

ABSTRACT
Synthesis of 2′-5′-oligoadenylates (2-5A) by oligoadenylate synthetase (OAS) is an important innate cellular response that limits viral replication by activating the latent cellular RNase, RNase L, to degrade single-stranded RNA. Some rotaviruses and coronaviruses antagonize the OAS/RNase L pathway through the activity of an encoded 2H phosphoesterase domain that cleaves 2-5A. These viral 2H phosphoesterases are phylogenetically related to the cellular A kinase anchoring protein 7 (AKAP7) and share a core structure and an active site that contains two well-defined HΦ(S/T)Φ (where Φ is a hydrophobic residue) motifs, but their mechanism of substrate binding is unknown. Here, we report the structures of a viral 2H phosphoesterase, the C-terminal domain (CTD) of the group A rotavirus (RVA) VP3 protein, both alone and in complex with 2-5A. The domain forms a compact fold, with a concave β-sheet that contains the catalytic cleft, but it lacks two α-helical regions and two β-strands observed in AKAP7 and other 2H phosphoesterases. The cocrystal structure shows significant conformational changes in the R loop upon ligand binding. Bioinformatics and biochemical analyses reveal that conserved residues and residues required for catalytic activity and substrate binding comprise the catalytic motifs and a region on one side of the binding cleft. We demonstrate that the VP3 CTD of group B rotavirus, but not that of group G, cleaves 2-5A. These findings suggest that the VP3 CTD is a streamlined version of a 2H phosphoesterase with a ligand-binding mechanism that is shared among 2H phosphodiesterases that cleave 2-5A.
IMPORTANCE The C-terminal domain (CTD) of rotavirus VP3 is a 2H phosphoesterase that cleaves 2′-5′-oligoadenylates (2-5A), potent activators of an important innate cellular antiviral pathway. 2H phosphoesterase superfamily proteins contain two conserved catalytic motifs and a proposed core structure. Here, we present structures of a viral 2H phosphoesterase, the rotavirus VP3 CTD, alone and in complex with its substrate, 2-5A. The domain lacks two α-helical regions and β-strands present in other 2H phosphoesterases. A loop of the protein undergoes significant structural changes upon substrate binding. Together with our bioinformatics and biochemical findings, the crystal structures suggest that the RVA VP3 CTD domain is a streamlined version of a cellular enzyme that shares a ligand-binding mechanism with other 2H phosphodiesterases that cleave 2-5A but differs from those of 2H phosphodiesterases that cleave other substrates. These findings may aid in the future design of antivirals targeting viral phosphodiesterases with cleavage specificity for 2-5A.
INTRODUCTION
One of the earliest steps in the battle between a virus and its host is the detection of pathogen-associated molecular patterns by cellular sensors. For RNA viruses, these patterns may be found in RNA products synthesized during viral transcription and replication. A number of cytoplasmic cellular molecules have been described that sense foreign RNA and respond by triggering innate pathways, resulting in the production of type I interferons (IFNs) (reviewed in references 1 and 2). Oligoadenylate synthetases (OASs) are cytoplasmic sensors that, upon binding of double-stranded RNA (dsRNA), synthesize 2′-5′-oligoadenylates (2-5As), which are the signaling molecules that activate the latent RNase (RNase L) (reviewed in references 3 to 5). Activated RNase L cleaves viral and cellular single-stranded RNA, inducing autophagy and apoptosis and limiting viral replication. In some cell types, the small RNAs produced by RNase L cleavage are sensed by retinoic acid-inducible gene I (RIG-I)-like receptors, amplify IFN production, and thereby upregulate expression of OAS and hundreds of other genes that contribute to the establishment of an antiviral state. RNA viruses have evolved a variety of mechanisms to evade and antagonize this important antiviral pathway (reviewed in references 4 and 5). One such mechanism, which has been demonstrated by group A rotaviruses (RVA) and group 2 betacoronaviruses, is cleavage of 2-5A by a virus-encoded phosphodiesterase (PDE) (6, 7).
Rotaviruses are major causes of gastroenteritis in humans and animals (8). Of the eight proposed rotavirus species (RVA to RVH), RVA is responsible for the deaths of more than 450,000 infants and young children annually (9, 10). Segment 3 of the RVA dsRNA genome encodes VP3, an 835-amino-acid minor virion component, and is linked to virulence in animal models of infection (8, 11–13). While the N-terminal ∼690 amino acids of RVA VP3 have mRNA capping functions, the C-terminal ∼150 amino acids possess 2′,5′-PDE activity that cleaves 2-5A (6, 14). This VP3 C-terminal domain (CTD) is predicted to be structurally homologous to members of the 2H phosphoesterase superfamily (6, 14, 15). The VP3 CTD, which also is predicted to be present in RVB and RVG strains, may contribute to the virulence and success of the RVA species (6, 14). The biological potential of the RVA VP3 CTD has been underscored by studies in which this domain complemented viral growth and pathogenesis in mice infected with recombinant mouse hepatitis virus (MHV) encoding a defective form of the virulence determinant ns2 (6).
2H phosphoesterase superfamily members hydrolyze phosphoester linkages through a mechanism involving two highly conserved active-site HΦ(S/T)Φ motifs, where Φ is a hydrophobic amino acid (15, 16). While the histidines in the catalytic motifs are proposed to contribute directly to catalysis, the conserved serines/threonines are thought to stabilize substrate in the binding cleft (17). Phylogenetically, the RVA VP3 CTD, coronavirus ns2, torovirus putative nsp9, and cellular A kinase anchoring protein 7 (AKAP7) belong to the eukaryotic-viral LigT-like family (group II) (15). The RVA VP3 CTD, MHV ns2, and AKAP7 all possess 2′,5′-PDE activity, for which the histidines in the two HΦ(S/T)Φ motifs are required (6, 18, 19). Although the structure of a cellular AKAP7 has been characterized (20), the structures of viral PDEs have been narrowly studied.
Here, we present the crystal structures of the viral 2H PDE encoded by RVA VP3 alone and in complex with a 2-5A substrate. The domain forms a concave antiparallel β-sheet that contains the catalytic cleft. The structure is similar to that of cellular AKAP7 but lacks two β-strands and two interspersed α-helices, suggesting a revision of the minimal core structure required for PDE activity. The structure of RVA VP3 CTD in complex with 2-5A reveals the residues involved in substrate recognition and catalysis, which are further validated by using mutational and enzymatic analysis. We found that, like the RVA VP3 CTD, the RVB VP3 CTD, but not the RVG VP3 CTD, is a functional PDE that cleaves 2-5A. Our studies provide a structural platform for further dissection of the biology of rotaviruses and other RNA viruses that antagonize the OAS-RNase L pathway.
MATERIALS AND METHODS
Cloning of rotavirus VP3 CTDs into pLIC1HMN and pLIC1HM.
