anergy, these findings support the idea that latent HIV-1 infection is associated with a stable, unresponsive phenotype of the host T cells.
FIG 5.

Latently HIV-1 infected T cells exhibit an altered, distinct kinetic NF-κB activation profile. An altered NF-κB response is a hallmark of T cell anergy. (A) Kinetic NF-κB profiles for parental Jurkat cells and latently HIV-1 infected CA5 T cells were established for a period of 360 min following PMA stimulation using TransAm assays. Kinetic profiles are shown for NF-κB p65 and NF-κB p50. (B to F) With the characteristic overshooting NF-κB peak in the latently infected T cells occurring within the first 120 min to replace the sinus wave-shaped normal NF-κB response seen in uninfected cells, we monitored the kinetic NF-κB responses in four additional latently infected T cell lines over this period. T cells latently infected with J-LAI-A, J-LAI-B, J-LAI-C, or J-LAI-E were generated using LTR chimeric viruses that would reflect the prototypical transcription factor binding site compositions of subtypes A, B, C, and E, all within the backbone of the molecular HIV-1 LAI clone. All cells were generated using the J2574 GFP reporter T cell population. We had demonstrated previously that each of these viruses establishes a different level of latency at the population level (35). Shown are NF-κB kinetic profiles for unstimulated controls (C) and following TNF-α stimulation (TNF) for the parental J2574 T cells (B), LAI-A infection (high level of latency establishment) (C), LAI-B infection (reference virus) (D), LAI-C infection (high level of latency establishment) (E), and LAI-E infection (very low level of latency establishment) (F). Independently of the latency establishment phenotype of the virus, all latently infected T cell lines exhibited the same altered kinetic NF-κB activation profile.
Generation of a protein-protein interaction network (PIN) signature describing host cell control of latent HIV-1 infection.
Having produced experimental evidence that latent HIV-1 infection is associated with stable phenotypic changes of the infected host cells, we asked whether it is possible to identify and describe changes to the host cells due to latent HIV-1 infection events at a system-wide level. Since we demonstrated previously that certain kinases have a gatekeeper function for the control of latent HIV-1 infection (40), we employed kinome array analysis to characterize kinome-wide changes to the host cell that would stabilize latent HIV-1 infection.
To optimize data quality, we combined two methods of kinome analysis. Kinase substrate peptide-based analysis (PamGene) would allow us to measure differences in the kinase activity profile between uninfected control T cells and latently HIV-1 infected T cells. Antibody microarray-based kinome analysis (Kinexus) would allow us to quantify the presence and the phosphorylation state of the kinases in these two cell types. Importantly, the two methods cover partially different kinomic space, which also allowed us to maximize the total kinomic space assessed.
Due to the relatively large amounts of input material required for kinome array analysis and the dependence of the output data quality on the purity of the input material, the rarity of latently infected T cells in ex vivo patient material or even in models of latent HIV-1 infection in primary T cells precludes the use of primary T cell material at this time. Since it had been demonstrated previously that, at least with respect to kinase control of latent HIV-1 infection, data generated in T cell lines can be predictive for the outcome of experiments in primary T cell models of latent HIV-1 infection (39, 40), we used latently HIV-1 infected T cell lines for these studies. For the ensuing proof-of-concept studies, we chose to work with latently HIV-1 infected CA5 T cells.
Comparison of the kinase expression and phosphorylation states found in the lysates of Jurkat T cells and CA5 T cells by using Kinexus antibody arrays revealed differences in a total of 37 spots (see Table S1A in the supplemental material). PamGene substrate phosphorylation array analysis suggested that the phosphorylation status of 18 substrate peptides was significantly altered by the kinase activity present in CA5 T cell lysates from that in lysates from the parental Jurkat cells (see Table S1B in the supplemental material). By use of these peptides, 30 upstream kinases were identified by scoring potential kinases based on their prevalence in the top five kinase scoring lists for each phosphopeptide as mapped in the Kinexus upstream kinase database (www.phosphonet.ca). To maximize the kinomic space covered by the two methods, a data set comprising altered kinases detected by the Kinexus antibody array, altered peptide substrates, and the top five kinase scoring lists for each positive phosphopeptide as mapped in the Kinexus upstream kinase database was generated. This list was uploaded into MetaCore, and a one-step shortest-pathway protein-protein interaction network was generated (Fig. 6).
FIG 6.

Kinome analysis-derived protein-protein interaction network describing phenotypic changes of CA5 T cells enabling the stability of latent HIV-1 infection. Lysates of Jurkat and latently HIV-1 infected CA5 T cells were loaded onto Kinexus kinase antibody arrays, and the expression levels and phosphorylation states of a total of 37 kinases and phosphatases were determined. Only spots with altered expression or activity in CA5 T cells relative to that in parental Jurkat cells that had a Z′ ratio of >1.2 were included. The same lysates were loaded onto PamGene chips, which revealed a total of 18 substrate peptides to be differentially phosphorylated by the kinase activity present in latently infected CA5 T cells. Using the Kinexus upstream kinase database, a total of 30 kinases were predicted to exert this phosphorylation activity. The accumulated set of kinases was uploaded on MetaCore to generate the one-step interaction network presented, which is predicted to control latent HIV-1 infection. Detailed information on the regulation of the various altered kinases can be found in Table S1 in the supplemental material.
GO process analysis revealed enrichment of altered kinases/phosphatases that were associated with intracellular signal transduction (61%; P = 1.2e−53), regulation of cellular metabolism (65%; P = 9.3e−45), regulation of programmed cell death (54%; P = 5.591e−44), and, of particular interest for an immortalized T cell line-based model system of HIV-1 latency, cell cycle regulation (38%; P = 4.756e−33). Interestingly, at the molecular level, an anergic state is reflected in changes to signal transduction pathways controlling cell proliferation, cytokine induction, cell signaling, and metabolism.
PIM-1 and CDK2 inhibition affect latent HIV-1 infection in a synergistic manner.
