

Abstract
Group A Streptococcus (GAS) can cause life-threatening invasive infections, including necrotizing fasciitis. There are no effective treatments for severe invasive GAS infections. The platelet-activating factor (PAF) acetylhydrolase SsE produced by GAS is required for invasive GAS to evade innate immune responses and to invade soft tissues. This study determined whether the enzymatic activity of SsE is critical for its function in GAS skin invasion and inhibition of neutrophil recruitment and whether SsE is a viable target for immunotherapy for severe invasive GAS infections. An isogenic derivative of M1T1 strain MGAS5005 producing SsE with an S178A substitution (SsES178A), an enzymatically inactive SsE mutant protein, was generated. This strain induced higher levels of neutrophil infiltration and caused smaller lesions than MGAS5005 in subcutaneous infections of mice. This phenotype is similar to that of MGAS5005 sse deletion mutants, indicating that the enzymatic activity of SsE is critical for its function. An anti-SsE IgG1 monoclonal antibody (MAb), 2B11, neutralized the PAF acetylhydrolase activity of SsE. Passive immunization with 2B11 increased neutrophil infiltration, reduced skin invasion, and protected mice against MGAS5005 infection. However, 2B11 did not protect mice when it was administered after MGAS5005 inoculation. MGAS5005 induced vascular effusion at infection sites at early hours after GAS inoculation, suggesting that 2B11 did not always have access to infection sites. Thus, the enzymatic activity of SsE mediates its function, and SsE has the potential to be included in a vaccine but is not a therapeutic target. An effective MAb-based immunotherapy for severe invasive GAS infections may need to target virulence factors that are critical for systemic survival of GAS.
INTRODUCTION
Group A Streptococcus (GAS) is a major human pathogen that commonly causes pharyngitis and superficial skin infections (1). This pathogen can also cause severe invasive infections, such as necrotizing fasciitis, streptococcal toxic shock syndrome, pneumonia, and bacteremia. Streptococcal necrotizing fasciitis results from GAS infection of the subcutaneous tissue, which progresses rapidly, causes necrosis of the fascia and subcutaneous tissue, and leads to systemic infection (2). Annually in the United States, there are more than 10 million cases of streptococcal pharyngitis and about 10,000 cases of invasive GAS infections (3.5 cases per 100,000 persons), with a fatality rate of 13.7%, and invasive infections are most frequently caused by serotype M1, M3, and M12 GAS strains (3).
While antibiotic treatment is effective to treat pharyngitis patients, it is not effective for treating severe invasive GAS infections (4). Prompt surgical debridement, fluid and electrolyte management, and analgesia are mainstays of therapy for necrotizing fasciitis (5–7). Clindamycin, hyperbaric oxygen therapy, and intravenous immunoglobulin are used as adjunctive treatments of severe invasive GAS infections (4–7). New strategies to treat severe GAS infections are desirable.
Severe invasive GAS isolates are usually more virulent than pharyngitis isolates (8, 9). Hypervirulence of some invasive GAS isolates is attributable to natural mutations in the two-component regulatory system CovRS (also known as CsrRS) or to their capacity to acquire CovRS mutations during infection (8–11). CovRS negatively regulates many virulence factors (12–15), including the capsule synthase HasA (12), streptolysin S (15), DNase Sda1 (16), interleukin-8/CXC chemokine peptidase SpyCEP (17), and platelet-activating factor (PAF) acetylhydrolase SsE (18, 19). Many CovRS-regulated virulence factors evade neutrophil responses. Natural CovRS mutations are selected by neutrophils to enhance the expression of multiple virulence factors and downregulate the production of the nonspecific protease SpeB, maximizing the potential of GAS to evade neutrophil responses and resulting in hypervirulence (11, 16, 20–22).
