

ABSTRACT
High-grade tumors in the brain are among the deadliest of cancers. Here, we took a promising oncolytic virus, vesicular stomatitis virus (VSV), and tested the hypothesis that the neurotoxicity associated with the virus could be eliminated without blocking its oncolytic potential in the brain by replacing the neurotropic VSV glycoprotein with the glycoprotein from one of five different viruses, including Ebola virus, Marburg virus, lymphocytic choriomeningitis virus (LCMV), rabies virus, and Lassa virus. Based on in vitro infections of normal and tumor cells, we selected two viruses to test in vivo. Wild-type VSV was lethal when injected directly into the brain. In contrast, a novel chimeric virus (VSV-LASV-GPC) containing genes from both the Lassa virus glycoprotein precursor (GPC) and VSV showed no adverse actions within or outside the brain and targeted and completely destroyed brain cancer, including high-grade glioblastoma and melanoma, even in metastatic cancer models. When mice had two brain tumors, intratumoral VSV-LASV-GPC injection in one tumor (glioma or melanoma) led to complete tumor destruction; importantly, the virus moved contralaterally within the brain to selectively infect the second noninjected tumor. A chimeric virus combining VSV genes with the gene coding for the Ebola virus glycoprotein was safe in the brain and also selectively targeted brain tumors but was substantially less effective in destroying brain tumors and prolonging survival of tumor-bearing mice. A tropism for multiple cancer types combined with an exquisite tumor specificity opens a new door to widespread application of VSV-LASV-GPC as a safe and efficacious oncolytic chimeric virus within the brain.
IMPORTANCE Many viruses have been tested for their ability to target and kill cancer cells. Vesicular stomatitis virus (VSV) has shown substantial promise, but a key problem is that if it enters the brain, it can generate adverse neurologic consequences, including death. We tested a series of chimeric viruses containing genes coding for VSV, together with a gene coding for the glycoprotein from other viruses, including Ebola virus, Lassa virus, LCMV, rabies virus, and Marburg virus, which was substituted for the VSV glycoprotein gene. Ebola and Lassa chimeric viruses were safe in the brain and targeted brain tumors. Lassa-VSV was particularly effective, showed no adverse side effects even when injected directly into the brain, and targeted and destroyed two different types of deadly brain cancer, including glioblastoma and melanoma.
INTRODUCTION
Patients diagnosed with high-grade gliomas in the brain have a poor prognosis and generally succumb to the tumor within a year (1, 2). Similarly, melanoma metastases in the brain also are a death sentence, with a similar or worse prognosis than glioma (3). Surgery, radiation, and chemotherapeutics can extend life modestly but often at the expense of quality of life. One approach to eliminating tumors is with oncolytic viruses, viruses that target and either destroy the tumor directly or generate an immune response against the infected tumor.
One virus that is effective at targeting tumors is vesicular stomatitis virus (VSV) (4). VSV is a rapidly replicating negative-strand enveloped RNA virus. The broad tropism of its envelope glycoprotein, G, facilitates the rapid targeting and infection of many types of cells, including cancer cells (5). VSV effectively targets tumors due to mutations in cancer cells that lead to deficits in interferon (IFN)-based antivirus immunity (4, 6). However, VSV bearing the native VSV G protein generates a detrimental infection of normal neurons (7, 8, 9, 10, 11, 12) that can lead to potential adverse consequences, including neurological dysfunction or death (13).
Although multiple strategies for attenuating VSV bearing the native G protein have met with some success (14, 15, 16, 17, 18, 19, 20, 21), the neurotropism inherent to any VSV bearing the native G protein remains a major obstacle to safety. This raises the question of whether substituting binding glycoproteins from other nonrelated viruses might result in chimeric viruses with reduced neuronal infection and neuron death. Ebola and Marburg virus are members of the Filoviridae family and have a single negative-strand RNA genome; Lassa fever virus and lymphocytic choriomeningitis virus (LCMV) are members of the Arenaviridae family with an ambisense single-strand RNA genome. Both virus families have an envelope and show broad tissue tropism attributable to wide expression of their respective receptors or coreceptors, including the Niemann-Pick C1 cholesterol transporter (22, 23) and T-cell immunoglobulin and mucin domain protein (TIM-1) for filoviruses and α-dystroglycan (24) for arenaviruses. The low-density lipoprotein (LDL) receptor may be the native VSV receptor (25).
Although all these native viruses are dangerous, with Ebola, Marburg, and Lassa viruses requiring biohazard level 4 containment, chimeric viruses that express some of their genes, particularly the glycoprotein gene, appear to be well tolerated within the brain (26, 27). We postulated that if a glycoprotein, particularly one with a broad-spectrum binding potential from another virus, were substituted for the VSV G, we might avoid the neurotoxicity associated with VSV but retain the ability of the virus to target and destroy multiple types of cancer within the brain. Previous work showed that lentivirus vectors pseudotyped with the Ebola virus glycoprotein showed no transduction in the brain, whereas pseudotyping with LCMV, VSV, or Mokola virus (rabies virus relative) (28) did lead to central nervous system (CNS) transfection. Similarly, a rabies virus with the Ebola virus glycoprotein substituted for the rabies virus glycoprotein was safe within the brain (29). Here, we tested a series of chimeric VSVs bearing the glycoprotein of five other viruses to identify an optimal candidate for oncolytic virotherapy in the brain. Substitution of the Lassa virus glycoprotein for the VSV glycoprotein yielded excellent results, eliminating the neurotoxicity of native VSV while retaining a strong ability to target and kill tumor cells within the brain.
MATERIALS AND METHODS
Viruses and cells.
The VSV-wtG (where wt is “wild type”) included the Indiana serotype for the G protein. Chimeric VSVs expressing the glycoprotein from the Josiah strain of Lassa fever virus (VSV-LASV-GPC), the SAD B19 strain of rabies virus (VSV-RABV-G), the Armstrong 53b strain of lymphocytic choriomeningitis virus (VSV-LCMV-GPC), the Mayinga/Zaire strain of Ebola virus (VSV-EBOV-GP), or the Musoke strain of Marburg virus (VSV-MARV-GP) were used as described previously (8, 30, 31, 32, 33); in all the chimeric viruses used here, the native VSV G protein gene was deleted, and the glycoprotein gene from one of the other viruses was substituted for VSV in the same gene position from which the VSV G protein gene had been deleted. As per convention, the glycoprotein precursor gene from Lassa virus and LCMV is denoted by GPC, and the glycoprotein gene from Ebola and Marburg viruses is denoted by GP. The primary VSV-EBOV-GP used lacked the mucin-like sequence of the native molecule. A green fluorescent protein (GFP) reporter gene was engineered into the first genome position of these VSVs. We also used VSV-EBOV-GP and VSV-LASV-GPC that did not express any reporter genes (34); this VSV-EBOV-GP contained the full-length Ebola virus GP, including the mucin-like domain. For in vitro experiments, viruses were propagated on Vero cells, purified, and concentrated using sucrose gradient centrifugation (35). For in vivo experiments, viruses were propagated on Vero cells and filter purified according to a protocol described previously (36). Plaque titers for all viruses were determined on Vero cells prior to experiments.
Human glioma U87 and U118 were obtained from ATCC (Manassas, VA), mouse glioma CT2A was a gift from T. Seyfried (Boston College, Chestnut Hill, MA), and human melanoma were provided by Yale SPORE in Skin Cancer. Normal human glial cells were derived from human temporal lobectomies (37). Normal human embryonic neurons were purchased from Sciencell (Carlsbad, CA). The human cancer cell lines SJSA-1, BT-549, T-47D, HCT116, SW480, T24, and RT4 were kindly provided by S. Mella (Yale University Cancer Center), and the DU-145 line was from B. Gullen (Yale University). Stably transfected tumor cells expressing red fluorescent protein (RFP) (rU87 and rYUMAC) were generated as described earlier (38). Primary cultures of mouse brain were generated by dissociating the cortex of E17 mice for predominantly neuronal cultures and whole brain tissue of P1 mice for mixed neuronal/glia cultures. Cells were plated in minimum essential medium (MEM) (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) overnight before medium was replaced with Neurobasal/B27 medium (Invitrogen). Melanoma cells were maintained in Opti-MEM–5% FBS, melanocyte medium containing additional supplements listed elsewhere (38). Glioma cells, human glia, and fibroblasts were maintained in MEM-10% FBS (Invitrogen). All cultures were kept in a humidified atmosphere containing 5% CO2 at 37°C.
Mouse procedures.
Six- to 8-week-old male Swiss Webster mice received the following virus doses: intracranially (i.c.), 3.6 × 104 PFU in 1 μl into the right striatum (2 mm lateral and 0.5 mm rostral to Bregma at a 3-mm depth); intravenously (i.v.), 106 PFU in 100 μl via tail vein injection; intranasally (i.n.), 2.5 × 105 PFU in 25 μl in each nostril. Stereotactic application of virus or tumor cells was performed under full anesthesia using ketamine and xylazine (100 and 10 mg/kg of body weight, respectively). Uni- and bilateral intracranial glioma and melanoma xenografts were established in 4- to 6-week-old male CB17 SCID mice by injection of 5 × 104 cells each into the left and right striatum (37, 38). Fifteen days after tumor placement, mice received virus either via a unilateral intratumoral injection (3.6 × 104 PFU in 1 μl) or via tail vein injection (106 PFU in 100 μl). Mice were monitored daily and euthanized if one of the following conditions was observed: (i) weight loss of 25% or more, (ii) immobility, (iii) occurrence of adverse neurological symptoms, or (iv) reaching the end of the observation period of the survival study. For histologic analysis of early states of viral infection, mice were sacrificed at day 2 or 8 after viral inoculation.
