

ABSTRACT
Kaposi's sarcoma-associated herpesvirus (KSHV) encodes multiple viral proteins that activate extracellular signal-regulated kinase (ERK)–mitogen-activated protein kinase (MAPK) cascades. One of these viral proteins, ORF45, mediates sustained ERK-p90 ribosomal S6 kinase (RSK) activation during KSHV lytic replication and facilitates viral translation through the phosphorylation of a eukaryotic translation initiation factor, eIF4B. The importance of ERK-RSK activation for KSHV viral transcription has been shown; however, which transcription factor senses the sustained MAPK signaling and leads to viral transcription remains poorly understood. Here we show that the presence of ORF45 leads to the prolonged accumulation of c-Fos during the late stage of KSHV lytic replication through ERK-RSK-dependent phosphorylation and stabilization and that the depletion of c-Fos disrupts viral lytic transcription. Genome-wide screening revealed that c-Fos directly binds to multiple viral gene promoters and enhances viral transcription. Mutation of the ERK-RSK phosphorylation sites of c-Fos restrains KSHV lytic gene expression and virion production. These results indicate that the prolonged accumulation of c-Fos promotes the progression of viral transcription from early to late stages and accelerates viral lytic replication upon sustained ORF45-ERK-RSK activation during the KSHV lytic life cycle.
IMPORTANCE During KSHV lytic replication, transient activation and sustained activation of ERK-RSK induce viral immediate early (IE) transcription and late transcription, respectively. Studies have revealed that ERK-RSK activates several transcription factors involved in IE gene expression, including Ets, AP-1, CREB, and C/EBP, which lead to the transient ERK-RSK activation-dependent IE transcription. Whereas c-Fos acts as a sensor of sustained ERK-RSK activation, ORF45-ERK-RSK signaling mediates c-Fos phosphorylation and accumulation during late KSHV lytic replication, consequently promoting viral transcription through the direct binding of c-Fos to multiple KSHV promoters. This finding indicates that c-Fos mediates distinct viral transcriptional progression following sustained ERK-RSK signaling during the KSHV lytic life cycle.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) naturally infects humans, and over 95% of healthy persons have no symptoms. KSHV causes three types of malignancies in immunosuppressed patients: Kaposi's sarcoma, body cavity-based lymphoma, and multicentric Castleman's disease (1–3). KSHV establishes a latent infection in most infected cells, whereas a small proportion of cells develop lytic infection. The genetic profiles of KSHV-infected populations differ from those of uninfected populations, with host cell transcriptional remodeling observed in latently KSHV-infected cells (4) and global mRNA shutoff noted during lytic replication (5, 6).
A limited number of viral transcripts appear in the latent stage, whereas the viral genome produces all of the viral transcripts during the lytic phase (7). A key viral replication and transcription activator (RTA) switches latent infection to lytic replication (8). Studies have characterized multiple cellular signaling pathways and transcription factors required for RTA expression. Mitogen-activated stimuli or stresses initiate RTA expression through mitogen-activated protein kinase (MAPK) or stress-activated protein kinases (SAPK), respectively (9–14). Thereafter, RTA activates viral lytic transcription and replication. RTA binding sites in the KSHV genome and responsive KSHV promoters have been characterized (15, 16); these studies revealed that most viral gene expression is not directly activated by RTA and requires additional cellular transcription factors. Based on the analysis of elements in the RTA, ORF45, and K8 promoters, multiple transcription factors, including c-Fos, c-Jun, Sp1, CREB, C/EBP, c-Myc, and ATF-2, are required for the expression of immediate early (IE) genes (11–13, 17–19); most of these transcription factors are the direct or indirect targets of MAPK pathways (20, 21). Recently, nonconventional viral DNA elements, viral noncoding RNA, and viral proteins required for KSHV late transcription have been characterized (22–26). The viral lytic proteins ORF24, ORF31, and ORF34 assemble into a transcriptional activator complex (25), and ORF24 recruits RNA polymerase II to viral late promoters by replacing TATA box binding protein (TBP) (26) to mediate activation of KSHV late gene expression. Studies have also discovered that ORF24 homologs of the beta-gammaherpesvirus subfamily (Epstein-Barr virus [EBV], murine gammaherpesvirus 68 [MHV68], and cytomegalovirus [CMV]) function as TBP-like protein and are required for viral late gene expression (22, 27, 28). These findings indicate that viral proteins can serve as components of the viral transcription preinitiation complex during late stages of viral gene expression. However, the host transcription factors that are required for KSHV late lytic gene expression remain largely unknown.
Extracellular signal-regulated kinase (ERK)–MAPK is activated during KSHV infection with biphasic kinetics: transient activation is induced by viral glycoproteins independent of viral gene expression during primary infection (12, 29), whereas sustained activation is mediated by viral gene products such as vGPCR and ORF45 at the late stage during reactivation by mitogen-like stimuli, histone deacetylase (HDAC) inhibitors, or inducible RTA (30–32). The different kinetics of ERK-MAPK activation result in distinct outcomes depending on the threshold strength and duration (33, 34). Transient MAPK activation contributes to the rapid response to biological stimuli, such as mitogens and epidermal growth factor (EGF), to activate the transcription factors Ets, STAT, CREB, and other nuclear targets required to induce the expression of immediate early genes (IEGs). When MAPK signaling remains active for a long time, the IEG products are stabilized and alter the transcriptional program. During KSHV lytic replication in either primary infection or reactivation, transient ERK activation induced by viral particles, 12-O-tetradecanoylphorbol-13-acetate (TPA), or another stimulus activates IE expression and initiates lytic replication (11, 35–37); however, ORF45-null KSHV and an ORF45-F66A point mutant (which are deficient in sustained ERK-p90 ribosomal S6 kinase [RSK] activation) exhibit reduced lytic gene expression and progeny virion production (31, 38, 39), suggesting that sustained ERK-RSK activation is essential for late viral transcription. In fact, a recent study showed that ORF45 directly increases the recruitment of RNA polymerase II and enhances transcription through RSK2 (40), indicating that ORF45-RSK2 promotes late transcription.
Transient ERK activation does not result in c-Fos accumulation, whereas c-Fos is phosphorylated and stabilized by sustained ERK-RSK activation and acts as a sensor of sustained MAPK activation (41). The phosphorylation of c-Fos at Ser362 and Ser374 by sustained ERK-RSK activation recruits ERK to c-Fos for additional phosphorylation at Thr325 and Thr331 and primes AP-1-dependent transcription. c-Fos accumulation is observed during immediate early TPA-induced KSHV reactivation, is decreased 24 h after TPA treatment (14), and activates RTA expression (13). This c-Fos accumulation is due to TPA-induced transient ERK-MAPK activation, but not ORF45-mediated sustained ERK activation, during late KSHV replication (31). The accumulation and function of c-Fos during the KSHV late lytic life cycle remain poorly understood. In the present study, we reveal that prolonged c-Fos accumulation is induced by ORF45-mediated sustained ERK-RSK activation during late KSHV lytic replication. As a consequence, c-Fos accelerates viral transcription through multiple direct binding events at KSHV promoters.
MATERIALS AND METHODS
Chemicals and antibodies.
