

Abstract
Vitamin A deficiency (A−) remains a public health concern in developing countries and is associated with increased susceptibility to infection. Citrobacter rodentium was used to model human Escherichia coli infections. A− mice developed a severe and lethal (40%) infection. Vitamin A-sufficient (A+) mice survived and cleared the infection by day 25. Retinoic acid treatment of A− mice at the peak of the infection eliminated C. rodentium within 16 days. Inflammation levels were not different between A+ and A− mouse colons, although the A− mice were still infected at day 37. Increased mortality of A− mice was not due to systemic cytokine production, an inability to clear systemic C. rodentium, or increased pathogenicity. Instead, A− mice developed a severe gut infection with most of the A− mice surviving and resolving inflammation but not eliminating the infection. Improvements in vitamin A status might decrease susceptibility to enteric pathogens and prevent potential carriers from spreading infection to susceptible populations.
INTRODUCTION
Vitamin A deficiency is a significant problem in developing countries, where inadequate micronutrient intake remains a public health concern (1). The World Health Organization estimates that over 20% of preschool-aged children are clinically vitamin A deficient (1, 2). Vitamin A deficiency contributes to the higher prevalence of respiratory and diarrheal diseases as well as increased childhood mortality in those countries. Conversely, vitamin A supplementation is practiced as a means to reduce mortality in preschool-aged children by reducing the severity of infectious diseases (3).
Vitamin A and its active metabolite, retinoic acid (RA), are important regulators of T cell responses. RA inhibited gamma interferon (IFN-γ) production from T cells in vitro (4). In addition, T cells from vitamin A-deficient (A−) mice overproduced IFN-γ (5). RA also inhibited Th17 cells in vitro and in vivo (6). RA increased the expression of the gut homing receptors α4β7 and CCR9 on T cells (7), which recruit T cells to the gut mucosa. In vitro, RA inhibited interleukin-17 (IL-17) and induced expression of the transcription factor FoxP3, associated with regulatory T cells, and IL-10 production (8). Vitamin A and RA are key regulators of T cell cytokine production and gut homing.
Several lines of experimental evidence support a beneficial effect of vitamin A and RA on the host response to infection (9, 10). In the gut, the mechanisms that account for the anti-infective effects of vitamin A include support of B cell function and T cell-dependent B cell antibody responses (11). Gut infection of A− mice with Trichinella spiralis resulted in T cells that produced IFN-γ but not IL-4 and, as a result, reduced the rate of parasite clearance (9). Furthermore, RA treatment reduced colonic inflammation caused by dextran sodium sulfate and infection (12). The reduction in gastrointestinal (GI) inflammation with RA treatment was attributed to the inhibition of IL-17 and IFN-γ (12, 13). These data suggest that vitamin A and RA regulate T cell function to limit inflammation following chemical and infectious injury in the gut.
Citrobacter rodentium is a mouse pathogen that models human infections with enteropathogenic Escherichia coli and causes attaching and effacing lesions of the cecum and colon in mice (14). The natural route of C. rodentium transmission is fecal-oral. Resistant mouse strains, including C57BL/6 mice, are infected transiently with C. rodentium and clear the infection within 2 to 3 weeks (14). The acquired immune system was required for early protection from C. rodentium, as demonstrated in recombination-activating gene (Rag)-knockout (KO) mice lacking T and B cells that were unable to clear the infection (15). Robust IL-22 production from innate lymphoid cells and macrophages has been shown to induce protective Th17 responses that result in clearance (16, 17). T cells and B cells were essential for resolution of C. rodentium infection, since mice without T cells (CD4 KO) or B cells (Igμ KO) developed fatal infections (17). Host resistance to C. rodentium depends on IL-22 production from innate cells, T cells, B cells, and Th17 cells.
Here, we determined the effect of vitamin A deficiency on host resistance to C. rodentium infection in C57BL/6 mice, a normally resistant mouse strain. Because of the well-demonstrated inhibitory effects of RA on the differentiation of Th1 and Th17 cells, we predicted that a bacterial infection that required Th17 cell responses for resistance might be less severe in A− mice and exacerbated in RA-treated mice. Interestingly and contrary to expectations, A− mice developed a chronic infection with C. rodentium. The C. rodentium infection was lethal for 40% of A− mice, while none of the vitamin A-sufficient (A+) or RA-treated A− mice died prematurely from infection.
MATERIALS AND METHODS
Mice.
C57BL/6 mice were originally from Jackson Laboratories (Bar Harbor, MN) and bred at the Pennsylvania State University (University Park, PA) for experiments. Vitamin A-deficient (A−) and vitamin A-sufficient (A+) mice were generated as previously described (5, 18). Briefly, mice were fed a purified diet that did not contain any vitamin A (A−) or that contained 25 μg of retinyl acetate (vitamin A) per day (A+). At weaning, mice were continuously fed the A− or A+ diet until the end of the study. Serum retinol status was determined by ultrahigh-pressure liquid chromatography at 6 to 7 weeks of age in pooled samples. For some experiments, A− mice were treated with 37.5 μg of all-trans-RA (Sigma-Aldrich, St. Louis, MO) administered orally in 10 μl corn oil three times per week (19). For some experiments, mice were injected intraperitoneally (i.p.) with E. coli O111:B4 lipopolysaccharide (LPS) (6 mg/kg of body weight; Sigma-Aldrich). Experimental procedures were approved by the Office of Research Protection Institutional Animal Care and Use Committee of the Pennsylvania State University, University Park, PA.