A codon-optimized gene encoding RVA RRV VP3 (GenBank accession number KJ869109) or the RVA VP3 CTD R792A mutant and genes encoding RVB Bang117 VP3 (GenBank GU391303) and the CTD (amino acids 652 to 768) of RVG 03V0567 VP3 (GenBank JQ920005) were synthesized by Genscript (Piscataway, NJ). pLIC1HM was generated by modifying pET-42a(+) (Novagen, Darmstadt, Germany) to introduce a His6 tag, the malE gene (encoding maltose binding protein [MBP]), a tobacco etch virus (TEV) protease cleavage site, and a ligation-independent cloning site in place of the glutathione S-transferase (GST) tag and multiple cloning site located between the T7 promoter and T7 terminator. pLIC1HMN was generated by introducing an Asn10 linker between the MBP-encoding and TEV protease cleavage site-encoding sequences of pLIC1HM. Sequences encoding the RVA (amino acids 694 to 835), RVB (amino acids 648 to 763), and RVG VP3 CTDs or MHV ns2 were amplified by PCR and inserted into plasmid pLIC1HMN (RVA) or pLIC1HM (RVB, RVG, and RVA R792A) by ligation-independent cloning. Point mutations in RVA VP3 CTD pLIC1HMN, RVB VP3 CTD pLIC1HM, and RVG VP3 CTD pLIC1HM were engineered using round-the-horn PCR.
Expression and purification of VP3 CTDs.
For large-scale purification, recombinant protein expression was induced in Escherichia coli strain BL21(DE3)/pLysS (Life Technologies, Carlsbad, CA) transformed with RVA VP3 CTD pLIC1HMN at 20°C overnight at an optical density at 600 nm (OD600) of 0.6 to 0.8 with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG). The recombinant N-terminal His6-MBP-tagged RVA VP3 CTD was purified using Ni-nitrilotriacetic acid (NTA) agarose (Qiagen, Venlo, Netherlands). TEV protease cleavage, followed by incubation with Ni-NTA agarose, was used to remove the His6-MBP tag, His6-TEV, and uncleaved protein. Cleavage left Ser-Gly at the N terminus of the RVA VP3 CTD, which was concentrated and further purified by size exclusion chromatography on a Superdex-75 column (GE Healthcare, Little Chalfont, United Kingdom) in 10 mM HEPES, pH 7.2, 100 mM NaCl, and 1 mM dithiothreitol (DTT). The concentration of the purified RVA VP3 CTD was determined by the A280, using a molar extinction coefficient of 12,800 M−1 cm−1, calculated using Vector NTI, version 11, software (Invitrogen, Carlsbad, CA). Selenomethionine-substituted VP3 CTD was expressed in B834 (DE3) (Novagen, Darmstadt, Germany) using an Overnight Express Autoinduction System 2 (EMD Millipore, Billerica, MA) and purified as the native protein. For small-scale purification, expression of His6-MBP and His6-MBP-VP3 CTD fusion proteins was induced in BL21(DE3)/pLysS transformed with empty vector, RVA VP3 CTD pLIC1HMN, RVB VP3 CTD pLIC1HM, RVG VP3 CTD pLIC1HM, or mutants thereof at 18°C overnight at an OD600 of 0.6 to 0.8 with 1 mM IPTG. The recombinant His6-MBP-tagged proteins were affinity purified using Ni-NTA agarose and dialyzed against 20 mM Tris, pH 7.4, and 150 mM NaCl. Concentrations were estimated based on absorbance, using a conversion of 1 A280 unit as 1 mg/ml.
Synthesis and purification of 2-5A (p3A2).
2-5A was synthesized as described previously (21, 22). The unfractionated 2-5A mixture was clarified by centrifugation at 3,000 × g for 15 min and then separated from macromolecules with a Centriprep (molecular mass cutoff of 3,000 Da [Millipore, Billerica, MA]). The reaction mixture was analyzed on a Dionex PA100 (4 mm by 250 mm) analytical column (Dionex Inc., Sunnyvale, CA) interfaced with a System Gold High-Performance Liquid Chromatograph (HPLC; Beckman-Coulter, Pasadena, CA) under the control of the 32-Karat work station software. A (2′-5′)p3A2 (a 5′ triphosphate and two adenylyl residues connected by a 2′-5′ linkage) construct was purified from the 2-5A mixture by HPLC on a Dionex PA100 (22 mm by 250 mm) preparative column with a gradient of 10 mM to 800 mM NH4HCO3-(NH4)2CO3, pH 8.0. The peak fractions were collected and lyophilized twice after being dissolved in diethyl pyrocarbonate (DEPC)-treated water. The characterization of (2′-5′)p3A2 (>95% purity) was done by analytical HPLC and electrospray mass spectrometry (ES-MS) by direct infusion in negative ion mode on a Micromass Quattro Ultima mass spectrometer. The molecular ions were observed at M(−1) 835 Da and M(−2) 417 Da.
Crystallization.
Crystallization screens for the RVA VP3 CTD at concentrations of 10.4 mg/ml were carried out by hanging-drop vapor diffusion using a Mosquito crystallization robot (TTP LabTech, Melbourn, United Kingdom) and visualized using Rock Imager (Formulatrix) at 20°C. Native RVA VP3 CTD was crystallized under a condition with 14% polyethylene glycol (PEG) 3350, 0.2 M sodium malonate, pH 4, and 1 M NaI. Selenomethionine-substituted RVA VP3 CTD was crystallized under a condition with 21% PEG 3350 and 0.2 M sodium malonate, pH 5. The RVA VP3 CTD and 2-5A substrate were cocrystallized under a condition with 0.1 M HEPES sodium, pH 7.5, 2% (vol/vol) polyethylene glycol 400, and 2.0 M ammonium sulfate at 4°C.
Data collection and processing.
Crystals of the RVA VP3 CTD were cryoprotected by being soaked in crystallization buffer with 20% ethylene glycol and flash frozen in liquid nitrogen, and then they shipped to the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory. Diffraction data were collected on the 5.0.1 beamline, processed with IMOSFLM, scaled, and merged in SCALA, as implemented in the CCP4 suite (23). Space groups were confirmed using POINTLESS (24) (Table 1). Phases and initial electron density maps were calculated by SAD phasing using SHELX (25), and automated model building with experimental phases was performed using ARP/wARP (26) before iterative cycles of manual model building and refinement using COOT (27) and PHENIX (28). The structure of the VP3 CTD in complex with 2-5A was determined by molecular replacement using Phaser (29). Ligand interactions were analyzed using COOT and LIGPLOT (30) with donor-to-acceptor distances between 2.6 Å and 3.2 Å for H-bonding interactions and C-C distances between 3.4 Å and 4.5 Å for hydrophobic interactions. Structural figures were generated using the University of California–San Francisco (UCSF) Chimera program (31).
TABLE 1.
Crystallographic data collection and refinement statistics
| Parameter | Value for the parameter in:a |
||
|---|---|---|---|
| VP3 CTD | SeMet VP3 CTDb | VP3 CTD/2-5A | |
| Data collection | |||
| Space group | P21 | P21 | P43212 |
| Cell dimensions | |||
| a, b, c (Å) | 40.24, 68.37, 52.42 | 40.43, 68.03, 52.42 | 97.14, 97.14, 110.73 |
| α, β, γ (°) | 90.00, 106.88, 90.00 | 90.00, 107.09, 90.00 | 90.00, 90.00, 90.00 |
| Wavelength (Å) | 0.9774 | 0.9774 | 0.9765 |
| Resolution (Å) (range) | 31.86–1.35 (1.42–1.35) | 40.34–1.65 (1.74–1.65) | 35.0–3.10 (3.15–3.10) |
| Rmerge (%) | 3.9 (47.1) | 8.8 (33.1) | 14.6 (82.8) |
| I/σI | 19.2 (3.2) | 8.4 (3.0) | 23.6 (4.4) |
| Completeness (%) | 95.2 (93.5) | 99.3 (99.3) | 99.9 (100.0) |
| Multiplicity | 4.9 (4.9) | 3.4 (3.5) | 18.4 (18.4) |
| Anomalous completeness | 81.2 (75.4) | ||
| Anomalous multiplicity | 1.8 (1.8) | ||
| DelAnom correlation between half-sets | 0.156 (0.098) | ||
| Refinement | |||
| Resolution (Å) (range) | 31.86–1.35 | 34.50–3.10 | |
| No. of reflections | 111,847 | 9,763 | |
| Rwork/Rfree (%) | 19.16/20.59 | 26.06/27.24 | |
| Average B factor (Å2) | |||
| Total | 23.20 | 37.80 | |
| Malonate ion | 21.24 | 69.30 | |
| Ethylene glycol | 31.81 | 52.3 (2-5A) | |
| Water | 33.74 | 10.25 | |
| RMS deviations | |||
| Bond length (Å) | 0.008 | 0.004 | |
| Bond angle (°) | 1.274 | 1.141 | |
Numbers in parentheses correspond to values for the highest-resolution shell.
