

ABSTRACT
The more than 200 closely spaced annotated open reading frames, extensive transcriptional read-through, and numerous unpredicted RNA start sites have made the analysis of vaccinia virus gene expression challenging. Genome-wide ribosome profiling provided an unprecedented assessment of poxvirus gene expression. By 4 h after infection, approximately 80% of the ribosome-associated mRNA was viral. Ribosome-associated mRNAs were detected for most annotated early genes at 2 h and for most intermediate and late genes at 4 and 8 h. Cluster analysis identified a subset of early mRNAs that continued to be translated at the later times. At 2 h, there was excellent correlation between the abundance of individual mRNAs and the numbers of associated ribosomes, indicating that expression was primarily transcriptionally regulated. However, extensive transcriptional read-through invalidated similar correlations at later times. The mRNAs with the highest density of ribosomes had host response, DNA replication, and transcription roles at early times and were virion components at late times. Translation inhibitors were used to map initiation sites at single-nucleotide resolution at the start of most annotated open reading frames although in some cases a downstream methionine was used instead. Additional putative translational initiation sites with AUG or alternative codons occurred mostly within open reading frames, and fewer occurred in untranslated leader sequences, antisense strands, and intergenic regions. However, most open reading frames associated with these additional translation initiation sites were short, raising questions regarding their biological roles. The data were used to construct a high-resolution genome-wide map of the vaccinia virus translatome.
IMPORTANCE This report contains the first genome-wide, high-resolution analysis of poxvirus gene expression at both transcriptional and translational levels. The study was made possible by recent methodological advances allowing examination of the translated regions of mRNAs including start sites at single-nucleotide resolution. Vaccinia virus ribosome-associated mRNA sequences were detected for most annotated early genes at 2 h and for most intermediate and late genes at 4 and 8 h after infection. The ribosome profiling approach was particularly valuable for poxviruses because of the close spacing of approximately 200 open reading frames and extensive transcriptional read-through resulting in overlapping mRNAs. The expression of intermediate and late genes, in particular, was visualized with unprecedented clarity and quantitation. We also identified novel putative translation initiation sites that were mostly associated with short protein coding sequences. The results provide a framework for further studies of poxvirus gene expression.
INTRODUCTION
Poxviruses comprise a large family that infect vertebrates and invertebrates, including species that are highly pathogenic for humans as well as for wild and domesticated animals (1, 2). Nevertheless, attenuated poxviruses are being employed as recombinant vectors for vaccines and potential cancer therapeutics. The cytoplasmic site of replication, the large linear double-stranded DNA genome with covalently closed ends, and the encoding of most, if not all, proteins required for DNA and RNA synthesis are distinguishing features of poxviruses. Studies with poxviruses have led to fundamental and far-reaching discoveries, such as the structure and biosynthesis of the 5′ cap (3, 4) and the 3′ poly(A) sequence (5, 6) on mRNA and numerous proteins that counteract host defenses (7, 8). Vaccinia virus (VACV), the prototype poxvirus that was used as the vaccine to eradicate smallpox, has a 200-kbp genome with more than 200 annotated open reading frames (ORFs) that are selectively expressed at early, intermediate, or late times of infection due to stage-specific viral promoters and transcription factors that work in concert with the multisubunit viral DNA-dependent RNA polymerase. Recent genome-wide transcription analyses (high-throughput sequencing of RNA transcripts, or RNA-Seq) revealed extraordinary complexity, with numerous unpredicted RNA start sites (9, 10) that parallel similar findings with other viruses and eukaryotic cells (11–14). Moreover, extensive transcriptional read-through leading to overlapping and complementary RNAs has contributed to difficulties in understanding VACV gene expression.
Recent advances in genome-wide ribosome profiling have provided a powerful new technique to quantify active protein translation with sensitivity and resolution comparable to those of RNA-Seq (12, 15, 16). In ribosome profiling, only the mRNA fragments protected by ribosomes are isolated and analyzed by next-generation sequencing. We have adapted genome-wide ribosome profiling to further investigate VACV gene expression. The precise identification of ribosome occupancy allowed us to map and quantify the translated region of overlapping mRNAs and identify additional putative translation initiation sites, some of which have alternative start codons.
MATERIALS AND METHODS
Cell culture and virus infection.
HeLa S3 cells were cultured in minimum essential medium with the spinner modification (Quality Biological) and supplemented with 5% equine serum (HyClone) at 37°C in a 5% CO2 atmosphere. The preparation, purification, and titration of the VACV Western Reserve (WR) strain (American Type Culture Collection [ATCC] VR-1354) were carried out as described previously (17). HeLa cells were infected with 20 PFU per cell at 1 × 107 cells/ml for 30 min and then diluted 100-fold. At desired time points, an aliquot of cells was pretreated with 2 μg/ml of harringtonine, 100 μg/ml of cycloheximide, or 50 μM lactimidomycin and harvested for subsequent mRNA or ribosome profiling. Lactimidomycin was isolated from Streptomyces amphibiosporus (18).
Ribosome profiling and mRNA sequencing.
Ribosome footprinting was carried out as described elsewhere with minor modifications (16). Briefly, HeLa cells pretreated with translational inhibitors were lysed and treated with DNase (Ambion), and the lysate was clarified. A portion of the lysate was removed for mRNA isolation and subsequent mRNA sequencing (mRNA-Seq) analysis. For mRNA-Seq, the isolated mRNA was fragmented by digestion with RNase III (New England Biolabs). The fragmented RNAs were resolved by denaturing polyacrylamide gel electrophoresis in a 15% gel with urea, and fragments between 50 and 80 nucleotides (nt) were extracted. The remainder of the lysate was digested with RNase I (Ambion), after which the ribosomes were pelleted by ultracentrifugation. The ribosome-protected RNA fragments were isolated and separated by electrophoresis as described above except that fragments between 28 and 34 nt were extracted. The purified RNA fragments were then subjected to a series of enzyme treatments and molecular manipulations as described previously (16) to generate libraries for deep sequencing. The final libraries were purified from primers and unligated adaptors by electrophoresis and size selection of 150- to 200-bp fragments in a 4% agarose gel. These purified libraries were run on a single-read flow cell to generate 95-nt reads on a HiSeq 2000 system.
Computational analysis.
Sequencing reads were trimmed to remove adaptors and aligned to the VACV genome using Tophat2 (19) using default settings except that two alignments for a single sequence in the inverted terminal repeat region were allowed. The numbers of reads mapped to annotated ORFs were counted. Reads between 28 and 34 nt in length were used for further analysis. VACV reads covering each base and VACV genome annotation were visualized with MochiView (20).
Identification of putative translation initiation sites.
