

ABSTRACT
Herpes simplex virus 1 (HSV-1) causes one of the most prevalent herpesviral infections in humans and is the leading etiological agent of viral encephalitis and eye infections. Our understanding of how HSV-1 interacts with the host at the cellular and organismal levels is still limited. We and others previously reported that, upon infection, HSV-1 rapidly and efficiently downregulates CD1d cell surface expression and suppresses the function of NKT cells, a group of innate T cells with critical immunoregulatory function. The viral protein kinase US3 plays a major role in this immune evasion mechanism, and its kinase activity is required for this function. In this study, we investigated the cellular substrate(s) phosphorylated by US3 and how it mediates US3 suppression of CD1d recycling. We identified the type II kinesin motor protein KIF3A as a critical kinesin factor in the cell surface expression of CD1d. Interestingly, KIF3A is phosphorylated by US3 both in vitro and in infected cells. Mass spectrometry analysis of purified KIF3A showed that it is phosphorylated predominantly at serine 687 by US3. Ablation of this phosphorylation abolished US3-mediated downregulation of CD1d expression, suggesting that phosphorylation of KIF3A is the primary mechanism of HSV-1 suppression of CD1d expression by US3 protein. Understanding of the precise mechanism of viral modulation of CD1d expression will help to develop more efficient vaccines in the future to boost host NKT cell-mediated immune responses against herpesviruses.
IMPORTANCE Herpes simplex virus 1 (HSV-1) is among the most common human pathogens. Little is known regarding the exact mechanism by which this virus evades the human immune system, particularly the innate immune system. We previously reported that HSV-1 employs its protein kinase US3 to modulate the expression of the key antigen-presenting molecule CD1d to evade the antiviral function of NKT cells. Here we identified the key cellular motor protein KIF3A as a cellular substrate phosphorylated by US3, and this phosphorylation event mediates US3-induced immune evasion.
INTRODUCTION
Natural killer T (NKT) cells are a group of innate, immune-cell-like T cells restricted by the major histocompatibility complex (MHC) class I-like CD1d molecule and have critical immunoregulatory functions in diverse immune responses, including antimicrobial immunity (1–3). Numerous viruses have evolved elegant mechanisms by which to evade and/or suppress the function of NKT cells during the acute, chronic, or latent phase of infection (4, 5). One major mechanism of viral modulation of NKT cell function is to downregulate the expression of CD1d, the key antigen-presenting molecule required for NKT cell development and activation, on the antigen-presenting cell surface (4, 5).
Herpesviruses are highly prevalent in humans and generally have large DNA genomes. Most herpesviruses enter a latent stage after the acute infection stage and therefore have prolonged interaction with host immune systems. As a result of coevolution, most, if not all, herpesviruses have developed intricate mechanisms by which to modulate host immune systems for the benefit of viral survival (6). Because of the importance of NKT cells in antiviral immunity, viruses in all three major herpesvirus families (alpha-, beta-, and gammaherpesviruses) have evolved viral mechanisms to evade CD1d and NKT cell function (7–12). We and others have shown that herpes simplex virus 1 (HSV-1), the prototype alphaherpesvirus, has evolved to downregulate CD1d expression in antigen-presenting cells and evade NKT cell function (10, 13, 14). Furthermore, we have demonstrated that the viral protein kinase US3 is a major viral protein that downregulates cell surface expression of CD1d by suppressing its recycling (8).
CD1d protein is synthesized in the endoplasmic reticulum (ER) and transported to the cell surface via the secretory pathway. Once there, CD1d cycles between the cell surface and endosomal compartment through multiple rounds of endocytosis and recycling steps while surveying lipid antigens for NKT cell recognition (3, 9, 15). Cellular endosomal trafficking is microfilament or microtubule based for short- or long-range transport, respectively. Both transport mechanisms are active movements powered by motor proteins, including dyneins, kinesins, and myosins (16, 17). The outbound trafficking of endosomes, including their recycling, is powered by myosins and kinesins for short- and long-range movements, respectively (16, 18). Kinesins are generally composed of a motor domain, a coiled-coil domain, and a substrate-binding domain that binds target vesicles (18). N-, M-, and C-kinesins contain the motor domains in the amino terminus, middle, and carboxyl terminus, respectively. While N- and C-kinesins drive microtubule plus- or minus-end-directed transport, the M-kinesins depolymerize microtubules (16, 18). Many studies have shown that the phosphorylation and dephosphorylation of kinesins is a rapid and efficient mechanism by which to regulate microtubule-based transport in diverse cellular processes, including mitosis and axonal transport (16, 18).
In this study, we further investigated the molecular mechanism by which US3 modulates CD1d intracellular trafficking and identified the cellular motor protein KIF3A as the main target of US3 phosphorylation. We also found that phosphorylation of KIF3A is required for the US3-mediated downregulation of CD1d.
MATERIALS AND METHODS
Viruses, cells, antibodies, and DNA constructs.
The wild-type HSV-1 F and HSV-1 US3Δ (R7041) strains were generous gifts from Bernard Roizman (University of Chicago, Chicago, IL). The HSV-1 KOS strain expressing green fluorescent protein (GFP)-fused VP26 protein was generously provided by Prashant Desai (Johns Hopkins University, Baltimore, MD). The HeLa.CD1d cell line has been described previously (8). A rabbit monoclonal antibody (MAb) against KIF3A and a rabbit polyclonal antibody against KIF3B were purchased from Abcam. A mouse anti-V5 MAb was obtained from Invitrogen. A mouse MAb against myc was purchased from BioXcell. A goat anti-glutathione S-transferase (GST) polyclonal antibody was obtained from GE. A mouse MAb against human CD1d CD1d51.1 was obtained from Steven Porcelli (Albert Einstein College of Medicine, Bronx, NY). MAbs against Grp94 and LAMP1 were from Stressgen and BD-Pharmingen, respectively. A phycoerythrin-conjugated anti-CD63 MAb was purchased from BD-Pharmingen. Rabbit anti-US3 sera were generously provided by Bernard Roizman (University of Chicago, Chicago, IL) and Yasushi Kawaguchi (University of Tokyo, Tokyo, Japan). A mouse anti-HSV-1 ICP4 MAb was obtained from Virusys. A rabbit anti-protein kinase A (PKA) substrate (RRXS*/T*) MAb (catalog no. 9624) was from Cell Signaling.