SeMet, selenomethionine.
FRET-based assay of 2-5A cleavage.
Purified proteins (10 μM MBP or 1 μM MBP-VP3 CTD or mutants thereof) were diluted into assay buffer (20 mM HEPES, pH 7.2, 10 mM MgCl2, 1 mM dithiothreitol) and incubated at 30°C with 10 μM (2′-5′)p3A3 for the time indicated in Fig. 6. Reactions were stopped by heat inactivation at 95°C for 3 min. Residual (2′-5′)p3A3 was measured using a fluorescence resonance energy transfer (FRET)-based RNase L activity assay (32). Assays were performed in triplicate. Replicate values were analyzed in comparison to MBP and to MBP-RVA VP3 CTD using two-way analysis of variance (ANOVA) in GraphPad Prism, version 6.0.
FIG 6.

2-5A degradation activity by wild-type and mutant VP3 CTDs. 2-5A was incubated with the indicated proteins, and reaction products were assayed for the capacity to activate RNase L using a FRET-based assay. (A) Activity of RVA VP3 CTD and mutants thereof predicted to be involved in catalysis (H718 and H797) and ligand binding (L758 and R792). (B) Activity of the RVA VP3 CTD containing RVG-like point mutations (T799N and T799R), of RVB and RVG VP3 CTDs and catalytic histidine mutants thereof, and of an RVG VP3 CTD mutant (N742T) with an RVA-like second HΦ(S/T)Φ motif. Positive control, no 2-5A; negative control, no PDE. Measurements were taken in triplicate. Error bars represent standard deviations. *, P < 0.001, in comparison to RVA VP3 CTD; #, P < 0.001, in comparison to MBP.
Molecular modeling.
Homology models of the RVB CAL-1 VP3 CTD amino acids 649 to 763 (GenBank ACD39823) and RVG 03V0567 VP3 amino acids 653 to 768 (GenBank JQ920005) were generated using Modeller (33) in Chimera, with the coordinates of the RVA VP3 CTD as the template.
Protein structure accession numbers.
Coordinates for the RVA VP3 CTD alone and in complex with 2-5A have been deposited in the Protein Data Bank (PDB) under accession numbers 4RPT and 4YE2.
RESULTS
Crystal structure of the RVA VP3 CTD.
To investigate the structural basis for viral 2′,5′-PDE activity, the RVA VP3 CTD (residues 694 to 835) of simian strain RRV was cloned and expressed in E. coli. The VP3 CTD crystallized in the P21 space group, with two molecules in the asymmetric unit, and diffracted to 1.35-Å resolution. The VP3 CTD structure exhibits an α/β-fold with three α-helices (α1 to α3) and seven β-strands (β1 to β7), which form an antiparallel, concave, twisted β-sheet (Fig. 1A and 2C). This finding is consistent with a recently reported structure for the VP3 CTD of another simian RVA (strain SA11) (34). The two HΦ(S/T)Φ motifs, consisting of H718, L719, T720, and F721 from the β2-strand and H797, I798, T799, and L800 from the β5-strand, are located adjacent to one another at the base of the catalytic cleft. The electrostatic potential of the groove is predominantly positive, establishing a structural platform for binding to the negatively charged 2-5A substrate (Fig. 1B). In the crystal structure, a well-ordered, hydrophilic ethylene glycol is bound at the center of the catalytic site in each VP3 CTD molecule in the asymmetric unit. Although the disposition of the vicinal oxygen atoms of the ethylene glycol is slightly different in each monomer, these oxygen atoms engage in H-bonding interactions involving the histidine and threonine residues from the HΦ(S/T)Φ motifs. Except for minor changes in the flexible N-terminal loop, the two RVA VP3 CTD molecules in the asymmetric unit exhibit identical structural features.
FIG 1.

Crystal structure of the RVA VP3 CTD. (A) Ribbon representation of RVA VP3 CTD, with β-strands, α-helices, and loops shown in cyan, green, and gray, respectively. HΦ(S/T)Φ motifs are shown in yellow ball-and-stick representation. Bound ethylene glycol is shown in pink ball-and-stick representation. Blue atoms, N; red atoms, O. (B) Electrostatic surface potential of the RVA VP3 CTD oriented as in panel A. Positively and negatively charged residues are shown in blue and red, respectively.
FIG 2.

Two-dimensional topology diagrams of a consensus 2H phosphoesterase domain (A), the AKAP7 central domain (B), and the RVA VP3 CTD (C) and the predicted topology of the RVB and RVG VP3 CTDs (D). Light gray arrows indicate β-strands, dark gray rectangles indicate regions containing α-helices (and sometimes 310 helices), and dashed lines indicate loops. Strands and helices are numbered starting from the N terminus of each protein. Asterisks represent the location of HΦ(S/T)Φ motif histidine and serine or threonine residues. The figure is not drawn to scale.
HΦ(S/T)Φ motifs and substrate binding.
To gain structural insight into the mechanism of 2-5A recognition for VP3, we carried out extensive cocrystallization and soaking experiments with AMP, a nonhydrolyzable 2-5A analog, and a 2-5A substrate (p3A2) that was synthesized in-house and contains a 5′ triphosphate and two adenosines connected by a 2′-5′ linkage. The VP3 CTD was cocrystallized with 2-5A at 4°C in the P43212 space group with two molecules in the asymmetric unit and yielded clear density corresponding to the 5′ monophosphate and adenosine and to the 5′ monophosphate of the 2′ adenosine at the catalytic cleft in one of the two molecules (Fig. 3A). However, the electron densities for the sugar and base moieties of the second adenosine were absent, suggesting that the second base was not stabilized by any specific interactions with VP3 CTD. Based on LIGPLOT analysis, the 5′ monophosphate of the 5′ adenosine in 2-5A interacts with the residues T720, H797, and T799 of the HΦ(S/T)Φ motifs via hydrogen bonds (Fig. 3B and 4). H718 further stabilizes the ligand through hydrophobic interactions. In addition, residues L758, I762, and I795 form hydrophobic interactions with the sugar and base moieties of 2-5A. Of note, R792 of the flexible loop adjacent to the catalytic site undergoes significant structural changes, resulting in formation of a cation-π interaction with the adenine base of the substrate. The simulated omit density map clearly shows density at the 2′ phosphate that connects the two adenosines by a 2′-5′ linkage. The side chain of R792 also forms a hydrogen bond with the 2′ phosphate, suggesting that this moiety is important for positioning the 2-5A substrate in the catalytic cleft (Fig. 3C).
FIG 3.