The number and location of 5′ ends of ribosome-protected (Ribo)-harringtonine reads were used to identify peaks. The following rules were used to define a translation initiation site from the Ribo-harringtonine peak: (i) the region must have mRNA, Ribo-cycloheximide, and Ribo-harringtonine reads; (ii) the position must have at least 1,000 normalized reads from the harringtonine data; (iii) if the position was located within an annotated ORF, a peak must have read numbers at least 25% of the highest peak within that ORF; and (iv) the site must be a local maximum within 7 surrounding nucleotides. For a peak preceded by AAA, the first AUG within the following 9 nt (including the last A of the AAA) was designated a translation initiation site. If no AUG was found within the following 9 nt, the first near-cognate start codon (CUG, UUG, GUG, ACG, AGG, AAG, AUC, AUU, or AUA) was designated a putative initiation site. For a peak not preceded by AAA, an AUG located 12 nt downstream of the peak was designated an initiation site; an AUG following or prior to the 12-nt codon within 3 nt was designated an initiation site if the AUG was not found exactly 12 nt downstream. If no AUG was found in this region, a near-cognate start codon was designated a putative initiation site using the same procedure as for AUG.
Identification of ORFs associated with translation initiation sites.
The ORFs were defined, without encoding-length restriction, based on the presence of an AUG or near-cognate codon at the start and UAA, UAG, or UGA as a stop codon and on a ribosome footprint covering the entire potential encoding regions of the ORFs.
Normalization of VACV reads.
The raw sequence data for VACV ORFs were normalized using ORF length and total reads of each sample.
Accession number.
The sequencing data were deposited at the National Center for Biotechnology Information Sequence Read Archive under accession number SRP056975.
RESULTS
Experimental approach.
RNA sequences protected by ribosomes from RNase I digestion (ribosome footprint profiles) represent a snapshot of transcripts being actively translated, and the numbers of such reads provide an estimate of the density of ribosomes on a particular mRNA at a given time (21). HeLa cell suspension cultures were infected at a high multiplicity of purified VACV in order to synchronize gene expression. Previous RNA-Seq studies had shown that early mRNAs are present at 2 h after VACV infection, whereas predominantly intermediate and late mRNAs are present at 4 h and later times (22). Fragments of 50 to 80 nt generated by RNase III digestion of total polyadenylated mRNA and 28- to 34-nt RNA segments protected by ribosomes from RNase I digestion were prepared from VACV-infected cells at prereplicative (2 h) and postreplicative (4 and 8 h) times for deep sequencing (Fig. 1). Cycloheximide was added prior to mRNA or ribosome isolation in order to inhibit peptide synthesis and freeze ribosomes (23). Harringtonine and lactimidomycin, which bind to assembled 80S ribosome and free 60S ribosomes, respectively, were added to stall ribosomes at translation initiation sites (24, 25).
FIG 1.

Schematic diagram of the experimental approach. HeLa cells were infected with VACV WR at a multiplicity of 20 PFU/cell. Cycloheximide, harringtonine, or lactimidomycin was added at 2, 4, and 8 h postinfection, and mRNAs protected by ribosomes from digestion with RNase I were isolated and deep sequenced (ribosome profiling). Total mRNA in the presence of cycloheximide was simultaneously isolated, fragmented by digestion with RNase III, and deep sequenced (mRNA profiling).
Ribosome profiling resolved mRNA segments undergoing translation despite extensive transcriptional read-through.
Using RNA-Seq, we recently reported that the percentage of viral mRNA increased to 55% of the total infected cell mRNA by 4 h after infection (22), which was consistent with earlier nucleic acid hybridization studies (26). In the present experiments, the percentage of viral mRNA was greater than 30% at 2 h and 70 to 75% at 4 and 8 h. The ribosome-associated viral RNA reads accounted for more than 40% of the total at 2 h and 70 to 80% of the total at 4 and 8 h. Using 100,000 normalized reads as an arbitrary lower cutoff, 104 of 118 previously annotated early gene sequences were detected in ribosome-associated mRNAs at 2 h, and 90 of the 93 annotated intermediate and late gene sequences were detected at 4 and 8 h. These data indicated that VACV usurpation of cellular protein synthesis was largely mediated at the mRNA level although some translational enhancement was possible.
Viral genome-wide mRNA and ribosome footprint profiles were compared, as shown in Fig. 2. Read counts were aligned along the VACV genome at single-nucleotide resolution, with those above and below the horizontal line corresponding to transcription from opposite strands in the rightwards and leftwards directions, respectively. As previously shown (22), mRNA reads were located mostly on opposite strands near the two ends of the VACV genome at the early stage (2 h) but covered the genome more broadly at later stages (4 and 8 h). At 2 h, the ribosome footprints were similar to but sharper than the mRNA patterns (Fig. 2). At 4 h, the footprint pattern was much more discrete than the mRNA patterns and mostly corresponded to the boundaries of annotated ORFs on both DNA strands (Fig. 2). The 8-h footprint pattern appeared relatively unchanged from the preceding time (Fig. 2). The reproducibility of ribosome profiling in two completely independent experiments was demonstrated by dot blot comparisons of the number of reads that were mapped to the previously individually annotated ORFs (Fig. 3A). The r values of 0.95, 0.99, and 0.83 were calculated for the 2-, 4-, and 8-h times, respectively.
FIG 2.

Comparison of VACV genome-wide transcriptome and ribosome footprint maps at 2, 4, and 8 h of infection. The number of read counts per nucleotide is displayed over the genome-wide map of VACV ORFs. The reads above the line map to the upper DNA strand, and reads below the line map to the lower DNA strand. High read counts are off scale for display purpose. The VACV HindIII restriction endonuclease map is shown at the bottom. Abbreviations: mRNA, mRNA profiling; Ribo, ribosome profiling. The hours after infection are indicated.
FIG 3.

Reproducibility and cluster analysis. (A) Ribosome profiling with cycloheximide (Ribo-CHX). Read counts were mapped to the annotated ORFs of VACV from two independent experiments at 2, 4, and 8 h postinfection. Each dot represents the read count for an individual mRNA. The correlation coefficients (r) are noted on each plot. (B) Cluster analysis of early gene expression. Cluster analysis was performed on the normalized read counts of ribosome footprints for each early gene at 2, 4, and 8 h after infection. Line plots of the read counts corresponding to three clusters are shown.