C. rodentium infection.
C. rodentium strain ICC169 (nalidixic acid resistant) and bioluminescent strain ICC180 (kanamycin resistant) were kind gifts of Gad Frankel (London School of Medicine and Dentistry, London, United Kingdom). C. rodentium ICC169 was cultured in Luria-Bertani (LB; EMD Chemicals, Inc., Gibbstown, NJ) broth containing 50 μg/ml nalidixic acid (Sigma-Aldrich), while C. rodentium ICC180 was cultured in LB broth containing 100 μg/ml kanamycin (Sigma-Aldrich). Mice 8 to 10 weeks of age were infected by oral gavage with 5 × 109 CFU of the C. rodentium strain in 200 μl unless otherwise noted. For studies looking at in vivo infection kinetics, 5 × 109 CFU of C. rodentium ICC180 was used. Animals were imaged every other day using the IVIS50 small-animal imaging system (Xenogen Corp., Alameda, CA, USA). Images were analyzed by Living Image software (PerkinElmer, Waltham, MA). Additional groups of mice were injected intravenously (i.v.) with 104 to 108 CFU of C. rodentium strain ICC169. Feces and other tissues were collected, homogenized, and plated in serial dilutions on LB agar plates containing nalidixic acid.
For most experiments, mice were housed 1 per cage from the time of infection to prevent transmission from mouse to mouse. Natural transmission experiments were performed as previously described (20). Briefly, A−, RA day 0, and A+ mice were infected via oral gavage with 5 × 109 CFU. Three days postinfection (p.i.), each infected “seed” mouse was cohoused with two naive A+ mice.
Histology.
Distal colon sections were fixed in 10% formalin, sectioned, and stained with hematoxylin and eosin (Pennsylvania State University Animal Diagnostic Laboratory, University Park, PA). Specimens were coded and evaluated in a blinded fashion by a board-certified laboratory animal veterinarian with training in pathology. Crypt measurements were taken at 100× using the cellSens software (Olympus Corp., Center Valley, PA, USA). Sections were scored on a scale from 0 to 4 (0, none; 1, minimal; 2, mild; 3, moderate; 4, extensive) for severity of inflammation, epithelial sloughing, distention of muscularis, and edema. Total histology scores were generated by adding the scores for each category together, generating a value from 0 to 16 for each sample.
Flow cytometry.
Colonic intraepithelial lymphocytes (IELs) were isolated as previously described and stained for flow cytometry (21). Cells were counted and stained with phycoerythrin (PE)-Cy5 T cell receptor β (TCRβ) (BD Pharmingen, San Jose, CA, USA), fluorescein isothiocyanate (FITC) CD8β (eBioscience, San Diego, CA, USA), PE-Cy7 CD8α (BioLegend, San Diego, CA, USA), or PE-Texas Red CD4 (Invitrogen, Carlsbad, CA, USA). Cells were analyzed on an FC500 benchtop cytometer (Beckman Coulter, Brea, CA, USA), and data were analyzed using FlowJo 7.6.1 software (Tree Star, Ashland, OR, USA).
ELISA.
Cytokine production in the serum was measured for tumor necrosis factor alpha (TNF-α), IL-1β, and IFN-γ levels by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions (BD Biosciences, Minneapolis, MN).
Statistical analysis.
Statistical analyses were performed using GraphPad Prism software (GraphPad, La Jolla, CA, USA). Two-tailed Student t tests were used for serum retinol analysis. One-way analysis of variance (ANOVA) with Tukey's post hoc test was used to compare the systemic C. rodentium loads, for bioluminescence quantification, and for cell population analysis. Two-way ANOVA with Bonferroni's post hoc test was used to compare CFU, histology scores, and crypt lengths. Log rank tests were used for the survival curves and ratios of serum cytokine producers. For all analyses, P < 0.05 was used as the limit for significance.
RESULTS
A− mice are more susceptible to C. rodentium infection than A+ mice.
A+ and A− mice were generated as previously described (5, 18). As expected (22), 6- to 7-week-old A− mice had significantly lower serum retinol levels than did A+ mice (Fig. 1A). Bacterial fecal shedding in A+ mice peaked around day 10 and cleared within 25 days postinfection (Fig. 1B). The infection in A− mice followed the same kinetics as that in A+ mice until day 14. A+ and A− mice had similar numbers of C. rodentium bacteria in the cecum and feces at day 10 postinfection (cecum data not shown and Fig. 1A). After day 14, the A− mice continued to shed high numbers of C. rodentium bacteria in their feces, whereas the A+ mice began to clear the infection (Fig. 1B). All of the infected mice showed a small amount of weight loss (5% of starting weight) within the first 2 to 4 days of infection, but all of the A+ mice recovered and no A+ mice died following C. rodentium infection (Fig. 1C). Conversely, some of the A− mice failed to recover and instead lost significantly more of their initial body weight (10 to 20%), which resulted in the premature lethality of 40% of the A− mice (Fig. 1C). The A− mice that survived did not lose any more weight than the A+ mice, had normal exploratory behaviors (data not shown), and otherwise were undistinguishable from their A+ infected counterparts. A subset of A− mice died prematurely following weight loss while the remaining A− mice resumed normal behaviors but did not clear C. rodentium.
FIG 1.