Analysis of the PIN generated from the kinome data revealed that PIM-1 and CDK2 would converge into common downstream pathways with relevance for the stability of HIV-1 latency. For example, while the phosphorylation of NF-κB p65 on serine 276 by CDK2 activates the transcription of gene promoters (58), PIM-1 activity controls NF-κB signaling by stabilizing RelA/p65 (59). By extension, the convergence of these signal transduction pathways at the level of transcription factor control would suggest that a combination of CDK2 and PIM-1 inhibitors should have an additive or synergistic effect as HIV-1 reactivation inhibitors in this system. Based on the kinase inhibitor activity atlas provided by Anastassiadis et al. (52), PIM inhibitor IV (PIMi IV) and AS601245 would be the most potent and specific PIM-1 inhibitors currently available (AS601245 is listed as a JNK inhibitor, but in the kinase activity screen, it exerted only weak JNK-inhibitory activity but very potent PIM-1-inhibitory activity [see Table S2 in the supplemental material]). We have demonstrated previously that both PIMi IV and AS601245 inhibit HIV-1 reactivation in a model of HIV-1 latency in primary T cells (40, 41). Guided by the PIN, we combined either PIMi IV (Fig. 7A) or AS601245 (Fig. 7B) with roscovitine in dose-matrix experiments. In a concentration-dependent manner, either compound combination exerted synergistic effects and efficiently stabilized the latent state of the infection events by rendering the host cells refractory to the stimulatory signal (here PMA). These results provide an example of the utility of PINs generated from kinome data for the discovery of synergistic drug combinations that target latent HIV-1 infection.
FIG 7.

Protein-protein interaction networks reveal synergistic drug effects. The PIN depicted in Fig. 6 revealed the convergence of PIM-1 activity and CDK2 activity. In two dose-matrix experiments, we thus tested whether the PIN prediction would translate into the ability of PIM-1 inhibitor IV–roscovitine (A) and AS601245–roscovitine (B) combinations to act in concert as inhibitors of PMA-induced HIV-1 reactivation. For this purpose, roscovitine was titrated over a concentration range from 0.01 to 10 μM, whereas the two reported PIM-1 inhibitors were cross-titrated over a concentration range from 0.01 to 10 μM. PMA-induced reactivation was then measured as the percentage of GFP-positive cells 48 h poststimulation. Cell viability was not affected by the treatment.
Effects of IκB expression levels on the establishment of HIV-1 latency.
In agreement with the finding that the NF-κB response is kinetically altered in latently HIV-1 infected T cells, the highest altered spot on the Kinexus chip was IκB, which was found to be increased from 3,177 AU in control cells to 7,639 arbitrary units (AU) in latently infected CA5 T cells, with a Z′ ratio of 1.87 (see Table S1 in the supplemental material). To test whether IκB overexpression would have a direct effect on the establishment or control of HIV-1 latency, we first generated a population of J2574 reporter T cells that would overexpress IκB (J2574-IκB cells). The inhibitory effect of IκB transduction on stimulation-dependent NF-κB activation could be directly demonstrated by comparing the effects of PMA on the inducible GFP expression driven by the HIV-1 LTR in the parental J2574 control T cells and J2574-IκB cells. In the absence of Tat expression, in either cellular system, GFP expression is strictly correlated with the availability of NF-κB. As seen in Fig. 8A, both baseline GFP expression and PMA-induced GFP expression were lower in the J2574-IκB cell population than in the parental J2574 cell population, indicating that IκB was expressed in all cells and was physiologically active. The results generated for 30 individual paired infection cultures by infecting J2574 and J2574-IκB cells with increasing amounts of virus are shown in Fig. 8B to D. When we infected J2574 and J2574-IκB cells with HIV-1 NL4-3 over a wide MOI range, surprisingly, we did not observe a significant difference in the infection levels achieved as a function of the viral input (Fig. 8B). Unexpectedly, the silent infection reservoir, as measured on day 4 postinfection, was larger in the control cell population than in the IκB-overexpressing cell population (Fig. 8C). On day 14 postinfection, the latent HIV-1 reservoir in the J2574-IκB cell infection cultures was much larger than that in the corresponding J2574 cell infection cultures (Fig. 8D). While in control cells the stable latent HIV-1 infection reservoir had massively declined in relationship to the silent infection reservoir on day 4, a finding consistent with our previous data (17, 35), in J2574-IκB cells the silent reservoir seemed to completely transition into a stable reservoir (Fig. 8D). This suggests that the presence of NF-κB is essential for the transition of initially silent infection events into an active state or that, conversely, mechanisms that render host cells unresponsive and prevent increased NF-κB activity promote the establishment of latent HIV-1 infection. The data further emphasize the usefulness of kinome analysis as a tool to identify molecular control mechanisms or drug targets.
FIG 8.
Host cell IκB expression levels affect HIV-1 latency establishment. On the kinase antibody arrays, IκBa was detected as the highest altered spot in latently HIV-1 infected CA5 T cells. To test for a possible influence of IκB on HIV-1 latency control, we retrovirally transduced J2574 GFP reporter T cells to express IκB. (A) Following infection of the parental J2574 cells and J2574-IκB cells over a wide MOI range, we compared the levels of HIV-1 expression in infected J2574 cells and J2574-IκB cells, under baseline and PMA-activated conditions, to demonstrate that IκB is overexpressed and physiologically active in J2574-IκB cells and that it reduces HIV-1 expression, measured using GFP mean channel fluorescence as a surrogate marker. (B) The infection levels of J2574 cells and J2574-IκB cells as a function of viral input were determined on day 3 postinfection. (C) The establishment of a silent infection reservoir in the paired infection cultures was compared on day 4 postinfection. (D) The establishment of a stable latent HIV-1 reservoir in the paired infection cultures was compared on day 14 postinfection. Each data set represents a summary of results for 30 paired infection cultures generated at different MOIs.
A role for Dyrk1 in HIV-1 latency control.
In accordance with our conceptual hypothesis that HIV-1 latency could be stabilized by an anergy-like, unresponsive state of the host cells, and with the support of kinomic data, we decided to focus on nodes involved in G0 arrest and G0/G1 transition control. One such primary hit was increased phosphorylation of dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1a) substrates by lysates from latently HIV-1 infected T cells as measured on the PamGene kinase substrate chip. Alterations of the expression and/or activity of a series of other cell cycle-associated proteins were also suggested by our kinome analysis data (see Table S1 in the supplemental material).