CovRS-regulated virulence factors that critically contribute to the hypervirulence of invasive GAS are potential targets for the development of therapeutics for the treatment of severe invasive GAS infections. We target SsE to test this potential. SsE is a protective antigen and is required for the skin invasion and dissemination of hypervirulent M1T1 GAS in a mouse model of necrotizing fasciitis (23). SsE has potent PAF acetylhydrolase activity and critically contributes to the inhibition of neutrophil recruitment by GAS (19, 24). If the enzymatic activity of SsE is critical for its functions, an inhibitory antibody of SsE is likely protective and can have the potential to be developed as a therapeutic agent for treatment of invasive GAS infection. In this study, we tested the essentiality of the SsE activity for GAS function, generated a neutralizing monoclonal antibody (MAb), and evaluated the protection and therapeutic value of the inhibitory MAb. We found that a derivative of M1T1 strain MGAS5005 that produced an inactive SsE mutant displayed the phenotype of an sse deletion mutant of MGAS5005 and that the inhibitory anti-SsE MAb protected mice against MGAS5005 infection when it was administered prior to, but not after, GAS inoculation.
MATERIALS AND METHODS
Declaration of ethical approvals.
All animal procedures were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (25). The protocols for the experiments were approved by the Institutional Animal Care and Use Committee at Montana State University ([MSU] permit numbers 2011-57 and 2014-45). Blood was collected from healthy donors in accordance with a protocol approved by the Institutional Review Board at MSU (protocol number BL120513). Written informed consent was provided by study participants and/or their legal guardians.
Materials.
The following chemicals and reagents for cell fusion, hybridoma selection, and cell culture were from HyClone Laboratory, Inc.: sodium pyruvate, nonessential amino acids, HEPES, l-glutamine, penicillin-streptomycin, fetal bovine serum, and RPMI 1640 medium. Polyethylene glycol (PEG) was from EMD Chemicals, Inc. Hypoxanthine aminopterin thymidine (HAT), dimethyl sulfoxide (DMSO), and 2-mercaptoethanol were from Sigma. HB basal medium T000 and HB101 lyophilized supplement T151 were from Irvine Scientific. Protein G agarose was purchased from Pierce. An isotype control MAb, MOPC-21, was purchased from BioXcell. A mouse monoclonal antibody isotyping enzyme-linked immunosorbent assay (ELISA) kit was from Eagle Bioscience. Recombinant SsE protein and anti-SsE antiserum were described previously (23).
Bacterial strains and growth.
MGAS5005 (15), its sse deletion mutant MGAS5005 Δsse (18), and a nonspontaneous revertant of the MGAS5005 Δsse mutant, MGAS5005 Δsse-sse (18), have been described previously. These strains and a derivative strain of MGAS5005 Δsse, MGAS5005 Δsse-sse with an S-to-A change at position 178 encoded by sse (MGAS5005 Δsse-sseS178A) and producing inactive SsES178A mutant protein, were grown in Todd-Hewitt broth (Difco Laboratories, Detroit, MI) supplemented with 0.2% yeast extract (THY broth). An MGAS5005 srtA mutant was grown in THY broth containing 150 mg of spectinomycin per liter. Tryptose agar with 5% sheep blood (Becton Dickinson, Cockeysville, MD) and THY agar were used as solid media. GAS bacteria used for mouse infection experiments were grown to the exponential growth phase, harvested by centrifugation, and washed three times with and resuspended in pyrogen-free Dulbecco's phosphate-buffered saline (DPBS). Inocula and viable GAS bacteria from mouse tissues were determined by plating.
Generation of MGAS5005 Δsse-sseS178A.