The KIF3A gene was subcloned by PCR from a cDNA construct from Transomics Inc. into the pTracer-EF/V5-HisA (Life Technologies Inc.) and pEBG (Addgene, Inc.) vectors. The pEBG.US3 construct was subcloned from the pLPCX.US3 construct (8). pTracer-US3KA and pEBG-US3KA were generated by mutating the HSV-1 US3 gene in a pEBG.US3 construct by PCR with previously described primers (8). For the in vitro phosphorylation assay, the US3 gene was subcloned into pGEX-4X-1 (GE Healthcare) from pLPCX.US3 (8). The C-terminal peptides (amino acids 531 to 702 of KIF3A for GST-KIF3A.CT and amino acids 796 to 904 of HSV-1 glycoprotein B [gB] for GST-gBct) were subcloned from KIF3A cDNA (Addgene) and pLPCX.gB (8) plasmids, respectively, into a pGEX.4X-1 vector (GE Healthcare). ATPase-mutated dominant negative mutant KIF3A.T107N in peGFP-C1 was kindly provided by Stefan Linder (University Medical Center Eppendorf, Hamburg, Germany) (19).
shRNA knockdown of KIF3A.
We designed two oligonucleotides for each KIF gene as described in a previous report (20) (CCGGGCCTGTTTGAACACATTCTAACTCGAGTTAGAATGTGTTCAAACAGGCTTTTTG and CCGGCGTCAGTCTTTGATGAAACTACTCGAGTAGTTTCATCAAAGACTGACGTTTTTG for KIF3A and CCGGGCCTGTTTGAATAAGAACAAACTCGAGTTTGTTCTTATTCAAACAGGCTTTTTG and CCGGCCTGCCTCTTACAACGTAGAACTCGAGTTCTACGTTGTAAGAGGCAGGTTTTTG for KIF3B) with the Mission short hairpin RNA (shRNA) search software (Sigma). They were chemically synthesized, annealed, and cloned into the lentiviral expression vector pLKO.1 (Sigma). The lentiviral-vector-bearing shRNAs were confirmed by DNA sequencing. Forty-eight hours after cotransfection with a gag/pol, Rev, and vesicular stomatitis virus G protein lentiviral packaging plasmid (Sigma) into 293T cells, the lentivirus-containing supernatant was harvested and used for infection of HeLa.CD1d cells in the presence of 8 μg/ml Polybrene (Sigma).
Transient transfection, coimmunoprecipitation, GST pulldown, immunofluorescence, SDS-PAGE, Phos-tag gel electrophoresis, Western blotting, virus infection, and flow cytometry.
Transient transfections of 293T and HeLa cells were performed with Bio-T (Bioland Co.) or polyethylenimine (Polysciences, Inc.) in accordance with the manufacturer's instructions. Other procedures, including coimmunoprecipitation, SDS-PAGE, Western blotting, immunofluorescence, virus infection, and flow cytometry, were performed essentially as described previously (8). For immunoprecipitation and GST pulldown, HeLa.CD1d or 293T cells were infected or transfected with plasmid DNA and lysed in TBS (20 mM Tris HCl, pH 7.4, 150 mM NaCl) with a protease inhibitor cocktail (Roche). The supernatant was immunoprecipitated with the antibodies indicated and protein A (Sigma)- or protein G-Sepharose beads (GE Healthcare). Precipitated protein was boiled in SDS-PAGE sample buffers and loaded onto an SDS-PAGE gel. GST-tagged proteins were purified with glutathione-Sepharose beads (GE Healthcare) in accordance with the manufacturer's instructions. Phos-tag gel electrophoresis of phosphorylated proteins was performed in accordance with the manufacturer's procedures (Wako Pure Chemical Industries, Ltd.).
Purification of KIF3A and MS/MS.
A plasmid expressing GST-fused KIF3A was cotransfected with or without pLPCX.US3.myc plasmid in 293T cells. Forty-eight hours later, cells were harvested and purified with glutathione-Sepharose beads (GE Healthcare) in accordance with the manufacturer's procedure. The proteins were analyzed separately by multidimensional liquid chromatography coupled with tandem mass spectrometry (MS/MS) at the Taplin Biological Mass Spectrometry Facility, Harvard Medical School (Boston, MA). The MS/MS spectra were run against a sequence database by the program SEQUEST and associated software packages for the identification of the proteins.
In vitro phosphorylation assay of KIF3A by US3 protein.
GST-KIF3Act, mutant GST-KIF3Act, and GST-gBct were purified from transiently transfected 293T cells or Escherichia coli with glutathione-Sepharose beads (GE Healthcare) in accordance with the manufacturer's instructions. Purified GST fusion proteins were verified for purity by Coomassie blue staining. One microgram of GST-fused substrate proteins was added to 0.2 μg of purified GST or GST-US3 protein and incubated in a buffer containing 50 mM Tris HCl (pH 9.0), 20 mM MgCl2, 1 mM dithiothreitol, 0.1% NP-40, 10 μM ATP, and 0.5 μCi/μl of [γ-32P]ATP (PerkinElmer) at 30°C for 30 min. GST fusion proteins were again purified with glutathione-Sepharose beads, and aliquots of the reaction product were analyzed by SDS-PAGE and subjected to autoradiography.
RESULTS
HSV-1 protein kinase US3 specifically downregulates the surface expression of CD1d and late endosome/lysosome proteins.