Dyrk1 is an integral part of the dimerization partner, RB-like, E2F, and multivulval class B (DREAM) complex, which is considered the master coordinator of cell cycle-dependent gene expression (60). MuvB binds p130–E2F4–dimerization partner (DP) to form the DP, RB-like protein, E2F, and MuvB (DREAM) complex to repress all cell cycle-dependent gene expression, thereby arresting anergic cells in the G0 phase (61). Association of the MuvB core with p130–E2F4, in turn, is dependent on Dyrk1a-mediated phosphorylation of the MuvB subunit LIN52. Inhibition of Dyrk1a activity would disrupt the DREAM complex and thereby reduce the ability of cells to enter quiescence or to remain arrested in G0 (Fig. 9A, adapted from www.reactome.org). Increased Dyrk1a activity, as we found on the PamGene STK substrate chip for CA5 T cells, is thus compatible with the idea that latently infected T cells exhibit a G0/anergy-associated phenotype that renders them unresponsive. Dyrk1a can be targeted with high specificity using either (1Z)-1-(3-ethyl-5-hydroxy-2(3H)-benzothiazolylidene)-2-propanone (INDY) or proINDY (62). The data for INDY/proINDY effects on latent HIV-1 infection are shown in Fig. 9B and C. As predicted by kinome analysis, addition of the Dyrk1 inhibitor INDY modestly mobilized latent HIV-1 infection but efficiently primed the host cells to respond to a second stimulus, here bryostatin, a clinically relevant PKC activator (63, 64), to trigger synergistic HIV-1 reactivation. These data demonstrate that kinome analysis can be used to identify new drug targets that can be pharmacologically addressed to trigger HIV-1 reactivation.
FIG 9.

Dyrk1 inhibitors alter host cell control of latent HIV-1 infection. Dyrk1 was discovered as a seed node on the PamGene kinase substrate chip. (A) Dyrk1a controls the DREAM complex, which in turn controls G0 cell cycle exit and quiescence, suggesting that in this system, inhibition of Dyrk1 could alter host cells with latent HIV-1 infection to release infection events from their latent state. (B and C) The addition of Dyrk1 inhibitor INDY (B) or preINDY (C) in a concentration-dependent manner primed latent HIV-1 infection for reactivation by a second, synergistically acting activator, here bryostatin.
T cells from HIV-1-infected patients on suppressive ART exhibit a stable altered kinome signature.
Due to the scarcity of latently HIV-1 infected T cells in vivo and the lack of reported biomarkers that would allow for the isolation of these cells, we currently have no tools to generate a kinome profile of primary, latently HIV-1 infected T cells. However, if our hypothesis that either HIV-1 infection or exposure to HIV-1 or to the inflammatory environment during active infection results in stable phenotypic changes to immune cells is correct, we should be able to detect such permanent phenotypic changes even in peripheral blood mononuclear cells (PBMCs) from HIV-1-infected donors on fully suppressive ART. Consistent with this idea are the findings that the T cell response in HIV-1-infected individuals, even following ART onset, is impaired and that immune reconstitution is incomplete (65, 66). Specifically, the idea that permanent kinomic signature changes occur as a result of HIV-1 infection or exposure to an infection environment has not been investigated, but if such changes are detectable, they would make a strong case that kinomic changes such as those seen for latently HIV-1 infected T cell lines could also support the in vivo stability of the latent HIV-1 reservoir.
To test this possibility, we obtained PBMCs from 9 healthy controls and 8 HIV-infected patients with CD4 counts of <300 who had been on fully suppressive antiretroviral therapy for 5 to 18 months. This choice of patients was driven by reports that the T cell populations of patients with CD4 counts of <300 exhibit clear signs of immune senescence, such as loss of CD28. The immunological status of the T cell population was determined by staining each sample for HLA-DR (activation marker) and CD28 (aging marker) in the context of CD3/CD4/CD8 subpopulation markers. As shown in Fig. 10A, relative to those from healthy controls, T cells from HIV-1 patients exhibited clear signs of baseline immune activation (HLA-DR upregulation) (67) and immune senescence (CD28 loss on a subset of CD8 T cells) (68). Other sample characteristics are summarized in Table S3 in the supplemental material.
FIG 10.

T cells from HIV-1 infected patients on suppressive ART exhibit a stable altered kinome signature. (A) To address whether a stably altered kinomic signature would be present in T cells from HIV-1 patients on ART, we initially immune-phenotyped PBMCs from 9 healthy controls and 8 gender-, race-, and age-matched HIV-1-infected patients for the expression of the activation marker HLA-DR and the possible loss of CD28 as an immunological senescence marker. (B) Cell lysate materials from the 9 control samples and from the 8 patient samples were pooled to produce one control sample and one HIV-1 patient sample. By this means, individual variations among donors are minimized. The samples were loaded onto Kinexus kinase antibody arrays, and expression levels and phosphorylation states of a total of 510 kinases and phosphatases were determined. Spots with altered expression or activity in HIV-1 patient samples relative to healthy-control samples that had a Z′ ratio of >1.2 were included to generate a one-step interaction network. For the purpose of better visualization, only kinases, phosphatases, and transcription factors are shown. Detailed information on the regulation of the various altered kinases can be found in Table S4 in the supplemental material. (Inset) Depiction of the equivalent CA5 T cell PIN, for easier comparability. Black arrows indicate kinases/phosphatases present in both networks.
For the ensuing kinome analysis, the T cell materials from healthy controls and from HIV-1 patients were pooled to provide one control sample and one HIV-1 patient sample. Based on our experience with similar studies, by this means we reduce background noise resulting from individual donor variation but maintain signals underlying important changes common to all donors (see also reference 69). Each sample was then loaded onto individual Kinexus antibody microarrays. Surprisingly, despite full and extended suppression of active viral replication in all patients, we found a massively altered kinome signature in the patient material. The list of 66 altered kinases/phosphatases obtained from these experiments is shown in Table S4 in the supplemental material, and the shortest-pathway protein-protein interaction network describing these differences relative to healthy donors is shown in Fig. 10B. Although we used only Kinexus antibody arrays to generate these data, the similarity between the protein-protein interaction network generated for HIV patient material and the PIN we generated for latently HIV-1 infected T cells (Fig. 6) is apparent, with a high degree of overlap for the most connected network hubs.
The most apparent changes in the kinome profile at the level of protein expression are the decrease in calcium/calmodulin-dependent protein kinases (CAMK) and caspase family members. CAMK are part of calcium-triggered signaling cascades involved in immune response, inflammation, and memory consolidation. In CD4 memory T cells, CAMK is required to link T-cell antigen receptor (TCR) signaling to the production of IL-2, gamma interferon (IFN-γ), and IL-4. Caspases are involved in the activation signaling cascades that control apoptosis execution. On the other end of the spectrum, the data indicate a potential increase in signal transducer and activator of transcription (STAT) and AKT expression and possibly pathway activity. The kinome array also suggests an increased level of protein phosphorylation activity in samples from HIV patients.