A− mice are more susceptible to C. rodentium infection than A+ mice. (A) Serum retinol analysis from groups of 6- to 7-week-old A+ and A− mice. Each data point represents the serum retinol values in pooled samples from a different litter of mice (n = 10 to 13 litters). (B) C. rodentium CFU in the feces. Data are shown as means ± standard errors of the means and come from 1 representative of 5 independent experiments with n being 3 to 5/group. (C) Survival of A− and A+ mice during C. rodentium infection with data combined from 5 experiments (n = 9 to 15/group). (D) Whole-body imaging of A+, A−, and RA day 0 treated mice infected with bioluminescent C. rodentium and imaged. The image shows 1 mouse from a total of 3 to 4 mice/treatment group. Means of the bioluminescence from all the mice are shown in Fig. S1 in the supplemental material. (A) Two-tailed Student t test; (B) two-way ANOVA with Bonferroni post hoc tests; (C) log rank test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To visualize the intestinal passage of C. rodentium in vivo, we made use of a bioluminescent C. rodentium strain and live-animal imaging. Between day 2 and day 6 postinfection, low levels of C. rodentium bioluminescence were detected in the upper parts of the gastrointestinal (GI) tract of A+ mice (Fig. 1D). Just before the peak in fecal shedding (Fig. 1B), the intensity of bioluminescence increased and moved into the lower GI tract of A+ mice (Fig. 1D). After day 10 of infection, the intensity decreased in A+ mice, and by day 14, when fecal shedding of C. rodentium was on the decline in A+ mice, less bioluminescence was detected in the intact A+ animals (Fig. 1D). The kinetics of C. rodentium transit in the A+ mice was similar to that reported previously in wild-type mice (23). The bioluminescence in the A− mice matched that of the A+ mice at day 2 (Fig. 1D). However, as early as day 4, the A− mice showed bioluminescence throughout the GI tract, which persisted in intensity through day 14 (Fig. 1D). Unlike the A+ mice, the intensity of bioluminescence in A− mice between day 4 and day 14 was high throughout the GI tract and significantly higher than in A+ mice (Fig. 1D; see also Fig. S1 in the supplemental material). Thus, the transit of C. rodentium through the GI tract occurred more rapidly and persisted longer in A− than A+ mice.
T cell frequencies are lower in the vitamin A-deficient gut.
To determine if vitamin A status causes differences in gut mucosal immune cell populations, the intraepithelial lymphocyte (IEL) populations of the colon were characterized. IELs are in direct contact with the intestinal epithelium and a source of T cells from the colon (24). In addition, the T cells in the IEL are in close contact with the microbiota and enteric pathogens in the colon. Fewer total IEL cells were isolated from the colons of uninfected A− than A+ mice (Fig. 2A). C. rodentium infection resulted in a significant increase in the number of cells in the colon of both A− and A+ mice (Fig. 2A). Interestingly, although the A+ mice resolved the infection by day 37, as shown in Fig. 1B, the numbers of T cells (TCRβ+) did not return to baseline by day 37 p.i. (Fig. 2A and B). The total numbers of IELs and TCRβ+ T cells in the colon of A− mice increased following infection but never reached the levels present in infected A+ mice (Fig. 2A). The numbers of TCRβ+, TCRβ+/CD8α+, and TCRβ+/CD8αβ+ T cells in the colon IEL compartment were lower in A− than in A+ mice, both before and after infection (Fig. 2A to E). Therefore, although the increases in total cell numbers and T cell subpopulations were similar in A+ and A− mice, the A− mice consistently had fewer T cells in the colonic IEL compartment than did A+ mice. Differences in T cell numbers and populations between A+ and A− mice were not associated with clearance of C. rodentium in A+ mice and persistence of C. rodentium in A− mice.
FIG 2.

T cells in the IELs of the colon from A+ and A− mice before and after infection. (A to E) Cell numbers in the colon IELs. (A) Immune cells. (B) TCRβ+ T cells. (C) CD8α+ T cells. (D) CD8αβ+ T cells. (E) CD4+ T cells. Values are the means ± standard errors of the means from combined data from 2 to 3 independent experiments with n being 6 to 15/group. Two-way ANOVA with Bonferroni post hoc tests. Asterisks indicate significant differences between groups at specific days by posttest: *, P < 0.05; **, P < 0.01; ***, P < 0.001. (F and G) Histology scores (F) and crypt length (G) of colons during infection. Values with different letters are significantly different from each other (P < 0.05).
Exacerbated inflammation and epithelial hyperplasia in A− mice following C. rodentium infection.
Histopathology sections of the colon were evaluated before and after infection for signs of inflammation, tissue damage, and hyperplasia, including measurements of crypt length. Before infection, A+ and A− mice had low histopathology scores that did not differ with vitamin A status (Fig. 2F; see also Fig. S2 in the supplemental material). Crypt lengths also did not differ between uninfected A+ and A− mice (Fig. 2G). After infection, the histopathology scores of A+ mouse colons did not change significantly, either at peak infection (day 10) or after resolution of infection (day 37) (Fig. 2F; see also Fig. S2). In A− mice, histopathology scores were significantly higher at day 10 postinfection than both baseline A− scores and scores for A+ mice at day 10 (Fig. 2F; see also Fig. S2). By day 37, the histopathology scores of A+ and A− mice were the same in spite of the fact that A+ mice had cleared the infection while A− mice had not (Fig. 2F; see also Fig. S2). Crypt length in the A+ mice was not affected by infection (Fig. 2G). Crypt length in the A− mice increased significantly after infection, and crypts were significantly longer on both day 10 and day 37 than at baseline and compared to the A+ mice at all time points (Fig. 2G). Overall, although A− mice exhibited more inflammation than A+ mice at day 10 postinfection, the effect was not present at day 37 even though A− mice continued to harbor C. rodentium and A+ mice did not.