After being given an anesthetic overdose, mice were transcardially perfused with 4% paraformaldehyde. Brains were harvested and stored in 4% paraformaldehyde, dehydrated in 30% sucrose solution, and cut into 20- to 30-μm coronal sections with a cryostat. For detection of infectious virus in designated mice after short (2 days) and long (>60 days) exposure to VSV-LASV-GPC, tissue samples were collected under sterile conditions from brain, lung, blood, and liver after euthanasia. Tissues were mechanically homogenized in phosphate-buffered saline (PBS) using a microcentrifuge tube tissue grinder. Part of the resultant tissue suspension was plated onto BHK monolayers and assessed for the presence of GFP-positive cells 24 h later.
To test the capability of VSV-LASV-GPC to induce antibody production, adult Swiss Webster mice received an intranasal and intramuscular primary VSV-LASV-GPC inoculation (at concentrations listed above) followed by a boost 4 weeks later. Two weeks later, mice were euthanized and bled, and serum was collected. Antibody-containing serum was diluted 1:50 to 1:10,000. Brain sections from noninfected transgenic mice expressing GFP in hypocretin neurons were used to target GFP. Brain sections of alpha/beta interferon receptor (IFN-α/β)-R-knockout mice infected with VSV-LASV-GPC were used to study virus reporter gene expression. rU87 and rYUMAC cells were tested for pathogens before tumor grafting and found to be pathogen free. All animal experiments adhered to institutional guidelines and were approved by the Yale University Animal Care and Use Committee.
Rat procedures.
The safety of intracranial VSV-LASV-GPC in rats was tested. Stereotactic coordinates and applied virus dose equaled those in rat tumor models described in the following. Syngeneic brain tumor models were established via stereotactic injection of 50,000 cells in 3 μl suspension into the right striatum (0.7 mm rostral of Bregma, 3.5 mm lateral, 5 mm deep) of 7- to 8-week-old Lewis rats (rat CNS-1 glioma). Seven days after tumor placement, rats received a single intracranial injection of 3 μl suspension containing 1.2 × 105 PFU of VSV-LASV-GPC into the area of the tumor. Rats with gliomas were euthanized at 3 days postinoculation (dpi) and brains were harvested. Other rats were given intracranial injection of VSV-LASV-GPC in safety studies.
In vitro experiments.
Viral infection of mixed neuronal/glial cultures and U87, U118, and melanoma tumor cultures was monitored by GFP expression of infected cells. Cultures were assessed for the presence of cytopathic effects before and after virus application. Cytopathic effects were noted as the appearance of rounding, blebbing, and syncytium formation. For analysis of infection characteristics of chimeric VSVs on mixed neuronal/glial cultures, the morphology of infected cells was used as a guide for identifying neuronal or glial infection by the virus. Identification of cell type was later corroborated by immunohistochemistry for NeuN and glial fibrillary acidic protein (GFAP) as described previously (39). Mixed human brain cultures were established by first plating a glial monolayer. Two days later, human neurons were seeded onto the glial monolayer. After 7 days in culture and morphological confirmation of neuron process outgrowth, cultures were inoculated with VSV-wtG or VSV-LASV-GPC (multiplicity of infection [MOI] of 1). GFP expression was quantified 24 h later.
Plaque assay was used to assess viral replication. Serial virus dilutions were used on monolayers of cells. A 0.5% agar solution in MEM was used as a semisolid overlay. For IFN pretreatment, cultures were incubated for 8 h with recombinant hybrid interferon type I IFN-α A/D (Sigma-Aldrich; catalog no. I4401) at a concentration of 100 IU/ml. To test the generalization of the oncolytic effect of VSV-LASV-GPC to other human cancer types, 8 human tumor cell types were infected at an MOI of 3 (primary inoculation). Two hours later, inoculum was removed and cultures were washed 3 times with PBS before the addition of fresh medium. Twenty-four hours later, infection rates were determined by GFP expression. To assess the capability of VSV-LASV-GPC to propagate in these tumor cultures, the supernatant from 24 h postinoculation (hpi) was filtered to remove cellular debris and transferred onto an uninfected monolayer of the same tumor designation (secondary inoculation). In some experiments, plaque size was measured (n = 60 plaques/cell type/virus) and means and standard errors of the means (SEMs) were determined as an approach to compare infection and replication of different chimeric viruses on different cell types, as described previously (21, 38).
Image analysis.
Virus-infected cultures and histological sections of mouse brain were analyzed using a fluorescence microscope (IX 71; Olympus Optical, Tokyo, Japan). A fluorescence stereomicroscope (SZX12; Olympus Optical) was used for whole-brain scanning before sectioning.
Quantitative reverse transcription (RT)-PCR.
Mouse neuronal cultures and U87 glioma cells were cultured in 6-well plates. VSV-LASV-GPC or VSV-wtG was added at an MOI of 1, and cultures were incubated for 20 min at 4°C to test virus binding or for 30 min at 37°C to test virus binding and internalization, as described elsewhere (37). Experiments were performed in duplicate. Cells were washed five times with PBS prior to RNA isolation using TRIzol reagent (Invitrogen, Carlsbad, CA). A Stratascript reverse transcriptase kit (Stratagene) was used for cDNA generation. TaqMan gene expression assays (Applied Biosystems, Foster City, CA) were used to quantify the expression of β-actin and VSV genomes using an ICycler iQ real-time PCR system (Bio-Rad, Hercules, CA). For specific VSV genome detection (excluding viral mRNA), primers were designed to yield a product that spanned the junction between N and P genes. PCR samples were measured in triplicates, normalized to β-actin expression, and compared to the expression of VSV-LASV-GPC binding to neurons as a reference (threshold cycle [ΔΔCT] method).
Statistical analysis.
Statistical significance was determined by Student's t test, analysis of variance (ANOVA), and chi-square test. P values of <0.05 were considered statistically significant. Survival studies were nonblinded. Mice were allocated to the experimental or control group based on cage number. Power analysis was employed to determine the group size for survival experiments of tumor-bearing mice and of virus-injected mice.
RESULTS
Chimeric viruses infect glioma.
Five chimeric VSVs were studied in which the VSV glycoprotein was replaced (8, 30, 31, 32, 33) by the glycoprotein genes from other viruses, including Lassa fever virus (LASV), rabies virus (RABV), LCMV, Ebola virus (EBOV), and Marburg virus (MARV). These chimeric viruses were first compared with a control VSV that retained the normal VSV glycoprotein (VSV-wtG) using in vitro tests. All chimeric viruses and VSV-wtG also encoded GFP in the first genomic position (Fig. 1A) and were examined first in human glioblastoma (GBM) to determine whether these viruses would infect human glioma. All six viruses infected U87 and U118 glioma cells in vitro 24 h postinoculation (hpi) (Fig. 1B).
FIG 1.

Productive infection of glioma cultures by chimeric VSVs. (A) Schematic shows the gene order, relative size of the genes, and substitution of the VSV glycoprotein (G) gene with glycoprotein genes from 5 other viruses. Glycoproteins from Lassa virus and LCMV are denoted by GPC; those from Ebola and Marburg viruses are denoted GP. (B) In initial experiments, we found that all chimeric viruses infected two human gliomas, U87 and U118, using an MOI of 0.1 and studied at 24 h postinoculation; infection is demonstrated by the GFP reporter. A phase-contrast image is shown below each corresponding image of viral infection indicated by GFP. (C) Representative virus plaques are shown, and relative plaque size is indicated by the GFP fluorescence for U118 glioma, a melanoma (501mel), Vero cells, and control human brain cells. Scale bar in millimeters. Plaques were allowed to develop for 20 to 24 h. In comparison of different viruses on a particular cell type, measurements were all done at the same time. (D) Plaque size was measured as an indicator of virus infection and replication and shown relative to the native VSV-wt. The size of each black circle shows the mean size of 60 randomly chosen and measured plaques 24 h after inoculation. SEM is shown by the black line on the upper right side of each circle.
To compare relative levels of infection and replication for each of the six viruses on several tumor cell types, we performed a quantitative plaque size analysis on glioma, melanoma, and normal human brain cells at 24 hpi (Fig. 1C and D). Plaque size gives insight into the ability of a virus to both infect and replicate within particular cell types and allows intervirus comparisons independent of viral titer as measured on any particular standard cell line, which may be more susceptible to one virus than another. All five chimeric viruses showed robust plaques of relatively similar size on U87 glioma cells; the largest plaques were generated by VSV-wtG (P < 0.01, ANOVA with Tukey-Kramer post hoc test), showing it manifested the fastest replication and infection rate, as expected (Fig. 1D). Within the chimeric viruses, VSV-LASV-GPC showed the largest plaque size on U118 glioma cells (P < 0.01), which were universally less susceptible to the chimeric viruses than U87 glioma. On U373 glioma cells, VSV-MARV-GP and VSV-RABV-G showed slightly smaller plaques than the other three chimeric viruses. On melanoma cells, VSV-MARV-GP showed slightly larger plaques than the other chimeric viruses. All viruses also showed some infection of normal cells. Although VSV-wt showed the largest plaques on both glioma and melanoma, it also showed large plaques on normal brain tissue, an undesirable phenotype. Typical of VSV-infected cells (37, 40), infected cells died, as confirmed by staining with membrane-impermeant dyes (not shown).