The correspondence of ribosome footprints to ORFs was better appreciated by zooming in on the genome-wide map. An expansion of the region corresponding to the HindIII D fragment of the VACV genome, which contains 13 previously annotated ORFs of which 7 are expressed early (27, 28), is shown in Fig. 4A. The VACV ORFs are identified by the common nomenclature in which a letter represents the HindIII restriction fragment and is followed by the gene number in that fragment; L or R may be added to indicate leftwards or rightwards transcription. In the tables shown later, ORFs were also identified by the ORF numbers in the complete genome sequence of the WR strain of VACV. Inspection of the mRNA profiles at 2 h demonstrated a general correspondence of reads to early-stage ORFs. However, the overlap with downstream ORFs previously led to some uncertainty regarding early and late gene expression in several places within the genome (22, 29). In comparison, the ribosome footprints stopped abruptly at the ends of the ORFs (Fig. 4A), clearly distinguishing between expressed ORFs and read-through transcription. A further expansion of the region of D9 to D10 shows the difference between the mRNA and ribosome footprint profiles more precisely (Fig. 4C). Similar examples were found throughout the genome, as illustrated by comparison of the mRNA and footprint profiles of the early I3 mRNA (Fig. 4B).
FIG 4.

Comparison of mRNA and ribosome footprint reads in expanded regions of the VACV genome. (A) Transcription and ribosome footprint maps of the HindIII D region of the VACV genome at 2, 4, and 8 h of infection. The annotated ORFs D1 to D13 in this region are displayed, with green and red indicating early and postreplicative expression, respectively. (B and C) Transcripts and ribosome footprints corresponding to the I3L and D9R ORFs at 2 h of infection. Note that L and R following the ORF number indicate leftwards and rightwards transcription, respectively.
In striking contrast to the 2-h time point, the total mRNA reads covered the entire DNA segment at 4 and 8 h, making it difficult to see correlations with annotated ORFs and impossible to distinguish early and late gene expression (Fig. 4A). Nevertheless, the ribosome footprints corresponded closely to the start and end positions of annotated late ORFs and revealed large differences in their expression levels. Thus, many more ribosomal reads were mapped to D13 and D8 than to D2, D3, D6, D10, and D11. Few reads were still associated with the early ORFs in the HindIII D genome fragment at 4 and 8 h, indicating tight regulation. An exception was the early D7 ORF in which there was still a considerable number of ribosome-associated reads at 4 h although expression was mostly gone at 8 h. The presence of D7 mRNA at 4 h after infection had previously been shown by Northern blotting (30). The D7 ORF encodes a subunit of the RNA polymerase that is packaged in virus particles (31). An unusual feature of the early D7 mRNA, which may be related to its continued translation at 4 h, is the presence of a 5′ poly(A) leader made by the VACV RNA polymerase reiteratively copying a TTT sequence in the promoter, which is more typical of RNAs made at intermediate and late times (31). Surprisingly, at 4 and 8 h some ribosome-protected reads were aligned with a segment of DNA containing two previously unannotated short ORFs that are antisense to the early D9 ORF (Fig. 4A).
Abundance and translational efficiencies of mRNAs.
The ability to resolve the ribosome footprints of even closely spaced ORFs with overlapping transcripts allowed us to analyze and quantify the synthesis of viral mRNA and proteins corresponding to all annotated ORFs over time (see Table S1 in the supplemental material). Although a few examples of proteins that are expressed throughout infection due to tandem early and postreplicative promoters were previously found (32, 33), there have been no systematic studies. We carried out cluster analysis using the ribosome-protected reads for the set of early proteins to discern expression patterns. The proteins in cluster 1 continued to increase, and those in cluster 2 remained steady from 2 to 8 h, whereas those in cluster 3 declined (Fig. 3B). Thus, the genes of cluster 3 are likely to have stringent early promoters, whereas some of those in clusters 1 and 2 could have dual early/intermediate or early/late promoters though other factors such as mRNA stability may be involved. Table S1 in the supplemental material includes the cluster designations of the early ORFs.
The 10 most highly translated ORFs at 2, 4, and 8 h are shown in rank order in Table 1. At 2 h, the majority of highly translated mRNAs encoded proteins involved in host interactions, DNA replication, and transcription. The most highly expressed protein was the secreted epidermal growth factor (C11) for which there are two identical copies of the ORF, one at each end of the genome. Interestingly, the role of the third most highly translated protein is unknown though it is conserved in most orthopoxviruses. In fact, no role has yet been established for 11 of the 50 mRNAs most highly translated at 2 h (data not shown). Of the most highly translated mRNAs at 4 h, most are classified as virion-associated, with intermediate or late promoters (Table 1). Both the H5R and N1L genes, which have transcriptional and host interaction functions, have early and late promoters (although at that time intermediate and late were not distinguishable) (34, 35). There were only minor changes in the rankings between 4 and 8 h (Table 1). The H5R gene was the only one ranked among the top 10 at each of the three time points.
TABLE 1.
Highly translated viral mRNAs and their encoded protein function categories at 2, 4, and 8 h post-VACV infection
| Time of expression and VACV WR ORFa | VACV Copenhagen ORF | Temporal expressionb | Functional category | No. of readsc |
|---|---|---|---|---|
| 2 h | ||||
| 009/210 | C11R | E | Host interaction | 21,247,140 |
| 103 | H5R | E/Ld | Transcription | 19,521,388 |
| 18 | E | Unknown | 18,505,965 | |
| 59 | E3L | E | Host interaction | 16,992,412 |
| 72 | I3L | E | DNA replication | 12,105,947 |
| 89 | L2R | E | Virion association | 8,753,364 |
| 29 | N2L | E | Host interaction | 6,987,681 |
| 200 | B19R | E | Host interaction | 6,446,599 |
| 172 | A46R | E | Host interaction | 5,931,269 |
| 41 | F2L | E | DNA replication | 5,731,347 |
| 4 h | ||||
| 70 | I1L | I | Virion association | 31,221,695 |
| 133 | A14L | L | Virion association | 27,674,145 |
| 56 | F17R | L | Virion association | 18,595,167 |
| 131 | A12L | I | Virion association | 17,138,138 |
| 103 | H5R | E/L | Transcription | 15,370,215 |
| 132 | A13L | L | Virion association | 12,605,197 |
| 139 | A19L | I | Virion association | 11,987,935 |
| 137 | A17L | L | Virion association | 11,388,463 |
| 28 | N1L | E/Ld | Host interaction | 11,342,780 |
| 157 | A34R | I | Virion association | 9,677,241 |
| 8 h | ||||
| 133 | A14L | L | Virion association | 32,690,917 |
| 70 | I1L | I | Virion association | 24,134,410 |
| 167 | A42R | I | Virion association | 18,693,389 |
| 56 | F17R | L | Virion association | 17,013,444 |
| 131 | A12L | I | Virion association | 16,613,835 |
| 28 | N1L | E/L | Host interaction | 14,260,755 |
| 103 | H5R | E/L | Transcription | 13,848,524 |
| 132 | A13L | L | Virion association | 12,915,930 |
| 139 | A19L | I | Virion association | 12,450,056 |
| 137 | A17L | L | Virion association | 10,427,057 |
For each time period post-VACV infection, the top 10 most highly translated mRNAs are listed.