12-O-Tetradecanoylphorbol-13-acetate (TPA), sodium butyrate (NaB), doxycycline, and cycloheximide (CHX) were purchased from Sigma-Aldrich (St. Louis, MO). Rabbit antibodies detecting the phosphorylated forms of ERK1/2 (Thr202/Tyr204) and c-Fos (Ser32), anti-c-Jun, anti-ERK1/2, anti-c-Fos, anti-Flag, and antihemagglutinin (anti-HA) antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA). Phospho-c-Fos Ser362 and Thr232 polyclonal antibodies were purchased from ImmunoWay Biotechnology Company (Newark, DE) and Santa Cruz Biotechnology, Inc. (Dallas, TX), respectively. The rat anti-LANA antibody was purchased from Advanced Biotechnologies, Inc. (Columbia, MD). Mouse anti-RTA, anti-ORF45, anti-ORF64, and anti-ORF65 antibodies have been described previously (31, 38).
Cells.
HEK293 and 293T cells were cultured at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics. BCBL1 cells were cultured in RPMI 1640 medium with 10% FBS and antibiotics. Bac16-harboring 293T cells were freshly established as described previously (38, 42). Doxycycline-inducible rKSHV.219-positive iSLK.219 cells and wild-type Bac16 and ORF45-null Stop45-harboring iSLK+ cells were maintained in DMEM supplemented with 10% FBS and antibiotics after selection with G418, puromycin, or hygromycin for 2 weeks (39, 43).
Plasmids.
ORF45 and wild-type and kinase-dead RSK2- and ERK2-expressing plasmids were described previously (32). Wild-type c-Fos was amplified by high-fidelity PCR from BCBL1 cDNA and was inserted into the pKH3 vector (FosWT). The double mutation of c-Fos serine 362 and serine 374 to alanine residues (FosDA) and the constitutive activation of c-Fos by the conversion of threonine 325 and threonine 331 to aspartic acid residues (FosTD) were carried out using QuikChange mutagenesis (Stratagene, La Jolla, CA). ORF45-luc and ORF50-luc promoter reporters contain 1 kb of ORF45 and ORF50 promoter sequences in the pGL3 vector (Promega, Madison, WI).
shRNAs.
Stable c-Fos-transduced cells were established with the pLKO.1-puro lentiviral knockout system using the standard procedure. The sequences of the two c-Fos short hairpin RNAs (shRNAs) were GCGGAGACAGACCAACTAGAA and TCTGCTTTGCAGACCGAGATT. The nonspecific shRNA control was the Mission pLKO.1-puro nontarget shRNA control (Sigma-Aldrich, St. Louis, MO).
Chromatin immunoprecipitation (ChIP).
The cells were fixed with 1% formaldehyde for 10 min at room temperature and quenched with 125 mM glycine. After the cells were collected and washed twice with cold phosphate-buffered saline (PBS), 1 × 107 cells were suspended in 1 ml of cold lysis buffer (150 mM NaCl, 50 mM Tris-HCl [pH 7.5], 5 mM EDTA, 0.5% NP-40, 1% Triton X-100, and 1× protease inhibitor cocktail). After eight rounds of sonication (10 s each), the cells were centrifuged at 12,000 × g at 4°C for 10 min and precleaned with control agarose beads, and then the precleaned cell lysates were incubated with a specific antibody overnight at 4°C. Fifty microliters of pretreated protein G beads (Life Technologies, CA) was added, and samples were incubated at 4°C for an additional 2 h and then washed five times and eluted with 150 μl of Tris-EDTA buffer containing 1% SDS. Cross-linked complexes were digested with proteinase K at 56°C for 2 h and then reversed at 65°C overnight. DNA was then extracted and analyzed using real-time PCR.
Real-time PCR.
Total RNA was extracted using the TRIzol reagent (Life Technologies, CA) and was reverse transcribed with Moloney murine leukemia virus (M-MLV) reverse transcriptase (Roche). Genomic DNA was extracted using a standard procedure. cDNAs and DNAs were evaluated by real-time PCR using a SYBR green real-time PCR master mix kit (Roche). The primers used for the real-time PCR array for 92 KSHV transcripts were described previously (44). The primers used for the real-time PCR array for 87 KSHV promoters in the ChIP assay were described in previous studies (15, 16) or designed using Primer3Plus (45), and the sequences of the primer pairs are available upon request.
Detection of virions.
The supernatants from Bac16-harboring 293T cells were collected and centrifuged at 12,000 × g at 4°C for 10 min. Two hundred microliters of supernatants was digested with 100 U DNase I for 20 min at 37°C. Following this, 20 μg/ml proteinase K and 1% SDS were added, and samples were incubated at 56°C for an additional 30 min. Viral DNA was extracted with a standard procedure and analyzed using real-time PCR.
RESULTS
Prolonged c-Fos accumulation during late KSHV lytic replication.
To detect the timing and activation of c-Fos during the KSHV lytic life cycle, the levels of c-Fos and c-Jun were measured during whole lytic replication, which was activated by sodium butyrate or doxycycline-inducible RTA (TPA was not used because it directly induces transient c-Fos accumulation). Following KSHV reactivation in Bac16-harboring 293T cells induced by the HDAC inhibitor sodium butyrate (NaB), a high level of c-Fos accumulated throughout the late lytic life cycle, peaking at 72 h after the maximal level of ORF45 expression and the appearance of ORF64 expression at 48 h, whereas only a low level of c-Fos was induced in KSHV-free 293T cells under the same treatment. The c-Jun levels were only minimally altered in both cell lines under similar conditions (Fig. 1A). Furthermore, c-Fos accumulated only during the late stage of the lytic cycle when KSHV lytic replication was initiated by doxycycline-inducible RTA in iSLK.219 cells, with no c-Fos accumulation occurring in either KSHV-negative SLK cells under doxycycline and sodium butyrate treatment or cells with latent KSHV infection or at the immediate early stage of KSHV reactivation (Fig. 1B). c-Fos accumulation appeared at a stage after RTA and ORF45 expression while preceding the expression of ORF64. c-Jun expression remained constant in all of these cells. These results suggest that c-Fos accumulates substantially during the late KSHV lytic cycle.
FIG 1.
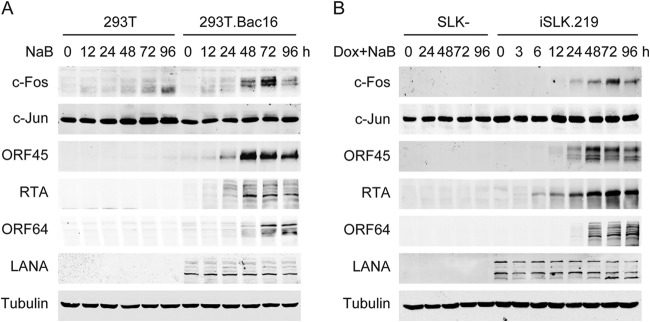
Prolonged c-Fos accumulation occurs during late KSHV lytic replication. (A) 293T and stable KSHV Bac16-harboring 293T cells were induced with 3 mM sodium butyrate (NaB). (B) KSHV-negative SLK- and RTA-inducible rKSHV.219-positive iSLK.219 cells were induced with 1 μg/ml doxycycline (Dox) and 1 mM NaB. At the time points indicated, the cells were collected, lysed, and analyzed by Western blotting.