Kinome analysis identified a distinct signature linking increased p27 KIP activity with decreased CDK2 expression. Cyclin-dependent kinase inhibitor 1B/p27 KIP has been reported to bind to and prevent the activation of cyclin E-CDK2 or cyclin D-CDK4 complexes and thus controls cell cycle progression at the G0/G1 transition. This would be consistent with our observations that CDK2 inhibition stabilizes latent HIV-1 infection (Fig. 3). Independently of the possible detailed implications of the kinome signature seen for PBMCs from HIV-1 patients, the data clearly demonstrate that even after therapeutic suppression of HIV-1 replication, primary T cells must be subject to long-term phenotypic changes that can be detected using kinome array analysis. By extension, this raises the possibility that latent HIV-1 infection in primary T cells from patients could be stabilized by phenotypic changes of the host cells similar to those we observed in T cell line models of latent HIV-1 infection.
DISCUSSION
Since its discovery, the remarkable stability of the latent HIV-1 reservoir has been the subject of many investigations (4, 70–76). Given that the individual half-life of memory T cells is limited to ∼50 days in HIV-1-infected patients (8), the stability of the latent HIV-1 reservoir cannot be a function of the actual lifetime of an individual cell but must be dependent on mechanisms that ensure the stability of the immunological memory. Accordingly, homeostatic proliferation of latently HIV-1 infected memory T cells has been reported as a major factor contributing to the stability of the HIV-1 reservoir (9, 12). By demonstrating that HIV integration into specific genes can promote the proliferation of latently HIV-infected T cells, a recent report by Wagner et al. added a second proliferation mechanism that would further slow the decay of the viral reservoir during ART (13). Cell proliferation in the absence of HIV-1 reactivation should be a feasible concept, since latent HIV-1 infection in T cell lines that proliferate constantly remains in a stable state (14–17).
While these findings explain how the pool of latently infected cells can be physically maintained given the half-life limitations of memory T cells, the question of why the latent reservoir maintains its stability despite the constant possibility of exposure to cognate antigen has not been addressed to date. Unless it is assumed either that (i) latently infected cells recognize only extremely rare antigens or that (ii) only HIV-specific T cells host latent HIV-1 infection events, and the onset of ART would thus withdraw cognate antigen, depletion of the reservoir should be continuous due to constant encounters with cognate antigen, which would trigger HIV-1 reactivation.
In theory, latent HIV-1 integration events could be protected by an increased transcriptional activation threshold generated through restrictive chromatin modifications at the latent viral promoter (LTR). Indeed, the formation of a repressive histone code has been associated with HIV-1 latency. While chromatin and histone modifications have been reported for some latent HIV-1 LTRs (77–79), the nature of these modifications and their downstream effects are not unique to the latent LTR but are shared with many cellular gene promoters that are transcriptionally silent in quiescent T cells but are induced upon T cell activation. For example, Rafati et al. reported that for latent HIV-1 infection events, the two nucleosomes that are found at the LTR are actively repositioned away from their predicted DNA binding sites as a function of the presence of BAF or PBAF, respectively, so as to possibly restrict the access of activating transcription factors to the LTR (80). Similar findings have been reported previously for many inactive but inducible cellular promoters (for recent reviews, see references 81 and 82). It thus seems unlikely that these types of promoter modifications underlie the recalcitrance of the population-wide pool of latent infection events to reactivation following exposure of their host cell to cognate antigen. Recognition of cognate antigen is the strongest biological T cell stimulus, and it is clear that TCR engagement should overcome such a potentially increased activation threshold, if it can induce cellular gene expression controlled by similar mechanisms.
We thus hypothesized that stable phenotypic changes of the host cells of latent HIV-1 infection events that render the cells unresponsive to activation are a prerequisite for maintaining latent HIV-1 infection.
Such phenotypic changes of host cells seem a distinct possibility in the setting of a chronic viral infection, such as that established by HIV-1. While the underlying molecular mechanisms of T-cell dysfunction that result from chronic viral infections are not well understood, functional impairment of antigen-specific T cells (e.g., T cell anergy) is a defining characteristic of many chronic infections. A now-classic example of changes caused by chronic infection that affect the ability of T cells to respond to stimulation is the discovery that chronic infection with lymphocytic choriomeningitis virus (LCMV) of mice would result in PD-1 upregulation. The interaction of PD1-L with PD-1 would have an immune-suppressive effect, and the interruption of this interaction with specific antibodies would enhance T-cell responses (83). Subsequently, Day et al. found that PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression (84). Of note, defective HIV-1-specific CD8+ T cell polyfunctionality, proliferation, and cytotoxicity are actually not restored by ART (85), suggesting that at least in some cell subpopulations, these changes are stable, even in the absence of active infection.
Much less is known about the effect of chronic viral infection on CD4+ T cells, but it is clear that CD4+ T cells are also affected by changes induced during chronic infections. Han et al. demonstrated that when antigen presentation was extended beyond the expansion phase, primed CD4+ T cells exhibited reduced memory functionality in terms of their proliferative capacity and cytokine expression potential. Upon antigen removal, the cells regained the ability to proliferate but remained unable to produce high levels of IL-2 and TNF-α (86). Translating these findings into an HIV-1 setting, we propose that persistent HIV-1 antigen exposure by itself could induce a dysfunctional state in CD4+ T cells that would be only partially reversible upon antigen removal following ART initiation.
Our results demonstrate that latently HIV-1 infected T cells are indeed phenotypically altered in ways that are consistent with reported T cell unresponsiveness such as that seen in T cell anergy. We described stable changes in the kinetic NF-κB response in latently infected T cells (Fig. 5) and demonstrated that interventions that reproduce T cell anergy promoted the establishment of latent infection (Fig. 1 and 2) or stabilized latent infection events (Fig. 4). The full extent of these phenotypic changes was highlighted by kinome analysis of latently infected T cells, which revealed massive changes in the baseline kinase activity profile from that of uninfected control cells (Fig. 6). Detailed analysis of some key altered kinases revealed that these phenotypic changes of the host cells are essential for the stabilization of latent HIV-1 infection (Fig. 8 and 9). Beyond providing a tool for dissecting cellular mechanisms that stabilize latent HIV-1 infection, we demonstrate that kinome array analysis can be used to identify novel drug targets that can be addressed with experimental or FDA-approved drugs to trigger HIV-1 reactivation using synergistically acting drug combinations (Fig. 9).