Our previous results showed that, upon infection, HSV-1 employs its viral protein kinase US3 to rapidly and efficiently downregulate cell surface CD1d expression by suppressing its recycling (8, 13). To dissect the cellular mechanism by which HSV-1 and US3 suppress the recycling pathway, we examined the specificity with which either HSV-1 or US3 modulates membrane trafficking. To better track HSV-1-infected cells and sensitively detect the cell surface expression of membrane proteins, we infected HeLa.CD1d cells with a GFP-expressing HSV-1 strain at a low multiplicity of infection (MOI) and compared the expression of CD1d on the surface of infected GFP-positive cells to that on the surface of uninfected GFP-negative cells. At an MOI of 0.5 and 12 h postinfection, approximately 8 to 9% of the HeLa.CD1d cells were infected and GFP positive (Fig. 1A). HSV-1 inhibits CD1d expression primarily by suppressing CD1d recycling (8, 13). Endocytic recycling of cellular membrane proteins has been categorized into rapid and slow recycling mechanisms via early/sorting endosomes and the endocytic recycling compartment, respectively (17). We therefore examined the expression of representative cell surface proteins by using different recycling routes upon viral infection. The transferrin receptor (TfR, CD71) goes primarily through the rapid recycling pathway (17). On the other hand, membrane proteins residing in late endosomes/lysosomes go through the endocytic recycling compartment and employ the slow recycling pathway (17, 21). After HSV-1 infection, no CD71 downregulation was detected, whereas CD1d was clearly downregulated (Fig. 1B). Interestingly, two other late endosome/lysosome-resident proteins, LAMP1 and CD63, were not downregulated but rather upregulated (Fig. 1B, lower panels), consistent with our earlier results obtained with human primary dendritic cells (13). It is possible that the increased expression of these two proteins is important for the viral replication and/or some cellular function. We then examined the mechanism and specificity of US3-mediated downregulation of CD1d expression. HeLa.CD1d cells were transiently transfected with plasmids expressing GFP only or US3 and GFP as previously described (8). GFP was used to track transfected cells. Flow cytometry analysis showed that expression of US3 clearly downregulated CD1d but did not downregulate CD71 (Fig. 1C), similar to the result of HSV-1 infection (above). Interestingly, the other two late endosome/lysosome proteins, CD63 and LAMP1, were also downregulated, with CD63 being downregulated more substantially (Fig. 1C). Control transfection of GFP did not downregulate either CD71 or late endosome/lysosome protein CD1d, CD63, or LAMP1 at a detectable level (Fig. 1C). These results suggest that US3 specifically downregulates cell surface expression of late endosome/lysosome proteins.
FIG 1.

CD1d downregulation by HSV-1 expressing GFP (A, B) or US3 protein (C). HeLa.CD1d cells were infected with HSV-1/GFP for 12 h (A, B) or transfected with plasmids expressing GFP only or US3 and GFP (C). The gating strategy for cells infected with HSV-1/GFP (A, B) is shown in panel A. A GFP-expressing plasmid was used as a control for transfected cells (C). GFP-positive and GFP-negative cells were designated infected or transfected and uninfected or untransfected cells, respectively. Expression levels of indicated cell surface proteins were then analyzed by flow cytometry.
KIF3A is a critical motor protein for the cell surface expression of CD1d.
US3 is one of the early late (γ1) genes expressed during HSV-1 infection, and its expression can be detected at approximately 3 to 4 h postinfection (8, 22). Concomitant with US3 expression, the virus rapidly suppresses cell surface expression of CD1d as early as 5 to 6 h postinfection (8, 10, 13, 14). We hypothesized that a rapid shutdown of cytoskeleton-based intracellular vesicular trafficking is a major mechanism of suppression of CD1d expression. Little is known about the cytoskeleton-based intracellular transport of CD1d-containing vesicles. One elegant study showed that the cytoplasmic tail of CD1d interacts with Rho-associated coiled-coil kinase 1 (ROCK1) and ROCK2, which regulate actin fiber polymerization by phosphorylating LIM kinase (23). In animal cells, actin filaments support short-range movements of vesicles, whereas microtubules support long-range movements (16). As most intracellular CD1d proteins reside in the late endosome/lysosome compartment (13, 24, 25), we reasoned that long-range transport is at least as critical as short-range transport for intracellular trafficking of CD1d-containing vesicles and cell surface CD1d expression. The outbound long-range intracellular transport of late endosome/lysosome vesicles to the cell surface is powered mostly by the superfamily of kinesin motor proteins (16, 18, 21). To better understand the roles of kinesins in US3-mediated CD1d downregulation, we first investigated the role of a kinesin(s) that associates with cellular late endosomes/lysosomes. A previous biochemical study demonstrated that KIF3A is the kinesin protein most abundantly associated with late vesicles in mammalian cells (26). Antibodies against KIF3A, but not other kinesins, specifically inhibit the movement of these late vesicles along microtubules (26). Furthermore, an independent study showed that small interfering RNA knockdown or expression of a dominant negative mutant form of KIF3A specifically blocked the plus-end-directed transport of cellular cargos from late endosomes to the cell periphery (27). We therefore examined the role of KIF3A in the cell surface expression of CD1d. A dominant negative mutant form of KIF3A with a mutation in its ATPase domain (T107N) has been previously generated (19). We expressed this dominant negative mutant KIF3A protein in HeLa.CD1d cells and found that its suppression of KIF3A function indeed led to a clear downregulation of CD1d expression (Fig. 2A and B).
FIG 2.

Identification of KIF3A as a critical kinesin motor for CD1d expression. (A, B) A dominant negative mutant form of cellular KIF3A (T107N, GFP fused) was transiently expressed in HeLa.CD1d cells. Cells expressing the KIF3A dominant negative mutant protein were gated as GFP-positive cells (A), and cell surface expression of CD1d was analyzed by flow cytometry and CD1d expression levels of GFP-positive transfected cells and GFP-negative untransfected cells were compared (B). A GFP-expressing plasmid was used as a control. (C, D) shRNA knockdown of KIF3A and KIF3B was performed by lentiviral transduction, and the expression of KIF3A, KIF3B, and the control (ctrl) β-actin was assayed by Western blotting (immunoblotting [IB]) (C), while cell surface expression of CD1d was assayed by flow cytometry (D).
To further confirm that KIF3A and its heterotrimer complex kinesin II are critical for cell surface expression of CD1d, we knocked down KIF3A protein expression with shRNAs and measured the impact on cell surface expression of CD1d. In mammalian cells, the major kinesin II complex is composed of KIF3A paired with KIF3B and KAP (18, 28). Little is known about the relative turnover rates of the KIF3A, KIF3B, and KAP proteins in kinesin II complexes. We therefore designed two separate shRNAs constructs for either KIF3A or KIF3B as previously described (20) in order to achieve efficient knockdown of the kinesin II complexes. shRNA constructs for both the KIF3A and KIF3B genes were found to decrease the expression levels of the targeted proteins (Fig. 2C). Interestingly, when the KIF3A protein is significantly knocked down, KIF3B protein levels are also reduced and vice versa, suggesting that lower levels of one KIF3 protein subunit may lead to instability of the complex and therefore decrease the total levels of the complex and other subunits. Upon the knockdown of either KIF3A or KIF3B, we observed that CD1d expression levels were significantly downregulated, suggesting that microtubules and KIF3A-based long-range trafficking are critical for CD1d cell surface expression (Fig. 2D). Interestingly, when KIF3A was knocked down more efficiently by shKIF3A-2 shRNA, the CD1d expression level was reduced more than it was by shKIF3A-1 shRNA (compare Fig. 2C, left panels, and D, middle panels), suggesting a dosage effect of the KIF3A expression level on CD1d expression.