The data described above show that all chimeric viruses did infect tumor cells with variation between the different viruses. A critical aspect of any potential oncolytic virus, particularly within the brain, is whether the virus will infect normal brain cells, particularly neurons, and whether the level of infection will be reduced compared with that of VSV that retained the normal wild-type glycoprotein. We tested the relative infection of all six viruses on brain cultures that included neurons and glia. The relative infection of mouse neurons versus glia is shown in Fig. 2A, examined at a time when uninfected cells were still apparent. VSV-wtG showed the greatest relative level of neuronal infection (around 90% neurons and 10% glia). VSV-LASV-GPC and VSV-LCMV-GPC showed the least neuronal infection relative to astrocyte infection.
FIG 2.

Chimeric viruses show reduced neuron infection and safety in the brain. (A) Mouse brain cultures were inoculated with VSV-wtG and VSV chimeras (MOI of 5). At 24 hpi, expression of GFP by neurons or glia was analyzed in duplicate cultures, and the relative fraction of neuronal or glial infection is displayed. Noninfected cells were not included. (B) Stereotactic viral injection (3.6 × 104 PFU) into the right striatum of adult Swiss Webster mice with VSV-wtG and VSV-ΔM51 strains (n = 6 each) resulted in strong neurotoxicity and death. In contrast, all mice injected with VSV-LASV-GPC and VSV-EBOV-GP (n = 8 each) survived. (C) Similar intracranial injection of VSV-IFN (n = 7) or VSV-LASV-GPC (n = 4) into striatum. Two additional mice received VSV-IFN in the hypothalamus; both died in 3 days (not included on graph). (D) Human neuronal cultures inoculated with virus (MOI of 1) show significantly reduced infection with VSV-LASV-GPC and VSV-EBOV-GP compared to the strong infection with VSV-wtG.
To move forward to in vivo analyses of safety and efficacy, we decided to test two viruses, one from the arenavirus family (Lassa virus and LCMV) and another from the filovirus family (Ebola and Marburg viruses). In selecting viruses for in vivo testing, we reasoned that VSV-LASV, -LCMV, and -EBOV had the greatest overall growth capacity (plaque size) across different human gliomas (P < 0.05) (Fig. 1D). Among these three, LASV and EBOV showed evidence of attenuated growth capacity (plaque size) compared to that of VSV-wtG in human brain and astrocytes (Fig. 1D). Although LASV and LCMV showed the lowest neuron-to-glia infectivity ratios (Fig. 2A), attenuation of infection in astrocytes is also important to safety in the brain. Together, the in vitro data point to VSV-LASV-GPC as having a good combination of glioma infectivity (Fig. 1C and D), low neuronal tropism (Fig. 2A), and reduced infection of normal glia where VSV-LASV-GPC showed less infection in brain and astrocyte cultures than VSV-wt or VSV-LCMV-GPC. VSV-EBOV-GP was selected as a second candidate, with the glycoprotein representative of the filovirus family to include for in vivo testing being another virus with broad glioma tropism, slightly greater infection of glioma than VSV-MARV-GP (Fig. 1D), and evidence of reduced neurotropism compared to that of VSV-wt.
To corroborate further the relative lack of infection of neurons of the chimeric viruses, we compared VSV-LASV-GPC and VSV-EBOV-GP to two VSVs that expressed the VSV glycoprotein in vivo; one of the VSVs was further attenuated by including an M51 mutation which enhances the antiviral innate immune response against the virus by reducing the ability of VSV to attenuate cellular antiviral responses (41). Injection of either VSV-wtG with or without the M51 mutation into the normal brain generated lethal consequences, with a median survival of 3.5 days for VSV-wtG (n = 6) and 8 days for the attenuated strain VSV-wtG with the M51 mutation (n = 6) (Fig. 2B) consistent with previous observations (42). This underlines the neurotoxicity of VSV with normal VSV-G protein, even when attenuated. In striking contrast, direct injections of VSV-LASV-GPC or VSV-EBOV-GP (3.6 × 104 PFU in 1 μl) into normal mouse brains exerted no adverse effect in >112 days (n = 8 each virus; P < 0.001; chi-square test) (Fig. 2B), and 100% of the intracerebrally inoculated mice survived. In another experiment, we compared VSV-LASV-GPC with VSV-IFN, a type I IFN-expressing virus modeled after one (43) currently in clinical trials for the treatment of liver cancer (NCT01628640). All mice (n = 9) receiving intracranial VSV-IFN died within 12 days, consistent with reports of this virus infecting brain tissue if blood-borne tumor cells bind to the meninges (44); all mice that received VSV-LASV-GPC (n = 4 here) survived with no adverse side effects (Fig. 2C). Similarly, VSV-LASV-GPC injected into the rat brain (n = 3) also showed no sign of neurotoxicity (>80 days). No VSV-LASV-GPC or infected cells were detected in the brain or elsewhere (liver, spleen, blood) by histological analysis or culture of inoculated mouse tissue at the conclusion of the experiment, suggesting the total elimination of the virus.
The injections of VSV-LASV-GPC and VSV-EBOV-GP were viable, as euthanasia at 2 dpi revealed limited infection of glia within the brain. However, by 8 dpi, few or no infected normal cells could be found. VSV-LASV-GPC and VSV-EBOV-GP were also injected intravenously (n = 5 each), and an additional set of mice were inoculated at combined intranasal/intramuscular/subcutaneous sites (n = 5, total 8 × 106 PFU VSV-LASV-GPC); none of these mice showed signs of viral pathogenicity, and no virus could be harvested from these mice 2 months postinoculation. We also tested intracranial injection of a different VSV-LASV-GPC that contained no GFP. In a 6-week time frame following brain infection, no adverse effects were identified in the infected mice (n = 5), showing that the attenuation mediated by inclusion of the GFP gene in the first genomic position was not critical for CNS safety.
Basis for selective infection.
To elucidate mechanisms whereby the chimeric viruses appeared to show strong infection of glioma but less infection of normal neurons and glia, we examined the innate immune response. In contrast to the strong protective effect of type I IFN on cultures of normal human neurons, glia, or fibroblasts (Fig. 3B), IFN had little effect in attenuating infection by VSV-LASV-GPC of U87 human glioma, as virtually all glioma cells were infected and GFP positive by 24 h postinoculation (Fig. 3A).
FIG 3.

Survival and oncoselectivity following infection with VSV-LASV-GPC are interferon dependent. (A and B) IFN-α protects nontumor cells from VSV-LASV-GPC infection. Cultures of human U87 glioma (A) and normal cells, including human glia, human fibroblasts, and human neurons (B), were treated with 100 IU/ml IFN-α for 8 h before being infected with VSV-LASV-GPC at an MOI of 0.1. In spite of IFN treatment, strong viral infection was seen on glioma cells at 24 hpi; in contrast, little VSV-LASV-GPC infection was found on nontumor normal human cells. Scale bar, 100 μm. (C) Cultures containing human neurons and glia were infected with VSV-wtG or VSV-LASV-GPC at an MOI of 1 in the presence or absence of 100 IU/ml IFN-α, respectively. IFN had little effect in reducing infection of neurons by VSV-wtG but substantially attenuated infection of neurons by VSV-LASV-GPC (P < 0.05). (D) Intracranial injection of VSV-wtG (n = 3) and VSV-LASV-GPC (n = 5) into IFN-α/β-R−/− mice resulted in death within 3 days (VSV-wtG) and 7 days (VSV-LASV-GPC). VSV-LASV-GPC injection in normal mice (IFN-α/β-R+/+) did not result in neurotoxicity or mortality in any mice. (E to G) Typical microscopic images of injected striatum sections showing VSV-LASV-GPC infection in IFN-α/β-R−/− and IFN-α/β-R+/+ mice. Scale bar, 100 μm. (H) Binding of VSV-wtG and VSV-LASV-GPC was assessed by qRT-PCR following incubation at 4°C, and binding and internalization at 37°C on neurons or U87 glioma cells (n = 3; SEM). (I) Quantification of viral replication in neurons and U87 glioma cells was assessed by plaque assay at 15 and 24 hpi. VSV-wtG produced equal titers of progeny in both neurons and glioma. (J) In contrast, VSV-LASV-GPC progeny production was strongly reduced in neurons compared to human glioma.
These data are consistent with the view that a primary mechanism underlying the selective VSV-LASV-GPC infection of cancer cells over normal glia is related to deficiencies in innate immunity. The lack of cancer cell protection by added IFN points to a deficient IFN response among VSV-LASV-GPC-susceptible tumors, similar to the mechanism suggested for the enhanced relative susceptibility of a number of tumor types to native VSV and to several other viruses, including Newcastle disease virus, reovirus, and myxoma virus (45, 46, 47). The observation that normal brain cells in SCID mice which are deficient in T- and B-cell antiviral defenses showed little infection and no adverse effects from VSV-LASV-GPC in the brain further supports the view that an innate immune mechanism is protective of normal brain cells.