It is worth mentioning that in addition to the kinases that we targeted in this analysis (Dyrk1a, PIM-1, CDK2), other, previously reported mechanisms involved in HIV-1 latency control were also represented in the altered kinome of latently HIV-1 infected cells. For example, HSP90 is listed as a major hub in the PIN describing host cell control of latent HIV-1 infection (Fig. 6). While we have not experimentally targeted the HSP90 node in our system, HSP90 has recently been reported to be of major importance for HIV-1 latency control (87, 88), further supporting the idea that kinome array analysis and PINs derived from these data are predictive beyond the limitations of our HIV-1 latency models.
However, the single most important finding of our study is that the data demonstrate that latent HIV-1 infection is a host cell phenomenon that goes beyond transcription factor restriction and possible restrictive changes at the histone and chromatin level, thereby adding another mechanism to the list of control mechanisms for latent HIV-1 infection. Our data suggest that HIV-1 latency is established and maintained in T cells that exhibit a dramatically altered phenotype that can be correlated with unresponsiveness or T cell anergy. Essentially, the data presented confirm our previous results showing that a gatekeeper kinase activity controls latent HIV-1 infection and even supersedes the effect of high-level NF-κB activation (40). These and other data further demonstrated that some kinase inhibitors can have identical effects on latent HIV-1 infection in T cell lines and primary T cells (40, 41). The similarity of kinomic changes observed in cell material from HIV-1 patients (Fig. 10) and in latently infected T cell lines (Fig. 6) suggests that the biological principle of phenotypic host cell changes controlling latent HIV-1 infection could indeed be transferable to the in vivo situation. However, in these experiments, the phenotypic changes observed cannot be directly related to effects of proviral integration but must result from other environmental factors. This suggests that an actual infection event may not drive the phenotypic changes observed in latently HIV-1 infected cell clones; rather, these changes may result from the overall inflammatory environment during the initial infection.
To move forward, it will be key to transfer the kinome analysis approach described here to primary ex vivo material from HIV-1 patients. At present, obtaining sufficient material poses a major challenge due to the scarcity of latently HIV-1 infected cells and the absence of biomarkers associated with latent HIV-1 infection, which would allow us to identify these cells and specifically investigate their status. However, if latent HIV-1 infection is associated with an unresponsive, dysfunctional T cell phenotype, as suggested by our data, a larger T cell population that could act as a surrogate population for latently HIV-1 infected T cells and could be used in such future studies may be identified.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded in part by NIH grants R01-AI104499 and R21-AI116188 to O.K. Parts of the work were performed in the UAB CFAR BSL-3 facilities and by the UAB CFAR Flow Cytometry Core/Joint UAB Flow Cytometry Core, which are funded by NIH/NIAID P30 AI027767 and by NIH 5P30 AR048311.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00571-15.
REFERENCES
- 1.Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, Kuo YH, Brookmeyer R, Zeiger MA, Barditch-Crovo P, Siliciano RF. 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 2.Chun TW, Davey RT Jr, Ostrowski M, Shawn Justement J, Engel D, Mullins JI, Fauci AS. 2000. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat Med 6:757–761. doi: 10.1038/77481. [DOI] [PubMed] [Google Scholar]
- 3.Chun TW, Engel D, Mizell SB, Ehler LA, Fauci AS. 1998. Induction of HIV-1 replication in latently infected CD4+ T cells using a combination of cytokines. J Exp Med 188:83–91. doi: 10.1084/jem.188.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, Quinn TC, Chaisson RE, Rosenberg E, Walker B, Gange S, Gallant J, Siliciano RF. 1999. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med 5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 5.Hellerstein M, Hanley MB, Cesar D, Siler S, Papageorgopoulos C, Wieder E, Schmidt D, Hoh R, Neese R, Macallan D, Deeks S, McCune JM. 1999. Directly measured kinetics of circulating T lymphocytes in normal and HIV-1-infected humans. Nat Med 5:83–89. doi: 10.1038/4772. [DOI] [PubMed] [Google Scholar]
- 6.Macallan DC, Wallace D, Zhang Y, De Lara C, Worth AT, Ghattas H, Griffin GE, Beverley PC, Tough DF. 2004. Rapid turnover of effector-memory CD4+ T cells in healthy humans. J Exp Med 200:255–260. doi: 10.1084/jem.20040341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vrisekoop N, den Braber I, de Boer AB, Ruiter AF, Ackermans MT, van der Crabben SN, Schrijver EH, Spierenburg G, Sauerwein HP, Hazenberg MD, de Boer RJ, Miedema F, Borghans JA, Tesselaar K. 2008. Sparse production but preferential incorporation of recently produced naive T cells in the human peripheral pool. Proc Natl Acad Sci U S A 105:6115–6120. doi: 10.1073/pnas.0709713105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ladell K, Hellerstein MK, Cesar D, Busch R, Boban D, McCune JM. 2008. Central memory CD8+ T cells appear to have a shorter lifespan and reduced abundance as a function of HIV disease progression. J Immunol 180:7907–7918. doi: 10.4049/jimmunol.180.12.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel MR, Ghattas G, Brenchley JM, Schacker TW, Hill BJ, Douek DC, Routy JP, Haddad EK, Sekaly RP. 2009. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med 15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyman O, Letourneau S, Krieg C, Sprent J. 2009. Homeostatic proliferation and survival of naive and memory T cells. Eur J Immunol 39:2088–2094. doi: 10.1002/eji.200939444. [DOI] [PubMed] [Google Scholar]