The codon of the sse gene in pGRV-sse (18) for the catalytic residue, Ser178, of SsE was mutated into a codon for Ala by site-directed mutagenesis using a QuikChange site-directed mutagenesis kit and primers 5′-GTTTTCCTTATGGGAGATGCAGCAGGTGGAGGTTTAGCC-3′ and 5′-GGCTAAACCTCCACCTGCTGCATCTCCCATAAGGAAAA-3′, resulting in the suicide plasmid pGRV-sseS178A. The mutated codon for the Ser-to-Ala replacement (underlined) was verified by DNA sequencing. pGRV-sseS178A was introduced by electroporation into the MGAS5005 Δsse mutant, which had a deletion of a 621-bp fragment of sse (18). The plasmid was inserted into the MGAS5005 Δsse genome through a homologous recombination event at one flanking fragment of the sseS178A mutant gene, resulting in spectinomycin-resistant merodiploid transconjugants that were selected by 150 μg/ml spectinomycin. One transconjugant was grown in THY broth without spectinomycin selection for eight passages (one passage represents an increase in the optical density at 600 nm [OD600] of 0.05 to 0.7) to allow the second crossover event to occur in the other flanking fragment, generating an MGAS5005 derivative that carried sseS178A. The culture was plated on THY agar plates after the last passage, and colonies obtained were spotted in parallel on THY agar plates with and without spectinomycin to identify strains that were sensitive to spectinomycin. Spectinomycin-sensitive strains were analyzed by PCR to identify strains that had a greater sse PCR product than MGAS5005 Δsse. The sse locus of an MGAS5005 sseS178A strain was sequenced to confirm the Ser-to-Ala replacement and to rule out spurious mutations.
Generation of an srtA mutant.
An MGAS5005 mutant strain defective in the sortase A gene (srtA::aad, or srtA mutant) was generated by insertion inactivation. An internal 512-bp fragment (bases 73 to 591) of the srtA gene was PCR amplified using primers 5′-ACCATGGTTAATTCTAGGTTTAGCATTGC-3′ and 5′-ACCATGGTGTATCATCGATAACATCAAC-3′ and cloned into pFWaad (26) at the NcoI site to yield the suicide plasmid pFWaad-srtA mutant. The suicide plasmid was introduced into MGAS5005 by electroporation, and srtA mutants were selected with spectinomycin and confirmed by PCR and DNA sequence analyses.
Generation of hybridomas.
BALB/c mice were immunized subcutaneously with 30 μg of SsE using alum as an adjuvant on day 1 and day 15. Four weeks later, the mice were boosted by intravenous (i.v.) injection of 30 μg of SsE. Spleens were collected 3 days after the i.v. booster and ground to obtain spleen cells; fusion was performed using a ratio of 2 spleen cells to 1 cell of myeloma cell line SP2/0, and cells were plated in 96-well plates. Hybridomas of myeloma and B cells were selected using the HAT medium, which contained HAT. An enzyme-linked immunosorbent assay (ELISA) was used to identify wells that contained SsE-specific antibodies. Our interest was inhibitory MAbs; therefore, we did not focus on a particular antibody isotype and subspecies and used polyclonal secondary antibodies for mouse IgG-IgM-IgA (conjugated with horseradish peroxidase [HRP]) in ELISAs to screen for SsE-specific antibody-producing hybridomas. Hybridomas in each positive well were subcloned through a process whereby cells were counted and diluted to 1,000, 100, 50, 10, and 5 cells/ml and plated on 96-well plates with feeder cells. After 2 weeks, subclones were checked visually under an inverted microscope for colonies of cells in wells. An ELISA was performed for each colony arising from a single cell to identify hybridoma clones with robust anti-Sse antibody production.
Production of MAb 2B11.
Hybridoma 2B11 clones producing anti-SsE-specific antibody were first grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% serum in T175 flasks in 10% CO2 at 37°C to a density of 6 × 105 cells/ml. The cells were spun down by centrifugation, and the cell pellet was resuspended in serum-free HB101 complete medium. The cells were transferred to 2,000-ml roller bottles containing 500 ml of serum-free HB101 complete medium at a density of 3 × 105 cells/ml. The culture in tightly capped bottles was slowly rolled at 37°C on a roller for 5 days. The culture was centrifuged to obtain supernatant that usually contained 5 to 10 mg of anti-SsE MAb 2B11/liter.
Purification of MAb 2B11.
MAb in the supernatant of the hybridoma culture was precipitated overnight at 4°C by ammonium sulfate at 50% saturation. MAb precipitates were pelleted by centrifugation and dissolved in endotoxin-free DPBS and dialyzed against 4 liters of DPBS at 4°C overnight. The dialyzed MAb sample was loaded onto a 10-ml protein G-Sephadex column preequilibrated with DPBS. The column was washed with 80 ml of DPBS. The MAb was eluted with 0.1 M glycine, pH 3.0, and collected in tubes containing 0.1 ml of 1 M Tris, pH 8.0. Fractions containing the MAb at >95% purity based on SDS-PAGE analysis were pooled and dialyzed against endotoxin-free DPBS.