US3 interacts specifically with KIF3A and phosphorylates KIF3A.
To determine whether US3 directly phosphorylates KIF3A, we first examined potential US3-KIF3A interaction by immunofluorescence analysis. HeLa.CD1d cells were cotransfected with plasmids expressing US3 and KIF3A. As reported previously (8, 29), US3 is expressed mostly in the cytosol and has a granular staining pattern with some nuclear localization, whereas KIF3A is expressed primarily in the cytosol. In the cytosol, US3 and KIF3A have a high degree of colocalization (Fig. 3A). We further examined whether US3 specifically interacts with KIF3A. Initial efforts to investigate the potential interaction by coimmunoprecipitation did not detect a positive KIF3A-US3 interaction. It has been reported that many kinase-substrate interactions are transient and mutation of the kinase motif to render a kinase-dead mutant preserves the interaction of kinases with their substrates (30). We therefore coexpressed a kinase-dead mutant form of US3 (US3K220A) (8, 29) with KIF3A. Indeed, a coprecipitation assay clearly showed a specific interaction between US3K220A and KIF3A (Fig. 3B). These results, together with the immunofluorescence results, suggest that US3 and KIF3A indeed interact with each other intracellularly.
FIG 3.

Phosphorylation of KIF3A by US3. (A) Colocalization of US3 and KIF3A in HeLa cells. HeLa cells were transfected with myc- or V5-tagged US3 and KIF3A, respectively. Colocalization of the two proteins was analyzed by costaining with anti-myc or anti-V5 antibodies. (B) Interaction between US3 and KIF3A. The GST-, V5-, or myc-tagged KIF3A and US3K220A (US3KA) proteins were coexpressed in 293T cells, and the US3/KIF3A complexes were purified by GST pulldown and analyzed by Western blotting (immunoblotting [IB]). Western blotting with anti-GST, anti-V5, and anti-myc antibodies was performed to verify the expression of tagged proteins, while Western blotting with anti-Grp94 antibodies was used to control the total protein amount of cell lysates. WCL, whole-cell lysate. (C) In vitro phosphorylation of KIF3A by US3. Purified GST or GST-fused C-terminal fragments of KIF3A (KIF3Act) and HSV-1 gB (gBct) were incubated with purified GST-US3 protein in the presence of [γ-32P]ATP. Phosphorylation of these proteins was analyzed by autoradiography (left). An equivalent reaction mixture without [γ-32P]ATP (unlabeled samples) was analyzed by Coomassie staining for examination of protein purity (right). (D) Coomassie blue staining of GST-fused US3 or kinase-dead mutant US3 (US3K220A) protein purified from transfected 293T cells. (E) Purified GST-fused KIF3A was coincubated with purified GST-fused or kinase-dead mutant US3 (US3KA) in the presence of [γ-32P]ATP. To compare phosphorylation by wild-type and mutant US3 proteins at comparable amounts of input protein, a smaller amount of wild-type US3 (1/20) was also used for the phosphorylation assay. The input US3, US3KA, and KIF3A proteins were detected by Western blotting with anti-myc or anti-KIF3A antibodies. (F, G) Analysis of US3 phosphorylation of KIF3A by the Phos-tag method. Plasmids expressing KIF3A and US3 were cotransfected into 293T cells, and KIF3A was purified by GST pulldown (F). For viral infection, HeLa.CD1d cells were infected with the HSV-1 F strain or US3-deficient virus (US3Δ) at an MOI of 5 for 12 h. Endogenous KIF3A was precipitated with anti-KIF3A antibodies (G). Phosphorylation of KIF3A was analyzed by Western blotting with anti-KIF3A antibodies after electrophoresis in the presence of Phos-tag. Western blotting with anti-ICP4 antibodies was performed to confirm the infection of HeLa.CD1d cells by US3Δ mutant HSV-1. U.I., uninfected.
We then examined whether US3 can directly phosphorylate KIF3A. Phosphorylation of cellular motor proteins to regulate intracellular trafficking has been reported in diverse cellular processes, and one major regulation mode is the phosphorylation of the C-terminal cargo-binding domains of N-kinesins (16, 31–33). The C terminus of KIF3A (KIF3Act, amino acids 531 to 702), which has been shown to bind to cargo vesicles (34), was cloned and expressed as a GST fusion protein. Purified GST-KIF3Act was incubated with GST-US3 purified from transiently transfected 293T cells. US3 has been reported to phosphorylate several viral and cellular proteins, including viral gB, at the C terminus (35–37). Therefore, the C terminus of gB (gBct, amino acids 796 to 904) was used as a positive control in the phosphorylation assay. The in vitro kinase assay showed that purified US3 kinase robustly phosphorylates the C terminus of KIF3A (Fig. 3C), suggesting that US3 interacts directly with KIF3A and phosphorylates it at the C terminus.
To confirm that KIF3A is phosphorylated directly by US3, we purified both wild-type US3 and the kinase-dead mutant form US3K220A (8, 35) and repeated the phosphorylation assay. The US3 kinase-dead mutant form was expressed in transfected cells at lower levels (Fig. 3D), presumably because the US3 protein enhances cellular translation, including its own expression (38). Nevertheless, even at a lower protein input, wild-type US3 potently phosphorylated KIF3A protein whereas kinase-dead US3 did not (Fig. 3E), suggesting that phosphorylation is directly by US3 and not any cellular kinase(s) copurified with the US3 kinase.