- 11.Surh CD, Sprent J. 2008. Homeostasis of naive and memory T cells. Immunity 29:848–862. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 12.Bosque A, Famiglietti M, Weyrich A, Goulston C, Planelles V. 2011. Homeostatic proliferation fails to efficiently reactivate HIV-1 latently infected central memory CD4+ T cells. PLoS Pathog 7:e1002288. doi: 10.1371/journal.ppat.1002288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagner TA, McLaughlin S, Garg K, Cheung CY, Larsen BB, Styrchak S, Huang HC, Edlefsen PT, Mullins JI, Frenkel LM. 2014. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 345:570–573. doi: 10.1126/science.1256304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jordan A, Bisgrove D, Verdin E. 2003. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J 22:1868–1877. doi: 10.1093/emboj/cdg188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kutsch O, Benveniste EN, Shaw GM, Levy DN. 2002. Direct and quantitative single-cell analysis of human immunodeficiency virus type 1 reactivation from latency. J Virol 76:8776–8786. doi: 10.1128/JVI.76.17.8776-8786.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones J, Rodgers J, Heil M, May J, White L, Maddry JA, Fletcher TM III, Shaw GM, Hartman JL IV, Kutsch O. 2007. High throughput drug screening for human immunodeficiency virus type 1 reactivating compounds. Assay Drug Dev Technol 5:181–189. doi: 10.1089/adt.2006.040. [DOI] [PubMed] [Google Scholar]
- 17.Duverger A, Jones J, May J, Bibollet-Ruche F, Wagner FA, Cron RQ, Kutsch O. 2009. Determinants of the establishment of human immunodeficiency virus type 1 latency. J Virol 83:3078–3093. doi: 10.1128/JVI.02058-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wange RL. 2000. LAT, the linker for activation of T cells: a bridge between T cell-specific and general signaling pathways. Sci STKE 2000(63):re1. doi: 10.1126/stke.2000.63.re1. [DOI] [PubMed] [Google Scholar]
- 19.Jaafoura S, de Goër de Herve MG, Hernandez-Vargas EA, Hendel-Chavez H, Abdoh M, Mateo MC, Krzysiek R, Merad M, Seng R, Tardieu M, Delfraissy JF, Goujard C, Taoufik Y. 2014. Progressive contraction of the latent HIV reservoir around a core of less-differentiated CD4+ memory T cells. Nat Commun 5:5407. doi: 10.1038/ncomms6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jenkins MK, Pardoll DM, Mizuguchi J, Quill H, Schwartz RH. 1987. T-cell unresponsiveness in vivo and in vitro: fine specificity of induction and molecular characterization of the unresponsive state. Immunol Rev 95:113–135. doi: 10.1111/j.1600-065X.1987.tb00502.x. [DOI] [PubMed] [Google Scholar]
- 21.Schwartz RH. 2003. T cell anergy. Annu Rev Immunol 21:305–334. doi: 10.1146/annurev.immunol.21.120601.141110. [DOI] [PubMed] [Google Scholar]
- 22.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. 2007. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 23.Mirshahidi S, Huang CT, Sadegh-Nasseri S. 2001. Anergy in peripheral memory CD4+ T cells induced by low avidity engagement of T cell receptor. J Exp Med 194:719–731. doi: 10.1084/jem.194.6.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fathman CG, Lineberry NB. 2007. Molecular mechanisms of CD4+ T-cell anergy. Nat Rev Immunol 7:599–609. doi: 10.1038/nri2131. [DOI] [PubMed] [Google Scholar]
- 25.Jenkins MK. 1992. The role of cell division in the induction of clonal anergy. Immunol Today 13:69–73. doi: 10.1016/0167-5699(92)90137-V. [DOI] [PubMed] [Google Scholar]
- 26.Wells AD, Walsh MC, Sankaran D, Turka LA. 2000. T cell effector function and anergy avoidance are quantitatively linked to cell division. J Immunol 165:2432–2443. doi: 10.4049/jimmunol.165.5.2432. [DOI] [PubMed] [Google Scholar]
- 27.Sun Y, Permar SR, Buzby AP, Letvin NL. 2007. Memory CD4+ T-lymphocyte loss and dysfunction during primary simian immunodeficiency virus infection. J Virol 81:8009–8015. doi: 10.1128/JVI.00482-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Younes SA, Yassine-Diab B, Dumont AR, Boulassel MR, Grossman Z, Routy JP, Sekaly RP. 2003. HIV-1 viremia prevents the establishment of interleukin 2-producing HIV-specific memory CD4+ T cells endowed with proliferative capacity. J Exp Med 198:1909–1922. doi: 10.1084/jem.20031598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Potter SJ, Lacabaratz C, Lambotte O, Perez-Patrigeon S, Vingert B, Sinet M, Colle JH, Urrutia A, Scott-Algara D, Boufassa F, Delfraissy JF, Theze J, Venet A, Chakrabarti LA. 2007. Preserved central memory and activated effector memory CD4+ T-cell subsets in human immunodeficiency virus controllers: an ANRS EP36 study. J Virol 81:13904–13915. doi: 10.1128/JVI.01401-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kannanganat S, Kapogiannis BG, Ibegbu C, Chennareddi L, Goepfert P, Robinson HL, Lennox J, Amara RR. 2007. Human immunodeficiency virus type 1 controllers but not noncontrollers maintain CD4 T cells coexpressing three cytokines. J Virol 81:12071–12076. doi: 10.1128/JVI.01261-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jain V, Hartogensis W, Bacchetti P, Hunt PW, Hatano H, Sinclair E, Epling L, Lee TH, Busch MP, McCune JM, Pilcher CD, Hecht FM, Deeks SG. 2013. Antiretroviral therapy initiated within 6 months of HIV infection is associated with lower T-cell activation and smaller HIV reservoir size. J Infect Dis 208:1202–1211. doi: 10.1093/infdis/jit311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee NS, Barber L, Kanchwala A, Childs CJ, Kataria YP, Judson MA, Mazer MA, Arce S. 2011. Low levels of NF-κB/p65 mark anergic CD4+ T cells and correlate with disease severity in sarcoidosis. Clin Vaccine Immunol 18:223–234. doi: 10.1128/CVI.00469-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sundstedt A, Sigvardsson M, Leanderson T, Hedlund G, Kalland T, Dohlsten M. 1996. In vivo anergized CD4+ T cells express perturbed AP-1 and NF-κB transcription factors. Proc Natl Acad Sci U S A 93:979–984. doi: 10.1073/pnas.93.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li W, Whaley CD, Mondino A, Mueller DL. 1996. Blocked signal transduction to the ERK and JNK protein kinases in anergic CD4+ T cells. Science 271:1272–1276. doi: 10.1126/science.271.5253.1272. [DOI] [PubMed] [Google Scholar]