ORF45 mediates c-Fos phosphorylation and accumulation.
Studies have shown that ORF45 causes sustained ERK-RSK activation during KSHV lytic replication (31, 32) and that c-Fos acts as a sensor of sustained ERK activation through phosphorylation and stabilization (41). We thus presumed that ORF45-mediated ERK-RSK activation contributes to c-Fos accumulation during the late stage of KSHV lytic replication. c-Fos accumulation was increased in cells that ectopically expressed ORF45 or ERK2-RSK2 (Fig. 2A, lanes 4 and 6), and expressing all three of these resulted in a more significant elevation of c-Fos accumulation as well as a shift in motility (Fig. 2A, lane 8). In contrast, expression of the kinase-dead mutant of ERK2 plus RSK2 or c-Fos containing mutations of Ser362 and Ser374 (the phosphorylation sites of RSK and ERK, respectively) abolished c-Fos accumulation and its motility shift (Fig. 2A, compare lanes 8 and 10 and lanes 8 and 9, respectively).
FIG 2.

ORF45 mediates c-Fos accumulation and phosphorylation. (A) Small amounts (10 ng/well) of wild-type (W) and double-mutated (M) c-Fos expression plasmids were cotransfected into 293T cells in 12-well plates with 100 ng/each of ORF45 and wild-type and kinase-dead (D) RSK2 and ERK constructs. Thirty-six hours later, the cells were extracted, and c-Fos accumulation was analyzed as indicated. (B) Large amounts (500 ng/well) of wild-type c-Fos construct were cotransfected as described above, and c-Fos phosphorylation was analyzed as indicated. (C) Wild-type Bac16 and ORF45-null STOP45-harboring iSLK+ cells were left uninduced or were induced with 1 μg/ml Dox and 1 mM NaB for 48 h and analyzed as indicated. (D) Whole-cell extracts were obtained from wild-type Bac16-harboring iSLK+ cells induced with 1 μg/ml Dox and 1 mM NaB, and the phosphorylation of c-Fos was detected by Western blotting. (E) Total RNA was extracted from wild-type Bac16- and ORF45-null STOP45-harboring iSLK+ cells that were untreated or treated with 1 μg/ml Dox and 1 mM NaB for 48 h, and c-Fos mRNA was detected using reverse transcription–real-time PCR.
Furthermore, c-Fos phosphorylation at Ser362, the phosphorylation site of RSK, was increased by ORF45 and RSK2 overexpression. The combination of ORF45, RSK2, and ERK2 augmented c-Fos phosphorylation at this site, and kinase-dead mutations of RSK2 and ERK2 significantly reduced it (Fig. 2B, top). The phosphorylated forms (marked by its motility shift) and rates of phosphorylation at both Ser32 and Thr232 were consistently dramatically elevated in the presence of ORF45 alone or with triple ORF45-ERK2-RSK2 overexpression, and kinase-dead ERK2 plus RSK2 similarly ablated c-Fos phosphorylation at these sites (Fig. 2B, middle).
To determine whether ORF45 mediates c-Fos accumulation during KSHV lytic replication, iSLK+ cells harboring wild-type Bac16 and ORF45-null STOP45 were left uninduced or were induced with 1 μg/ml doxycycline and 1 mM sodium butyrate. A high level of c-Fos accumulation was observed in Bac16-harboring cells under lytic induction. In contrast, a much lower level of c-Fos was observed in lytic STOP45-harboring cells, whereas the levels of RTA and pERK1 (p44) were similar but the pERK2 level was noticeably lower, as expected because ORF45 is known to affect ERK2 specifically (31) (Fig. 2C, compare lanes 2 and 4). Furthermore, the phosphorylation of c-Fos was detected in Bac16-harboring cells during lytic replication but not in uninduced cells (Fig. 2D). In addition, the mRNA level of c-Fos was only moderately increased during lytic replication but exhibited a negligible difference between Bac16- and STOP45-harboring cells (Fig. 2E). These results indicate that ORF45 induces c-Fos accumulation and phosphorylation through ERK and RSK activation.
To further investigate the stabilization of c-Fos during the KSHV life cycle, Bac16-harboring 293T cells were induced by TPA and sodium butyrate and followed by MG132 treatment. c-Fos was undetectable in latent cells despite MG132 treatment, whereas its levels were significantly increased in lytic cells and further augmented in the presence of MG132. However, the level of c-Fos was dramatically reduced by treatment with cycloheximide (CHX), an inhibitor of protein synthesis (Fig. 3A). Similarly, the accumulation of c-Fos was augmented in the presence of MG132 in BCBL1 cells that were induced by either TPA or sodium butyrate as well as in mock-treated cells (Fig. 3B). Furthermore, the half-life of c-Fos was determined following incubation with CHX for various lengths of time in both 293T and KSHV-infected 293T cells after sodium butyrate treatment for 3 days. The half-life of c-Fos was increased to more than 6 h in Bac16-harboring 293T cells undergoing KSHV late lytic replication, whereas in uninfected 293T cells, the half-life was approximately 1 h. This increase suggests that c-Fos stability was greatly enhanced during KSHV late lytic replication (Fig. 3C). Overall, these results suggest that c-Fos accumulates during KSHV lytic replication through posttranslational stabilization.
FIG 3.

c-Fos stability is enhanced during KSHV lytic replication. (A) Bac16-harboring 293T cells were induced with 20 ng/ml TPA and 0.3 mM NaB for 48 h, followed by an additional 4 h of incubation with either 20 μg/ml MG132 or 20 μg/ml cycloheximide (CHX). (B) BCBL1 cells were induced with either 20 ng/ml TPA or 1 mM NaB for 48 h, and then 20 μg/ml MG132 was added, followed by an additional 4 h of incubation. The cells were collected and lysed, and c-Fos and c-Jun were analyzed as indicated. (C) Both 293T cells and Bac16-harboring 293T cells were treated with 3 mM NaB for 3 days, after which the half-life of c-Fos was measured under each condition, following the addition of 40 μg/ml CHX and an additional incubation for the indicated length of time. Whole-cell extracts were analyzed with anti-c-Fos and anti-tubulin antibodies, and quantitation of c-Fos band intensity was analyzed using NIH ImageJ software and is shown as the mean values from duplicate experiments.
c-Fos is required for KSHV late lytic transcription.
To evaluate the role of c-Fos in KSHV lytic replication, stable c-Fos shRNAs were introduced into BCBL1 cells, resulting in greatly depleted c-Fos mRNA (Fig. 4A). The stable cells were treated with TPA or sodium butyrate. The expression of viral genes was then analyzed using real-time PCR (Fig. 4B to D) and Western blotting (Fig. 4E). The expression of the ORF45, RTA, and ORF64 lytic genes was significantly inhibited at the late stage in c-Fos depleted cells, whereas the expression of the LANA latent gene was unchanged. To further confirm the role of c-Fos in lytic viral expression after RTA expression, transient c-Fos depletion by shRNAs was performed in iSLK.219 cells, and lytic replication was initiated by doxycycline-inducible RTA under different time courses. The expression of IE genes (ORF45 and RTA) was barely affected throughout the lytic replication, while the expression of lytic genes (ORF64 and ORF65) was dramatically decreased at the late stage, with unchanged LANA expression (Fig. 4F). These data suggest that c-Fos is required for viral lytic gene expression.