In order to investigate whether US3 phosphorylates KIF3A in vivo, we examined the phosphorylation status of KIF3A in cells expressing US3 kinase. Since KIF3A is a relatively large protein (∼80 kDa), the size shift due to potential phosphorylation of KIF3A may not be readily detectable by regular SDS-PAGE. We therefore employed the recently developed Phos-tag method, which specifically increases the molecular mass shift of phosphorylated proteins for detection (39). In cells expressing US3, a portion of KIF3A was clearly shifted to a high-molecular-mass band (Fig. 3F), consistent with the expected KIF3A phosphorylation. More importantly, we purified the KIF3A protein from HSV-1-infected cells and found that a portion of the KIF3A protein was phosphorylated only in cells infected with wild-type HSV-1 strain F and not in cells infected with US3-deficient HSV-1 (Fig. 3G). These results suggest that US3 specifically phosphorylates KIF3A in HSV-1-infected cells.
HSV-1 US3 protein phosphorylates KIF3A at serine 687.
To understand how US3 phosphorylation of KIF3A modulates CD1d intracellular trafficking, we endeavored to identify the phosphorylation site(s) of KIF3A. KIF3A was expressed in 293T cells as a GST fusion protein in the presence or absence of US3 expression and purified to homogeneity by GST pulldown (Fig. 4A). MS/MS analysis was performed to identify the amino acid(s) specifically phosphorylated upon coexpression with US3. Two phosphorylated peptides were detected containing the amino acid sequences RSAKPETVIDSLLQ and RKQTPVPDKK, which correspond to amino acids 686 to 699 and 629 to 639, respectively, of the KIF3A protein (Fig. 4A). Since US3 is a PKA-like serine/threonine protein kinase (40), serine 687 (S687) is the apparent phosphorylation site on the first phosphopeptide, while the only threonine, T633, is most likely to be the phosphorylation site on the second phosphopeptide. To confirm that S687 and T633 can be phosphorylated by US3, we repeated the in vitro phosphorylation assay with the purified US3 protein as shown in Fig. 3C with a KIF3A protein with either of the two amino acids mutated. Mutation of either amino acid to alanine led to a lower phosphorylation signal level (Fig. 4B), suggesting that both amino acids can be phosphorylated by US3, consistent with the MS/MS results.
FIG 4.

Identification of KIF3A phosphorylation site by US3 kinase. (A) GST-KIF3A fusion protein was expressed in 293T cells in the presence or absence of US3 kinase, purified by GST pulldown, and analyzed by SDS-PAGE, followed by Coomassie blue staining. MS/MS identified two potential phosphorylation sites at the C terminus of KIF3A, serine 687 and threonine 633, as indicated by the arrows. M.W., molecular mass. (B) In vitro phosphorylation of GST or GST-fused wild-type (WT) or mutant C-terminal KIF3A protein by purified GST-fused US3 protein. The input GST and GST-fused KIF3A proteins were examined with Coomassie blue staining. (C) Detection of KIF3A phosphorylation with a phosphospecific antibody. GST-fused KIF3A was expressed in 293T cells in the presence or absence of US3 and purified by GST pulldown. A serine/threonine phosphospecific antibody (9624) detects KIF3A phosphorylated by US3. IB, immunoblotting; WCL, whole-cell lysate. (D) Mapping of KIF3A phosphorylation sites. Threonine 633 and serine 687 were mutated to alanines. Mutant or wild-type KIF3A was expressed as a V5-tagged protein in 293T cells in the presence or absence of US3. The V5-tagged KIF3A protein was purified by anti-V5 antibody immunoprecipitation (IP) and then analyzed by Western blotting with a phosphospecific antibody (9624). Total immunoprecipitated KIF3A and expressed GST-US3 proteins were measured by Western blotting with anti-KIF3A and anti-GST antibodies, respectively.
Currently, there is no phosphospecific antibody against phosphorylated S687 or T633 in KIF3A; therefore, we tested the reactivity of the anti-phospho-PKA substrate motif antibody (catalog no. 9624; Cell Signaling), which was generated with a phosphorylated peptide containing the PKA substrate motif (RRXS*/T*). We found that this antibody indeed specifically detected KIF3A phosphorylation by the US3 protein (Fig. 4C). To identify which of the two amino acids is phosphorylated by US3, an alanine was introduced to either of these two sites. Mutant KIF3A proteins were coexpressed with US3 as V5-tagged proteins and purified by anti-V5 antibody immunoprecipitation, followed by Western blotting with the 9624 antibody. Mutation of S687 (S687A) completely diminished the Western blotting signal (Fig. 4D), suggesting that the 9624 antibody was detecting KIF3A with S687 phosphorylated. Interestingly, mutation of T633 (T633A) did not reduce the phosphorylation level but rather increased it. We then mutated both T633 and S687 to alanines and coexpressed the resulting mutant KIF3A protein with US3. The phosphorylation signal was completely abolished (Fig. 4D), strongly suggesting that the phosphorylation signal detected by the 9624 antibody is specific for KIF3A phosphorylation at S687.
US3 phosphorylates KIF3A at S687 during HSV-1 infection, and this phosphorylation is required for US3-mediated CD1d downregulation.
We next analyzed KIF3A phosphorylation in HSV-1-infected cells. HeLa.CD1d cells were infected with HSV-1 strain F or a US3-deficient strain for 12 h. Endogenous KIF3A was then immunoprecipitated and blotted for the 9624 anti-phospho-PKA substrate antibody. KIF3A phosphorylation was detected only in cells infected with the wild-type F strain of HSV-1 and not in cells infected with the US3-deficient virus or in uninfected cells (Fig. 5A), suggesting that US3 phosphorylates KIF3A at serine 687 in HSV-1-infected cells.
FIG 5.