Molecular interactions of 2-5A with the RVA VP3 CTD. (A) Crystal structure of the RVA VP3 CTD in complex with a 2-5A substrate. The RVA VP3 CTD is represented in purple ribbon diagram. A simulated annealing omit difference map, contoured at the 3σ level and represented as a gray mesh, shows the binding of 2-5A to the RVA VP3 CTD. Bound ligand is shown in yellow ball-and-stick representation. (B) Molecular interactions between the RVA VP3 CTD and the observed moieties of 2-5A analyzed using LIGPLOT (30). The RVA VP3 CTD is oriented as in Fig. 2A, with side chains of interacting amino acids shown as sticks and labeled. The hydrogen bond and the cation-π interactions are indicated as black dashed lines and red dashed lines with double arrows, respectively. (C) Conformational changes of RVA VP3 CTD upon binding to 2-5A. The apo structure of VP3 CTD (cyan) is superimposed on that of the VP3 CTD/2-5A complex (purple and yellow, respectively). Significant structural changes are indicated with arrows. Blue atoms, N; red atoms, O; orange atoms, P.
FIG 4.

Detailed molecular interactions between the RVA VP3 CTD and the observed moieties of a 2-5A substrate, which is equivalent to 2′P-AMP, as determined using LIGPLOT. The amino acid residues and the ligand involved in the interactions are labeled. The carbon, nitrogen, and oxygen atoms are shown as black, blue and red circles, respectively. Hydrogen bond interactions are shown as green dashed lines between the respective donor and acceptor atoms, along with the bond distance. The van der Walls contacts are indicated by red arcs with spokes radiating toward the ligand atoms they contact. The contacted atoms are shown with spokes radiating back.
Structural homologs of the VP3 CTD.
A DALI search identified the AKAP7 central domain (PDB code 2VFK; Z = 12.5) (Fig. 5A) and the 2′-5′ RNA ligase from Thermus thermophilus (PDB code 1IUH; Z = 12.4) as the closest structural homologs of the RVA VP3 CTD, with root mean square (RMS) deviations of 3.0 Å and 2.7 Å, respectively, for the matching Cα atoms. Superposition of the VP3 CTD structure with that of AKAP7 showed that the HΦ(S/T)Φ motifs are structurally conserved and revealed three significant differences (Fig. 5B). First, while the region that corresponds to residues 706 to 717 in the VP3 CTD structure is predominantly a loop, in both AKAP7 and 2′-5′ RNA ligase structures, this region is longer and contains a well-defined α-helix (Fig. 2 and 5B and C). Second, while the bacterial 2′-5′ RNA ligase and AKAP7 form well-defined β-strands in the corresponding region, RVA VP3 CTD residues 750 to 753 do not. Third, the region between residues 803 and 816 is a loop in the VP3 CTD structure, but in the other two structures, the corresponding region is significantly longer and contains an α-helix and a β-strand.
FIG 5.

Structural homolog of the RVA VP3 CTD. AKAP7 (tan; PDB 2VFK) is shown alone (A) or superimposed on the RVA VP3 CTD (cyan) (B). Two loops of the RVA VP3 CTD, residues 706 to 717 and 803 to 816, are shown in dark blue. The corresponding AKAP7 residues, 100 to 131 and 230 to 261, which form helices and strands, are shown in orange. The R loop in each structure is indicated. (C) Structure-based alignment of RVA VP3 CTD and AKAP7 central domain. Blue highlighting indicates identical amino acids in the alignment. The secondary structure of the RVA VP3 CTD is shown in light blue below the sequences, and the secondary structure of the AKAP7 central domain is shown in gray above. Asterisks denote VP3 CTD residues that interact with 2-5A (Fig. 4). (D and E) Comparison of the catalytic sites of the RVA VP3 CTD (D; purple) and AKAP7 (E; tan). RVA VP3 CTD residues interacting with 2-5A (yellow ball-and-stick representation) are labeled. AKAP7 residues interacting with AMP (green ball-and-stick representation) are shown. Blue atoms, N; red atoms, O; orange atoms, P.
Another interesting difference between the RVA VP3 CTD and its homolog AKAP7 relates to the conformation of a loop near the catalytic cleft. In the AKAP7 structure, R219 in the R loop is involved in a cation-π interaction with the adenine base of AMP (Fig. 5C) (20). The R-loop arginine is highly conserved among 2H phosphoesterases that cleave 2-5A. The conformation of the R loop and the orientation of the R219 side chain do not appear to change between the apo and AMP-bound forms of AKAP7 (20). In contrast, the R loop and the structurally equivalent arginine in the RVA VP3 CTD, R792, undergo a significant conformational change upon binding to 2-5A (Fig. 3C). In comparison to its location in the apo RVA VP3 CTD structure, the R792 side chain in the CTD/2-5A complex has moved by ∼4Å and reoriented to form a cation-π interaction with the adenine base. Other residues of the RVA VP3 CTD R loop, M793 and F794, shifted 3.2 Å and 4.14 Å, respectively. Of note, the oxygen atom of M793 would provide steric hindrance for the base moiety of the 5′ adenosine in the apo structure. Upon VP3 CTD binding to the 2-5A substrate, M793 moves away from the catalytic cleft. In addition, F179 in AKAP7, which is involved in a π-π stacking interaction with the adenine base of AMP, is replaced by L758 in the VP3 CTD structure, which also provides this stabilizing interaction (Fig. 5C to E). These observations suggest that although there are some amino acid sequence changes in the VP3 CTD, it uses the same catalytic motifs for hydrolyzing the 2-5A substrate and similar interactions to stabilize substrate in the catalytic cleft. However, the RVA VP3 CTD introduces significant conformational changes in the R loop upon substrate binding.
Mutational analysis of substrate-binding residues by the RVA VP3 CTD.
To quantify contributions of RVA VP3 amino acids that interact with 2-5A, we purified the CTD and point mutants thereof as N-terminal maltose-binding protein (MBP) fusions and measured their 2-5A degradation activity. Purified proteins were incubated with 2-5A. Then, 2-5A degradation over time was quantified using a fluorescence resonance energy transfer (FRET)-based assay of RNase L activation (Fig. 6A) (32). The RVA VP3 CTD cleaved 2-5A with similar kinetics and efficiency to MHV ns2, suggesting that the crystallized domain is catalytically active. As expected, HΦ(S/T)Φ mutants H718A and H797A were inactive. While both L758A and L758F substitutions significantly diminished RVA VP3 CTD 2′,5′-PDE activity, the L758A substitution was the more detrimental of the two changes (Fig. 6A). As a case in point, the L758A mutant exhibited activity indistinguishable from that of MBP following 5 or 15 min of incubation with 2-5A, whereas the activity of the L758F mutant was significantly different from that of MBP following any length of incubation with 2-5A. The side chain of R792 forms H bonds and a cation-π interaction with substrate in the VP3 CTD/2-5A structure (Fig. 3 and 4). Surprisingly, replacing R792 with either isoleucine or alanine had a moderate effect, resulting in approximately half the level of activity at most time points (Fig. 6A).
2′,5′-PDE activity of VP3 CTDs from divergent rotavirus species.
To determine whether RVB (strain Bang117) and RVG (strain 03V0567) VP3 CTDs are functional 2′,5′-PDEs, we expressed and purified these domains and quantified their 2-5A cleavage activities. The RVB VP3 CTD was less active than the RVA VP3 CTD and exhibited delayed kinetics (Fig. 6B). In contrast, the RVG VP3 CTD demonstrated no significant activity at any point in the assay. As expected, mutation of the conserved histidine in the second HΦ(S/T)Φ motif (H736A) ablated activity for the RVB VP3 CTD.