FIG 4.

c-Fos shRNAs decrease late KSHV lytic gene expression. (A to E) Stable c-Fos shRNAs were transduced into BCBL1 cells, and the cells were selected with 2 μg/ml puromycin for 2 weeks. After c-Fos mRNA expression was validated by real-time PCR (A), the cells were induced with 20 ng/ml TPA or 1 mM NaB. At different time points, the cells were collected and total RNAs were extracted, reverse transcribed, and subjected to real-time PCR for ORF45 (B), ORF64 (C), and ORF50 (D) expression. Whole-cell extracts were subjected to Western blotting as indicated (E). (F) Transient c-Fos shRNAs were transduced into iSLK.219 cells with double retrovirus infection, and then the cells were induced with 1 μg/ml doxycycline and 1 mM NaB. The cells were collected at different time points, lysed, and analyzed by Western blotting as indicated. NS, nonspecific shRNA control.
Genome-wide screening of c-Fos binding to KSHV promoters.
To characterize the role of c-Fos in KSHV transcription, c-Fos binding to KSHV promoters was screened by a genome-wide ChIP assay (Fig. 5A and B). Bac16-harboring 293T cells were induced with TPA and NaB. c-Fos accumulation was observed from the IE to the late stage, with similar levels at 8 h in the presence of low ORF45 expression and at 48 h with high ORF45 expression (Fig. 5A), which represent TPA-induced rapid c-Fos accumulation and ORF45-induced prolonged c-Fos accumulation during IE and the late stage of KSHV reactivation, respectively. The c-Fos binding to KSHV promoters was analyzed at both 8 h and 48 h using a genome-wide ChIP assay with an anti-c-Fos antibody and real-time PCR array including 87 KSHV promoters. The enrichments of ChIP DNA over control IgG were calculated and normalized to that of β-actin promoters as previously described (15, 16). Eight strong binding promoter sites with both types of c-Fos were observed, including IE genes (K4.1, ORF19, and ORF57), DE genes (ORF37 and ORF61), late genes (ORF62), latent genes (K12), and undetermined genes (ORF63), according to the annotation of KSHV 2.0 (7). Several promoter sites showed minimal binding to both types of c-Fos, including K8.1, microRNA (miRNA), and ORF73 (Fig. 5B). However, a subset of promoter binding sites likely failed to be detected because the primer pairs that were used in this study amplify only core promoter regions; in addition, the possibility that the suboptimal conditions used in our analysis may have limited antibody accessibility to c-Fos in certain chromatin sites cannot be excluded.
FIG 5.

Genome-wide screening for c-Fos binding to KSHV promoters. (A and B) Bac16-harboring 293T cells were induced with 20 ng/ml TPA and 0.3 mM NaB. (A) c-Fos accumulation was confirmed by Western blotting as indicated. (B) At 8 h or 48 h, the cells were collected and fixed. After sonication and preclearing, the same amount of cellular DNA lysates was subjected to a ChIP assay with an anti-c-Fos antibody, with normal IgG used as the negative control. The whole panel of cellular and viral DNAs was extracted and analyzed by real-time PCR in triplicate in two independent experiments, the relative DNA levels were normalized to control IgG, and the relative fold enrichment values were calculated relative to that of β-actin promoters. The results are shown as a heat map. #, rapid (8 h) versus prolonged (48 h), P < 0.05. The positive c-Fos binding under both conditions is indicated in red. (C and D) Control or HA-c-Fos constructs were transfected into Bac16-harboring 293T cells for 48 h, and then the cells were left uninduced (C) or induced as described above for 48 h (D). ChIP assays were performed with an anti-HA antibody, and viral DNAs were analyzed by real-time PCR in triplicate in two independent experiments. (E) Control, wild-type c-Fos (FosWT), constitutively active c-Fos (FosTD), or double phosphorylation site-mutated c-Fos (FosDA) constructs were cotransfected into HEK293 cells with ORF45-luc and ORF50-luc promoter reporters in the absence or presence of ORF45 and RSK2 overexpression. Thirty-six hours later, the dual luciferase activity was measured, and the data are shown as the means from three independent experiments performed in duplicate.
Notably, the binding of c-Fos to viral promoters was significantly different with rapid c-Fos accumulation (at 8 h) and prolonged c-Fos accumulation (at 48 h). Under conditions of prolonged accumulation, c-Fos exhibited reduced binding to two loci (ORF19 and ORF35), whereas under conditions of rapid accumulation, c-Fos exhibited increased binding to the ORF54 promoter site (P < 0.05). Similarly, the genome-wide binding activity of c-Fos showed that its prolonged accumulation led to different degrees of binding to KSHV promoters compared to its binding under conditions in which it rapidly accumulated. These data indicate that rapid and prolonged c-Fos activation lead to distinct promoter binding profiles during the IE stage and late stage of KSHV lytic replication, respectively.
To confirm the binding of c-Fos to the KSHV promoters, ectopic HA-tagged c-Fos was introduced into Bac16-harboring 293T cells, and ChIP assays were performed with an anti-HA antibody. Fifteen positive promoters binding to either type of c-Fos, the RTA promoter, and 4 negative promoters were analyzed using real-time PCR. Eleven of the 15 promoters and the RTA promoter showed high binding and 4 showed moderate binding to c-Fos under both mock and prolonged activation, and 4 negative promoters showed only weak binding (Fig. 5C and D). To validate the different functions of c-Fos in KSHV lytic transcription, ORF45 and ORF50 promoter-driven luciferase reporters were cotransfected with a wild-type construct (FosWT), a constitutively active construct (FosTD), or a phosphorylation site-mutated construct (FosDA). Without stimuli, the wild-type c-Fos and constitutively active FosTD activated the ORF50 promoter, whereas mutated FosDA exhibited reduced activity; ORF45-RSK overexpression augmented the activities of the ORF50 promoter mediated by wild-type c-Fos and constitutively active FosTD but not by mutated FosDA (Fig. 5E). However, none of the c-Fos constructs activated the ORF45 promoter regardless of ORF45-RSK activation, indicating that c-Fos does not activate ORF45 expression. These results suggest that c-Fos directly binds to KSHV promoters to activate the transcription of multiple viral genes.
c-Fos promotes KSHV lytic transcription.
To investigate c-Fos-dependent viral transcription, ectopic wild-type and mutated c-Fos were introduced into Bac16-harboring 293T cells with or without TPA and NaB treatment, and then the genome-wide viral transcripts were analyzed using a KSHV transcript real-time PCR array. Without TPA and NaB treatment, c-Fos moderately stimulated the transcription of a series of viral genes; however, quite a few viral genes were not activated by c-Fos under this condition compared to after TPA and NaB treatment, which induced a whole panel of viral transcription (Fig. 6A, lane 2 versus 4). In contrast, under TPA and NaB treatment, c-Fos overexpression significantly promoted viral transcription (Fig. 6A, lane 5 versus 4). These results suggest that c-Fos can promote KSHV lytic transcription but is not sufficient to initiate the whole panel of viral transcription, indicating that the activation of c-Fos and/or a cooperative partner(s) is required for efficient KSHV reactivation.