Isotyping of anti-SsE MAb 2B11.
The isotype of MAb 2B11 was determined using an ELISA ISO13-K01 kit from Eagle Biosciences. Each of the eight wells in strips from the kit were coated with antibodies specific for IgG1, IgG2a, IgG2b, IgG3, IgA, IgM, and kappa or lambda light chains. Purified MAb 2B11 was added to each well of the strip, followed by 50 μl of HRP-conjugated goat anti-mouse IgG, IgA, and IgM. Strips were incubated for 1 h with rotational mixing. Following incubation, wells were washed four times with 300 μl of wash buffer, and 100 μl of 3,3′,5,5′-tetramethylbenzidine (TMB) substrate was added and incubated for 20 min. The substrate reaction was stopped by the addition of 100 μl of stop solution, resulting in the appearance of yellow color, and the isotype was discernible by the yellow pigment.
Neutralization of SsE PAF acetylhydrolase activity by MAb 2B11.
SsE in 40 mM Tris-HCl, pH 8.0, was incubated with 2B11 at various concentrations in wells of a 96-well plate at room temperature for 5 min, and the PAF acetylhydrolase activity of the reaction mixtures was then assayed using a 2-thio-PAF assay as previously described (24). Briefly, the SsE-2B11 solutions were mixed with 30 μl of solution containing 0.88 mM 2-thio-PAF and 1.26 mM 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB). Absorbance at 414 nm (A414) of the reaction mixtures was recorded every 9 s with a SpectraMax 384 Plus spectrophotometer (Molecular Devices, Sunnyvale, CA) and used to determine the rates of hydrolysis of 2-thio-PAF. The decrease produced by 2B11 in the slope of the A414-versus-time plots was used to calculate neutralization of the SsE activity by 2B11. The PAF acetylhydrolase activity in the culture supernatant of GAS strains was similarly measured.
Phagocytosis and survival of GAS in blood.
For phagocytosis assays, MGAS5005 and its mutants from the exponential growth phase in THY broth were washed with DPBS and labeled with 0.75 μg/ml fluorescein isothiocyanate (FITC) in DPBS at 37°C for 20 min. The labeled bacteria were washed and resuspended at 1 × 109 CFU/ml in DPBS. A fluorescence-assisted cell sorting (FACS)-based whole-blood phagocytosis assay was performed as previously described (27, 28). Briefly, 10 μl of the labeled bacteria was mixed with 100 μl of nonimmune heparinized human blood and incubated with gentle shaking at 37°C for 20 min. The samples were immediately processed using an Immunolyse kit (Beckman Coulter) according to the manufacturer's protocol and analyzed by flow cytometry. The percentage of polymorphonuclear leukocytes (PMNs) with fluorescent bacteria was used as a measurement of phagocytosis efficiency. For GAS survival in blood, about 106 CFU of unlabeled bacteria in 20 μl of DPBS was added into 0.5 ml of the same nonimmune heparinized human blood and rotated from end to end at 37°C for 1 h. Viable GAS bacteria in the blood samples were quantified by plating.
Histological analyses.
At 24 h after subcutaneous inoculation of 0.2 ml of MGAS5005 suspension at an OD600 of 1.0 in DPBS in three BALB/c mice treated with MAb MOPC-21 or 2B11 at 6 h prior to GAS inoculation, skin samples were excised with a wide margin around the infection site after the skin was peeled off and fixed in 10% neutral buffered formalin for 24 h. The samples were dehydrated with ethanol, cleared with xylene, and infiltrated with paraffin using a Tissue Embedding Console System (Sakura Finetek, Inc.). The paraffin blocks were processed to obtain 4-μm sections, which were stained with hematoxylin and eosin (H&E) or with a tissue Gram stain kit from Richard-Allan Scientific according to the manufacturer's protocol. The stained slides were examined using a Nikon Eclipse 80i microscope.
Mouse infections.