To test whether the intrinsic IFN system is essential for the resistance of the brain to VSV-LASV-GPC, we injected VSV-LASV-GPC into the brains of transgenic mice (n = 5) lacking the type I IFN receptor. Although IFN-α/β-R−/− mice with intracerebral infection from VSV-LASV-GPC survived longer than those with VSV-wtG (n = 3) (Fig. 3D to G), all these mice ultimately died within a week of CNS inoculation. These data support the view that an innate IFN-mediated immune response is critical for both short- and long-term survival after VSV-LASV-GPC infection of the brain.
The findings described above motivated a comparison of the binding of VSV-wtG and VSV-LASV-GPC to glioma cells and neurons. As judged by quantitative RT-PCR, both viruses bound similarly to glioma cells and to neurons at 4°C (Fig. 3H), a cold temperature at which endocytosis is inhibited. At 37°C, approximately 2 logs more virus became cell associated than at 4°C, indicating effective and similar rates of binding plus internalization in both cell types and for both viruses. We then examined the ability of infected glioma cells and neurons to generate progeny virions. On gliomas, both VSV-wtG (Fig. 3I) and VSV-LASV-GPC (Fig. 3J) showed substantial virus replication. In contrast, replication of VSV-LASV-GPC was greatly attenuated in neurons, by 4.5 logs relative to VSV-wtG, which showed robust replication in neuron cultures (Fig. 3I and J). Together, these data suggest that although both VSV-wtG and VSV-LASV-GPC bind to glioma cells and neurons, VSV-LASV-GPC replicates poorly in neurons. In neurons, a block to replication appears most likely to occur at the point of endosomal escape and uncoating, which may in part be dependent on glycoprotein mediation or possibly a subsequent step.
VSV-LASV-GPC targets and destroys brain gliomas.
As the studies described above showed no adverse effect of VSV-LASV-GPC or VSV-EBOV-GP in the CNS, we next tested whether the virus might target and destroy brain tumors. Fifteen days after implant of red fluorescent human glioma into the SCID mouse brain, VSV-LASV-GPC (106 PFU in 100 μl) was injected intravenously (tail vein). The virus crossed the blood-brain barrier in the area of tumors and showed a highly selective tumor infection and complete destruction of tumor cells within the brain. Mice with brain tumors survived only if treated with VSV-LASV-GPC, with no adverse effect as of 80 days (Fig. 4). Mice with brain tumors that did not receive virus all died from the tumor (median survival of 29 days; n = 8; Fig. 4A and C). The chimeric virus VSV-EBOV-GP also targeted brain tumors after intravenous inoculation (Fig. 4A and D); VSV-EBOV-GP extended life minimally (median survival of 34 days; n = 8). Histological analysis showed large tumors in mice not treated with virus (Fig. 4C) and selective but incomplete infection of the tumor treated with VSV-EBOV-GP (Fig. 4D). In contrast, few tumor cells if any were found following treatment with VSV-LASV-GP (Fig. 4E), suggesting highly selective infection and destruction of glioma. This is consistent with low-magnification examination of tumor-bearing brain from mice treated with virus where no red tumors were seen in the brain of mice receiving intravenous VSV-LASV-GPC (Fig. 4B). In another short-term experiment, mice showed near-complete infection of bilateral tumor masses 8 days postinoculation, with little infection outside the tumor area, suggesting a rapid and selective VSV-LASV-GPC anti-tumor action (n = 4).
FIG 4.

Intravenous VSV-LASV-GPC destroys brain glioma and prolongs life indefinitely. CB17 SCID mice with unilateral striatal xenografts of human RFP-expressing rU87 glioma were treated with a single intravenous injection of either VSV-LASV-GPC, VSV-EBOV-GP (each 100 μl of 106 PFU), or saline (control) 15 days after tumor placement. (A) Kaplan-Meier curve showing modest survival benefit of VSV-EBOV-GP-treated mice compared to untreated control (median survival of 29 versus 34 days; n = 8 each group). In contrast, VSV-LASV-GPC-treated mice showed complete survival throughout the observation period (80 days, n = 8). Histological analysis showed no residual viable tumor mass, only scar tissue and tumor cavity (E). (B) The panel displays representative brains for each group. Brains from untreated control mice showed massive expansion of the tumor mass (pink), causing a significant midline shift of the longitudinal cerebral fissure. Large tumor mass and midline shift is also seen in brains from mice treated with VSV-EBOV-GP, indicating limited therapeutic effect. In contrast, brains from VSV-LASV-GPC-treated mice (top row) showed no visible expansion of the brain at the end of the observation period. (C) In control mouse, red glioma expanded substantially. (D) Mouse treated with VSV-EBOV-GP showed selective infection in tumor, but the infection was not complete. (E) Intravenous VSV-LASV-GPC completely eliminated tumor by 79 days. The majority of VSV-LASV-GPC-treated brains showed an empty tumor cavity, indicating successful oncolysis. (F) Syngeneic rat brain tumor model was established by stereotactically grafting RFP-expressing CNS-1 rat glioma into Lewis rats into the right striatum. VSV-LASV-GPC was applied to the area of the tumor 7 days after tumor placement; rats were euthanized 3 days later. VSV-LASV-GPC caused widespread oncolysis in the central portions of CNS-1 tumors with dead or dying cells, resulting in fading of the RFP and GFP signals and a morphological shift to cell debris (open arrowheads in panel G) from the whole-cell architecture seen in noninfected CNS-1 glioma (white arrowheads in panel H). Infection was restricted to the tumor. These data support the view that VSV-LASV-GPC not only targets human glioma but can also selectively infect rodent glioma within the rodent brain.
Using a syngeneic rodent tumor model, we tested whether VSV-LASV-GPC would infect rat glioma in immunocompetent rats. VSV-LASV-G showed selective strong cytolytic infection of rat CNS-1 glioma tumors after intracerebral inoculation, with little detectable infection of normal brain (Fig. 4F to H), demonstrating that VSV-LASV-GPC selectively infects glioma not only in mice but also in immunocompetent rats.
A major problem with high-grade gliomas in humans is tumor cell migration within the brain; thus, neurosurgical removal or focused radiation may eliminate the main tumor body but is generally unable to eliminate the large number of cells migrating from the tumor into the brain. To model this, we implanted the left and right side of the SCID mouse brain with human glioma (in striatum). Fifteen days later, VSV-LASV-GPC was stereotactically injected unilaterally, only into the tumor on the right side of the brain. Eight days later, the brains were examined; we chose an 8-day postinoculation survival as a probable point when the injected tumor might be completely infected or destroyed but when the contralateral tumor might still show an ongoing infection. VSV-LASV-GPC had completely destroyed the inoculated tumor on the right side of the brain, and the virus had migrated to the contralateral left tumor and begun the process of infection and destruction without infecting the intervening normal brain (Fig. 5A and B). Remarkably, VSV-LASV-GPC selectively destroyed the brain tumor with no adverse effects to the intervening SCID mouse brain. These observations are consistent with the view that the virus was suppressed in normal brain cells by a T- and B-cell-independent mechanism.
FIG 5.

VSV-LASV-G infects widespread gliomas within brain. CB17 SCID mice received bilateral xenografts of RFP-expressing rU87 glioma. Fifteen days later, the right-side tumor was injected stereotactically with 1 μl (3.6 × 104 PFU) of VSV-LASV-GPC. Brains were harvested 8 days later. (A) Analysis for expression of RFP (tumor) and GFP (virus infection) revealed a consistently bright fluorescent signal on the left noninjected side and faint or absent signal together with substantial dead cell glioma debris on the injected right side. These observations suggest successful ipsilateral tumor oncolysis and progressive infection contralateral to the side of virus inoculation. Images show a dorsal view of uncut brains of 4 mice taken with an Olympus fluorescence microscope. (B) Coronal sections from the striatum (from panel A above) showed little residual tumor or infected cells with faint RFP-positive tumor debris at the injected right-side tumor (microscopic images in panel B); the noninjected left side (L) showed VSV-LASV-GPC infection due to contralateral spread of the virus within the brain. The right side (R) showed only tumor remnants remaining after virus infection. Micrographs were made from a dorsal view of the whole brain.
VSV-LASV-GPC infects melanoma.
We next asked whether VSV-LASV-GPC was selective for gliomas or was able to target other types of metastatic brain cancer. Melanomas are the deadliest form of skin cancer, and one of the chief problems is metastasis into the brain (48). Red fluorescent human melanoma was injected into the left and right sides of the brain (Fig. 6A), similar to the glioma experiments described above. VSV-LASV-GPC was subsequently injected unilaterally into the right-side melanoma, and the mice were euthanized 8 days later. VSV-LASV-GPC not only caused complete destruction of the injected tumor mass on the right side (Fig. 6B, D, and F), it also crossed the midline and showed good infection of the noninjected contralateral melanoma (Fig. 6B, C, and E), with no neuron infection in the intervening brain. The partial infection of the contralateral melanoma was expected and was probably due to the short, 8-day period of postinoculation survival.
FIG 6.