FIG 6.

c-Fos promotes KSHV lytic replication. (A) Control, wild-type, or mutated c-Fos constructs were transfected into Bac16-harboring 293T cells for 48 h, and then the cells were left uninduced or were induced with TPA and NaB for 48 h. The total RNA was extracted and reverse transcribed, and viral cDNAs were analyzed by real-time PCR in duplicate in three independent experiments. The relative viral gene expression was normalized to GAPDH, and the results are shown as a heat map. The gene names in red indicate high c-Fos binding promoters, and the clusters of gene timing are marked at the right according to the KSHV 2.0 annotation (7). IE, immediate early; DE, delayed early. N/A, not annotated. (B) Control, wild-type, or mutated c-Fos constructs were transfected as described above, and the cells were collected, extracted, and analyzed by Western blotting as indicated. (C and D) Different amounts of wild-type or mutated c-Fos constructs were transfected into Bac16-harboring 293T cells for 4 days, and then the cells were collected and analyzed as indicated (C). The medium was collected, virion DNAs were extracted, and the genomic copy number of virion DNA was detected by real-time PCR in duplicate from triple independent experiments (D). *, P < 0.01.
In addition, mutated c-Fos did not exhibit this function; instead, it had a dominant negative effect on the transcription of a panel of viral genes (Fig. 6A, lane 6 versus 4), indicating that ERK-RSK-mediated c-Fos phosphorylation is required for c-Fos to function in KSHV transcription. In addition to binding promoters, c-Fos also activated transcription both adjacent to and distant from the binding loci; this finding suggests the possibility that secondary transactivation of viral and cellular targets of c-Fos is involved in KSHV viral transcription.
The transcription-promoting function of c-Fos and the dominant negative effect of mutated c-Fos were confirmed by the expression of lytic genes during KSHV lytic replication. As shown in Fig. 6B, none of the c-Fos constructs changed the expression of the latent genes LANA (ORF73) and LANA2 (K10.5), whereas wild-type c-Fos increased the expression of lytic genes (RTA, ORF45, and ORF64). The phosphorylation site-mutated c-Fos ablated the expression of all three lytic genes. To further investigate the role of c-Fos in viral replication, different amounts of both c-Fos constructs were transfected into Bac16-harboring cells. The expression of the viral lytic genes (ORF64, ORF50, and ORF45) was increased by c-Fos overexpression; in contrast, the expression of these genes was blocked by the c-Fos mutant in a dose-dependent manner (Fig. 6C). Consequently, virion production was inhibited by the c-Fos mutant (Fig. 6D). Together, all of these results suggest that c-Fos promotes KSHV lytic transcription and replication following its phosphorylation and stabilization by sustained ERK-RSK activation.
DISCUSSION
Studies have revealed that c-Fos is rapidly elevated during immediate early TPA-induced KSHV reactivation and activates AP1-dependent RTA expression during primary infection; the inhibition of ERK activation disrupts c-Fos accumulation and RTA expression (12–14). During late lytic replication, KSHV induces a sustained ERK-RSK activation through ORF45 (31, 32, 39), promoting viral lytic translation (46), mTORC1 activation (47), and transcription from the HIV long terminal repeat (LTR) (40). In addition to cytoplasmic ORF45-RSK activation (46, 47), ORF45 and its homologs colocalize with ERK-RSK in the nucleus and induce the phosphorylation of nuclear components to facilitate viral lytic gene expression (31, 48, 49). Consistent with c-Fos acting as a sensor for sustained ERK-RSK activation, ORF45 mediates prolonged c-Fos accumulation through ERK-RSK activation, which leads to c-Fos phosphorylation and stabilization during the KSHV late lytic stage. Then, the c-Fos that is present for a prolonged period directly binds to the KSHV promoters and accelerates lytic transcription and promotes lytic replication.
Our studies show that c-Fos is essential but not sufficient for KSHV lytic replication; ectopic c-Fos expression alone was not able to activate the full panel of viral lytic transcription (Fig. 6). c-Fos alone was also not sufficient to cause strong RTA induction, because c-Fos phosphorylation by ORF45-mediated ERK-RSK activation (Fig. 5E) or c-Jun cooperation is required for efficient RTA induction (11). Mutations of the c-Fos phosphorylation sites of ERK and RSK greatly reduced RTA induction and disrupted KSHV lytic replication, indicating that c-Fos activation and accumulation are synchronously induced by sustained ERK-RSK activation. In addition, c-Jun phosphorylation and activation are required for RTA induction and KSHV lytic replication even if c-Fos is ectopically expressed or constitutively activated (11, 13, 36). Thus, the accelerating effects of c-Fos on KSHV lytic transcription require upstream signaling for its activation and another synergistic partner(s) for heterodimerization.
Multiple transcription factors have been characterized for KSHV IE lytic transcription. During mitogen-like stimulus-induced KSHV reactivation, Ets, C/EBP, c-Fos, and c-Jun, all of which act as the substrates of MAPK pathways, activate RTA and K8 transcription (11, 12, 36, 50–52), while during HDAC inhibitor-induced KSHV reactivation, NF-Y and SP1 mediate the transcription of the IE genes ORF45 and RTA (19, 53). In addition, XBP-1 transactivates RTA expression linked to plasma cell differentiation (54, 55). However, studies have revealed only 34 viral promoters that respond to RTA induction through 19 RTA binding sites in the KSHV genome (15, 16), indicating that host transcription factors are involved in the progression of the delayed and late lytic transcription program. We recently revealed that sustained ERK-RSK activation by ORF45 is essential for late gene expression (39). In the present study, we further defined that ORF45 contributes to c-Fos accumulation through sustained ERK-RSK activation, consequently accelerating most viral lytic transcription following ORF45 expression through c-Fos binding to multiple KSHV genomic loci. Disruption of c-Fos led to significant inhibition of viral gene transcription. The transcriptional activation of lytic gene expression by c-Fos involves two layers: it first directly activates viral promoters and secondarily activates expression through RTA. The importance of c-Fos beyond RTA expression was validated by the finding that ORF64 and ORF65 were reduced by depleting c-Fos in iSLK.219 cells following doxycycline-induced RTA expression, while the expression of ORF45 and RTA was barely affected in these cells (Fig. 4F). Because this prolonged c-Fos accumulation strongly occurs following ORF45 expression, it primarily accelerates the viral lytic transcription at the delayed and late stages if TPA and other mitogen-like stimuli are not present.