Immunocompetent, hairless female mice (strain Crl:SKH1-hrBR) (5 weeks old) were from Charles River Laboratory. BALB/c mice were bred at the Animal Resource Center at Montana State University using breeding mice from Jackson Laboratories. In the comparison of MGAS5005, MGAS5005 Δsse, MGAS5005 Δsse-sse, and MGAS5005 Δsse-sseS178A, groups of five hairless and BALB/c female mice were inoculated subcutaneously with 0.2 ml of GAS suspensions at an OD600 of 1.0. In one series of experiments to evaluate the protective effect of MAb 2B11 in GAS infection, groups of 5-week-old, female BALB/c mice were injected intraperitoneally (i.p.) with 0.5 ml of DPBS alone or with 250 mg of MAb 2B11 or control MAb MOPC-21 and, 6 h later, injected with 0.2 ml of MGAS5005 suspension in DPBS at an OD600 of 0.9 (an inoculum of about 108 CFU). In another series of experiments, mice were first injected with 0.2 ml of MGAS5005 at an OD600 of 0.9 and then 6 h later injected intraperitoneally with 250 mg of 2B11 or MOPC-21 in 0.5 ml of DPBS. In each series of experiments, mice in groups of 10 mice were monitored daily for 14 days to determine survival rates, and mice in groups of 5 mice were euthanized at 24 h after GAS inoculation to collect skin samples for measurement of lesion size and neutrophil recruitment and to obtain the liver and spleen for measurement of GAS loads. Skin lesions were recognized by the boundary of inflammation after the skin around the infection site was peeled off, and lesion sizes were measured by analyzing lesion pictures using the area measurement tool of the Adobe Acrobat, version 9, software program (Adobe Systems, Inc.). Numbers of recruited neutrophils at infection sites in the skin were determined by a myeloperoxidase assay, as described previously (19). GAS loads in organs were determined by plating.
Vascular effusion assay.
MGAS5005-induced vascular effusion in skin was assessed using a Miles assay (29). Groups of five BALB/c mice were injected subcutaneously with 0.2 ml of MGAS5005 suspension at an OD600 of 0.9, and 0.1 ml of 2% Evans blue dye was injected into the tail vein at 6 h and 20 h after GAS inoculation. The mice were sacrificed at 30 min after the Evans blue injection to peel off skin for examining vascular effusion.
Other analyses.
Western blotting was performed as described previously (18). An endotoxin test for purified 2B11 MAb was performed using the limulus amebocyte lysate Pyrogent Plus (Lonza, Walkersville, MD) according to the manufacturer's protocol. Statistical analyses were performed using the Prism software program (Graph-Pad Software, Inc.). Survival data were analyzed using a log rank (Mantel-Cox) test. The data shown in Fig. 2B and C were analyzed using a one-way analysis of variance (ANOVA) Newman-Keuls test, and other data of lesion sizes, neutrophil levels, GAS in human blood, and GAS loads were analyzed using a two-tailed Mann-Whitney t test.
FIG 2.

Essentiality of the enzymatic activity of SsE for its function. (A) Comparison of skin lesions at day 2 after subcutaneous inoculation of immunocompetent hairless mice with 0.2 ml of MGAS5005, MGAS5005 Δsse, MGAS5005 Δsse-sse, and MGAS5005 Δsse-sseS178A at OD600 values of 0.9. (B) Lesion sizes (solid symbols) and neutrophil recruitment (open symbols) at 24 h after subcutaneous inoculation of MGAS5005, MGAS5005 Δsse-sse, and MGAS5005 Δsse-sseS178A in BALB/c mice. ns, not significant for the difference in lesion sizes between MGAS5005 and MGAS5005 Δsse-sse; ***, P < 0.001, for the difference in PMN numbers (open circles) between MGAS5005 Δsse-sseS178A and the other strains. (C) Phagocytosis of MGAS5005 (wt), MGAS5005 Δsse (Δsse), MGAS5005 Δsse-sseS178A (sseS178A), and MGAS5005 srtA mutant (srtA) bacteria by human PMNs. FITC-labeled bacteria (107 CFU) were incubated with 100 μl of nonimmune heparinized human blood at 37°C for 20 min. Red blood cells were lysed using an Immunolyse kit, and percentages of PMNs with associated bacteria were determined by flow cytometry. ***, P < 0.001, for the difference in the values for phagocytosis between the srtA mutant and the other strains. (D) GAS survival in human blood. The umbers of the indicated strains inoculated (solid squares) and numbers of viable GAS bacteria in nonimmune blood after 1 h of incubation (open circles) are shown. ns, not significant; **, P < 0.01, for the difference in GAS numbers between the inoculum and incubated samples of each strain.