RA treatment of A− mice results in clearance of C. rodentium.
To determine if RA could rescue the severe infection in A− mice, A− mice were orally dosed with RA. RA dosing began either 2 weeks before infection (RA −day 14) or on the day of infection (RA day 0) (Fig. 3). Once started, RA dosing continued 3 times weekly throughout the remainder of the experiment. Treatment of A− mice with RA, either 2 weeks before (RA −day 14) or on the day of (RA day 0) infection, resulted in fecal shedding of C. rodentium similar to that of A+ mice (Fig. 1B and 3). In a separate experiment, the RA day 0 mice were also infected with bioluminescent C. rodentium. The bioluminescence in the RA day 0 mice was of a high intensity and resembled that in A− rather than A+ mice (Fig. 1D; see also Fig. S1 in the supplemental material). Interestingly, by day 14 the intensity of bioluminescence in the RA day 0 group was lower and resembled that in the A+ rather than the A− mice (Fig. 1D; see also Fig. S1).
FIG 3.
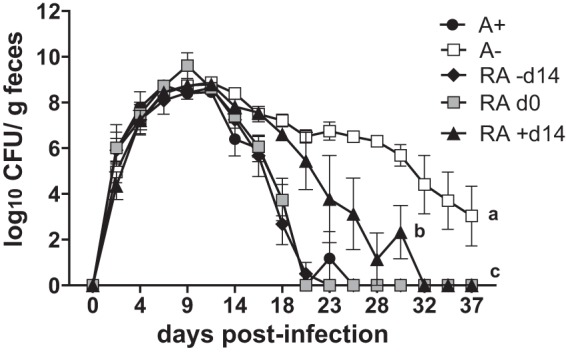
RA treatment of A− mice eliminates C. rodentium infection. CFU in the feces of A+ mice, A− mice, A− mice treated with RA for 2 weeks before infection (RA −day 14), A− mice treated with RA on the day of infection (RA day 0), and A− mice treated with RA starting on day 14 of infection (RA +day 14). Values are the means ± standard errors of the means from one representative of 2 independent experiments (n = 3 to 5/group). Two-way ANOVA with Bonferroni post hoc tests. Values with different letters are significantly different from each other (P < 0.01 to 0.0001).
To determine whether RA could be used therapeutically in A− mice with an established C. rodentium infection, the RA treatment of A− mice was started on day 14 postinfection (RA +day 14). Day 14 of infection occurs just after the peak of fecal shedding and before A+ mice show a rapid decline in C. rodentium shedding (Fig. 1B). Treatment of A− mice with RA, three times weekly, beginning on day 14, resulted in a gradual decline in fecal shedding of C. rodentium (Fig. 3), which was significant as early as 4 days following the start of RA treatment, compared to untreated A− mice (day 18 postinfection, Fig. 3). As observed in earlier experiments, untreated A− mice maintained a persistent infection while the RA +day 14 treated mice cleared the infection 18 days after the start of the RA treatment (day 32 postinfection, Fig. 3). Thus, RA treatment of A− mice cleared the C. rodentium infection, which otherwise was persistent.
Increased C. rodentium CFU in the liver and spleen of A− mice.
At day 14 postinfection, mice that had been infected with the bioluminescent strain of C. rodentium were euthanized to determine which organs harbored C. rodentium. In the A+ group, bioluminescence was detected in the colon but not in other parts of the GI tract (see Fig. S3A in the supplemental material). In addition, A+ mice did not have visible bioluminescence in their spleen or liver at day 14 postinfection (see Fig. S3B). A− mice had high levels of bioluminescence in the upper portions of the GI tract that were significantly higher than those in A+ mice (see Fig. S3C). A− mice also had visible bioluminescence at day 14 in both the spleen and the liver (see Fig. S3B). The bioluminescence in the spleen and liver of RA day 0 mice was more similar to that in A+ mice than to that in A− mice at day 14 postinfection (Fig. 4A; see also Fig. S3B). Quantification of the bioluminescence showed that A− mice had higher bioluminescence in the spleen, liver, and small intestine than did either the A+ or the RA day 0 mice (Fig. 4A).
FIG 4.

Systemic spread of C. rodentium following GI infection. Mice were sacrificed at day 14 postinfection with the bioluminescent strain of C. rodentium and at day 10 postinfection for culturing. (A) Spleen and liver were imaged and quantitated. Sample images are shown in Fig. S3 in the supplemental material. Values are from the images of n = 3 to 4 mice/treatment group. (B) CFU of C. rodentium recovered in the spleen and liver of day 10 infected mice. Values are individual CFU and are the means ± standard errors of the means combined from three independent experiments with n = 9 to 15/group. One-way ANOVA with Tukey posttests. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Next, we determined whether viable C. rodentium was detectable in the spleen and liver of our mice following gastric infection with C. rodentium. Cultures of the spleen and liver at day 10 and day 14 postinfection showed that A+ mice had detectable C. rodentium in their internal organs (Fig. 4B; also day 14 data not shown). A− mice had significantly more C. rodentium bacteria in the spleen and liver than did A+ mice (Fig. 4B), while the spleen and liver of RA day 0 mice did not differ from those of A+ mice but were significantly different from those of A− mice (Fig. 4B). At day 37 of infection, the A− mice no longer had C. rodentium present in the small intestine, spleen, and liver, even though they continued to shed C. rodentium in the feces (data not shown).