VSV-LASV-GPC targets intracranial melanoma. RFP-expressing rYUMAC melanoma cells were injected bilaterally into the striatum of CB17 SCID mice (see control, left column in panel A) as a model for melanoma metastasis. Fifteen days later, the right-side tumor was injected stereotactically with 1 μl (3.6 × 104 PFU) VSV-LASV-GPC. At 8 dpi, brains were harvested and analyzed for expression of RFP (tumor) and GFP (virus infection). Here, a dorsal view of the whole brain (n = 4) is shown, with red indicating the melanoma and green (GFP) the presence of virus. (B) Coronal sections from the striatum showed little residual tumor or infected cells with faint RFP-positive tumor debris at the injected right-side tumor; the noninjected left side showed VSV-LASV-GPC infection due to contralateral spread of the virus with minimal infection of intervening normal brain. (C and D) VSV-LASV-GPC injected into the right melanoma completely eliminated it and then began to infect and destroy the noninfected left melanoma. (E and F) Higher magnification showing the initial signs of infection in the left melanoma and the complete loss of melanoma cells on the injected side. (G, H, and I) Panel G shows red melanoma cells migrating away from the main tumor body, panel H shows GFP-labeled virus-infected cells, and panel I shows the merged image, demonstrating that only melanoma cells were infected by the virus.
A primary complication with both glioma and melanoma is the migration of cancer cells away from the main tumor body and into the brain. VSV-LASV-GPC showed substantial potential for attacking single migrating melanoma cells without infecting the normal neurons or glia in between (Fig. 6G to I). The virus completely eliminated all detectable tumor cells in the main tumor body, and no melanoma cells could be detected migrating away from the right tumor, suggesting the virus eliminated all traces of the right melanoma within 8 days.
Because VSV-LASV-GPC infected two unrelated brain tumor types, glioma and melanoma, we also tested it on other types of human cancer cells that sometimes metastasize into the brain. VSV-LASV-GPC infected (Fig. 7, middle column) and replicated in (Fig. 7, right column showing secondary inoculation by supernatant) colon, prostate, breast, bone, and bladder cancer cells (Fig. 7), suggesting its oncolytic potential was not restricted to glioma and melanoma within the brain. We also tested a virus-resistant sarcoma with upregulated interferon-stimulated genes (49) and found that, similar to other viruses, including VSV, Sindbis, and cytomegalovirus, VSV-LASV-GPC infection was highly attenuated in these cells (not shown).
FIG 7.
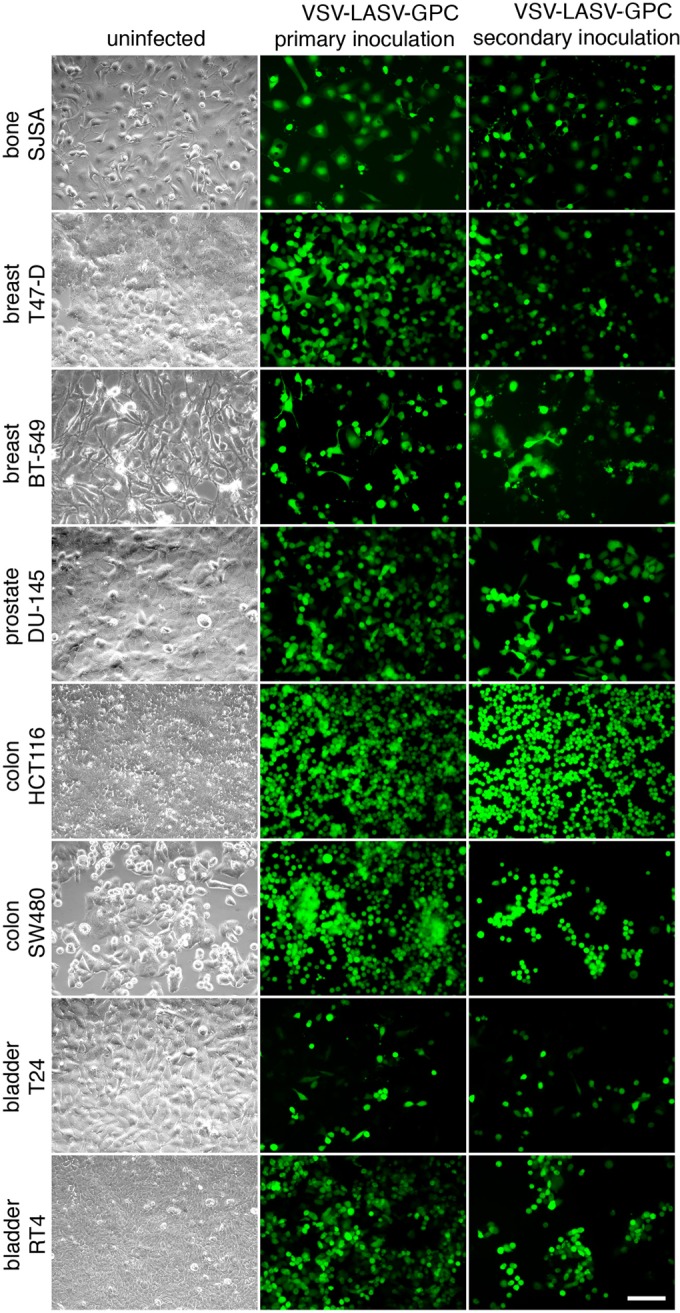
Infection of VSV-LASV-GPC generalizes to a wide variety of human tumors. Eight human tumor lines (see phase-contrast images in left column) originating from bone, breast, prostate, colon, and bladder cancer were infected with VSV-LASV-GPC at an MOI of 3 (primary inoculation). Two hours later, virus inoculum was removed and cells were washed 3 times. At 24 h after infection, all tumor lines showed infection as indicated by GFP expression (middle column). To test for viral propagation in these tumors, supernatant was filtered (0.22 μm) and transferred to uninfected tumor dishes (secondary inoculation; right column). Twenty-four hours later, positive GFP expression indicates transfer of viral progeny produced by tumors infected during primary inoculation.
Immune system activation.
In addition to a direct oncolytic attack on cancer cells, oncolytic viruses can also initiate an attack by the immune system on tumor cells. To test whether VSV-LASV-GPC would evoke a humoral immune response, mice were inoculated intranasally and intramuscularly with this virus. VSV-LASV-GPC generated high titer antisera to VSV-LASV-GPC-infected cells (Fig. 8, top row) and also to the GFP transgene expressed by the virus (Fig. 8, bottom row). Antisera with dilutions out to 1:10,000 generated positive immunostaining of GFP-expressing neurons from transgenic mice with no virus present, suggesting the potential for a secondary systemic immune response against VSV-LASV-GPC-infected gliomas as reported for wild-type VSVs (50). It is interesting to note that a chimeric VSV-LCMV-GPC was recently shown to be only weakly immunogenic with regard to the generation of neutralizing antibodies against the virus (27). VSV-LASV-GPC appeared to completely eliminate human brain tumors in our experiments; the strong immune response against tumors initiated by virus infection could serve to augment tumor eradication in the event of incomplete direct tumor destruction (51). Chimeric VSVs that generate a strong immune response may be beneficial in terms of evoking a strong secondary immune response against tumor-related antigens.
FIG 8.
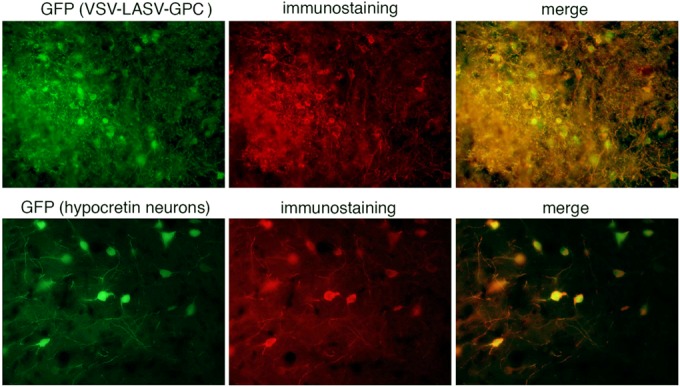
VSV-LASV-GPC induces humoral response and antibody production against virus and GFP transgene. Serum from mice injected and boosted with VSV-LASV-GPC intranasally (5 × 105 PFU in 50 μl) and intramuscularly (2 × 105 PFU in 20 μl) was diluted and used for immunostaining of cells infected with VSV-LASV-GPC. The top row displays example images of sections of brains from IFN-α/β-R−/− mice showing widespread infection with VSV-LASV-GPC. Antibodies raised in VSV-LASV-GPC-immunized mice labeled infected cell bodies and processes. The same serum was used to test for labeling of a GFP transgene. Sections of transgenic mice expressing GFP in hypocretin neurons were used. In the absence of VSV-LASV-GPC, the antiserum selectively labeled GFP-positive neurons red, indicating that VSV-LASV-GPC evokes an immune response to nonviral antigens associated with the virus. Control nonimmune serum immunostaining was negative.