The mRNAs are listed in rank order in terms of the normalized number of reads mapped to the ORF.
The L-stage promoter was not distinguished from the intermediate one.
As a measure of relative translational efficiency, we compared the ratio of mRNA reads to ribosome-protected reads that were mapped to each annotated ORF. At 2 h after infection, there was a good correlation (r = 0.92), indicating that synthesis of early proteins is regulated largely by mRNA abundance. Interestingly, half of the ORFs among the 15 with the highest relative translational efficiencies have unknown functions. The 15 ORFs with the lowest relative translational efficiencies were pseudogenes. At 4 and 8 h, calculations of relative translational efficiency were not meaningful because of extensive transcriptional read-through.
Translation initiation sites of annotated ORFs.
Ribosome profiling, while providing significant new information regarding the translation of viral mRNAs, left important questions that could only be answered by accurate determination of translational initiation sites, which were experimentally identified as the AUG or cognate sequence close to the 5′ peaks of harringtonine or lactimidomycin reads, as detailed in Materials and Methods. As noted earlier, treatments with these translational inhibitors can result in the accumulation of ribosomes at or near the translation initiation sites. For early mRNAs at 2 h, we observed accumulation of ribosome-protected reads near the start of most annotated VACV early ORFs, which is illustrated by the A31R ORF (Fig. 5A). Although there was a greater number of ribosome-protected reads near the start of the mRNA with all three drugs, lactimidomycin treatment led to the greatest concentration of reads near the initiation sites, as previously reported with other systems (36, 37). Table S2 in the supplemental material lists the translation initiation sites determined for the annotated ORFs. The same initiation sites were identified with both inhibitors for nearly every early mRNA. The distance from the 5′ nucleotide of the first protected fragment to the start codon was usually 12 to 13 nt with harringtonine but frequently shorter with lactimidomycin (see Table S2). For postreplicative intermediate or late mRNAs analyzed with harringtonine, the 5′ nucleotide of the first protected fragment was most often close to or even within the first AUG codon (Fig. 5B; see also Table S2). However, postreplicative mRNA fragments containing the translation initiation site could not be identified with lactimidomycin. The short size of the fragment isolated with harringtonine and the inability to isolate the translation initiation fragment with lactimidomycin are likely related to the 5′ poly(A) leader that is formed by reiterative copying of a TTT sequence, which is part of the promoter and overlaps the start codon of the ORF of intermediate and late genes (10, 38–42). We suspect that, after lactimidomycin treatment, the 5′ poly(A) leader was not sufficiently protected from RNase I digestion by the ribosome and that the resulting translation initiation fragment was smaller than 28 nt and therefore was not recovered during the gel purification step.
FIG 5.

Identification of VACV translation initiation sites. (A) Ribosome footprints corresponding to A31R mRNA after treatment with cycloheximide, harringtonine, or lactimidomycin at 2 h of infection. The Ribo-harringtonine 5′ end shows only the first nucleotide of the ribosome footprint with the translation initiation site labeled by an asterisk. (B) Distance from the 5′ end of the ribosome footprint to the start codon for early L2R mRNA and late D10R mRNA. Multiple A residues were manually added to the sequence upstream of the D10R ORF to mimic the nontemplated A residues formed by RNA polymerase slippage. The ATG start codons are underlined. (C) Correlation between the number of reads associated with the translation initiation (TI) peak and the ribosome footprint (Ribo) for each individual mRNA indicated by a dot at 2, 4, and 8 h postinfection. (D) Logos of translation initiation sites and their surrounding nucleotides of the 20 early genes with the highest and the 20 early genes with the lowest relative translation efficiencies (RTE).
There was a high correlation of the translation initiation site reads and ribosome footprint reads of individual annotated VACV ORFs at 2, 4, and 8 h postinfection (Fig. 5C), suggesting that translation initiation is the major regulatory step for VACV protein synthesis at both early and postreplicative stages. Analysis of the translation initiation sites and their surrounding nucleotides for 20 early genes with highest and lowest relative translation efficiencies indicates that those with the highest efficiencies adhere more closely to the Kozak sequence, i.e., A/G at −3 and G/A at +4 (Fig. 5D) (43). A similar analysis for intermediate and late genes was not performed because most of the mRNAs have a 5′ poly(A) sequence immediately preceding the start codon.
Although most translation initiation sites were mapped at the predicted initiating methionine, we found some that were at a downstream methionine, thereby shortening the previously predicted protein size by 4 to 38 amino acids (see Table S2 in the supplemental material). The thymidylate kinase (VACWR174; A48R) would be 23 amino acids shorter, consistent with the start of the homologous region of other poxvirus thymidylate kinases. The VACWR001 ORF (C23L) was annotated to encode a 244-amino-acid protein homologous to a chemokine binding protein of other orthopoxviruses. However, ribosome profiling indicated that only a portion of the ORF was translated and that the translation initiation site was 23 nt upstream of the annotated ORF start codon (Fig. 6A; see also Table S2). The ORF from the start codon determined here is predicted to encode a 57-amino-acid peptide, which is supported by experimental observations that the protein is approximately 7.5 kDa (44, 45). Furthermore, the upstream AUG corresponds to the annotated translation initiation sites of most other orthopoxviruses, indicating that VACV WR as well as some other strains of VACV suffered a frameshift mutation preventing expression of the full-length protein. Additional misannotated ORFs are shown in Table S2 in the supplemental material.
FIG 6.

Representative novel translation units. Panels show mRNA reads and ribosome (Ribo)-protected reads aligned along portions of the VACV genome represented as filled arrows. Asterisks indicate the 5′ ends of the harringtonine peaks preceding the translation initiation sites. (A) RNA was obtained after adding cycloheximide or harringtonine at 2 h after infection, and the ribosome-protected reads were mapped along the C23L (VACWR001) ORF. The long filled arrow designated C23L is the annotated ORF. The short filled arrow designated cC23L represents the corrected ORF following the translation initiation site determined in this study (VACWR001). (B) Identification of translation initiation sites for two uORFs (uG2.1R and uG2.2R) upstream of the G2R (VACWR080) ORF. The sequences of the short ORFs are shown. (C) Identification of a translation initiation site for a downstream frameshifting ORF (dORF) in D4R (VACWR109). (D) Identification of translation initiation site for multiple truncated ORFs (tORFs) in E7R (VACWR062). (E) Identification of a translation initiation site for a novel ORF antisense to A5R (VACWR124). (F) Identification of a translation initiation site for an unannotated ORF between C19L (VACWR008) and C11R (VACWR009). The nucleotide sequence of the ORF is shown.
Characterization of nonannotated putative translation initiation sites.