Mortality of A− mice following infection with C. rodentium is not due to overproduction of systemic cytokines.
To determine whether A− mice died due to cytokine overproduction, serum cytokine levels were determined before (day 0) and at the peak (day 10) of infection in A+ and A− mice (Table 1). None of the A+ mice had detectable levels of any cytokines before infection. At day 0, several of the A− mice had detectable levels of TNF-α, IL-1β, or IFN-γ in their serum (Table 1). Infection resulted in detectable TNF-α, IL-1β, and IFN-γ in the serum of both A+ and A− mice. There were significantly more A− mice with IL-1β in the serum (10 of 19) than A+ mice (1 of 13) at day 10 postinfection (Table 1). Serum IFN-γ was higher (but not significantly different) in A− mice than in A+ mice at day 10 postinfection (P = 0.0817). To measure the capacity for cytokine production, LPS was injected i.p. into A− and A+ mice to measure serum cytokine response. A− mice produced significantly more TNF-α than did A+ mice following LPS injection, while the IL-1β responses were the same in A+ and A− mice (Fig. 5A; also IL-1β data not shown). Overall, A− mice were more likely to have IL-1β detectable in the serum after C. rodentium infection and produced more TNF-α after LPS than did A+ mice.
TABLE 1.
Serum cytokines in A+ and A− mice
| Day of C. rodentium infection | No. of mice with detectable level of cytokine/total no. of mice |
|||||
|---|---|---|---|---|---|---|
| TNF-α |
IL-1β |
IFN-γ |
||||
| A+ | A− | A+ | A− | A+ | A− | |
| 0 | 0/0 | 1/8 | 0/0 | 4/8 | 0/0 | 3/6 |
| 10 | 2/13 | 4/19 | 1/13 | 10/19a | 0/13 | 4/19 |
A− value is significantly different than A+ value (P < 0.05).
FIG 5.
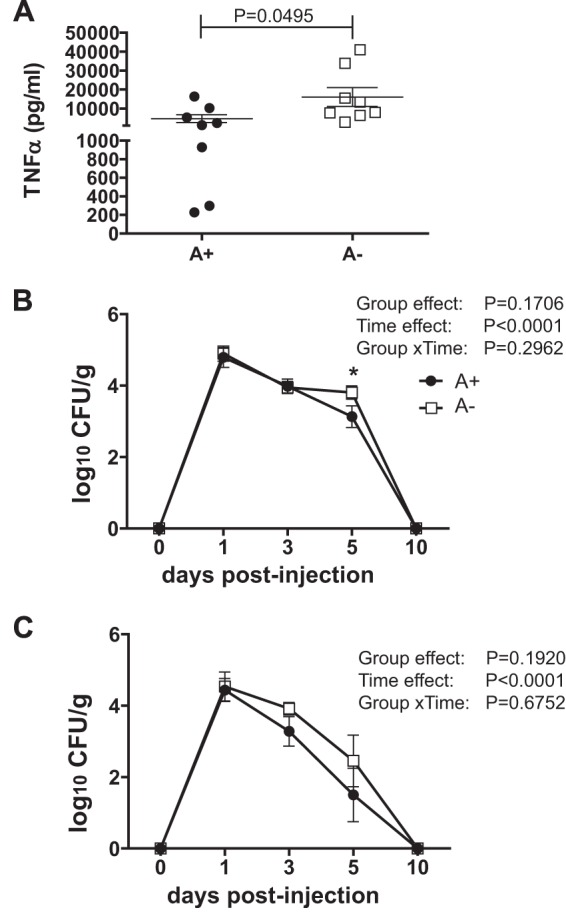
Effect of vitamin A status on LPS response and i.v. challenge with C. rodentium. (A) Serum TNF-α in A− and A+ mice treated with LPS. (B and C) CFU in the spleen (B) and liver (C) of mice following i.v. injection of C. rodentium. Values are the means ± standard errors of the means combined from two to three independent experiments with n being 8 to 9/group/time point. (A) Two-tailed Student t test; (B and C) two-way ANOVA with Bonferroni post hoc tests. Asterisks indicate significant differences between groups at specific days by posttest (*, P < 0.05).
We hypothesized that A− mice were dying following oral challenge with C. rodentium because of systemic spread. We attempted to determine the 50% lethal dose (LD50) of i.v. injected C. rodentium in A+ mice. All of the A+ mice survived an i.v. dose of 108 CFU of C. rodentium and readily cleared the bacteria. A− mice also survived an i.v. injection of 108 CFU of C. rodentium. A+ mice had the highest number of organisms in the spleen and liver 1 day post-i.v. infection with 108 CFU, which declined at day 3 and again at day 5 and was completely cleared by day 10 postinfection (Fig. 5B and C). The group × time interaction showed no differences in the clearance of i.v. injected C. rodentium between A+ and A− mice (Fig. 5B and C). In addition, i.v. injection was not lethal and did not induce a systemic cytokine response in either A+ or A− mice (data not shown).
Host vitamin A status does not alter C. rodentium infectivity.