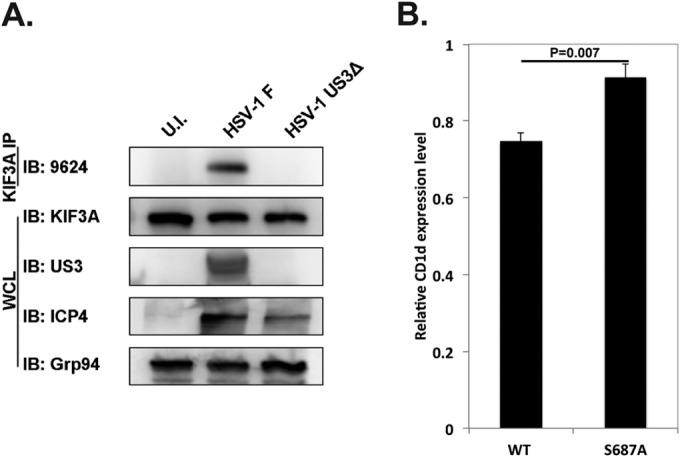
KIF3A phosphorylation at serine 687 during HSV-1 infection is essential for US3-mediated CD1d downregulation. (A) HeLa.CD1d cells were infected with wild-type (WT, strain F) or US3-deficient mutant HSV-1 (US3Δ) at an MOI of 5 for 12 h. Endogenous KIF3A protein was purified by immunoprecipitation with anti-KIF3A antibodies, and phosphorylation of KIF3A was analyzed with a phosphospecific antibody (9624). IP, immunoprecipitation; IB, immunoblotting; WCL, whole-cell lysate; U.I., uninfected. (B) HeLa.CD1d cells were cotransfected with US3 and GFP-tagged wild-type or S687A mutant KIF3A, and the cell surface expression of CD1d was analyzed by flow cytometry. The relative CD1d expression level is presented as the ratio of the CD1d expression level (measured as the mean fluorescence intensity of CD1d staining) in GFP-positive transfected cells to the CD1d expression level in GFP-negative untransfected cells. Statistics were calculated from three independent experiments.
To investigate whether S687 phosphorylation of KIF3A by US3 is required for US3-mediated CD1d downregulation, we cotransfected US3 with either wild-type or S687A mutant KIF3A into HeLa.CD1d cells. The S687A mutation completely abolished US3-mediated CD1d downregulation (Fig. 5B), suggesting that phosphorylation at S687 is required for US3 to shut down intracellular trafficking and cell surface expression of CD1d. This result also suggests that in these transfected cells, although US3 phosphorylates serine 687 of endogenous KIF3A and nullifies its function in CD1d exocytosis, S687A mutant KIF3A may evade this and stay functional in transporting late endosomes/lysosomes to the cell surface, thereby retaining CD1d expression.
DISCUSSION
HSV-1 and US3-mediated CD1d downregulation.
We and other groups have demonstrated that HSV-1, upon infection, rapidly and efficiently downregulates a critical antigen-presenting molecule, CD1d, thereby inhibiting the function of NKT cells (8, 10, 13, 14). In addition to downregulating key antigen-presenting molecules such as CD1d and MHC class I/II for the sake of immune evasion (41, 42), HSV-1 reprograms the entire cellular machinery for the benefit of its own replication. Therefore, it is conceivable that the virus precisely modulates the cell surface expression of diverse membrane proteins, depending on their role(s) in cellular metabolism and viral replication. This may explain why we observed specific downregulation of CD1d expression but not other cellular molecules, including CD71, CD63, and Lamp1 (Fig. 1, left). When the US3 protein is expressed ectopically in uninfected cells, it downregulates both CD1d and other late endosomal/lysosomal proteins, such as CD63 and Lamp1. This result suggests that US3, on its own, targets the trafficking of multiple (if not all) late endosomal/lysosomal proteins. The results of viral infection and ectopic expression of US3 together also suggest that during viral infection, other viral genes may have an impact on the trafficking of other late endosomal/lysosomal proteins, including CD63 and Lamp1.
Regulation of KIF3A function by US3 phosphorylation.
By phosphorylating KIF3A at serine 687, US3 modifies the function of KIF3A and inhibits KIF3A-based exocytosis of late endosomes/lysosomes. Ablation of this phosphorylation by the mutation of S687 to alanine abolished the US3-mediated downregulation of CD1d expression on the cell surface (Fig. 5B). This is the first report of KIF3A phosphorylation at S687 by a viral protein kinase. Little is known about how this phosphorylation might modify the function of KIF3A and kinesin II. Phosphorylation of different kinesins inhibits kinesin-based transport by interfering with their interactions with either cargo proteins/cargo adaptor proteins or microtubules (18). In vitro assays have shown that cellular calcium calmodulin kinase II can phosphorylate KIF3A at S687 and this phosphorylation is required for the interaction of KIF3A with tubulin, possibly by the disruption of an autoinhibitory interaction between the N and C termini of KIF3A (31). On the other hand, the C termini of KIF3A and KIF3B have been demonstrated to interact with Rip11/FIP5, the adaptor protein for cargo endosomes (34). Interestingly, consistent with the structures of kinesin-based motor complexes, most phosphorylation events at the C termini of kinesins disrupt their association with cargo proteins/cargo adaptor proteins, whereas phosphorylation at the N-terminal motor domains disrupts their association with microtubules (18). It is therefore tempting to speculate that US3 phosphorylation of KIF3A at the C terminus S687 disrupts the interaction between kinesin II motor and cargo adaptor proteins, possibly Rip11/FIP5 (34) or dynactin, another recently reported adaptor protein (43).
Functions of kinesins in CD1d exocytosis.
Kinesins are encoded by a large gene family composed of 44 members in humans (18, 44). Kinesins transport diverse cargoes, including different membranous organelles and large protein complexes. Motor-cargo combinations for individual kinesins have a high degree of specificity, although there is redundancy in the function of individual kinesins (18). CD1d expression and exocytosis in antigen-presenting cells are expected to involve multiple cellular organelles, including the ER, the trans-Golgi network, early or recycling endosomes, and late endosomes/lysosomes (3, 15). Therefore, multiple kinesin motors may contribute to CD1d cell surface expression. To investigate the roles of individual kinesins in the cell surface expression of CD1d, we performed a survey of kinesins in human epithelial cells by expressing dominant negative mutant forms of individual kinesins in CD1d-expressing cells. Indeed, we discovered that multiple kinesins play a role in CD1d expression, whereas functional inhibition of KIF3A led to the lowest expression of CD1d (R. Xiong and W. Yuan, unpublished results). Little is known regarding how these diverse kinesins can be regulated by protein phosphorylation and how these phosphorylations may contribute to the regulation of CD1d expression during diverse immune responses. HSV-1 protein kinase US3, which efficiently downregulates CD1d expression (8), may provide a useful tool with which to dissect the function and regulation of kinesins that are important for CD1d expression.