Modeling RVB and RVG CTD structures.
The VP3 CTDs of RVB and RVG, respectively, exhibit only 17.2% and 19.1% sequence identity with the CTD of RVA and are each ∼25 amino acids shorter (Fig. 7A). To predict structural differences among VP3 CTDs from different rotavirus species, we generated homology models for RVB and RVG VP3 CTDs using Modeller (Fig. 7B and C) (33). The homology models show a very similar disposition of the catalytic HΦ(S/T)Φ motifs (and HΦNΦ for RVG). However, there are noticeable differences in comparison to the RVA VP3 CTD: the loop between the β1- and β2-strands is nine amino acids shorter, the length of the α1-helix is reduced by six amino acids, and the β7 -sheet is absent in the modeled structures (Fig. 7). Coupled with 2-5A cleavage assays (Fig. 6B), the models suggest that these three regions of the VP3 CTD are dispensable for activity, at least in the case of RVB.
FIG 7.

Homology models of RVB and RVG VP3 CTD. (A) Sequence alignment of VP3 CTDs from RVA, RVB, and RVG strains. The HΦ(S/T)Φ motifs and gaps in RVB and RVG sequences are indicated with black and red boxes, respectively. Colored columns indicate amino acid identity, with dark blue representing the highest identity. The secondary structure of the RRV VP3 CTD is shown above the sequences. VP3 residues that interact with 2-5A (Fig. 4) are denoted by black asterisks. (B and C) Ribbon representations of homology models of the RVB VP3 CTD (B; orange) and the RVG VP3 CTD (C; pink), with histidine and threonine (or asparagine) residues from the HΦ(S/T)Φ motifs shown in ball-and-stick representation and labeled. (D) Structural superimposition of the RVA VP3 CTD structure (cyan) with RVB (orange) and RVG (pink) VP3 CTD models. Red arrows indicate major structural differences.
Catalytic motif requirements for 2′,5′-PDE activity.
RVG VP3 sequences aligning with the second predicted HΦ(S/T)Φ motif are HΦNΦ and HΦRΦ (Fig. 7A) (35). To determine the effects of these catalytic-site alterations on PDE activity, we replaced the threonine in the second HΦ(S/T)Φ motif of RVA VP3 CTD (T799) with asparagine or arginine. We also engineered the corresponding N742T mutation in RVG VP3 PDE, changing HΦNΦ to HΦTΦ. The 2-5A degradation capacities of mutant VP3 CTDs over time were quantified using a FRET-based assay (Fig. 6B). Replacing threonine in the second HΦ(S/T)Φ motif with either asparagine or arginine significantly decreased RVA VP3 CTD 2′,5′-PDE activity, with essentially no activity observed for the arginine substitution mutant. Nonetheless, an N742T mutation was unable to confer efficient 2′,5′-PDE activity on the RVG VP3 CTD.
Identification of amino acids conserved among 2H PDEs that cleave 2-5A.
To identify amino acids conserved among 2H PDEs that cleave or are predicted to cleave 2-5A, we aligned sequences from a variety of RVA and RVB VP3 CTDs, torovirus putative nsp9 proteins, coronavirus ns2 proteins, and AKAP7 proteins (Fig. 8). We then mapped the level of conservation among the aligned proteins onto the structure of the RVA VP3 CTD/2-5A complex (Fig. 9). The most highly conserved amino acids mapped to (i) the first half of the β1-strand and the loop that precedes it, (ii) the β2- and β5-strands, which each contain an HΦ(S/T)Φ motif, and (iii) the loop preceding the β5-strand (Fig. 8). In the three-dimensional structure, these conserved residues cluster in the catalytic motifs and an adjacent region along one side of the cleft, forming a putative substrate recognition platform (Fig. 9). Only five amino acids were identical for all sequences in the alignment: P697, H718, R792, H797, and T799 (Fig. 8 and 9). In addition to the catalytic motif histidines and threonine, these exceptionally well-conserved residues include the R-loop arginine, which stacks with the substrate adenine base, and a structurally adjacent proline, which may be involved in maintaining local protein conformation or in substrate recognition.
FIG 8.

Sequence alignment of viral and cellular 2H phosphoesterase superfamily members known or predicted to bind and degrade 2-5A. Amino acid sequences of RVA and RVB VP3 CTDs, coronavirus ns2, Berne and Breda virus pp1a (predicted nsp9), and AKAP7 were aligned using the T-Coffee multiple sequence alignment server (45, 46). Shown is a subset of the larger alignment. Species of origin, viral strain and/or protein name, and GenBank/PDB accession number are indicated. Colored columns indicate amino acid identity, with dark blue representing the highest identity. The secondary structure of the RRV VP3 CTD is shown in light blue above the sequences, and the secondary structure of the AKAP7 central domain is shown in gray below. A region of the alignment representing the C termini of the proteins, in which no identical or similar residues were observed, has been omitted. Si, simian; Av, avian; Hu, human; Bo, bovine; Eq, equine.
FIG 9.

Conservation among 2H phosphoesterases known or predicted to cleave 2-5A. Surface representation of the RVA VP3 CTD with bound 2-5A (yellow ball-and-stick representation) (Fig. 3), colored by sequence-based amino acid conservation (Fig. 8). The most and least highly conserved positions are shown in maroon and cyan, respectively, as indicated by the scale bar. The molecules are oriented as in Fig. 1A (A) and rotated 180° about the y axis (B). (C) View of the catalytic cleft. A 90% transparent surface reveals the underlying ribbon drawing and side chains of highly conserved amino acids, several of which are shown and labeled. Blue atoms, N; red atoms, O; orange atoms, P.
DISCUSSION
Successful viral replication depends on the capacity to evade intrinsic cellular antiviral defenses. 2H phosphoesterases are found in eukaryotes, bacteria, archaea, and DNA and RNA viruses and mediate a variety of functions via cleavage of nucleoside-containing substrates (15). Evasion of antiviral effects of the OAS/RNase L pathway through degradation of 2-5A by a 2H phosphoesterase is a strategy employed by at least two distinct RNA viruses, rotaviruses and coronaviruses (6, 7). Based on a close phylogenetic relationship with rotavirus, coronavirus, and torovirus 2H 2′,5′-PDE domains, AKAP7 or a related cellular 2H phosphoesterase is thought to have been acquired by these RNA viruses through recombination (15, 18). The high-resolution structure of the RVA VP3 CTD presented herein together with that recently reported for another RVA VP3 CTD (34) further support this idea, based on structural homology with AKAP7, and suggests that after the domain was usurped, it was streamlined, likely to accommodate the relatively small genomic space of a virus.
The streamlined nature of the RVA VP3 CTD and its efficient enzymatic activity suggest a new minimal core topology for 2H phosphoesterases. Two α-helical regions and the β-strands that follow α2 and α4 in AKAP7 and in the consensus 2H phosphoesterase structure are replaced by simple loops in the RVA VP3 CTD, without obvious impact on enzymatic activity levels (Fig. 2, 5B, and 6). Considering the 2′,5′-PDE activity and predicted structure of the RVB VP3 CTD (Fig. 2D and 6B, and 7), an even more minimal structure than that reported here may form the 2H phosphoesterase core. Based on sequence alignment (Fig. 8), coronavirus ns2 and torovirus nsp9 may have an overall topology more similar to that of AKAP7 than to that of the RVA VP3 CTD, perhaps with a longer α2-helix and a shorter α3-helix. Broadening the constraints on predicted topology may permit future identification of novel 2H phosphoesterase superfamily members.