RESULTS
Essentiality of the enzymatic activity of SsE for its function.
To determine whether the enzymatic activity of SsE is required for its function in skin invasion and inhibition of neutrophil infiltration, MGAS5005 Δsse-sseS178A, a derivative mutant of MGAS5005 producing an enzymatically inactive SsES178A mutant protein, was constructed. The mutated sseS178A gene was knocked into the genome of MGAS5005 Δsse at the sse locus, resulting in MGAS5005 Δsse-sseS178A. Since construction of mutants of M1T1 GAS frequently introduces spontaneous mutations that downregulate the production of the M protein, a major virulence factor, and, in turn, alters infections (30), we checked the production of the M protein by Western blotting. As shown in Fig. 1A, MGAS5005 Δsse, MGAS5005 Δsse-sseS178A, and MGAS5005 Δsse-sse all produced the M protein at levels that were similar to the level in MGAS5005. The S178A replacement did not alter the levels of SsE production as Western blotting detected similar levels of SsE or SsES178A in the culture supernatants of MGAS5005, MGAS5005 Δsse-sse, and MGAS5005 Δsse-sseS178A, whereas SsE was not detected in the MGAS5005 Δsse supernatant; all the strains secreted the Spy19 protein at similar levels (Fig. 1A). As expected, the culture supernatants of MGAS5005 Δsse and MGAS5005 Δsse-sseS178A had no detectable PAF acetylhydrolase activity, whereas it was detected in the culture supernatants of MGAS5005 and MGAS5005 Δsse-sse (Fig. 1B). Thus, an isogenic strain of MGAS5005 was successfully generated to produce an enzymatically inactive SsES178A mutant protein.
FIG 1.
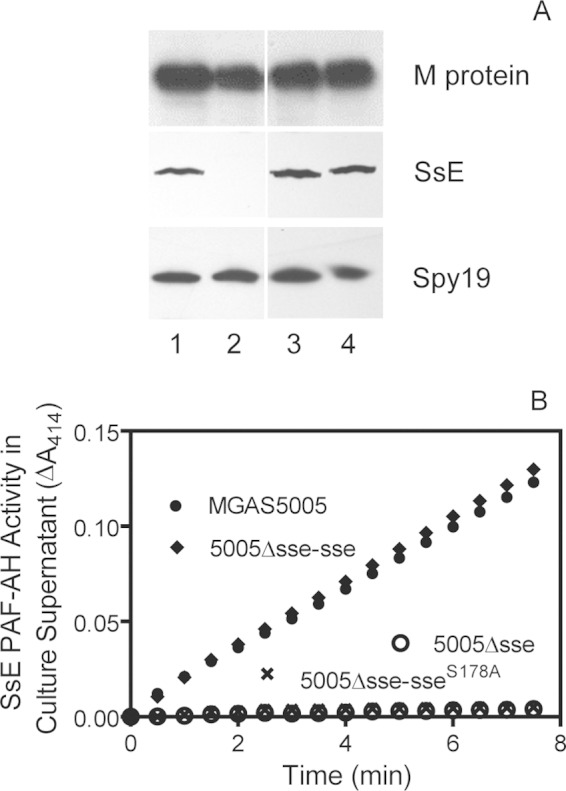
Production of enzymatically inactive SsES178A by MGAS5005 Δsse-sseS178A. (A) Western blots showing that MGAS5005 Δsse-sseS178A had similar levels of SsES178A in its culture supernatant as the positive-control strains. Blots for the M protein and Spy19 are included as controls. Lane 1, MGAS5005; lane 2, MGAS5005 Δsse; lane 3, MGAS5005 Δsse-sse; lane 4, MGAS5005 Δsse-sseS178A. (B) PAF acetylhydrolase (AH) activity measured by the 2-thio-PAF acetylhydrolase assay in 10 μl of culture supernatant of MGAS5005, MGAS5005 Δsse, MGAS5005 Δsse-sse, and MGAS5005 Δsse-sseS178A.