- 35.Duverger A, Wolschendorf F, Zhang M, Wagner F, Hatcher B, Jones J, Cron RQ, van der Sluis RM, Jeeninga RE, Berkhout B, Kutsch O. 2013. An AP-1 binding site in the enhancer/core element of the HIV-1 promoter controls the ability of HIV-1 to establish latent infection. J Virol 87:2264–2277. doi: 10.1128/JVI.01594-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jeeninga RE, Hoogenkamp M, Armand-Ugon M, de Baar M, Verhoef K, Berkhout B. 2000. Functional differences between the long terminal repeat transcriptional promoters of human immunodeficiency virus type 1 subtypes A through G. J Virol 74:3740–3751. doi: 10.1128/JVI.74.8.3740-3751.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Opijnen T, Jeeninga RE, Boerlijst MC, Pollakis GP, Zetterberg V, Salminen M, Berkhout B. 2004. Human immunodeficiency virus type 1 subtypes have a distinct long terminal repeat that determines the replication rate in a host-cell-specific manner. J Virol 78:3675–3683. doi: 10.1128/JVI.78.7.3675-3683.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peden K, Emerman M, Montagnier L. 1991. Changes in growth properties on passage in tissue culture of viruses derived from infectious molecular clones of HIV-1LAI, HIV-1MAL, and HIV-1ELI. Virology 185:661–672. doi: 10.1016/0042-6822(91)90537-L. [DOI] [PubMed] [Google Scholar]
- 39.Shishido T, Wolschendorf F, Duverger A, Wagner F, Kappes J, Jones J, Kutsch O. 2012. Selected drugs with reported secondary cell-differentiating capacity prime latent HIV-1 infection for reactivation. J Virol 86:9055–9069. doi: 10.1128/JVI.00793-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolschendorf F, Bosque A, Shishido T, Duverger A, Jones J, Planelles V, Kutsch O. 2012. Kinase control prevents HIV-1 reactivation in spite of high levels of induced NF-κB activity. J Virol 86:4548–4558. doi: 10.1128/JVI.06726-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duverger A, Wolschendorf F, Anderson JC, Wagner F, Bosque A, Shishido T, Jones J, Planelles V, Willey C, Cron RQ, Kutsch O. 2014. Kinase control of latent HIV-1 infection: PIM-1 kinase as a major contributor to HIV-1 reactivation. J Virol 88:364–376. doi: 10.1128/JVI.02682-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheadle C, Vawter MP, Freed WJ, Becker KG. 2003. Analysis of microarray data using Z score transformation. J Mol Diagn 5:73–81. doi: 10.1016/S1525-1578(10)60455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Telander DG, Malvey EN, Mueller DL. 1999. Evidence for repression of IL-2 gene activation in anergic T cells. J Immunol 162:1460–1465. [PubMed] [Google Scholar]
- 44.Howe CJ, LaHair MM, Robinson PJ, Rodriguez-Mora O, McCubrey JA, Franklin RA. 2003. Models of anergy in the human Jurkat T cell line. Assay Drug Dev Technol 1:537–544. doi: 10.1089/154065803322302790. [DOI] [PubMed] [Google Scholar]
- 45.Chunder N, Wang L, Chen C, Hancock WW, Wells AD. 2012. Cyclin-dependent kinase 2 controls peripheral immune tolerance. J Immunol 189:5659–5666. doi: 10.4049/jimmunol.1202313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, Inagaki M, Delcros JG, Moulinoux JP. 1997. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem 243:527–536. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- 47.Payton M, Chung G, Yakowec P, Wong A, Powers D, Xiong L, Zhang N, Leal J, Bush TL, Santora V, Askew B, Tasker A, Radinsky R, Kendall R, Coats S. 2006. Discovery and evaluation of dual CDK1 and CDK2 inhibitors. Cancer Res 66:4299–4308. doi: 10.1158/0008-5472.CAN-05-2507. [DOI] [PubMed] [Google Scholar]
- 48.Contreras X, Barboric M, Lenasi T, Peterlin BM. 2007. HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog 3:1459–1469. doi: 10.1371/journal.ppat.0030146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Contreras X, Schweneker M, Chen CS, McCune JM, Deeks SG, Martin J, Peterlin BM. 2009. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J Biol Chem 284:6782–6789. doi: 10.1074/jbc.M807898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fujinaga K, Cujec TP, Peng J, Garriga J, Price DH, Grana X, Peterlin BM. 1998. The ability of positive transcription elongation factor B to transactivate human immunodeficiency virus transcription depends on a functional kinase domain, cyclin T1, and Tat. J Virol 72:7154–7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taube R, Lin X, Irwin D, Fujinaga K, Peterlin BM. 2002. Interaction between P-TEFb and the C-terminal domain of RNA polymerase II activates transcriptional elongation from sites upstream or downstream of target genes. Mol Cell Biol 22:321–331. doi: 10.1128/MCB.22.1.321-331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. 2011. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol 29:1039–1045. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anandasabapathy N, Ford GS, Bloom D, Holness C, Paragas V, Seroogy C, Skrenta H, Hollenhorst M, Fathman CG, Soares L. 2003. GRAIL: an E3 ubiquitin ligase that inhibits cytokine gene transcription is expressed in anergic CD4+ T cells. Immunity 18:535–547. doi: 10.1016/S1074-7613(03)00084-0. [DOI] [PubMed] [Google Scholar]
- 54.Bodor J, Spetz AL, Strominger JL, Habener JF. 1996. cAMP inducibility of transcriptional repressor ICER in developing and mature human T lymphocytes. Proc Natl Acad Sci U S A 93:3536–3541. doi: 10.1073/pnas.93.8.3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tenbrock K, Juang YT, Tolnay M, Tsokos GC. 2003. The cyclic adenosine 5′-monophosphate response element modulator suppresses IL-2 production in stimulated T cells by a chromatin-dependent mechanism. J Immunol 170:2971–2976. doi: 10.4049/jimmunol.170.6.2971. [DOI] [PubMed] [Google Scholar]
- 56.Clavijo PE, Frauwirth KA. 2012. Anergic CD8+ T lymphocytes have impaired NF-κB activation with defects in p65 phosphorylation and acetylation. J Immunol 188:1213–1221. doi: 10.4049/jimmunol.1100793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoffmann A, Levchenko A, Scott ML, Baltimore D. 2002. The IκB-NF-κB signaling module: temporal control and selective gene activation. Science 298:1241–1245. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 58.Prasad RC, Wang XL, Law BK, Davis B, Green G, Boone B, Sims L, Law M. 2009. Identification of genes, including the gene encoding p27Kip1, regulated by serine 276 phosphorylation of the p65 subunit of NF-κB. Cancer Lett 275:139–149. doi: 10.1016/j.canlet.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nihira K, Ando Y, Yamaguchi T, Kagami Y, Miki Y, Yoshida K. 2010. Pim-1 controls NF-κB signalling by stabilizing RelA/p65. Cell Death Differ 17:689–698. doi: 10.1038/cdd.2009.174. [DOI] [PubMed] [Google Scholar]
- 60.Sadasivam S, DeCaprio JA. 2013. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat Rev Cancer 13:585–595. doi: 10.1038/nrc3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Litovchick L, Florens LA, Swanson SK, Washburn MP, DeCaprio JA. 2011. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev 25:801–813. doi: 10.1101/gad.2034211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ogawa Y, Nonaka Y, Goto T, Ohnishi E, Hiramatsu T, Kii I, Yoshida M, Ikura T, Onogi H, Shibuya H, Hosoya T, Ito N, Hagiwara M. 2010. Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A. Nat Commun 1:86. doi: 10.1038/ncomms1090. [DOI] [PubMed] [Google Scholar]