Using the same criteria that allowed us to identify the translation initiation sites of the 162 previously annotated ORFs, we detected an additional 596 initiation sites at AUG or near-cognate start codons (AUU, AUC, AUA, ACG, AGG, AAG, CUG, GUG, or UUG) (see Table S3 in the supplemental material). New translation initiation sites were more frequent at 4 and 8 h than at 2 h (Fig. 7A) and generally were associated with short ORFs (Fig. 7B). However, because the criteria for defining a translation initiation site are somewhat arbitrary and depend on the use of translation inhibitors, we consider these initiation sites as putative until confirmed by other means. In Table 2, we listed a subset of the ORFs that start with AUG and potentially encode proteins of at least 25 amino acids. Those associated with smaller ORFs and cognate start codons might have regulatory roles. The putative translation initiation sites fell into groups associated with the following: (i) upstream ORFs (uORFs) located in the 5′ untranslated region of mRNA (n = 24), (ii) truncated non-frameshifting ORFs (tORFs) (n = 134, including 9 with non-frameshifting upstream translation initiation sites), (iii) downstream frameshifting ORFs (dORFs) (n = 316), and (iv) noncoding-region ORFs (nORFs) (n = 115). Because of the short 5′ untranslated regions of early mRNAs and the presence of the 5′ poly(A) sequence of intermediate and late mRNAs, few uORFs were identified. However, two prominent peaks within the unusually long 320-nt untranslated region of G2R (VACWR080) met the criteria for translation initiation sites and were designated uG2.1R and uG2.2R (Fig. 6B). The majority of the relatively long ORFs associated with the putative translation initiation sites fell into the tORF category (Table 2). Three tORFs within E7R (VACWR63) are shown in Fig. 6D. The dORFs were usually short, as shown in Fig. 6C for one within D4R (VACWR109). There were two varieties of nORFs: antisense (Fig. 6E) and those between annotated ORFs (Fig. 6F). Previously, we showed that there are several shorter isoforms of A25 proteins with N-terminal truncations (10). At least two of these truncated proteins have corresponding translation initiation sites, identified in Table S3 in the supplemental material.
FIG 7.
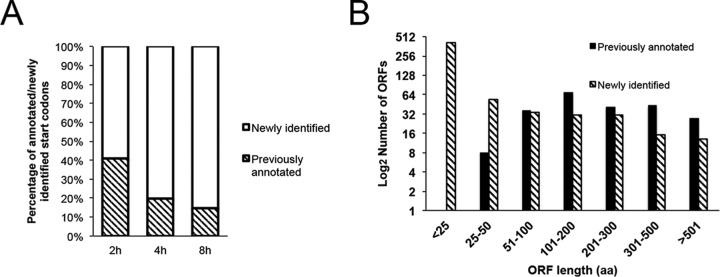
Comparison of the previously annotated and newly identified translation initiation sites. (A) Percentages of annotated and newly identified putative translation initiation sites at 2, 4, and 8 h of infection. (B) Size distribution of ORF lengths in amino acids (aa) associated with previously annotated and newly identified putative translation initiation sites.
TABLE 2.
ORFs associated with newly identified translation initiation sites
| TIS (nt)a | Strand | Read count | ORF length (aa)e | ORF typeb | Associated ORFc | Expression timingd |
|---|---|---|---|---|---|---|
| 4398 | − | 11,854 | 57 | cORF | 001 (C23L) | E |
| 10518 | − | 2,264 | 75 | tORF | 13 | PR |
| 17588 | − | 2,794 | 201 | tORF | 23 (C5L) | PR |
| 19282 | − | 8,367 | 201 | tORF | 25 (C3L) | PR |
| 22821 | − | 1,550 | 170 | tORF | 29 (N2L) | PR |
| 24221 | − | 1,260 | 447 | tORF | 30 (M1L) | PR |
| 27685 | − | 22,052 | 26 | dORF | 35 (K4L) | PR |
| 33845 | − | 1,503 | 295 | tORF | 43 (F4L) | PR |
| 34242 | − | 6,881 | 97 | tORF | 44 (F5L) | PR |
| 35417 | − | 6,975 | 76 | cORF | 46 (F7L) | E |
| 35663 | + | 1,003 | 33 | nORF | PR | |
| 38817 | − | 3,507 | 338 | tORF | 50 (F11L) | PR |
| 42113 | − | 4,315 | 48 | tORF | 53 (F14L) | PR |
| 42315 | − | 2,737 | 25 | tORF | 53.5 (F14.5) | PR |
| 43072 | − | 2,734 | 42 | tORF | 55 (F16L) | PR |
| 48073 | − | 5,450 | 97 | tORF | 59 (E3L) | E |
| 49329 | + | 2,985 | 310 | cORF | 61 (E5R) | E |
| 49398 | + | 3,430 | 287 | tORF | 61 (E5R) | E |
| 52222 | + | 8,181 | 153 | tORF | 63 (E7R) | PR |
| 52243 | + | 3,244 | 146 | tORF | 63 (E7R) | PR |
| 52276 | + | 8,194 | 135 | tORF | 63 (E7R) | PR |
| 59240 | − | 2,578 | 29 | dORF | 68 (O1L) | E |
| 59340 | − | 1,380 | 664 | tORF | 68 (O1L) | PR |
| 64458 | − | 1,503 | 63 | tORF | 74 (I5L) | PR |
| 79355 | + | 1,070 | 179 | tORF | 91 (L4R) | PR |
| 81353 | + | 2,204 | 323 | tORF | 95 (J3R) | PR |
| 83399 | + | 2,686 | 29 | dORF | 98 (J6R) | PR |
| 88545 | − | 3,524 | 73 | tORF | 101 (H3L) | E |
| 91919 | + | 23,869 | 187 | tORF | 103 (H5R) | PR |
| 104029 | + | 2,614 | 209 | tORF | 114 (D9R) | E |
| 110994 | − | 4,437 | 31 | dORF | 120 (A2L) | PR |
| 115895 | − | 1,043 | 32 | tORF | 126 (A7L) | PR |
| 117574 | − | 2,267 | 30 | dORF | 126 (A7L) | PR |
| 118061 | + | 1,894 | 262 | tORF | 127 (A8R) | E |
| 123568 | − | 6,787 | 49 | tORF | 132 (A13L) | PR |
| 126224 | + | 5,584 | 488 | tORF | 138 (A18R) | PR |
| 127877 | − | 25,602 | 68 | tORF | 139 (A19L) | PR |
| 131219 | + | 5,414 | 1156 | cORF | 144 (A24R) | PR |
| 131876 | − | 1,293 | 29 | nORF | PR | |
| 137695 | − | 2,130 | 47 | dORF | 148 (A25L) | PR |
| 142279 | + | 1,115 | 51 | tORF | 154 (A31R) | E |
| 143165 | − | 1,120 | 254 | tORF | 155 (A32L) | PR |
| 147704 | + | 1,833 | 257 | cORF | 163 (A39R) | PR |
| 151371 | + | 2,340 | 74 | tORF | 169 | PR |
| 154612 | − | 1,368 | 231 | tORF | 173 (A47L) | E |
| 154775 | + | 1,124 | 204 | cORF | 174 (A48R) | E |
| 158807 | + | 1,410 | 29 | dORF | 178 (A52R) | E |
| 163219 | + | 8,677 | 37 | dORF | 181.5 | PR |
| 175192 | + | 1,723 | 33 | dORF | 197 (B15R) | PR |
| 179114 | + | 4,622 | 347 | tORF | 200 (B19R) | PR |
| 190314 | + | 12,001 | 57 | cORF | (C23L) | E |
TIS, translation initiation site; position of start codon.
cORF, correction of annotated ORF; dORF, downstream frameshifting ORF; nORF, previous unannotated noncoding region ORF; tORF, downstream truncated ORF or upstream nonframeshifting ORF.