ACKNOWLEDGMENTS
This work was supported by an NIH R01 grant (AI91987), an NIH U01 grant (GM111849), and funding from the Harry Lloyd Charitable Trust and the Margaret Early Medical Research Trust to W.Y. The project described here was supported in part by NIH grant P30CA014089 to the University of Southern California Norris Comprehensive Cancer Center from the National Cancer Institute.
We thank Jae U. Jung, Lucio Comai, Shou-Jiang Gao, and Pinghui Feng at the University of Southern California for valuable discussions and critical readings of the manuscript. We also thank Yan Yuan at the University of Pennsylvania and Fanxiu Zhu at Florida State University for providing valuable reagents.
REFERENCES
- 1.Tupin E, Kinjo Y, Kronenberg M. 2007. The unique role of natural killer T cells in the response to microorganisms. Nat Rev Microbiol 5:405–417. doi: 10.1038/nrmicro1657. [DOI] [PubMed] [Google Scholar]
- 2.Bendelac A, Savage PB, Teyton L. 2007. The biology of NKT cells. Annu Rev Immunol 25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- 3.Brigl M, Brenner MB. 2004. CD1: antigen presentation and T cell function. Annu Rev Immunol 22:817–890. doi: 10.1146/annurev.immunol.22.012703.104608. [DOI] [PubMed] [Google Scholar]
- 4.Juno JA, Keynan Y, Fowke KR. 2012. Invariant NKT cells: regulation and function during viral infection. PLoS Pathog 8:e1002838. doi: 10.1371/journal.ppat.1002838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tessmer MS, Fatima A, Paget C, Trottein F, Brossay L. 2009. NKT cell immune responses to viral infection. Expert Opin Ther Targets 13:153–162. doi: 10.1517/14712590802653601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brulois K, Jung JU. 2014. Interplay between Kaposi's sarcoma-associated herpesvirus and the innate immune system. Cytokine Growth Factor Rev 25:597–609. doi: 10.1016/j.cytogfr.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanchez DJ, Gumperz JE, Ganem D. 2005. Regulation of CD1d expression and function by a herpesvirus infection. J Clin Invest 115:1369–1378. doi: 10.1172/JCI24041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rao P, Pham HT, Kulkarni A, Yang Y, Liu X, Knipe DM, Cresswell P, Yuan W. 2011. Herpes simplex virus 1 glycoprotein B and US3 collaborate to inhibit CD1d antigen presentation and NKT cell function. J Virol 85:8093–8104. doi: 10.1128/JVI.02689-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan W, Kang SJ, Evans JE, Cresswell P. 2009. Natural lipid ligands associated with human CD1d targeted to different subcellular compartments. J Immunol 182:4784–4791. doi: 10.4049/jimmunol.0803981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raftery MJ, Winau F, Kaufmann SH, Schaible UE, Schonrich G. 2006. CD1 antigen presentation by human dendritic cells as a target for herpes simplex virus immune evasion. J Immunol 177:6207–6214. doi: 10.4049/jimmunol.177.9.6207. [DOI] [PubMed] [Google Scholar]
- 11.Banovic T, Yanilla M, Simmons R, Robertson I, Schroder WA, Raffelt NC, Wilson YA, Hill GR, Hogan P, Nourse CB. 2011. Disseminated varicella infection caused by varicella vaccine strain in a child with low invariant natural killer T cells and diminished CD1d expression. J Infect Dis 204:1893–1901. doi: 10.1093/infdis/jir660. [DOI] [PubMed] [Google Scholar]
- 12.Han J, Rho SB, Lee JY, Bae J, Park SH, Lee SJ, Lee SY, Ahn C, Kim JY, Chun T. 2013. Human cytomegalovirus (HCMV) US2 protein interacts with human CD1d (hCD1d) and down-regulates invariant NKT (iNKT) cell activity. Mol Cells 36:455–464. doi: 10.1007/s10059-013-0221-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan W, Dasgupta A, Cresswell P. 2006. Herpes simplex virus evades natural killer T cell recognition by suppressing CD1d recycling. Nat Immunol 7:835–842. doi: 10.1038/ni1364. [DOI] [PubMed] [Google Scholar]
- 14.Liu J, Glosson NL, Du W, Gervay-Hague J, Brutkiewicz RR. 2013. A Thr/Ser dual residue motif in the cytoplasmic tail of human CD1d is important for the down-regulation of antigen presentation following a herpes simplex virus 1 infection. Immunology 140:191–201. doi: 10.1111/imm.12127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gumperz JE. 2006. The ins and outs of CD1 molecules: bringing lipids under immunological surveillance. Traffic 7:2–13. doi: 10.1111/j.1600-0854.2005.00364.x. [DOI] [PubMed] [Google Scholar]
- 16.Soldati T, Schliwa M. 2006. Powering membrane traffic in endocytosis and recycling. Nat Rev Mol Cell Biol 7:897–908. doi: 10.1038/nrm2060. [DOI] [PubMed] [Google Scholar]
- 17.Maxfield FR, McGraw TE. 2004. Endocytic recycling. Nat Rev Mol Cell Biol 5:121–132. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- 18.Hirokawa N, Noda Y, Tanaka Y, Niwa S. 2009. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol 10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- 19.Wiesner C, Faix J, Himmel M, Bentzien F, Linder S. 2010. KIF5B and KIF3A/KIF3B kinesins drive MT1-MMP surface exposure, CD44 shedding, and extracellular matrix degradation in primary macrophages. Blood 116:1559–1569. doi: 10.1182/blood-2009-12-257089. [DOI] [PubMed] [Google Scholar]