c-Fos accumulation distinguishes transient and sustained ERK-RSK activation during the IE and late KSHV lytic stages, and this accumulation is mediated by distinct MAPK signaling pathways driven by extracellular stimuli or viral products that activate IE or late transcription, respectively. A genome-wide c-Fos ChIP assay revealed that c-Fos exhibits a differential pattern of binding to the KSHV genome between the rapid c-Fos accumulation mediated by TPA induction and the prolonged c-Fos accumulation mediated by ORF45; both the global binding pattern and the c-Fos binding at specific sites were significantly different (Fig. 5B). This implied that there is a difference in output of KSHV lytic transcription between rapid and prolonged c-Fos accumulation. The rapid and sustained ERK-MAPK activations lead to distinct patterns of substrates and gene expression (33, 34, 41), and ORF45, ERK, and RSK form multiprotein complexes that prevent phosphate groups from being exposed to phosphatases (32), indicating that ORF45-mediated ERK-RSK activation may target a specific output distinct from that triggered by TPA or extracellular stimuli. Presumably, ORF45-RSK changes the c-Fos binding affinity for viral promoters or the cooperative partner(s) via the following mechanism. First, ORF45-RSK likely colocalizes with c-Fos in the nuclear compartment and directs c-Fos to specific targets. A recent study has shown that ORF45-RSK2 increases the recruitment of RNA polymerase II to the HIV-1 LTR and activates transcription, whereas active RSK2 does not (40), suggesting that sustained RSK activation by ORF45 preferentially targets c-Fos or another nuclear substrate(s) to distinct viral promoters. Second, a type of methylated AP-1 site is preferentially bound by the c-Jun/c-Fos heterodimer (56), and GpC methylation is altered during KSHV reactivation (57); thus, methylation of the AP-1 sites in the KSHV promoter may alter the affinity with c-Fos. Third, prolonged c-Fos accumulation presents at the late stage of lytic replication without any change in the c-Jun level (Fig. 1), whereas TPA induces rapid c-Fos accumulation, as well as elevated c-Jun expression and phosphorylation (11, 14). Thus, the c-Fos-Jun heterodimer is probably different between TPA treatment and ORF45 overexpression. Finally, the c-Fos/c-Jun association with CREB binding protein (CBP), which alters histone modifications, is obstructed by LANA (58). The replication-associated LANA expression may affect the c-Fos-Jun interaction with DNA elements and components of transcription. Alternatively, ectopic c-Fos expression is not able to strongly activate the ORF35 promoter that exhibits binding to the c-Fos-Jun heterodimer and act as a cooperative factor with RTA to induce ORF35 expression (59). All of these studies indicate that the activity and promoter selectivity of the c-Fos-Jun heterodimer are influenced by the recruitment and binding of other viral and cellular factors.
Based on the combined profiles of c-Fos binding and transcription, the expression of the KSHV lytic genes can be divided into c-Fos-dependent and -independent clusters (Fig. 5B and 6A). Several strong binding sites and multiple moderate binding sites for c-Fos have been characterized in a KSHV genome-wide ChIP assay, and these bindings in the KSHV promoters activated the transcription of the KSHV lytic genes, including IE, DE, and late genes. In contrast, other clusters of promoters do not bind to and are not directly activated by c-Fos; for example, the expression of ORF45 is not reduced by c-Fos depletion when the level of RTA expression is not affected (Fig. 4F), and ectopic c-Fos expression does not activate the ORF45 promoter (Fig. 5E), indicating that there is c-Fos-independent ORF45 expression, which is secondarily affected through RTA or other factors. However, this cluster of viral lytic genes that are not activated by c-Fos still requires sustained ERK-RSK activation by ORF45 (39), indicating that other transcription factors are involved in the late transcription. In fact, a recent study has revealed that the transcription factor Elk-1 is phosphorylated by nuclear ERK-RSK activation by the ORF45 homolog of rhesus monkey rhadinovirus (48). Further characterization of the other substrates of the sustained ERK-RSK activation may elucidate the details of late lytic transcription.
In summary, we have revealed that ORF45-mediated ERK-RSK activation contributes to prolonged c-Fos accumulation during the late KSHV lytic life cycle. As a consequence, c-Fos activates a panel of lytic genes by directly binding to the KSHV promoters and subsequently facilitates lytic replication. In addition, it is presumed that prolonged c-Fos accumulation attenuates inflammation via inhibition of the NF-κB pathway and tumor necrosis factor alpha (TNF-α) expression (60–62). Therefore, c-Fos accumulation benefits viral transcription and replication during the late KSHV lytic life cycle.
ACKNOWLEDGMENTS
We thank all members of our laboratory for helpful assistance.
This work was supported by Natural Science Foundation of China grants (81371792 and U1301121) and the Guangdong Innovative Research Team Program (no. 2009010058) to E.K. and by National Institutes of Health grant R01DE016680 to F.Z. D.A. was supported by National Institutes of Health grant F31CA183250.
REFERENCES
- 1.Dittmer DP, Damania B. 2013. Kaposi sarcoma associated herpesvirus pathogenesis (KSHV)—an update. Curr Opin Virol 3:238–244. doi: 10.1016/j.coviro.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mesri EA, Cesarman E, Boshoff C. 2010. Kaposi's sarcoma and its associated herpesvirus. Nat Rev Cancer 10:707–719. doi: 10.1038/nrc2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ganem D. 2010. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Invest 120:939–949. doi: 10.1172/JCI40567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mercier A, Arias C, Madrid AS, Holdorf MM, Ganem D. 2014. Site-specific association with host and viral chromatin by Kaposi's sarcoma-associated herpesvirus LANA and its reversal during lytic reactivation. J Virol 88:6762–6777. doi: 10.1128/JVI.00268-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Covarrubias S, Richner JM, Clyde K, Lee YJ, Glaunsinger BA. 2009. Host shutoff is a conserved phenotype of gammaherpesvirus infection and is orchestrated exclusively from the cytoplasm. J Virol 83:9554–9566. doi: 10.1128/JVI.01051-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glaunsinger B, Chavez L, Ganem D. 2005. The exonuclease and host shutoff functions of the SOX protein of Kaposi's sarcoma-associated herpesvirus are genetically separable. J Virol 79:7396–7401. doi: 10.1128/JVI.79.12.7396-7401.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arias C, Weisburd B, Stern-Ginossar N, Mercier A, Madrid AS, Bellare P, Holdorf M, Weissman JS, Ganem D. 2014. KSHV 2.0: a comprehensive annotation of the Kaposi's sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog 10:e1003847. doi: 10.1371/journal.ppat.1003847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng H, Liang Y, Sun R. 2007. Regulation of KSHV lytic gene expression. Curr Top Microbiol Immunol 312:157–183. doi: 10.1007/978-3-540-34344-8_6. [DOI] [PubMed] [Google Scholar]