DISCUSSION
Although VSV has shown considerable promise in selectively targeting and destroying many different types of tumors both outside and within the brain, the possibility of adverse neuronal effects within the brain potentially leading to motor dysfunction (52), behavioral disturbances (12), or death (13, 52) has been a block to forward progress. Here, we show that eliminating the native glycoprotein from the VSV genome and substituting glycoproteins from other viruses greatly reduces infection and cytolysis of neurons. All five chimeric VSVs tested showed considerably reduced neuron tropism and replication compared with the natural VSV glycoprotein. Not only were the two chimeric viruses tested safe within the brain, but direct injection of either VSV-LASV-GPC or VSV-EBOV-GP into the brains of SCID mice lacking the normal complement of B and T immune cells generated no adverse effect, and the innate immune system within the brain eliminated the virus. Importantly, after intravenous inoculation, the chimeric viruses VSV-LASV-GPC and VSV-EBOV-GP were able to cross the blood-brain barrier and selectively infect brain tumors with little or no infection of normal neurons or glia and no virally mediated adverse effects. VSV-LASV-GPC completely eliminated brain tumors and prolonged the lives of tumor-bearing mice indefinitely. VSV containing the Ebola virus glycoprotein also crossed the blood-brain barrier and selectively targeted the brain tumor but showed only partial infection of the glioma and only a modest survival benefit in mice bearing brain tumors.
Vesicular stomatitis virus safety in the brain.
The most problematic aspect of using VSV either as an oncolytic virus or as a vaccine vector against more dangerous viruses, including Ebola virus (34), HIV, and other pathogenic viruses, has been the concern about adverse effects of the virus within the brain (42, 52). VSV neurotoxicity can be reduced by generating peripheral immunity in advance of intracerebral inoculation (42) or by administering exogenous type I interferon or via intracerebral viral vectors that generate interferon (53). Attenuated VSVs have been constructed by a number of molecular alterations, including reduction of cytoplasmic amino acids in the G protein, mutations in the M gene, particularly at M51, and adding genes upstream of viral genes to reduce the expression of viral genes (11, 14, 16, 17), but most of these resultant VSVs still retain negative side effects in the brain due to neuronal infection leading to neuron death. Even a VSV that expresses interferon and is currently in clinical trials (VSV-IFN), although attenuated, is problematic and can be lethal if it gains access to the brain. None of these previous recombinant strategies directly eliminate the lethal neurotropism of the virus within the brain conferred by the native VSV glycoprotein. In contrast, we show in the current study that all five G protein chimeric viruses used showed reduced neuron infection, and the two tested in vivo showed complete safety within the brain. Even substitution of the glycoprotein from rabies virus, which has well-known neuronal targeting, still showed reduced infection of neurons compared with that of the VSV glycoprotein and has been suggested as safe in the rodent brain (8), underlining the importance of eliminating the VSV glycoprotein to block adverse effects in the brain.
Neither VSV-EBOV-GP nor VSV-LASV-GPC injected intracerebrally into normal mice, immunocompromised SCID mice, or rats evoked any adverse action, whereas similar concentrations of other VSVs with the native G, including attenuated VSV-CT9-M51 (38), VSV-wtG, VSV-mIFN, and VSV-M51 (Fig. 2B), were lethal. VSV-LASV-GPC was previously shown safe in rodents after intraperitoneal injection, but intracranial safety was not investigated (34). Our results here show that both VSV-LASV-GPC and VSV-EBOV-GP are safe in the rodent brain. The GFP reporter gene in both VSV-LASV-GPC and VSV-wtG provides some attenuation to both viruses by reducing expression of other VSV genes; in spite of this, VSV expressing the wild-type glycoprotein was still lethal within the brain even when carrying the attenuating GFP gene, whereas VSV-LASV-GPC and VSV-EBOV-GP were safe in the brain, suggesting that safety in the brain is due to the absence of the neurotoxic VSV glycoprotein. This view is further supported by our data showing safety of VSV-LASV-GPC in chimeric viruses that did not express the GFP gene. The lack of VSV-LASV-GPC infection of neurons in the rodent brain parallels our in vitro studies with human neurons showing reduced infection, suggesting the virus may also be safe in the human brain. This would be consistent with the finding that VSV-EBOV-GP exhibited no adverse consequence when injected into the primate brain (26).
Mechanisms of selectivity.
Our data suggest the advantage of the Lassa virus glycoprotein is that it allows a selective block to replication in neurons but not in a wide variety of human tumors. This block to neuron infection appears not to be at the binding or internalization steps but at a step further downstream in the life cycle, possibly at the uncoating/endosomal escape step in neurons, given the importance of the glycoprotein to this process (54).
Similar to wild-type VSV, type I IFN is important to the selectivity and safety of VSV-LASV-GPC; mice lacking type I IFN receptor succumbed to virus inoculation. We reported previously that whereas type II and III IFN may also contribute to immunity in the brain (39), type I IFN is necessary for survival with virus infection. The impairment of innate immunity characteristic of oncogenically transformed cells allows VSV-LASV-GPC to replicate rapidly, with cytolytic consequences for tumor cells. The addition of IFN to glioma had little to no effect on the infectivity of VSV-LASV-GPC but provided additional protection to normal neurons and glia. The exquisite tumor selectivity of VSV-LASV-GPC is thus a consequence of both reduced neurotropism (via substitution of the LASV-GPC for the VSV G protein) and virus susceptibility to endogenous IFN in normal cells. The virus is able to very selectively infect and destroy tumors whether entering from the bloodstream into the brain or by traveling from an infected tumor to a distant locus of tumor cell growth in the contralateral brain, all without substantive infection of the intervening healthy brain tissue. The fact that the innate immune system appears able to contain VSV-LASV-GPC suggests that the virus may prove safe to treat the increased incidence of cancers found in patients with compromised adaptive immune systems, for instance with AIDS.
Both VSV-LASV-GPC and VSV-EBOV-GP infected and killed glioma in vitro, and both infected and killed glioma in vivo after intravenous inoculation. However, VSV-LASV-GPC completely eliminated tumor cells and prolonged life indefinitely, whereas VSV-EBOV showed incomplete tumor infection in histological analysis and only a very modest survival benefit. These data support the view that in vitro studies of this sort are inferential and that, ultimately, in vivo experiments are needed. There are several factors that might underlie the reduced efficacy of VSV-EBOV-GP compared with VSV-LASV-GPC in vivo, including in vivo transformation of tumors to forms that are more resistant to infection by some viruses (55) and the existence of a greater number of cells and mechanisms for defeating virus infections even in immunocompromised mice that might be differentially activated or attenuated by viral glycoproteins. Furthermore, we cannot rule out the possibility that some types of tumors may show a greater in vivo susceptibility to VSV-EBOV-GP than found with gliomas here.
Vaccine vector.
VSV is a promising vaccine platform, and even single inoculations generate strong cellular and humoral immunity (56, 57, 58). Importantly, the risk of neurotoxicity can be reduced or eliminated by substitution of the Lassa virus glycoprotein for the VSV glycoprotein. VSV-LASV-GPC may prove beneficial as a vaccine vector, given its potent immunogenicity. Immunogenic viral vectors could be generated against other pathogenic organisms by substituting a gene coding for a pathogen protein in place of the GFP gene while retaining the LASV-GPC in place of the neurotoxic VSV glycoprotein. Use of the Lassa virus glycoprotein in place of the VSV glycoprotein may provide a safer vector for immunization against a number of pathogenic microorganisms compared with wild-type VSV.
Lassa virus glycoprotein.
That genes from potentially lethal viruses such as Ebola, Marburg, Lassa, or rabies virus can be integrated to generate a chimeric virus that is not only safe but potentially beneficial in selectively attacking and killing brain cancer is remarkable. This raises the question of whether other genetic combinations of two or more unrelated viruses might have similar beneficial effects. Lassa virus is a member of the Old World group of arenaviruses that tend to bind to similar receptors. In contrast, related arenaviruses from the New World group, including Sabia, Machupo, and others, bind to different receptors, for instance, transferrin receptor 1 (59), and thereby merit experimental attention related to their oncolytic potential. There are undoubtedly other virus glycoproteins unrelated to the 6 compared here that may prove useful to target tumor cells without the adverse neuronal side effects of VSV G. Small-molecule inhibitors such as ST-193 that block viruses pseudotyped with Lassa virus glycoprotein but not VSV or LCMV glycoproteins provide a further safety factor to control infection by VSV-LASV-GPC if needed (60). Lassa virus and LCMV share a binding protein on the cell surface, but unlike LCMV, the Lassa virus glycoprotein requires binding to sialyltransferase ST3GAL4-dependent LAMP1 within lysosomes to mediate cell infection (61), underlining potential differences in cell tropism between the two chimeric viruses.
VSV-LASV-GPC targets multiple cancers.
VSV-LASV-GPC is effective not only against gliomas (which arise within the brain) but also against melanoma, a cancer that arises in the skin but metastasizes into the brain, resulting in death within a few months of entering the brain. We also find a strong viral infection in other types of human cancer cells, including prostate, breast, colon, and bladder, which can sometimes metastasize into the brain. The broad infectivity and cytolysis of multiple types of cancer cells suggest that VSV-LASV-GPC may also be effective in targeting other types of brain tumors not tested here, including meningioma, astrocytoma, ependymoma, and oligodendroglioma, and this possibility merits further testing. The elimination of neurotropism by substitution of the Lassa virus glycoprotein for the VSV glycoprotein would provide an increased level of brain safety, even if the virus were used to target peripheral cancers.