- 20.Sathish N, Zhu FX, Yuan Y. 2009. Kaposi's sarcoma-associated herpesvirus ORF45 interacts with kinesin-2 transporting viral capsid-tegument complexes along microtubules. PLoS Pathog 5:e1000332. doi: 10.1371/journal.ppat.1000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grant BD, Donaldson JG. 2009. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol 10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benetti L, Roizman B. 2006. Protein kinase B/Akt is present in activated form throughout the entire replicative cycle of deltaU(S)3 mutant virus but only at early times after infection with wild-type herpes simplex virus 1. J Virol 80:3341–3348. doi: 10.1128/JVI.80.7.3341-3348.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gallo RM, Khan MA, Shi J, Kapur R, Wei L, Bailey JC, Liu J, Brutkiewicz RR. 2012. Regulation of the actin cytoskeleton by Rho kinase controls antigen presentation by CD1d. J Immunol 189:1689–1698. doi: 10.4049/jimmunol.1101484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jayawardena-Wolf J, Benlagha K, Chiu YH, Mehr R, Bendelac A. 2001. CD1d endosomal trafficking is independently regulated by an intrinsic CD1d-encoded tyrosine motif and by the invariant chain. Immunity 15:897–908. doi: 10.1016/S1074-7613(01)00240-0. [DOI] [PubMed] [Google Scholar]
- 25.Sugita M, Cao X, Watts GF, Rogers RA, Bonifacino JS, Brenner MB. 2002. Failure of trafficking and antigen presentation by CD1 in AP-3-deficient cells. Immunity 16:697–706. doi: 10.1016/S1074-7613(02)00311-4. [DOI] [PubMed] [Google Scholar]
- 26.Bananis E, Nath S, Gordon K, Satir P, Stockert RJ, Murray JW, Wolkoff AW. 2004. Microtubule-dependent movement of late endocytic vesicles in vitro: requirements for dynein and kinesin. Mol Biol Cell 15:3688–3697. doi: 10.1091/mbc.E04-04-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown CL, Maier KC, Stauber T, Ginkel LM, Wordeman L, Vernos I, Schroer TA. 2005. Kinesin-2 is a motor for late endosomes and lysosomes. Traffic 6:1114–1124. doi: 10.1111/j.1600-0854.2005.00347.x. [DOI] [PubMed] [Google Scholar]
- 28.Scholey JM. 2013. Kinesin-2: a family of heterotrimeric and homodimeric motors with diverse intracellular transport functions. Annu Rev Cell Dev Biol 29:443–469. doi: 10.1146/annurev-cellbio-101512-122335. [DOI] [PubMed] [Google Scholar]
- 29.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J Virol 82:6172–6189. doi: 10.1128/JVI.00044-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. 2004. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A 101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phang HQ, Hoon JL, Lai SK, Zeng Y, Chiam KH, Li HY, Koh CG. 2014. POPX2 phosphatase regulates the KIF3 kinesin motor complex. J Cell Sci 127:727–739. doi: 10.1242/jcs.126482. [DOI] [PubMed] [Google Scholar]
- 32.Liang Y, Pang Y, Wu Q, Hu Z, Han X, Xu Y, Deng H, Pan J. 2014. FLA8/KIF3B phosphorylation regulates kinesin-II interaction with IFT-B to control IFT entry and turnaround. Dev Cell 30:585–597. doi: 10.1016/j.devcel.2014.07.019. [DOI] [PubMed] [Google Scholar]
- 33.Häfner J, Mayr MI, Mockel MM, Mayer TU. 2014. Pre-anaphase chromosome oscillations are regulated by the antagonistic activities of Cdk1 and PP1 on Kif18A. Nat Commun 5:4397. doi: 10.1038/ncomms5397. [DOI] [PubMed] [Google Scholar]
- 34.Schonteich E, Wilson GM, Burden J, Hopkins CR, Anderson K, Goldenring JR, Prekeris R. 2008. The Rip11/Rab11-FIP5 and kinesin II complex regulates endocytic protein recycling. J Cell Sci 121:3824–3833. doi: 10.1242/jcs.032441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kato A, Arii J, Shiratori I, Akashi H, Arase H, Kawaguchi Y. 2009. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J Virol 83:250–261. doi: 10.1128/JVI.01451-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imai T, Sagou K, Arii J, Kawaguchi Y. 2010. Effects of phosphorylation of herpes simplex virus 1 envelope glycoprotein B by Us3 kinase in vivo and in vitro. J Virol 84:153–162. doi: 10.1128/JVI.01447-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wisner TW, Wright CC, Kato A, Kawaguchi Y, Mou F, Baines JD, Roller RJ, Johnson DC. 2009. Herpesvirus gB-induced fusion between the virion envelope and outer nuclear membrane during virus egress is regulated by the viral US3 kinase. J Virol 83:3115–3126. doi: 10.1128/JVI.01462-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chuluunbaatar U, Roller R, Feldman ME, Brown S, Shokat KM, Mohr I. 2010. Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev 24:2627–2639. doi: 10.1101/gad.1978310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kinoshita E, Kinoshita-Kikuta E, Koike T. 2009. Separation and detection of large phosphoproteins using Phos-tag SDS-PAGE. Nat Protoc 4:1513–1521. doi: 10.1038/nprot.2009.154. [DOI] [PubMed] [Google Scholar]
- 40.Benetti L, Roizman B. 2004. Herpes simplex virus protein kinase US3 activates and functionally overlaps protein kinase A to block apoptosis. Proc Natl Acad Sci U S A 101:9411–9416. doi: 10.1073/pnas.0403160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ahn K, Meyer TH, Uebel S, Sempe P, Djaballah H, Yang Y, Peterson PA, Fruh K, Tampe R. 1996. Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. EMBO J 15:3247–3255. [PMC free article] [PubMed] [Google Scholar]
- 42.Neumann J, Eis-Hubinger AM, Koch N. 2003. Herpes simplex virus type 1 targets the MHC class II processing pathway for immune evasion. J Immunol 171:3075–3083. doi: 10.4049/jimmunol.171.6.3075. [DOI] [PubMed] [Google Scholar]
- 43.Kodani A, Salome Sirerol-Piquer M, Seol A, Garcia-Verdugo JM, Reiter JF. 2013. Kif3a interacts with dynactin subunit p150 glued to organize centriole subdistal appendages. EMBO J 32:597–607. doi: 10.1038/emboj.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maliga Z, Junqueira M, Toyoda Y, Ettinger A, Mora-Bermudez F, Klemm RW, Vasilj A, Guhr E, Ibarlucea-Benitez I, Poser I, Bonifacio E, Huttner WB, Shevchenko A, Hyman AA. 2013. A genomic toolkit to investigate kinesin and myosin motor function in cells. Nat Cell Biol 15:325–334. doi: 10.1038/ncb2689. [DOI] [PubMed] [Google Scholar]