Structures of the RVA VP3 CTD have implications for the mechanisms of 2′,5′-PDE substrate binding and catalysis. The location of the two HΦ(S/T)Φ motifs and the predominantly electropositive potential of the groove formed by the β-sheet in the RVA VP3 CTD identify it as the substrate-binding cleft (Fig. 1). In the VP3 CTD/2-5A cocrystal structure, the HΦ(S/T)Φ motif histidines (H718 and H797) and threonines (T720 and T799) interact with bound 2-5A (Fig. 3 and 4). The lack of activity observed for the RVA VP3 CTD H718A and H797A mutants (Fig. 6A) is consistent with an essential role for these residues in catalysis and with previous observations (6, 14). The reduced enzymatic activity of the RVA VP3 CTD T799N mutant and the ablated activity of the T799R mutant (Fig. 6B) support the proposed role of HΦ(S/T)Φ threonines in substrate stabilization. While asparagine may permit substrate binding, the length of the arginine side chain likely would result in clashes with bound 2-5A. The localization of amino acids conserved among 2H phosphoesterases that cleave 2-5A in the base and one side of the binding cleft (Fig. 8 and 9) implies a functional role for these residues in substrate recognition and/or molecular orientation. Bound 2-5A contacts several of the amino acids that are highly conserved among these 2H phosphoesterases, including four (H718, R792, H797, and T799) of the five residues that are absolutely conserved (Fig. 3B, 4, 8, and 9).
The RVA VP3 CTD and AKAP7 appear to engage substrate similarly, but movement of the R loop upon substrate binding is observed only for the RVA VP3 CTD. In the AKAP7/AMP structure, the adenine base is held in position by two strong interactions, a π-π stacking with phenylalanine and a cation-π interaction with the R-loop arginine (Fig. 5E) (20). The phenylalanine in AKAP7 is replaced by leucine in the RVA VP3 CTD, which would preclude a strong stacking interaction with the base (Fig. 5D). However, steric stacking or hydrophobic stabilization by leucine appears to be sufficient for comparable cleavage activity as the activity levels of the RVA VP3 CTD and MHV ns2 were indistinguishable in our assay (Fig. 6A), and MHV A59 ns2 and the strain WA VP3 CTD previously have been shown to have activity similar to that of AKAP7 (6, 18, 19). In the structure of the complex with 2-5A, RVA VP3 CTD L758 interacts hydrophobically with an adenine base of the bound ligand (Fig. 3B and 4). Consistent with this structural observation, replacing L758 with alanine, which likely would not stabilize adenine, was more detrimental to 2′-5′-PDE activity than substitution with phenylalanine, which likely would form a π-π stack similar to that observed for AKAP7 with bound AMP (Fig. 6A) (20). In the apo VP3 CTD crystal structure, the R loop and R792 exhibit significantly different positioning and side chain orientation than those of apo AKAP7. This is also true for the apo VP3 CTD structure of RVA strain SA11 (34). To engage the base of the 5′ adenosine and the phosphate moieties of the second adenosine, this loop changes conformation and moves toward the substrate and catalytic site by several angstroms (Fig. 3C). In the structure of the RVA VP3 CTD/2-5A complex, R792 forms a cation-π interaction with a 2-5A adenine base (Fig. 3B). Based on this observation, we anticipated that R792 interactions may be critical for RVA VP3 CTD 2′,5′-PDE activity. However, our FRET data demonstrated that this enzyme is still capable of substrate cleavage, albeit with delayed kinetics and diminished overall levels, when isoleucine or alanine is substituted for arginine (Fig. 6A). Since 2-5A is degraded in minutes by cellular phosphodiesterases in cells and in serum (36–39), moderate changes in activity in vitro may translate to significant effects in the RNase L response in vivo. Based on the diminished activity of R792A and R792I mutants and the absolute conservation of the R-loop arginine, the cation-π interaction contributes importantly to substrate stabilization for 2′,5′-PDEs (Fig. 6A and 8).
While the molecular interactions described above appear to stabilize the 2′ P-AMP region of 2-5A, the absence of observed density for the 2′ adenosine suggests a lack of specific interactions between the VP3 CTD and this region of the substrate. Density also is lacking for the β and γ phosphates of the 5′ adenosine of the 2-5A substrate, which likely were hydrolyzed during the prolonged crystallization process, leaving a bound p1A2 ligand. One would not necessarily expect to observe specific interactions between the VP3 CTD and the β and γ phosphates since the enzyme cleaves 2′-5′-linked monophosphorylated adenosines in longer 2-5A substrates. The 2′ phosphate moiety of the 2-5A substrate is critical to trigger the conformational changes of the R loop, which include the movements of R792, M793, and F794. Without the M793 moving away from the catalytic cleft, the oxygen atom of M793 would clash with the base of the 5′ adenosine. Based on our observations, there is no specific recognition of the substrate's 2′-5′ linkage. However, the triggering of R-loop movement by the 2′ phosphate moiety of 2-5A described above may in part explain the preference for a 2′-5′-linked substrate. In addition, the conserved residues adjacent to those involved in catalysis and recognition of the 5′ adenosine, including D695 and P697 (Fig. 9), may aid in substrate recognition by providing steric boundaries in the binding cleft. The products of 2-5A hydrolysis by 2′,5′-PDEs are AMP and ATP (6, 7), which indicates that these enzymes cleave between the 2′ C of an adenine base and the α phosphate of a 2′-5′-linked AMP. The engagement of 2-5A via the 5′ adenosine suggests that this is the moiety of 2-5A that is initially recognized by the PDE. While it is unknown whether 2′,5′-PDEs are processive or distributive enzymes, in either case the substrate must translocate in order for cleavage to occur at the correct position between the two adjacent adenosine moieties.
Structures of several 2H phosphoesterases have been determined, but only a few have been resolved with substrate. Besides the structures of AKAP7 with AMP and, now, the dimer of RVA VP3 CTD with 2-5A, only the catalytic domain of mouse 2′,3′-cyclic nucleotide 3′-PDE with 2′-O-(sulfidophosphinato)adenine (PDB 2YOZ) and the E. coli 2′,3′-cyclic PDE ThpR with AMP (PDB 4QAK) have been published (20, 40, 41). Each of the 2′,3′-PDEs interacts with substrate through HΦ(S/T)Φ histidine and threonine residues and a π-π stacking interaction between the adenine base and a phenylalanine three amino acids downstream from the first HΦ(S/T)Φ threonine. These interactions orient the ligands distinctly from those of AKAP7 and the RVA VP3 CTD (Fig. 10). While the residue located three amino acids downstream from the first HΦ(S/T)Φ threonine tends to be aromatic for members of the archaeo-bacterial LigT family (group I) of 2H phosphoesterases (15), the equivalent position in 2′,5′-PDEs tends to be hydrophobic but not aromatic (Fig. 8). 2-5A would be incapable of binding in the site for 2′,3′-PDE substrates due to strong steric clashes with the protein. These observations suggest a clear difference in substrate positioning between 2′,3′-PDEs and 2′,5′-PDEs. Differences in ligand binding among 2H-PDEs may be exploited in the future to design antivirals that specifically inhibit substrate recognition by 2′,5′-PDEs though the effects of such inhibitors on cellular 2′,5′-PDE function would need to be determined.