In vitro testing suggested VSV-LASV-GPC showed a good combination of reduced neurotropism and broad infectivity across different gliomas. Similar to VSV-LASV-GPC, VSV-LCMV-GPC had a reduced neurotropism and showed a somewhat reduced infectivity of gliomas tested here. A recent paper (27) showed that VSV-LCMV also has the potential to target brain tumors; however, questions remain as to which glycoprotein will prove to be the most useful. The humoral response to VSV-LCMV was greatly reduced compared with that of VSV-wtG (27). We show here that VSV-LASV-GPC generated robust humoral immunity and antibody formation against virally expressed exogenous genes, including GFP. It remains an open question as to whether low viral immunogenicity of LCMV-GPC will ultimately represent an advantage, because it allows for repeat administration, or a disadvantage. The reduced humoral immune response to VSV-LCMV might lead to an incomplete elimination of the virus by the immune system, leading to chronic infection and/or a less potent stimulation of antitumor immunity. The generation of a strong immune response to infected cancer tissue is an important benefit to the efficacy of oncolytic viruses in immunocompetent hosts.
ACKNOWLEDGMENTS
We thank Sean Whelan, Heinz Feldmann, and Thomas Geisbert for chimeric VSVs, John Rose and Glen Barber for additional recombinant VSVs, Y. Yang for technical assistance, and Justin Paglino for manuscript comments.
Research support was provided by NIH CA161048, CA175577, NS083848, CA188359, and F31 AG041582.
C.C. is an investigator of HHMI. G.W. is currently at Section of Virology, Medical University, Innsbruck, Austria.
REFERENCES
- 1.Croteau D, Mikkelsen T, Rempel SA, Bogler O, Rosenblum M. 2001. New innovations and developments for glioma treatment. Clin Neurosurg 48:60–81. [PubMed] [Google Scholar]
- 2.Wrensch M, Minn Y, Chew T, Bondy M, Berger MS. 2002. Epidemiology of primary brain tumors: current concepts and review of the literature. Neuro Oncol 4:278–299. doi: 10.1093/neuonc/4.4.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sampson JH, Carter JH Jr, Friedman AH, Seigler HF. 1998. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg 88:11–20. doi: 10.3171/jns.1998.88.1.0011. [DOI] [PubMed] [Google Scholar]
- 4.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC. 2000. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nature Med 6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 5.Hastie E, Grdzelishvili V. 2012. Vesicular stomatitis virus as a flexible platform for oncolytic virotherapy against cancer. J Gen Virol 93:2529–2545. doi: 10.1099/vir.0.046672-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balachandran S, Barber GN. 2000. Vesicular stomatitis virus (VSV) therapy of tumors. IUBMB Life 50:135–138. doi: 10.1080/713803696. [DOI] [PubMed] [Google Scholar]
- 7.Johnson JE, Nasar F, Coleman JW, Price RE, Javadian A, Draper K, Lee M, Reilly PA, Clarke DK, Hendry RM, Udem SA. 2007. Neurovirulence properties of recombinant vesicular stomatitis virus vectors in non-human primates. Virology 360:36–49. doi: 10.1016/j.virol.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beier KT, Saunders A, Oldenburg IA, Miyamichi K, Akhtar N, Luo L, Whelan SP, Sabatini B, Cepko CL. 2011. Anterograde or retrograde transsynaptic labeling of CNS neurons with vesicular stomatitis virus vectors. Proc Natl Acad Sci U S A 108:15414–15419. doi: 10.1073/pnas.1110854108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cronin J, Zhang XY, Reiser J. 2005. Altering the tropism of lentiviral vectors through pseudotyping. Curr Gene Ther 5:387–398. doi: 10.2174/1566523054546224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muik A, Kneiske I, Werbizki M, Wilflingseder D, Giroglou T, Ebert O, Kraft A, Dietrich U, Zimmer G, Momma S, von Laer D. 2011. Pseudotyping vesicular stomatitis virus with lymphocytic choriomeningitis virus glycoproteins enhances infectivity for glioma cells and minimizes neurotropism. J Virol 85:5679–5684. doi: 10.1128/JVI.02511-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lun X, Senger DL, Alain T, Oprea A, Parato K, Stojdl D, Lichty B, Power A, Johnston RN, Hamilton M, Parney I, Bell JC, Forsyth PA. 2006. Effects of intravenously administered recombinant vesicular stomatitis virus (VSVdelta51) on multifocal and invasive gliomas. J Natl Cancer Inst 98:1546–1557. doi: 10.1093/jnci/djj413. [DOI] [PubMed] [Google Scholar]
- 12.Lundh B, Löve A, Kristensson K, Norrby E. 1988. Non-lethal infection of aminergic reticular core neurons: age-dependent spread of ts mutant vesicular stomatitis virus from the nose. J Neuropathol Exp Neurol 47:497–506. doi: 10.1097/00005072-198809000-00001. [DOI] [PubMed] [Google Scholar]
- 13.van den Pol AN, Dalton KP, Rose JK. 2002. Relative neurotropism of a recombinant rhabdovirus expressing a green fluorescent envelope glycoprotein. J Virol 76:1309–1327. doi: 10.1128/JVI.76.3.1309-1327.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts A, Buonocore L, Price R, Forman J, Rose JK. 1999. Attenuated vesicular stomatitis viruses as vaccine vectors. J Virol 73:3723–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flanagan EB, Zamparo JM, Ball LA, Rodriguez LL, Wertz GW. 2001. Rearrangement of the genes of vesicular stomatitis virus eliminates clinical disease in the natural host: new strategy for vaccine development. J Virol 75:6107–6114. doi: 10.1128/JVI.75.13.6107-6114.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahmed M, Cramer SD, Lyles DS. 2004. Sensitivity of prostate tumors to wild-type and M protein mutant vesicular stomatitis viruses. Virology 330:34–49. doi: 10.1016/j.virol.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 17.Ramsburg E, Publicover J, Buonocore L, Poholek A, Robek M, Palin A, Rose JK. 2005. A vesicular stomatitis virus recombinant expressing granulocyte-macrophage colony-stimulating factor induces enhanced T-cell responses and is highly attenuated for replication in animals. J Virol 79:15043–15053. doi: 10.1128/JVI.79.24.15043-15053.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clarke DK, Nasar F, Lee M, Johnson JE, Wright K, Calderon P, Guo M, Natuk R, Cooper D, Hendry RM, Udem SA. 2007. Synergistic attenuation of vesicular stomatitis virus by combination of specific G gene truncations and N gene translocations. J Virol 81:2056–2064. doi: 10.1128/JVI.01911-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooper D, Wright KJ, Calderon PC, Guo M, Nasar F, Johnson JE, Coleman JW, Lee M, Kotash C, Yurgelonis I, Natuk RJ, Hendry RM, Udem SA, Clarke DK. 2008. Attenuation of recombinant vesicular stomatitis virus-human immunodeficiency virus type 1 vaccine vectors by gene translocations and G gene truncation reduces neurovirulence and enhances immunogenicity in mice. J Virol 82:207–219. doi: 10.1128/JVI.01515-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wollmann G, Rogulin V, Simon I, Rose JK, van den Pol AN. 2010. Some attenuated variants of vesicular stomatitis virus show enhanced oncolytic activity against human glioblastoma cells relative to normal brain cells. J Virol 84:1563–1573. doi: 10.1128/JVI.02040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van den Pol AN, Davis JN. 2013. Highly attenuated recombinant vesicular stomatitis virus VSV-12′GFP displays immunogenic and oncolytic activity. J Virol 87:1019–1034. doi: 10.1128/JVI.01106-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. 2011. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krishnan A, Miller EH, Herbert AS, Ng M, Ndungo E, Whelan SP, Dye JM, Chandran K. 2012. Niemann-Pick C1 (NPC1)/NPC1-like1 chimeras define sequences critical for NPC1's function as a filovirus entry receptor. Viruses 4:2471–2484. doi: 10.3390/v4112471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kunz S. 2009. Receptor binding and cell entry of Old World arenaviruses reveal novel aspects of virus-host interaction. Virology 387:245–249. doi: 10.1016/j.virol.2009.02.042. [DOI] [PubMed] [Google Scholar]
- 25.Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. 2013. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Nat Acad Sci U S A 110:7306–7311. doi: 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mire CE, Miller AD, Carville A, Westmoreland SV, Geisbert JB, Mansfield KG, Feldmann H, Hensley LE, Geisbert TW. 2012. Recombinant vesicular stomatitis virus vaccine vectors expressing filovirus glycoproteins lack neurovirulence in nonhuman primates. PLoS Negl Trop Dis 6:e1567. doi: 10.1371/journal.pntd.0001567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muik A, Stubbert LJ, Jahedi RZ, Geiβ Y, Kimpel J, Dold C, Tober R, Volk A, Klein S, Dietrich U, Yadollahi B, Falls T, Miletic H, Stojdl D, Bell JC, von Laer D. 2014. Re-engineering vesicular stomatitis virus to abrogate neurotoxicity, circumvent humoral immunity, and enhance oncolytic potency. Cancer Res 74:3567–3578. doi: 10.1158/0008-5472.CAN-13-3306. [DOI] [PubMed] [Google Scholar]
- 28.Watson DJ, Kobinger GP, Passini MA, Wilson JM, Wolfe JH. 2002. Targeted transduction patterns in the mouse brain by lentivirus vectors pseudotyped with VSV, Ebola, Mokola, LCMV, or MuLV envelope proteins. Mol Ther 5:528–537. doi: 10.1006/mthe.2002.0584. [DOI] [PubMed] [Google Scholar]