WR strain ORF numbers are given; common ORF names are in parentheses.
E, early stage (2 h); PR, postreplicative state (4 or 8 h).
aa, amino acids.
DISCUSSION
Efforts to systematically analyze VACV gene expression by tiling microarrays (29, 46) and RNA-seq (22) have been frustrated by inefficient transcription termination, which leads to extensive overlapping of transcripts. Moreover, mapping of transcription initiation sites indicated that gene expression is more complex than previously deduced by analysis of large ORFs (9, 10). Ribosome footprinting, which allows mapping of actively translated segments of mRNA, had never been applied to studies of poxvirus gene expression. The recent combination of ribosome footprinting and RNA-Seq (12, 15, 16) led us to adapt this technique for analysis of VACV genome-wide expression.
In the first phase of our analysis, we aligned polyadenylated mRNA and ribosome footprint reads along the VACV genome with respect to the previously annotated ORFs. At early times the mRNA and ribosome footprint profiles generally matched well although the latter defined the ORFs more sharply. There was a good correlation (r = 0.92) between the numbers of mRNA reads to ribosome-protected reads for individual ORFs, indicating that synthesis of early proteins is regulated largely by mRNA abundance. The proteins synthesized most actively at 2 h have roles in host interactions, DNA replication, and mRNA synthesis. At postreplicative times, the mRNA reads nearly always overlapped neighboring ORFs, producing a pattern that was difficult to decipher. In contrast, the ribosome-protected reads clearly defined the starts and ends of ORFs, allowing the determination of relative translation levels. The latter indicated large variations in synthesis of individual proteins. However, the extensive transcriptional read-through prevented analysis of translational efficiencies at postreplicative times. At late times, the most highly translated mRNAs encoded virion-associated proteins, and relatively few had roles in host interactions. Two highly translated mRNAs that were in the top 10 at early and late times had previously been shown to have both early and late RNA start sites indicative of dual promoters (34, 35). Cluster analysis indicated that a subset of early mRNAs continued to increase at late times, suggesting the presence of additional genes with dual promoters, as had been previously reported for intermediate and late postreplicative genes (47, 48).
A further understanding of gene expression came from mapping translation initiation sites. The same sites were identified with the translation inhibitors harringtonine and lactimidomycin for nearly every early gene although the latter inhibitor has been reported to be more stringent (36, 37). For genes expressed prior to viral DNA replication, the distance from the 5′ end of the first ribosome-protected fragment to the translation initiation codon was less with lactimidomycin than with harringtonine. In addition, this distance was less for postreplicative genes than for early genes when harringtonine was used, and translation initiation sites for postreplicative genes could not be identified after lactimidomycin treatment. This anomaly is likely related to the unique 5′ poly(A) sequence, produced by slippage of the viral RNA polymerase on a TTT sequence that forms part of intermediate and late promoters and overlaps the initiation codon of intermediate (41) and late (39, 40) mRNAs. The run of consecutive A residues immediately preceding the AUG start codon may result in weaker RNase protection by ribosomes. Therefore, the translation initiation sites of postreplicative mRNAs were mapped only with harringtonine. Comparison of the experimentally defined translation initiation sites with those predicted from ORF analysis allowed us to correct some discrepancies in which a downstream AUG was actually used.
The initial annotation of the complete VACV genome considered unique ORFs of 65 or more amino acids (49). Subsequently, several smaller ORFs were found to encode conserved proteins with important roles in replication, including a 63-amino-acid subunit of the RNA polymerase (50), 53- and 49-amino-acid proteins that are important for virulence in animal models (51), a 42-amino-acid protein needed for viral membrane formation (52), and a 35-amino-acid protein required for virus entry into cells (53). However, the possibility of additional small ORFs embedded within larger ORFs either in or out of frame existed. Using the same criteria for mapping translation initiation sites at the start of previously annotated ORFs, we found many additional putative translation initiation sites, particularly within larger ORFs. The new translation initiation sites could arise by ribosome scanning past earlier start codons or from shorter mRNAs as we previously reported evidence for pervasive transcription initiation (10). The presence of noncoded A residues preceding many of the novel translation initiation sites was consistent with their origin from novel mRNAs arising by RNA polymerase slippage at a preceding TTT sequence. However, we caution that the criteria used to define initiation sites are somewhat arbitrary and involved the use of translation inhibitors, and therefore further proof that they code for proteins or have regulatory roles must await further experimentation. Because of the close packing of the major ORFs, there were few putative new translation units within intergenic regions. Overall, the read numbers of the novel translation initiation sites were lower than those at the start of larger previously annotated ORFs. Although AUG was the predominant predicted start codon, other near-cognate start codons were found, especially at the postreplicative stage. The genome map shown in Fig. 8 summarizes our ribosome profiling data, including annotated ORFs and previously unannotated ones that start with AUG and have 25 or more amino acid codons.
FIG 8.

VACV genome-wide expression map. The previously annotated ORFs of the WR strain of VACV are color coded as indicated and shown with arrows pointing in the direction of transcription. The annotated ORFs shown to be translated by ribosome profiling are indicated in gray. ORFs that start with AUG and are 25 amino acids or longer that were associated with new putative translation initiation sites are indicated in black. In many cases the additional new translation site is close to the previously annotated one so that the gray and black arrows appear similar in length.
Although questions remain regarding the synthesis and stability of peptides potentially encoded by the novel translation units and their possible structural or regulatory roles, large numbers of short ORFs and alternative start codons were found by ribosome profiling carried out with cytomegalovirus (12), Kaposi's sarcoma-associated herpesvirus (54), and bacteriophage lambda (55) as well as with uninfected eukaryotic cells (15, 16, 36). Pervasive gene expression can increase the coding capacity and expand the functional repertoire of genomes by generating additional RNAs and proteins. Furthermore, poxviruses acquired cellular genes during their evolution, and pervasive transcription and translation may help explain how the novel ORFs, which may have been randomly inserted into the poxvirus genome, were expressed initially. Presumably, natural selection would then result in enhancing promoter and translation initiation sites that allow optimal expression.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dan Bruno for assistance with library purification and next-generation sequencing.