FIG 10.
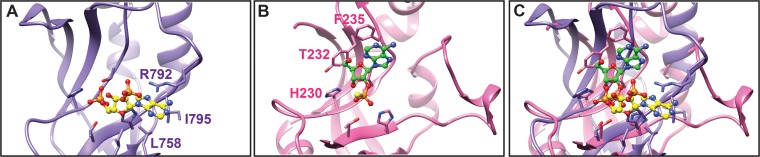
Comparison of substrate interactions. Ribbon drawings of the RVA VP3 CTD (purple) with bound 2-5A (yellow ball-and-stick representation) and of mouse 2′,3′-cyclic nucleotide 3′-PDE (pink) with bound 2′-O-(sulfidophosphinato)adenine (green ball-and-stick representation) alone (A and B) or as an overlay (C). 2′,3′-Cyclic nucleotide 3′-PDE residues that interact with the substrate, including the conserved phenylalanine (F235) that forms a π-π stack, are shown and labeled in pink. Blue atoms, N; red atoms, O; orange atoms, P.
It is unclear why the RVG VP3 CTD fails to cleave 2-5A efficiently in comparison to the RVA and RVB VP3 CTDs (Fig. 6B). RVB and RVG VP3 CTD sequences align similarly with the RVA VP3 CTD and conserve similar numbers of residues predicted to interact with substrate (Fig. 7A). Likewise, their modeled structures show similar folds and identical differences in comparison to the RVA VP3 CTD structure (Fig. 7B to D). N742, which is found in place of threonine in the second HΦ(S/T)Φ motif, is the only highly conserved residue that differs obviously among 2H phosphoesterases that cleave 2-5A. Substitution of the corresponding threonine (T799) in the RVA VP3 CTD diminished activity. However, an N742A substitution failed to render the RVG VP3 CTD an effective 2′,5′-PDE (Fig. 6B). Whereas RVA and RVB strains have been isolated primarily from mammalian hosts, RVG strains have been isolated only from birds (42). Differences in host immune responses or virus pathobiology may place less selective pressure on RVG strains to retain an RNase L antagonist function. Alternatively, these viruses may encode another RNase L antagonist, or the RVG VP3 CTD may sequester 2-5A during rotavirus infection or cleave a different substrate.
Since the 2′,5′-PDE domain exists at the C terminus of VP3 in divergent rotavirus species, we propose that this structural and functional domain was acquired by an ancestral rotavirus and subsequently lost from some species. A potential explanation for the absence of a VP3 CTD from some rotavirus species is that they evolved additional mechanisms to target similar innate antiviral pathways. For example, RVC strains, which lack a VP3 CTD, encode an 8-kDa dsRNA-binding protein that inhibits PKR activation (43). RVH viruses also encode a small protein with homology to dsRNA-binding proteins (44). These proteins may prevent RNase L activation by sequestering dsRNA during rotavirus infection. It is currently unclear what mechanisms RVD and RVF may use to antagonize the OAS-RNase L pathway. Based on the variety of known evasion strategies and the apparent importance of the response for viral replication (3–5), novel mechanisms of OAS/RNase L antagonism for RNA viruses, including rotaviruses and coronaviruses, likely remain to be elucidated.
ACKNOWLEDGMENTS
We thank Marco Morelli for providing the pLIC1HM and pLIC1HMN vectors.
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases at the NIH (to K.M.O. and J.T.P.), by NIH grants R37 AI36040 (to B.V.V.P.) and RO1-AI104887 (to S.R.W. and R.H.S.), and by Robert Welch Foundation grant Q1279 (to B.V.V.P.). The Berkeley Center for Structural Biology is supported in part by the NIH, National Institute of General Medical Sciences, and by the Howard Hughes Medical Institute. The Advanced LightSource is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract number DE-AC02-05CH11231.
REFERENCES
- 1.Goubau D, Deddouche S, Reis e Sousa C. 2013. Cytosolic sensing of viruses. Immunity 38:855–869. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li XL, Ezelle HJ, Hsi TY, Hassel BA. 2011. A central role for RNA in the induction and biological activities of type 1 interferons. Wiley Interdiscip Rev RNA 2:58–78. doi: 10.1002/wrna.32. [DOI] [PubMed] [Google Scholar]
- 3.Chakrabarti A, Jha BK, Silverman RH. 2011. New insights into the role of RNase L in innate immunity. J Interferon Cytokine Res 31:49–57. doi: 10.1089/jir.2010.0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silverman RH. 2007. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J Virol 81:12720–12729. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silverman RH, Weiss SR. 2014. Viral phosphodiesterases that antagonize double-stranded RNA signaling to RNase L by degrading 2-5A. J Interferon Cytokine Res 34:455–463. doi: 10.1089/jir.2014.0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang R, Jha BK, Ogden KM, Dong B, Zhao L, Elliott R, Patton JT, Silverman RH, Weiss SR. 2013. Homologous 2′,5′-phosphodiesterases from disparate RNA viruses antagonize antiviral innate immunity. Proc Natl Acad Sci U S A 110:13114–13119. doi: 10.1073/pnas.1306917110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao L, Jha BK, Wu A, Elliott R, Ziebuhr J, Gorbalenya AE, Silverman RH, Weiss SR. 2012. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe 11:607–616. doi: 10.1016/j.chom.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desselberger U. 2014. Rotaviruses. Virus Res 190:75–96. doi: 10.1016/j.virusres.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 9.Anonymous. 2013. Rotavirus vaccines WHO position paper: January 2013—recommendations. Vaccine 31:6170–6171. doi: 10.1016/j.vaccine.2013.05.037. [DOI] [PubMed] [Google Scholar]
- 10.Matthijnssens J, Otto PH, Ciarlet M, Desselberger U, Van Ranst M, Johne R. 2012. VP6-sequence-based cutoff values as a criterion for rotavirus species demarcation. Arch Virol 157:1177–1182. doi: 10.1007/s00705-012-1273-3. [DOI] [PubMed] [Google Scholar]
- 11.Feng N, Yasukawa LL, Sen A, Greenberg HB. 2013. Permissive replication of homologous murine rotavirus in the mouse intestine is primarily regulated by VP4 and NSP1. J Virol 87:8307–8316. doi: 10.1128/JVI.00619-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoshino Y, Saif LJ, Kang SY, Sereno MM, Chen WK, Kapikian AZ. 1995. Identification of group A rotavirus genes associated with virulence of a porcine rotavirus and host range restriction of a human rotavirus in the gnotobiotic piglet model. Virology 209:274–280. [DOI] [PubMed] [Google Scholar]
- 13.Wang W, Donnelly B, Bondoc A, Mohanty SK, McNeal M, Ward R, Sestak K, Zheng S, Tiao G. 2011. The rhesus rotavirus gene encoding VP4 is a major determinant in the pathogenesis of biliary atresia in newborn mice. J Virol 85:9069–9077. doi: 10.1128/JVI.02436-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ogden KM, Snyder MJ, Dennis AF, Patton JT. 2014. Predicted structure and domain organization of rotavirus capping enzyme and innate immune antagonist VP3. J Virol 88:9072–9085. doi: 10.1128/JVI.00923-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mazumder R, Iyer LM, Vasudevan S, Aravind L. 2002. Detection of novel members, structure-function analysis and evolutionary classification of the 2H phosphoesterase superfamily. Nucleic Acids Res 30:5229–5243. doi: 10.1093/nar/gkf645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hofmann A, Tarasov S, Grella M, Ruvinov S, Nasr F, Filipowicz W, Wlodawer A. 2002. Biophysical characterization of cyclic nucleotide phosphodiesterases. Biochem Biophys Res Commun 291:875–883. doi: 10.1006/bbrc.2002.6527. [DOI] [PubMed] [Google Scholar]