- 9.Ye F, Zhou F, Bedolla RG, Jones T, Lei X, Kang T, Guadalupe M, Gao SJ. 2011. Reactive oxygen species hydrogen peroxide mediates Kaposi's sarcoma-associated herpesvirus reactivation from latency. PLoS Pathog 7:e1002054. doi: 10.1371/journal.ppat.1002054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li X, Feng J, Sun R. 2011. Oxidative stress induces reactivation of Kaposi's sarcoma-associated herpesvirus and death of primary effusion lymphoma cells. J Virol 85:715–724. doi: 10.1128/JVI.01742-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang SE, Wu FY, Chen H, Shamay M, Zheng Q, Hayward GS. 2004. Early activation of the Kaposi's sarcoma-associated herpesvirus RTA, RAP, and MTA promoters by the tetradecanoyl phorbol acetate-induced AP1 pathway. J Virol 78:4248–4267. doi: 10.1128/JVI.78.8.4248-4267.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma-Walia N, Krishnan HH, Naranatt PP, Zeng L, Smith MS, Chandran B. 2005. ERK1/2 and MEK1/2 induced by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J Virol 79:10308–10329. doi: 10.1128/JVI.79.16.10308-10329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan H, Xie J, Ye F, Gao SJ. 2006. Modulation of Kaposi's sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection. J Virol 80:5371–5382. doi: 10.1128/JVI.02299-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen A, Brodie C, Sarid R. 2006. An essential role of ERK signalling in TPA-induced reactivation of Kaposi's sarcoma-associated herpesvirus. J Gen Virol 87:795–802. doi: 10.1099/vir.0.81619-0. [DOI] [PubMed] [Google Scholar]
- 15.Chen J, Ye F, Xie J, Kuhne K, Gao SJ. 2009. Genome-wide identification of binding sites for Kaposi's sarcoma-associated herpesvirus lytic switch protein, RTA. Virology 386:290–302. doi: 10.1016/j.virol.2009.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ellison TJ, Izumiya Y, Izumiya C, Luciw PA, Kung HJ. 2009. A comprehensive analysis of recruitment and transactivation potential of K-Rta and K-bZIP during reactivation of Kaposi's sarcoma-associated herpesvirus. Virology 387:76–88. doi: 10.1016/j.virol.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Li H, Chan MY, Zhu FX, Lukac DM, Yuan Y. 2004. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: cis-acting requirements for replication and ori-Lyt-associated RNA transcription. J Virol 78:8615–8629. doi: 10.1128/JVI.78.16.8615-8629.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Chong OT, Yuan Y. 2004. Differential regulation of K8 gene expression in immediate-early and delayed-early stages of Kaposi's sarcoma-associated herpesvirus. Virology 325:149–163. doi: 10.1016/j.virol.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 19.Chang PJ, Wang SS, Chen LY, Hung CH, Huang HY, Shih YJ, Yen JB, Liou JY, Chen LW. 2013. ORF50-dependent and ORF50-independent activation of the ORF45 gene of Kaposi's sarcoma-associated herpesvirus. Virology 442:38–50. doi: 10.1016/j.virol.2013.03.023. [DOI] [PubMed] [Google Scholar]
- 20.Roux PP, Blenis J. 2004. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68:320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cargnello M, Roux PP. 2011. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong-Ho E, Wu TT, Davis ZH, Zhang B, Huang J, Gong H, Deng H, Liu F, Glaunsinger B, Sun R. 2014. Unconventional sequence requirement for viral late gene core promoters of murine gammaherpesvirus 68. J Virol 88:3411–3422. doi: 10.1128/JVI.01374-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong D, Wu NC, Xie Y, Feng J, Tong L, Brulois KF, Luan H, Du Y, Jung JU, Wang CY, Kang MK, Park NH, Sun R, Wu TT. 2014. Kaposi's sarcoma-associated herpesvirus ORF18 and ORF30 are essential for late gene expression during lytic replication. J Virol 88:11369–11382. doi: 10.1128/JVI.00793-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borah S, Darricarrere N, Darnell A, Myoung J, Steitz JA. 2011. A viral nuclear noncoding RNA binds re-localized poly(A) binding protein and is required for late KSHV gene expression. PLoS Pathog 7:e1002300. doi: 10.1371/journal.ppat.1002300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brulois K, Wong LY, Lee HR, Sivadas P, Ensser A, Feng P, Gao SJ, Toth Z, Jung JU. 25 March 2015. The association of Kaposi's sarcoma-associated herpesvirus ORF31 with ORF34 and ORF24 is critical for late gene expression. J Virol doi: 10.1128/JVI.00272-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davis ZH, Verschueren E, Jang GM, Kleffman K, Johnson JR, Park J, Von Dollen J, Maher MC, Johnson T, Newton W, Jager S, Shales M, Horner J, Hernandez RD, Krogan NJ, Glaunsinger BA. 2015. Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol Cell 57:349–360. doi: 10.1016/j.molcel.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wyrwicz LS, Rychlewski L. 2007. Identification of herpes TATT-binding protein. Antiviral Res 75:167–172. doi: 10.1016/j.antiviral.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 28.Gruffat H, Kadjouf F, Mariame B, Manet E. 2012. The Epstein-Barr virus BcRF1 gene product is a TBP-like protein with an essential role in late gene expression. J Virol 86:6023–6032. doi: 10.1128/JVI.00159-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie J, Pan H, Yoo S, Gao SJ. 2005. Kaposi's sarcoma-associated herpesvirus induction of AP-1 and interleukin 6 during primary infection mediated by multiple mitogen-activated protein kinase pathways. J Virol 79:15027–15037. doi: 10.1128/JVI.79.24.15027-15037.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bais C, Santomasso B, Coso O, Arvanitakis L, Raaka EG, Gutkind JS, Asch AS, Cesarman E, Gershengorn MC, Mesri EA. 1998. G-protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature 391:86–89. doi: 10.1038/34193. [DOI] [PubMed] [Google Scholar]
- 31.Kuang E, Tang Q, Maul GG, Zhu F. 2008. Activation of p90 ribosomal S6 kinase by ORF45 of Kaposi's sarcoma-associated herpesvirus and its role in viral lytic replication. J Virol 82:1838–1850. doi: 10.1128/JVI.02119-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuang E, Wu F, Zhu F. 2009. Mechanism of sustained activation of ribosomal S6 kinase (RSK) and ERK by kaposi sarcoma-associated herpesvirus ORF45: multiprotein complexes retain active phosphorylated ERK AND RSK and protect them from dephosphorylation. J Biol Chem 284:13958–13968. doi: 10.1074/jbc.M900025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murphy LO, Blenis J. 2006. MAPK signal specificity: the right place at the right time. Trends Biochem Sci 31:268–275. doi: 10.1016/j.tibs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 34.Chalmers CJ, Gilley R, March HN, Balmanno K, Cook SJ. 2007. The duration of ERK1/2 activity determines the activation of c-Fos and Fra-1 and the composition and quantitative transcriptional output of AP-1. Cell Signal 19:695–704. doi: 10.1016/j.cellsig.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 35.Ford PW, Bryan BA, Dyson OF, Weidner DA, Chintalgattu V, Akula SM. 2006. Raf/MEK/ERK signalling triggers reactivation of Kaposi's sarcoma-associated herpesvirus latency. J Gen Virol 87:1139–1144. doi: 10.1099/vir.0.81628-0. [DOI] [PubMed] [Google Scholar]