This project was supported in part by grants from the National Institute of Allergy and Infectious Diseases (R00AI100919) and National Institute of General Medical Sciences (a subaward of P20 GM103418) to Z.Y., by a National Cancer Institute grant (CA106150) to B.S., and by intramural research funds from the National Institute of Allergy and Infectious Diseases to B.M.
The contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00528-15.
REFERENCES
- 1.Moss B. 2013. Poxviridae, p 2129–2159. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Rancaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Damon I. 2013. Poxviruses, p 2160–2184. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Rancaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Wei CM, Moss B. 1975. Methylated nucleotides block 5′-terminus of vaccinia virus mRNA. Proc Natl Acad Sci U S A 72:318–322. doi: 10.1073/pnas.72.1.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin SA, Paoletti E, Moss B. 1975. Purification of mRNA guanylyltransferase and mRNA (guanine 7-)methyltransferase from vaccinia virus. J Biol Chem 250:9322–9329. [PubMed] [Google Scholar]
- 5.Kates J, Beeson J. 1970. Ribonucleic acid synthesis in vaccinia virus. II. Synthesis of polyriboadenylic acid. J Mol Biol 50:19–23. [DOI] [PubMed] [Google Scholar]
- 6.Moss B, Rosenblum EN, Paoletti E. 1973. Polyadenylate polymerase from vaccinia virions. Nat New Biol 245:59–63. doi: 10.1038/newbio245059a0. [DOI] [PubMed] [Google Scholar]
- 7.Smith GL, Benfield CT, Maluquer de Motes C, Mazzon M, Ember SW, Ferguson BJ, Sumner RP. 2013. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J Gen Virol 94:2367–2392. doi: 10.1099/vir.0.055921-0. [DOI] [PubMed] [Google Scholar]
- 8.Haller SL, Peng C, McFadden G, Rothenburg S. 2014. Poxviruses and the evolution of host range and virulence. Infect Genet Evol 21:15–40. doi: 10.1016/j.meegid.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Z, Bruno DP, Martens CA, Porcella SF, Moss B. 2011. Genome-wide analysis of the 5′ and 3′ ends of vaccinia virus early mRNAs delineates regulatory sequences of annotated and anomalous transcripts. J Virol 85:5897–5909. doi: 10.1128/JVI.00428-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Z, Martens CA, Bruno DP, Porcella SF, Moss B. 2012. Pervasive initiation and 3′ end formation of poxvirus post-replicative RNAs. J Biol Chem 287:31050–31060. doi: 10.1074/jbc.M112.390054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arvey A, Tempera I, Tsai K, Chen HS, Tikhmyanova N, Klichinsky M, Leslie C, Lieberman PM. 2012. An atlas of the Epstein-Barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell Host Microbe 12:233–245. doi: 10.1016/j.chom.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stern-Ginossar N, Weisburd B, Michalski A, Le VT, Hein MY, Huang SX, Ma M, Shen B, Qian SB, Hengel H, Mann M, Ingolia NT, Weissman JS. 2012. Decoding human cytomegalovirus. Science 338:1088–1093. doi: 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson LS, Willert EK, Virgin HW. 2010. Redefining the genetics of murine gammaherpesvirus 68 via transcriptome-based annotation. Cell Host Microbe 7:516–526. doi: 10.1016/j.chom.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hangauer MJ, Vaughn IW, McManus MT. 2013. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet 9:e1003569. doi: 10.1371/journal.pgen.1003569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ingolia NT, Lareau LF, Weissman JS. 2011. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Earl PL, Cooper N, Wyatt LS, Moss B, Carroll MW. 2001. Preparation of cell cultures and vaccinia virus stocks. Curr Protoc Mol Biol Chapter 16:Unit 16.16. doi: 10.1002/0471142727.mb1616s43. [DOI] [PubMed] [Google Scholar]
- 18.Seo JW, Ma M, Kwong T, Ju J, Lim SK, Jiang H, Lohman JR, Yang C, Cleveland J, Zazopoulos E, Farnet CM, Shen B. 2014. Comparative characterization of the lactimidomycin and iso-migrastatin biosynthetic machineries revealing unusual features for acyltransferase-less type I polyketide synthases and providing an opportunity to engineer new analogues. Biochemistry 53:7854–7865. doi: 10.1021/bi501396v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Homann OR, Johnson AD. 2010. MochiView: versatile software for genome browsing and DNA motif analysis. BMC Biol 8:49. doi: 10.1186/1741-7007-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ingolia NT. 2014. Ribosome profiling: new views of translation, from single codons to genome scale. Nat Rev Genet 15:205–213. doi: 10.1038/nrg3645. [DOI] [PubMed] [Google Scholar]
- 22.Yang Z, Bruno DP, Martens CA, Porcella SF, Moss B. 2010. Simultaneous high-resolution analysis of vaccinia virus and host cell transcriptomes by deep RNA sequencing. Proc Natl Acad Sci U S A 107:11513–11518. doi: 10.1073/pnas.1006594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Obrig TG, Culp WJ, McKeehan WL, Hardesty B. 1971. The mechanism by which cycloheximide and related glutarimide antibiotics inhibit peptide synthesis on reticulocyte ribosomes. J Biol Chem 246:174–181. [PubMed] [Google Scholar]
- 24.Fresno M, Jimenez A, Vazquez D. 1977. Inhibition of translation in eukaryotic systems by harringtonine. Eur J Biochem 72:323–330. doi: 10.1111/j.1432-1033.1977.tb11256.x. [DOI] [PubMed] [Google Scholar]
- 25.Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, Liu JO. 2010. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol 6:209–217. doi: 10.1038/nchembio.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boone RF, Moss B. 1978. Sequence complexity and relative abundance of vaccinia virus mRNA's synthesized in vivo and in vitro. J Virol 26:554–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niles EG, Condit RC, Caro P, Davidson K, Matusick L, Seto J. 1986. Nucleotide sequence and genetic map of the 16-kb vaccinia virus HindIII D fragment. Virology 153:96–112. doi: 10.1016/0042-6822(86)90011-5. [DOI] [PubMed] [Google Scholar]
- 28.Lee-Chen GJ, Bourgeois N, Davidson K, Condit RC, Niles EG. 1988. Structure of the transcription initiation and termination sequences of seven early genes in the vaccinia virus HindIII D fragment. Virology 163:64–79. doi: 10.1016/0042-6822(88)90234-6. [DOI] [PubMed] [Google Scholar]