- 17.Hofmann A, Zdanov A, Genschik P, Ruvinov S, Filipowicz W, Wlodawer A. 2000. Structure and mechanism of activity of the cyclic phosphodiesterase of Appr>p, a product of the tRNA splicing reaction. EMBO J 19:6207–6217. doi: 10.1093/emboj/19.22.6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gusho E, Zhang R, Jha BK, Thornbrough JM, Dong B, Gaughan C, Elliott R, Weiss SR, Silverman RH. 2014. Murine AKAP7 has a 2′,5′-phosphodiesterase domain that can complement an inactive murine coronavirus ns2 gene. mBio 5(4):e01312-14. doi: 10.1128/mBio.01312-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao L, Birdwell LD, Wu A, Elliott R, Rose KM, Phillips JM, Li Y, Grinspan J, Silverman RH, Weiss SR. 2013. Cell-type-specific activation of the oligoadenylate synthetase-RNase L pathway by a murine coronavirus. J Virol 87:8408–8418. doi: 10.1128/JVI.00769-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gold MG, Smith FD, Scott JD, Barford D. 2008. AKAP18 contains a phosphoesterase domain that binds AMP. J Mol Biol 375:1329–1343. doi: 10.1016/j.jmb.2007.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang H, Zeqiraj E, Dong B, Jha BK, Duffy NM, Orlicky S, Thevakumaran N, Talukdar M, Pillon MC, Ceccarelli DF, Wan LC, Juang YC, Mao DY, Gaughan C, Brinton MA, Perelygin AA, Kourinov I, Guarne A, Silverman RH, Sicheri F. 2014. Dimeric structure of pseudokinase RNase L bound to 2-5A reveals a basis for interferon-induced antiviral activity. Mol Cell 53:221–234. doi: 10.1016/j.molcel.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jha BK, Polyakova I, Kessler P, Dong B, Dickerman B, Sen GC, Silverman RH. 2011. Inhibition of RNase L and RNA-dependent protein kinase (PKR) by sunitinib impairs antiviral innate immunity. J Biol Chem 286:26319–26326. doi: 10.1074/jbc.M111.253443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans P. 2006. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 25.Sheldrick GM. 2010. Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr D Biol Crystallogr 66:479–485. doi: 10.1107/S0907444909038360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morris RJ, Perrakis A, Lamzin VS. 2003. ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol 374:229–244. doi: 10.1016/S0076-6879(03)74011-7. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wallace AC, Laskowski RA, Thornton JM. 1995. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng 8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 31.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 32.Thakur CS, Xu Z, Wang Z, Novince Z, Silverman RH. 2005. A convenient and sensitive fluorescence resonance energy transfer assay for RNase L and 2′,5′ oligoadenylates. Methods Mol Med 116:103–113. doi: 10.1385/1-59259-939-7:103. [DOI] [PubMed] [Google Scholar]
- 33.Sali A, Blundell TL. 1993. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234:779–815. [DOI] [PubMed] [Google Scholar]
- 34.Brandmann T, Jinek M. 10 March 2015. Crystal structure of the C-terminal 2′,5′-phosphodiesterase domain of group A rotavirus protein VP3. Proteins doi: 10.1002/prot.24794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phan TG, Vo NP, Boros A, Pankovics P, Reuter G, Li OT, Wang C, Deng X, Poon LL, Delwart E. 2013. The viruses of wild pigeon droppings. PLoS One 8:e72787. doi: 10.1371/journal.pone.0072787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnston MI, Hearl WG. 1987. Purification and characterization of a 2′-phosphodiesterase from bovine spleen. J Biol Chem 262:8377–8382. [PubMed] [Google Scholar]
- 37.Kubota K, Nakahara K, Ohtsuka T, Yoshida S, Kawaguchi J, Fujita Y, Ozeki Y, Hara A, Yoshimura C, Furukawa H, Haruyama H, Ichikawa K, Yamashita M, Matsuoka T, Iijima Y. 2004. Identification of 2′-phosphodiesterase, which plays a role in the 2-5A system regulated by interferon. J Biol Chem 279:37832–37841. doi: 10.1074/jbc.M400089200. [DOI] [PubMed] [Google Scholar]
- 38.Schmidt A, Chernajovsky Y, Shulman L, Federman P, Berissi H, Revel M. 1979. An interferon-induced phosphodiesterase degrading (2′-5′) oligoisoadenylate and the C-C-A terminus of tRNA. Proc Natl Acad Sci U S A 76:4788–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Silverman RH, Wreschner DH, Gilbert CS, Kerr IM. 1981. Synthesis, characterization and properties of ppp(A2′p)nApCp and related high-specific-activity 32P-labelled derivatives of ppp(A2′p)nA. Eur J Biochem 115:79–85. [DOI] [PubMed] [Google Scholar]
- 40.Myllykoski M, Raasakka A, Lehtimaki M, Han H, Kursula I, Kursula P. 2013. Crystallographic analysis of the reaction cycle of 2′,3′-cyclic nucleotide 3′-phosphodiesterase, a unique member of the 2H phosphoesterase family. J Mol Biol 425:4307–4322. doi: 10.1016/j.jmb.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Remus BS, Jacewicz A, Shuman S. 2014. Structure and mechanism of E. coli RNA 2′,3′-cyclic phosphodiesterase. RNA 20:1697–1705. doi: 10.1261/rna.046797.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Attoui H, Becnel J, Belaganahalli S, Bergoin M, Brussaard CP, Chappell JD, Ciarlet M, del Vas M, Dermody TS, Dormitzer PR, Duncan R, Fang Q, Graham R, Guglielmi KM, Harding RM, Hillman B, Makkay A, Marzachì C, Matthijnssens J, Mertens PPC, Milne RG, Mohd Jaafar F, Mori H, Noordeloos AA, Omura T, Patton JT, Rao S, Maan M, Stoltz D, Suzuki N, Upadhyaya NM, Wei C, Zhou H. 2012. Family Reoviridae, p 541–637. In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed). 2011. Virus taxonomy: classification and nomenclature of viruses. Ninth report of the International Committee on Taxonomy of Viruses Academic Press, San Diego, CA. [Google Scholar]
- 43.Langland JO, Pettiford S, Jiang B, Jacobs BL. 1994. Products of the porcine group C rotavirus NSP3 gene bind specifically to double-stranded RNA and inhibit activation of the interferon-induced protein kinase PKR. J Virol 68:3821–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morelli M, Ogden KM, Patton JT. 25 February 2015. Silencing the alarms: innate immune antagonism by rotavirus NSP1 and VP3. Virology doi: 10.1016/j.virol.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di Tommaso P, Moretti S, Xenarios I, Orobitg M, Montanyola A, Chang JM, Taly JF, Notredame C. 2011. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res 39:W13–W17. doi: 10.1093/nar/gkr245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kemena C, Notredame C. 2009. Upcoming challenges for multiple sequence alignment methods in the high-throughput era. Bioinformatics 25:2455–2465. doi: 10.1093/bioinformatics/btp452. [DOI] [PMC free article] [PubMed] [Google Scholar]