Previous reports have demonstrated that natural infection by C. rodentium of cohoused uninfected mice occurs with several fewer log units of bacteria than with laboratory-grown C. rodentium (20). We hypothesized that vitamin A and RA were attenuating the infectivity (virulence) of C. rodentium and that C. rodentium that had been passaged through A− mice might be more virulent. A+, RA day 0, and A− seed mice were infected by gavage using laboratory-grown C. rodentium, and 3 days later each was cohoused with naive A+ mice, housed in groups of 2 with the seed mice. As described previously (20), all seed mice, despite their vitamin A status, infected all cohoused naive A+ mice within 3 days of exposure (data not shown). Culturing of the feces, spleen, and liver from the cage mates of A+, A−, and RA day 0 seed mice showed that there were no differences in the CFU of C. rodentium recovered in the spleen, liver, and feces via natural transmission from either A+, A−, or RA day 0 seed mice (Fig. 6). Thus, the data do not show an effect of vitamin A status and/or RA treatment on C. rodentium infectivity.
FIG 6.
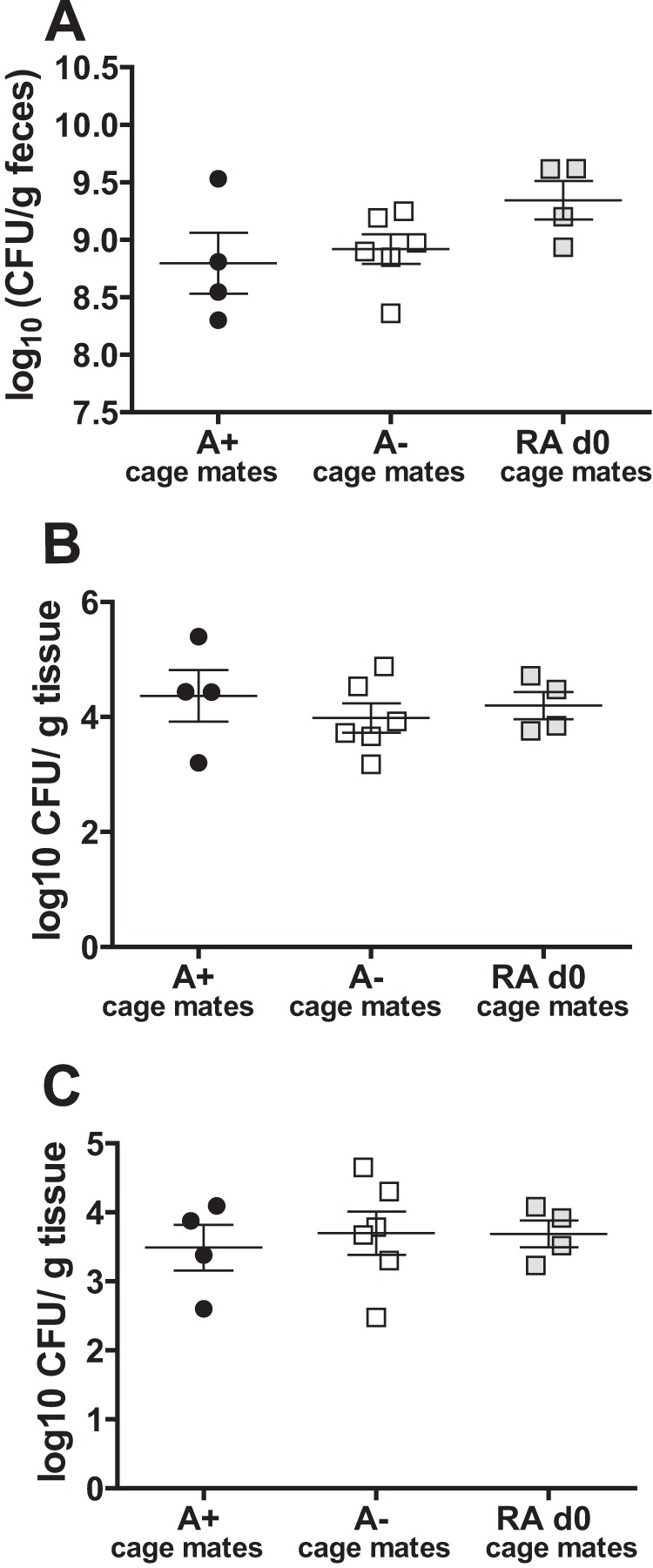
Natural transmission of C. rodentium is not affected by the host vitamin A status. CFU in the feces (A), spleen (B), and liver (C) of A+ cage mates of A−, RA day 0, and A+ laboratory-inoculated mice (n = 4 to 6/group). Values are the means ± standard errors of the means combined for two independent experiments. One-way ANOVA with Tukey posttests.
DISCUSSION
A− mice were found to be significantly more susceptible to C. rodentium infection than A+ mice. The increased susceptibility of A− mice included lethality of 40% of the A− mice by day 14. The kinetics of shedding of C. rodentium in A+ and A− mice were not different before day 14, after which the A+ mice proceeded to clear the infection while the A− mice either died or developed a persistent infection. The data suggest that early innate immune responses are adequate in A− mice during the initial phase of the infection, when fecal bacterial counts are increasing, and instead suggest that A− mice failed to generate an acquired immune response necessary to clear the infection. Our results unexpectedly showed that A− mice became chronic carriers of C. rodentium, while no longer showing symptoms (inflammation of the colon, diarrhea, etc.). Correcting vitamin A status or treating A− mice with RA effectively eliminated the infection and improved survival of A− mice, suggesting an added benefit not previously recognized for vitamin A and/or RA interventions.