- 29.Assarsson E, Greenbaum JA, Sundstrom M, Schaffer L, Hammond JA, Pasquetto V, Oseroff C, Hendrickson RC, Lefkowitz EJ, Tscharke DC, Sidney J, Grey HM, Head SR, Peters B, Sette A. 2008. Kinetic analysis of a complete poxvirus transcriptome reveals an immediate-early class of genes. Proc Natl Acad Sci U S A 105:2140–2145. doi: 10.1073/pnas.0711573105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee-Chen GJ, Niles EG. 1988. Transcription and translation mapping of the 13 genes in the vaccinia virus HindIII D fragment. Virology 163:52–63. doi: 10.1016/0042-6822(88)90233-4. [DOI] [PubMed] [Google Scholar]
- 31.Ahn B-Y, Jones EV, Moss B. 1990. Identification of the vaccinia virus gene encoding an 18-kilodalton subunit of RNA polymerase and demonstration of a 5′ poly(A) leader on its early transcript. J Virol 64:3019–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cochran MA, Puckett C, Moss B. 1985. In vitro mutagenesis of the promoter region for a vaccinia virus gene: evidence for tandem early and late regulatory signals. J Virol 54:30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kovacs GR, Moss B. 1996. The vaccinia virus H5R gene encodes late gene transcription factor 4: purification, cloning and overexpression. J Virol 70:6796–6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosel JL, Earl PL, Weir JP, Moss B. 1986. Conserved TAAATG sequence at the transcriptional and translational initiation sites of vaccinia virus late genes deduced by structural and functional analysis of the HindIII H genome fragment. J Virol 60:436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kotwal GJ, Hugin AW, Moss B. 1989. Mapping and insertional mutagenesis of a vaccinia virus gene encoding a 13,800-Da secreted protein. Virology 171:579–587. doi: 10.1016/0042-6822(89)90627-2. [DOI] [PubMed] [Google Scholar]
- 36.Lee S, Liu B, Lee S, Huang SX, Shen B, Qian SB. 2012. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc Natl Acad Sci U S A 109:E2424–E2432. doi: 10.1073/pnas.1207846109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao X, Wan J, Liu B, Ma M, Shen B, Qian SB. 2015. Quantitative profiling of initiating ribosomes in vivo. Nat Methods 12:147–153. doi: 10.1038/nmeth.3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bertholet C, Van Meir E, ten Heggeler-Bordier B, Wittek R. 1987. Vaccinia virus produces late mRNAs by discontinuous synthesis. Cell 50:153–162. doi: 10.1016/0092-8674(87)90211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwer B, Visca P, Vos JC, Stunnenberg HG. 1987. Discontinuous transcription or RNA processing of vaccinia virus late messengers results in a 5′ poly(A) leader. Cell 50:163–169. doi: 10.1016/0092-8674(87)90212-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patel DD, Pickup DJ. 1987. Messenger RNAs of a strongly expressed late gene of cowpox virus contains a 5′-terminal poly(A) leader. EMBO J 6:3787–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ahn B-Y, Moss B. 1989. Capped poly(A) leader of variable lengths at the 5′ ends of vaccinia virus late mRNAs. J Virol 63:226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baldick CJ Jr, Moss B. 1993. Characterization and temporal regulation of mRNAs encoded by vaccinia virus intermediate stage genes. J Virol 67:3515–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kozak M. 1992. Regulation of translation in eukaryotic systems. Annu Rev Cell Biol 8:197–225. doi: 10.1146/annurev.cb.08.110192.001213. [DOI] [PubMed] [Google Scholar]
- 44.Venkatesan S, Gershowitz A, Moss B. 1982. Complete nucleotide sequences of two adjacent early vaccinia virus genes located within the inverted terminal repetition. J Virol 44:637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patel AH, Gaffney DF, Subak-Sharpe JH, Stow ND. 1990. DNA sequence of the gene encoding a major secreted protein of vaccinia virus, strain Lister. J Gen Virol 71:2013–2021. doi: 10.1099/0022-1317-71-9-2013. [DOI] [PubMed] [Google Scholar]
- 46.Rubins KH, Hensley LE, Bell GW, Wang C, Lefkowitz EJ, Brown PO, Relman DA. 2008. Comparative analysis of viral gene expression programs during poxvirus infection: a transcriptional map of the vaccinia and monkeypox genomes. PLoS One 3:e2628. doi: 10.1371/journal.pone.0002628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang Z, Maruri-Avidal L, Sisler J, Stuart C, Moss B. 2013. Cascade regulation of vaccinia virus gene expression is modulated by multistage promoters. Virology 447:213–220. doi: 10.1016/j.virol.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Z, Reynolds SE, Martens CA, Bruno DP, Porcella SF, Moss B. 2011. Expression profiling of the intermediate and late stages of poxvirus replication. J Virol 85:9899–9908. doi: 10.1128/JVI.05446-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goebel SJ, Johnson GP, Perkus ME, Davis SW, Winslow JP, Paoletti E. 1990. The complete DNA sequence of vaccinia virus. Virology 179:247–266; 517–563. doi: 10.1016/0042-6822(90)90294-2. [DOI] [PubMed] [Google Scholar]
- 50.Amegadzie BY, Ahn BY, Moss B. 1992. Characterization of a 7-kilodalton subunit of vaccinia virus DNA-dependent RNA polymerase with structural similarities to the smallest subunit of eukaryotic RNA polymerase II. J Virol 66:3003–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Izmailyan R, Chang W. 2008. Vaccinia virus WR53.5/F14.5 protein is a new component of intracellular mature virus and is important for calcium-independent cell adhesion and vaccinia virus virulence in mice. J Virol 82:10079–10087. doi: 10.1128/JVI.00816-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maruri-Avidal L, Weisberg AS, Moss B. 2013. Direct formation of vaccinia viral membranes from the endoplasmic reticulum in the absence of the newly characterized L2-interacting A30.5 protein. J Virol 87:12313–12326. doi: 10.1128/JVI.02137-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Satheshkumar PS, Moss B. 2009. Characterization of a newly identified 35 amino acid component of the vaccinia virus entry/fusion complex conserved in all chordopoxviruses. J Virol 83:12822–12832. doi: 10.1128/JVI.01744-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arias C, Weisburd B, Stern-Ginossar N, Mercier A, Madrid AS, Bellare P, Holdorf M, Weissman JS, Ganem D. 2014. KSHV 2.0: a comprehensive annotation of the Kaposi's sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog 10:e1003847. doi: 10.1371/journal.ppat.1003847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu X, Jiang H, Gu Z, Roberts JW. 2013. High-resolution view of bacteriophage lambda gene expression by ribosome profiling. Proc Natl Acad Sci U S A 110:11928–11933. doi: 10.1073/pnas.1309739110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.