- 63.Wender PA, Cribbs CM, Koehler KF, Sharkey NA, Herald CL, Kamano Y, Pettit GR, Blumberg PM. 1988. Modeling of the bryostatins to the phorbol ester pharmacophore on protein kinase C. Proc Natl Acad Sci U S A 85:7197–7201. doi: 10.1073/pnas.85.19.7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DeChristopher BA, Loy BA, Marsden MD, Schrier AJ, Zack JA, Wender PA. 2012. Designed, synthetically accessible bryostatin analogues potently induce activation of latent HIV reservoirs in vitro. Nat Chem 4:705–710. doi: 10.1038/nchem.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gazzola L, Tincati C, Bellistri GM, Monforte A, Marchetti G. 2009. The absence of CD4+ T cell count recovery despite receipt of virologically suppressive highly active antiretroviral therapy: clinical risk, immunological gaps, and therapeutic options. Clin Infect Dis 48:328–337. doi: 10.1086/595851. [DOI] [PubMed] [Google Scholar]
- 66.Kelley CF, Kitchen CM, Hunt PW, Rodriguez B, Hecht FM, Kitahata M, Crane HM, Willig J, Mugavero M, Saag M, Martin JN, Deeks SG. 2009. Incomplete peripheral CD4+ cell count restoration in HIV-infected patients receiving long-term antiretroviral treatment. Clin Infect Dis 48:787–794. doi: 10.1086/597093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Biancotto A, Iglehart SJ, Vanpouille C, Condack CE, Lisco A, Ruecker E, Hirsch I, Margolis LB, Grivel JC. 2008. HIV-1 induced activation of CD4+ T cells creates new targets for HIV-1 infection in human lymphoid tissue ex vivo. Blood 111:699–704. doi: 10.1182/blood-2007-05-088435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Effros RB, Allsopp R, Chiu CP, Hausner MA, Hirji K, Wang L, Harley CB, Villeponteau B, West MD, Giorgi JV. 1996. Shortened telomeres in the expanded CD28− CD8+ cell subset in HIV disease implicate replicative senescence in HIV pathogenesis. AIDS 10:F17–F22. doi: 10.1097/00002030-199607000-00001. [DOI] [PubMed] [Google Scholar]
- 69.Kendziorski C, Irizarry RA, Chen KS, Haag JD, Gould MN. 2005. On the utility of pooling biological samples in microarray experiments. Proc Natl Acad Sci U S A 102:4252–4257. doi: 10.1073/pnas.0500607102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zack JA, Arrigo SJ, Weitsman SR, Go AS, Haislip A, Chen IS. 1990. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 61:213–222. doi: 10.1016/0092-8674(90)90802-L. [DOI] [PubMed] [Google Scholar]
- 71.Bukrinsky MI, Stanwick TL, Dempsey MP, Stevenson M. 1991. Quiescent T lymphocytes as an inducible virus reservoir in HIV-1 infection. Science 254:423–427. doi: 10.1126/science.1925601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 73.Stevenson M. 1997. Molecular mechanisms for the regulation of HIV replication, persistence and latency. AIDS 11(Suppl A):S25–S33. [PubMed] [Google Scholar]
- 74.Blankson JN, Finzi D, Pierson TC, Sabundayo BP, Chadwick K, Margolick JB, Quinn TC, Siliciano RF. 2000. Biphasic decay of latently infected CD4+ T cells in acute human immunodeficiency virus type 1 infection. J Infect Dis 182:1636–1642. doi: 10.1086/317615. [DOI] [PubMed] [Google Scholar]
- 75.Chun TW, Finzi D, Margolick J, Chadwick K, Schwartz D, Siliciano RF. 1995. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat Med 1:1284–1290. doi: 10.1038/nm1295-1284. [DOI] [PubMed] [Google Scholar]
- 76.Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ, Siliciano RF. 2003. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med 9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 77.Van Lint C, Emiliani S, Ott M, Verdin E. 1996. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J 15:1112–1120. [PMC free article] [PubMed] [Google Scholar]
- 78.Coull JJ, He G, Melander C, Rucker VC, Dervan PB, Margolis DM. 2002. Targeted derepression of the human immunodeficiency virus type 1 long terminal repeat by pyrrole-imidazole polyamides. J Virol 76:12349–12354. doi: 10.1128/JVI.76.23.12349-12354.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. 2006. NF-κB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J 25:139–149. doi: 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rafati H, Parra M, Hakre S, Moshkin Y, Verdin E, Mahmoudi T. 2011. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol 9:e1001206. doi: 10.1371/journal.pbio.1001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jiang C, Pugh BF. 2009. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet 10:161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bai L, Morozov AV. 2010. Gene regulation by nucleosome positioning. Trends Genet 26:476–483. doi: 10.1016/j.tig.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 83.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 84.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJ, Klenerman P, Ahmed R, Freeman GJ, Walker BD. 2006. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 85.Migueles SA, Weeks KA, Nou E, Berkley AM, Rood JE, Osborne CM, Hallahan CW, Cogliano-Shutta NA, Metcalf JA, McLaughlin M, Kwan R, Mican JM, Davey RT Jr, Connors M. 2009. Defective human immunodeficiency virus-specific CD8+ T-cell polyfunctionality, proliferation, and cytotoxicity are not restored by antiretroviral therapy. J Virol 83:11876–11889. doi: 10.1128/JVI.01153-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Han S, Asoyan A, Rabenstein H, Nakano N, Obst R. 2010. Role of antigen persistence and dose for CD4+ T-cell exhaustion and recovery. Proc Natl Acad Sci U S A 107:20453–20458. doi: 10.1073/pnas.1008437107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Anderson I, Low JS, Weston S, Weinberger M, Zhyvoloup A, Labokha AA, Corazza G, Kitson RA, Moody CJ, Marcello A, Fassati A. 2014. Heat shock protein 90 controls HIV-1 reactivation from latency. Proc Natl Acad Sci U S A 111:E1528–E1537. doi: 10.1073/pnas.1320178111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Serrao E, Wang CH, Frederick T, Lee CL, Anthony P, Arribas-Layton D, Baker K, Millstein J, Kovacs A, Neamati N. 2014. Alteration of select gene expression patterns in individuals infected with HIV-1. J Med Virol 86:678–686. doi: 10.1002/jmv.23872. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.