The increased mortality of A− mice following GI infection was not due to the systemic spread of C. rodentium, increased systemic cytokine production, or the virulence of C. rodentium. Others have reported mortality following gavage of C. rodentium in mice and have attributed that mortality to systemic spread and polymicrobial sepsis (15, 25, 26). Susceptible strains of mice (C3H/HeJ, C3H/HeOuJ, and C3H/HeN) develop a severe infection with 100% lethality due to bacterial translocation from the gut, cytokines in the serum, and crypt cell apoptosis (25). By comparison, C57BL/6 mice are relatively resistant to C. rodentium (15, 17, 25). For the first time, our data clearly show that C. rodentium does not grow following i.v. injection of large numbers of bacteria in either A+ or A− mice, indicating that vitamin A deficiency does not affect the ability to survive a systemic infection with C. rodentium. However, A− mice were more susceptible to oral infection with C. rodentium. We necropsied one recently deceased and one moribund A− mouse. Very little food was found in the stomach of either mouse, suggesting that the mice had stopped eating. The necropsy of A− mice failed to identify evidence of systemic infection (data not shown). It has been shown that LPS-induced proinflammatory cytokines such as TNF-α and IL-1β can result in anorexia (27). Our data are consistent with TNF-α- and IL-1β-induced anorexia, since the A− mice that died had lost significant amounts of weight (10 to 20% of original body weight). In addition, A− mice had higher TNF-α levels after LPS challenge and were more likely to have detectable IL-1β in their serum after infection than A+ mice. Surviving A− mice may have been just below the threshold of the response and therefore survived the infection-induced anorexia. Based on the inability of C. rodentium to grow following an i.v. injection and the necropsies of moribund and dead A− mice, we concluded that the mortality in our A− mice was not due to sepsis and/or the systemic spread of the infection.
A− mice had reduced numbers of total colonic IELs and all IEL T cell subsets in the colon before infection. Infection induced homing and expansion of T cells in the gut of both A+ and A− mice. Although the A− mice were persistently infected with C. rodentium, colonic inflammation as determined by histological score resolved and was not different at day 37 from that in A+ mice. This same phenomenon occurred in germfree mice monoassociated with C. rodentium (28). In germfree mice, C. rodentium infection was not cleared but the inflammatory response in the colon was nevertheless resolved (28). It therefore seems that vitamin A is not required to resolve inflammation in the colon and that, in the absence of vitamin A, T cells are able to arrive and respond to the infection. However, the acquired immune response in the A− mice was ineffective at eliminating the infection. The data suggest a requirement for vitamin A/RA to mount effective protective immunity after the initial infection. Since RA treatments effectively reduced C. rodentium numbers in already infected A− mice within 4 days of treatment, it will be of interest to identify the targets of RA in the GI tract for C. rodentium clearance.
In our study, A− mice developed persistent and sometimes fatal enteric infections. The cause of the premature lethality of A− mice was not due to sepsis and/or systemic spread of C. rodentium. Instead it seems that a high load of C. rodentium in the gut of A− mice results in infection-induced anorexia in 40% of the mice. When given early, vitamin A and/or RA interventions protected A− mice from the lethality following C. rodentium infection, and when administered later, RA cleared the persistent infections that occurred in surviving A− mice. Our work has important implications for the developing world, where vitamin A deficiency is prevalent. In particular, vitamin A-deficient humans and animals could be reservoirs for E. coli-like enteric pathogens. Vitamin A and RA treatments might be useful interventions to decrease morbidity and mortality from enteric infections. We show here a novel and unappreciated role for vitamin A/RA for eliminating persistent enteric infections and show added benefits to improving vitamin A status in the developing world.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by NIH T32 5CGR to K.L.M., the Pennsylvania State University College of Agriculture Science Competitive Graduate Student grant (K.L.M.), the National Institute of Neurologic and Stroke grant NS067563 (M.T.C.), the National Center for Complementary and Alternative Medicine and the Office of Dietary Supplements grant AT005378 (M.T.C.), and National Institute of Diabetes and Digestive and Kidney Diseases grant DK41479 (A.C.R.).
K.L.M., A.C.R., and M.T.C. designed the research; K.L.M., K.H.R., M.J.K., and J.W.D. conducted research; K.L.M., A.C.R., and M.T.C. analyzed research; K.L.M. and M.T.C. wrote the manuscript; M.T.C. had primary responsibility for final content.
The authors have nothing to declare in regard to competing financial interests.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00201-15.