- 36.Xie J, Ajibade AO, Ye F, Kuhne K, Gao SJ. 2008. Reactivation of Kaposi's sarcoma-associated herpesvirus from latency requires MEK/ERK, JNK and p38 multiple mitogen-activated protein kinase pathways. Virology 371:139–154. doi: 10.1016/j.virol.2007.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qin D, Feng N, Fan W, Ma X, Yan Q, Lv Z, Zeng Y, Zhu J, Lu C. 2011. Activation of PI3K/AKT and ERK MAPK signal pathways is required for the induction of lytic cycle replication of Kaposi's sarcoma-associated herpesvirus by herpes simplex virus type 1. BMC Microbiol 11:240. doi: 10.1186/1471-2180-11-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu FX, Li X, Zhou F, Gao SJ, Yuan Y. 2006. Functional characterization of Kaposi's sarcoma-associated herpesvirus ORF45 by bacterial artificial chromosome-based mutagenesis. J Virol 80:12187–12196. doi: 10.1128/JVI.01275-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fu B, Kuang E, Li W, Avey D, Li X, Turpin Z, Valdes A, Brulois K, Myoung J, Zhu F. 2015. Activation of p90 ribosomal S6 kinases by ORF45 of Kaposi's sarcoma-associated herpesvirus is critical for optimal production of infectious viruses. J Virol 89:195–207. doi: 10.1128/JVI.01937-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karijolich J, Zhao Y, Peterson B, Zhou Q, Glaunsinger B. 2014. Kaposi's sarcoma-associated herpesvirus ORF45 mediates transcriptional activation of the HIV-1 long terminal repeat via RSK2. J Virol 88:7024–7035. doi: 10.1128/JVI.00931-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. 2002. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol 4:556–564. doi: 10.1038/ncb822. [DOI] [PubMed] [Google Scholar]
- 42.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J Virol 86:9708–9720. doi: 10.1128/JVI.01019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J Virol Methods 174:12–21. doi: 10.1016/j.jviromet.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoo SM, Zhou FC, Ye FC, Pan HY, Gao SJ. 2005. Early and sustained expression of latent and host modulating genes in coordinated transcriptional program of KSHV productive primary infection of human primary endothelial cells. Virology 343:47–64. doi: 10.1016/j.virol.2005.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA. 2007. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35:W71–W74. doi: 10.1093/nar/gkm306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuang E, Fu B, Liang Q, Myoung J, Zhu F. 2011. Phosphorylation of eukaryotic translation initiation factor 4B (EIF4B) by open reading frame 45/p90 ribosomal S6 kinase (ORF45/RSK) signaling axis facilitates protein translation during Kaposi sarcoma-associated herpesvirus (KSHV) lytic replication. J Biol Chem 286:41171–41182. doi: 10.1074/jbc.M111.280982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang HH, Ganem D. 2013. A unique herpesviral transcriptional program in KSHV-infected lymphatic endothelial cells leads to mTORC1 activation and rapamycin sensitivity. Cell Host Microbe 13:429–440. doi: 10.1016/j.chom.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woodson EN, Anderson MS, Loftus MS, Kedes DH. 2014. Progressive accumulation of activated ERK2 within highly stable ORF45-containing nuclear complexes promotes lytic gammaherpesvirus infection. PLoS Pathog 10:e1004066. doi: 10.1371/journal.ppat.1004066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X, Zhu F. 2009. Identification of the nuclear export and adjacent nuclear localization signals for ORF45 of Kaposi's sarcoma-associated herpesvirus. J Virol 83:2531–2539. doi: 10.1128/JVI.02209-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu F, Harada JN, Brown HJ, Deng H, Song MJ, Wu TT, Kato-Stankiewicz J, Nelson CG, Vieira J, Tamanoi F, Chanda SK, Sun R. 2007. Systematic identification of cellular signals reactivating Kaposi sarcoma-associated herpesvirus. PLoS Pathog 3:e44. doi: 10.1371/journal.ppat.0030044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang SE, Wu FY, Yu Y, Hayward GS. 2003. CCAAT/enhancer-binding protein-alpha is induced during the early stages of Kaposi's sarcoma-associated herpesvirus (KSHV) lytic cycle reactivation and together with the KSHV replication and transcription activator (RTA) cooperatively stimulates the viral RTA, MTA, and PAN promoters. J Virol 77:9590–9612. doi: 10.1128/JVI.77.17.9590-9612.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang SE, Wu FY, Fujimuro M, Zong J, Hayward SD, Hayward GS. 2003. Role of CCAAT/enhancer-binding protein alpha (C/EBPalpha) in activation of the Kaposi's sarcoma-associated herpesvirus (KSHV) lytic-cycle replication-associated protein (RAP) promoter in cooperation with the KSHV replication and transcription activator (RTA) and RAP. J Virol 77:600–623. doi: 10.1128/JVI.77.1.600-623.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ye J, Shedd D, Miller G. 2005. An Sp1 response element in the Kaposi's sarcoma-associated herpesvirus open reading frame 50 promoter mediates lytic cycle induction by butyrate. J Virol 79:1397–1408. doi: 10.1128/JVI.79.3.1397-1408.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu F, Feng J, Harada JN, Chanda SK, Kenney SC, Sun R. 2007. B cell terminal differentiation factor XBP-1 induces reactivation of Kaposi's sarcoma-associated herpesvirus. FEBS Lett 581:3485–3488. doi: 10.1016/j.febslet.2007.06.056. [DOI] [PubMed] [Google Scholar]
- 55.Wilson SJ, Tsao EH, Webb BL, Ye H, Dalton-Griffin L, Tsantoulas C, Gale CV, Du MQ, Whitehouse A, Kellam P. 2007. X box binding protein XBP-1s transactivates the Kaposi's sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency. J Virol 81:13578–13586. doi: 10.1128/JVI.01663-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gustems M, Woellmer A, Rothbauer U, Eck SH, Wieland T, Lutter D, Hammerschmidt W. 2014. c-Jun/c-Fos heterodimers regulate cellular genes via a newly identified class of methylated DNA sequence motifs. Nucleic Acids Res 42:3059–3072. doi: 10.1093/nar/gkt1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Darst RP, Haecker I, Pardo CE, Renne R, Kladde MP. 2013. Epigenetic diversity of Kaposi's sarcoma-associated herpesvirus. Nucleic Acids Res 41:2993–3009. doi: 10.1093/nar/gkt033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lim C, Gwack Y, Hwang S, Kim S, Choe J. 2001. The transcriptional activity of cAMP response element-binding protein-binding protein is modulated by the latency associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J Biol Chem 276:31016–31022. doi: 10.1074/jbc.M102431200. [DOI] [PubMed] [Google Scholar]
- 59.Masa SR, Lando R, Sarid R. 2008. Transcriptional regulation of the open reading frame 35 encoded by Kaposi's sarcoma-associated herpesvirus. Virology 371:14–31. doi: 10.1016/j.virol.2007.08.023. [DOI] [PubMed] [Google Scholar]
- 60.Koga K, Takaesu G, Yoshida R, Nakaya M, Kobayashi T, Kinjyo I, Yoshimura A. 2009. Cyclic adenosine monophosphate suppresses the transcription of proinflammatory cytokines via the phosphorylated c-Fos protein. Immunity 30:372–383. doi: 10.1016/j.immuni.2008.12.021. [DOI] [PubMed] [Google Scholar]
- 61.Maruyama K, Sano G, Ray N, Takada Y, Matsuo K. 2007. c-Fos-deficient mice are susceptible to Salmonella enterica serovar Typhimurium infection. Infect Immun 75:1520–1523. doi: 10.1128/IAI.01316-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takada Y, Ray N, Ikeda E, Kawaguchi T, Kuwahara M, Wagner EF, Matsuo K. 2010. Fos proteins suppress dextran sulfate sodium-induced colitis through inhibition of NF-kappaB. J Immunol 184:1014–1021. doi: 10.4049/jimmunol.0901196. [DOI] [PubMed] [Google Scholar]