- 29.Papaneri AB, Wirblich C, Cann JA, Cooper K, Jahrling PB, Schnell MJ, Blaney JE. 2012. A replication-deficient rabies virus vaccine expressing Ebola virus glycoprotein is highly attenuated for neurovirulence. Virology 434:18–26. doi: 10.1016/j.virol.2012.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jae LT, Raaben M, Riemersma M, van Beusekom E, Blomen VA, Velds A, Kerkhoven RM, Carette JE, Topaloglu H, Meinecke P, Wessels MW, Lefeber DJ, Whelan SP, van Bokhoven H, Brummelkamp TR. 2013. Deciphering the glycosylome of dystroglycanopathies using haploid screens for Lassa virus entry. Science 340:479–483. doi: 10.1126/science.1233675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong AC, Sandesara RG, Mulherkar N, Whelan SP, Chandran K. 2010. A forward genetic strategy reveals destabilizing mutations in the ebolavirus glycoprotein that alter its protease dependence during cell entry. J Virol 84:163–175. doi: 10.1128/JVI.01832-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller EH, Obernosterer G, Raaben M, Herbert AS, Deffieu MS, Krishnan A, Ndungo E, Sandesara RG, Carette JE, Kuehne AI, Ruthel G, Pfeffer SR, Dye JM, Whelan SP, Brummelkamp TR, Chandran K. 2012. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J 31:1947–1960. doi: 10.1038/emboj.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. 2005. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garbutt M, Liebscher R, Wahl-Jensen V, Jones S, Möller P, Wagner R, Volchkov V, Klenk HD, Feldmann H, Ströher U. 2004. Properties of replication-competent vesicular stomatitis virus vectors expressing glycoproteins of filoviruses and arenaviruses. J Virol 78:5458–5465. doi: 10.1128/JVI.78.10.5458-5465.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cureton DK, Massol RH, Whelan SP, Kirchhausen T. 2010. The length of vesicular stomatitis virus particles dictates a need for actin assembly during clathrin-dependent endocytosis. PLoS Pathog 6:e1001127. doi: 10.1371/journal.ppat.1001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawson ND, Stillman EA, Whitt MA, Rose JK. 1995. Recombinant vesicular stomatitis viruses from DNA. Proc Natl Acad Sci U S A 92:4477–4481. doi: 10.1073/pnas.92.10.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ozduman K, Wollmann G, Piepmeier J, van den Pol AN. 2008. Systemic vesicular stomatitis virus selectively destroys multifocal glioma and metastatic carcinoma in brain. J Neurosci 28:1882–1893. doi: 10.1523/JNEUROSCI.4905-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wollmann G, Davis JN, Bosenberg MW, van den Pol AN. 2013. Vesicular stomatitis virus variants selectively infect and kill human melanomas but not normal melanocytes. J Virol 87:6644–6659. doi: 10.1128/JVI.03311-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van den Pol AN, Ding S, Robek MD. 2014. Long distance interferon signaling within the brain blocks virus spread. J Virol 88:3695–3704. doi: 10.1128/JVI.03509-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wollmann G, Robek MD, van den Pol AN. 2007. Variable deficiencies in the interferon response enhance susceptibility to vesicular stomatitis virus oncolytic actions in glioblastoma cells but not in normal human glial cells. J Virol 81:1479–1491. doi: 10.1128/JVI.01861-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waibler Z, Detje CN, Bell JC, Kalinke U. 2007. Matrix protein mediated shutdown of host cell metabolism limits vesicular stomatitis virus-induced interferon-alpha responses to plasmacytoid dendritic cells. Immunobiology 212:887–894. doi: 10.1016/j.imbio.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 42.Ozduman K, Wollmann G, Ahmadi SA, van den Pol AN. 2009. Peripheral immunization blocks lethal actions of vesicular stomatitis virus within the brain. J Virol 83:11540–11549. doi: 10.1128/JVI.02558-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Obuchi M, Fernandez M, Barber GN. 2003. Development of recombinant vesicular stomatitis viruses that exploit defects in host defense to augment specific oncolytic activity. J Virol 77:8843–8856. doi: 10.1128/JVI.77.16.8843-8856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yarde DN, Naik S, Nace RA, Peng KW, Federspiel MJ, Russell SJ. 2013. Meningeal myeloma deposits adversely impact the therapeutic index of an oncolytic VSV. Cancer Gene Ther 20:616–621. doi: 10.1038/cgt.2013.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phuangsab A, Lorence RM, Reichard KW, Peeples ME, Walter RJ. 2001. Newcastle disease virus therapy of human tumor xenografts: antitumor effects of local or systemic administration. Cancer Lett 172:27–36. doi: 10.1016/S0304-3835(01)00617-6. [DOI] [PubMed] [Google Scholar]
- 46.Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. 1998. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J 17:3351–3362. doi: 10.1093/emboj/17.12.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang F, Ma Y, Barrett JW, Gao X, Loh J, Barton E, Virigin HW, McFadden G. 2004. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nature Immunol 5:1266–1274. doi: 10.1038/ni1132. [DOI] [PubMed] [Google Scholar]
- 48.Carlino MS, Fogarty GB, Long GV. 2012. Treatment of melanoma brain metastases: a new paradigm. Cancer J 18:208–212. doi: 10.1097/PPO.0b013e31824b2890. [DOI] [PubMed] [Google Scholar]
- 49.Paglino JC, van den Pol AN. 2011. Vesicular stomatitis virus has extensive oncolytic activity against human sarcomas: rare resistance is overcome by blocking interferon pathways. J Virol 85:9346–9358. doi: 10.1128/JVI.00723-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Publicover J, Ramsburg E, Rose JK. 2004. Characterization of nonpathogenic, live, viral vaccine vectors inducing potent cellular immune responses. J Virol 78:9317–9324. doi: 10.1128/JVI.78.17.9317-9324.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wongthida P, Diaz RM, Pulido C, Rommelfanger D, Galivo F, Kaluza K, Kottke T, Thompson J, Melcher A, Vile R. 2011. Activating systemic T-Cell immunity against self tumor antigens to support oncolytic virotherapy with vesicular stomatitis virus. Hum Gene Ther 22:1343–1353. doi: 10.1089/hum.2010.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huneycutt BS, Plakhov IV, Shusterman Z, Bartido SM, Huang A, Reiss CS, Aoki C. 1994. Distribution of vesicular stomatitis virus proteins in the brains of BALB/c mice following intranasal inoculation: an immunohistochemical analysis. Brain Res 635:81–95. doi: 10.1016/0006-8993(94)91426-5. [DOI] [PubMed] [Google Scholar]
- 53.Wollmann G, Paglino JC, Maloney PR, Ahmadi SA, van den Pol AN. 2015. Attenuation of vesicular stomatitis virus infection of brain using antiviral drugs and an adeno-associated virus-interferon vector. Virology 475:1–14. doi: 10.1016/j.virol.2014.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rigaut KD, Birk DE, Lenard J. 1991. Intracellular distribution of input vesicular stomatitis virus proteins after uncoating. J Virol 65:2622–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grekova SP, Raykov Z, Zawatzky R, Rommelaere J, Koch U. 2012. Activation of a glioma-specific immune response by oncolytic parvovirus minute virus of mice infection. Cancer Gene Ther 19:468–475. doi: 10.1038/cgt.2012.20. [DOI] [PubMed] [Google Scholar]
- 56.Publicover J, Ramsburg E, Rose JK. 2005. A single-cycle vaccine vector based on vesicular stomatitis virus can induce immune responses comparable to those generated by a replication-competent vector. J Virol 79:13231–13238. doi: 10.1128/JVI.79.21.13231-13238.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buonocore L, Blight KJ, Rice CM, Rose JK. 2002. Characterization of vesicular stomatitis virus recombinants that express and incorporate high levels of hepatitis C virus glycoproteins. J Virol 76:6865–6872. doi: 10.1128/JVI.76.14.6865-6872.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schell JB, Rose NF, Bahl K, Diller K, Buonocore L, Hunter M, Marx PA, Gambhira R, Tang H, Montefiori DC, Johnson WE, Rose JK. 2011. Significant protection against high-dose simian immunodeficiency virus challenge conferred by a new prime-boost vaccine regimen. J Virol 85:5764–5772. doi: 10.1128/JVI.00342-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abraham J, Corbett KD, Farzan M, Choe H, Harrison SC. 2010. Structural basis for receptor recognition by New World hemorrhagic fever arenaviruses. Nat Struct Mol Biol 17:438–444. doi: 10.1038/nsmb.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Larson RA, Dai D, Hosack VT, Tan Y, Bolken TC, Hruby DE, Amberg SM. 2008. Identification of a broad-spectrum arenavirus entry inhibitor. J Virol 82:10768–10775. doi: 10.1128/JVI.00941-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jae LT, Raaben M, Herbert AS, Kuehne AI, Wirchnianski AS, Soh TK, Stubbs SH, Janssen H, Damme M, Saftig P, Whelan SP, Dye JM, Brummelkamp TR. 2014. Virus entry. Lassa virus entry requires a trigger-induced receptor switch. Science 344:1506–1510. doi: 10.1126/science.1252480. [DOI] [PMC free article] [PubMed] [Google Scholar]