REFERENCES
- 1.WHO. 2009. Global prevalence of vitamin A deficiency in populations at risk 1995–2005: WHO global database on vitamin A deficiency. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.West KP., Jr 2002. Extent of vitamin A deficiency among preschool children and women of reproductive age. J Nutr 132:2857S–2866S. [DOI] [PubMed] [Google Scholar]
- 3.Fawzi WW. 2000. Vitamin A supplementation: implications for morbidity and mortality in children. J Infect Dis 2000:S112–S133. doi: 10.1086/315921. [DOI] [PubMed] [Google Scholar]
- 4.Cantorna MT, Nashold FE, Hayes CE. 1994. In vitamin A deficiency multiple mechanisms establish a regulatory T helper cell imbalance with excess Th1 and insufficient Th2 function. J Immunol 152:1515–1522. [PubMed] [Google Scholar]
- 5.Carman JA, Smith SM, Hayes CE. 1989. Characterization of a helper T lymphocyte defect in vitamin A−deficient mice. J Immunol 142:388–393. [PubMed] [Google Scholar]
- 6.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. 2006. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 7.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. 2004. Retinoic acid imprints gut-homing specificity on T cells. Immunity 21:527–538. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 8.Elias KM, Laurence A, Davidson TS, Stephens G, Kanno Y, Shevach EM, O'Shea JJ. 2008. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood 111:1013–1020. doi: 10.1182/blood-2007-06-096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carman JA, Pond L, Nashold F, Wassom DL, Hayes CE. 1992. Immunity to Trichinella spiralis infection in vitamin A-deficient mice. J Exp Med 175:111–120. doi: 10.1084/jem.175.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ross AC, Stephensen CB. 1996. Vitamin A and retinoids in antiviral responses. FASEB J 10:979–985. [PubMed] [Google Scholar]
- 11.Mora JR, Iwata M, von Andrian UH. 2008. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol 8:685–698. doi: 10.1038/nri2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, Misiak A, Dungan LS, Sutton CE, Streubel G, Bracken AP, Mills KH. 2013. Retinoic acid expression associates with enhanced IL-22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med 210:1117–1124. doi: 10.1084/jem.20121588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spencer SP, Wilhelm C, Yang Q, Hall JA, Bouladoux N, Boyd A, Nutman TB, Urban JF Jr, Wang J, Ramalingam TR, Bhandoola A, Wynn TA, Belkaid Y. 2014. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science 343:432–437. doi: 10.1126/science.1247606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collins JW, Keeney KM, Crepin VF, Rathinam VA, Fitzgerald KA, Finlay BB, Frankel G. 2014. Citrobacter rodentium: infection, inflammation and the microbiota. Nat Rev Microbiol 12:612–623. doi: 10.1038/nrmicro3315. [DOI] [PubMed] [Google Scholar]
- 15.Bry L, Brenner MB. 2004. Critical role of T cell-dependent serum antibody, but not the gut-associated lymphoid tissue, for surviving acute mucosal infection with Citrobacter rodentium, an attaching and effacing pathogen. J Immunol 172:433–441. doi: 10.4049/jimmunol.172.1.433. [DOI] [PubMed] [Google Scholar]
- 16.Sonnenberg GF, Artis D. 2012. Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity 37:601–610. doi: 10.1016/j.immuni.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mundy R, MacDonald TT, Dougan G, Frankel G, Wiles S. 2005. Citrobacter rodentium of mice and man. Cell Microbiol 7:1697–1706. doi: 10.1111/j.1462-5822.2005.00625.x. [DOI] [PubMed] [Google Scholar]
- 18.Smith SM, Levy NS, Hayes CE. 1987. Impaired immunity in vitamin A-deficient mice. J Nutr 117:857–865. [DOI] [PubMed] [Google Scholar]
- 19.Ma Y, Chen Q, Ross AC. 2005. Retinoic acid and polyriboinosinic:polyribocytidylic acid stimulate robust anti-tetanus antibody production while differentially regulating type 1/type 2 cytokines and lymphocyte populations. J Immunol 174:7961–7969. doi: 10.4049/jimmunol.174.12.7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wiles S, Dougan G, Frankel G. 2005. Emergence of a ‘hyperinfectious’ bacterial state after passage of Citrobacter rodentium through the host gastrointestinal tract. Cell Microbiol 7:1163–1172. doi: 10.1111/j.1462-5822.2005.00544.x. [DOI] [PubMed] [Google Scholar]
- 21.Lefrancois L, Lycke N. 2001. Isolation of mouse small intestinal intraepithelial lymphocytes, Peyer's patch, and lamina propria cells. Curr Protoc Immunol Chapter 3:Unit 3.19. doi: 10.1002/0471142735.im0319s17. [DOI] [PubMed] [Google Scholar]
- 22.Restori KH, McDaniel KL, Wray AE, Cantorna MT, Ross AC. 2014. Streptococcus pneumoniae-induced pneumonia and Citrobacter rodentium-induced gut infection differentially alter vitamin A concentrations in the lung and liver of mice. J Nutr 144:392–398. doi: 10.3945/jn.113.186569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Basu R, O'Quinn DB, Silberger DJ, Schoeb TR, Fouser L, Ouyang W, Hatton RD, Weaver CT. 2012. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 37:1061–1075. doi: 10.1016/j.immuni.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meresse B, Malamut G, Cerf-Bensussan N. 2012. Celiac disease: an immunological jigsaw. Immunity 36:907–919. doi: 10.1016/j.immuni.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 25.Vallance BA, Deng W, Jacobson K, Finlay BB. 2003. Host susceptibility to the attaching and effacing bacterial pathogen Citrobacter rodentium. Infect Immun 71:3443–3453. doi: 10.1128/IAI.71.6.3443-3453.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh S, Dai C, Brown K, Rajendiran E, Makarenko S, Baker J, Ma C, Halder S, Montero M, Ionescu VA, Klegeris A, Vallance BA, Gibson DL. 2011. Colonic microbiota alters host susceptibility to infectious colitis by modulating inflammation, redox status, and ion transporter gene expression. Am J Physiol Gastrointest Liver Physiol 301:G39–G49. doi: 10.1152/ajpgi.00509.2010. [DOI] [PubMed] [Google Scholar]
- 27.Sarraf P, Frederich RC, Turner EM, Ma G, Jaskowiak NT, Rivet DJ III, Flier JS, Lowell BB, Fraker DL, Alexander HR. 1997. Multiple cytokines and acute inflammation raise mouse leptin levels: potential role in inflammatory anorexia. J Exp Med 185:171–175. doi: 10.1084/jem.185.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, Nunez G. 2012. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science 336:1325–1329. doi: 10.1126/science.1222195. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.