MGAS5005 Δsse-sseS178A was next compared with MGAS5005, MGAS5005 Δsse, and MGAS5005 Δsse-sse in terms of skin invasion and neutrophil response during subcutaneous infection in mice. Like MGAS5005 Δsse, MGAS5005 Δsse-sseS178A in subcutaneous infection of immunocompetent hairless mice was limited to inoculation sites, whereas, like MGAS5005, MGAS5005 Δsse-sse invaded soft tissue around inoculation sites (Fig. 2A). These results were confirmed with infection results in BALB/c mice. MGAS5005 Δsse-sseS178A caused small lesions of 112 ± 13 mm2, but MGAS5005 and MGAS5005 Δsse-sse led to greater lesions of 416 ± 70 mm2 and 395 ± 72 mm2, respectively, at 24 h after inoculation in BALB/c mice (Fig. 2B). In addition, the reintroduction of the wild-type (wt) sse gene, but not sseS178A, into MGAS5005 Δsse reduced neutrophil recruitment to the levels in MGAS5005 infection (Fig. 2B). Thus, the enzymatic activity of SsE is critical for the function of SsE in GAS skin invasion and inhibition of neutrophil recruitment.
SsE is a secreted protein, and MGAS5005 Δsse and MGAS5005 Δsse-sseS178A had normal M protein production. Therefore, these mutants are not expected to lose the resistance of MGAS5005 to phagocytosis by neutrophils. To rule out the possibility that the phenotype of these mutants indicated by the results shown in Fig. 2A and B might be due to other unknown mutations during the construction of the mutants, these mutants were compared with MGAS5005 with respect to phagocytosis by neutrophils and survival in nonimmune human blood. In an FACS-based phagocytosis assay using nonimmune human blood, percentages of neutrophils that had associated GAS were all about 40% for MGAS5005, MGAS5005 Δsse, and MGAS5005 Δsse-sseS178A, whereas 80% of neutrophils had associated MGAS5005 srtA mutant bacteria (Fig. 2C). SrtA anchors LPXTGE motif-containing proteins, including the antiphagocytic M protein, to the cell wall. These results indicate that the sse deletion and SsES178A replacement did not alter the resistance of MGAS5005 to phagocytosis by neutrophils. This conclusion is further supported by the survival of these bacteria in the same nonimmune human blood. The numbers of viable GAS bacteria were similar to the inoculum for MGAS5005, MGAS5005 Δsse, and MGAS5005 Δsse-sseS178A after 1 h of incubation in blood, whereas viable MGAS5005 srtA mutant counts dropped from 106 CFU of inoculum to 102 CFU (Fig. 2D).
Neutralizing anti-SsE MAb 2B11.
Since the enzymatic activity of SsE is critical for its function, neutralizing antibodies of SsE may be protective. To test this idea, five hybridoma clones that produced anti-SsE MAb were generated by standard hybridoma technology. Initial screening using the PAF acetylhydrolase assay found that the culture supernatant of clone 2B11, but not of four other clones, inhibited the PAF acetylhydrolase activity of SsE. Next, anti-SsE 2B11 MAb was produced from culture of the 2B11 clone in serum-free medium and purified by affinity chromatography. The purified MAb 2B11 had no endotoxin (data not shown). The antibody was IgG1 with a kappa light chain (Fig. 3A). MAb 2B11 showed dose-dependent inhibition of the PAF acetylhydrolase activity of SsE (Fig. 3B), and the inhibition of the SsE activity as a function of the concentration of 2B11 fitted to a one-site-specific binding model with a dissociation constant, KD, of 21 ± 4 nM (Fig. 3C).
FIG 3.
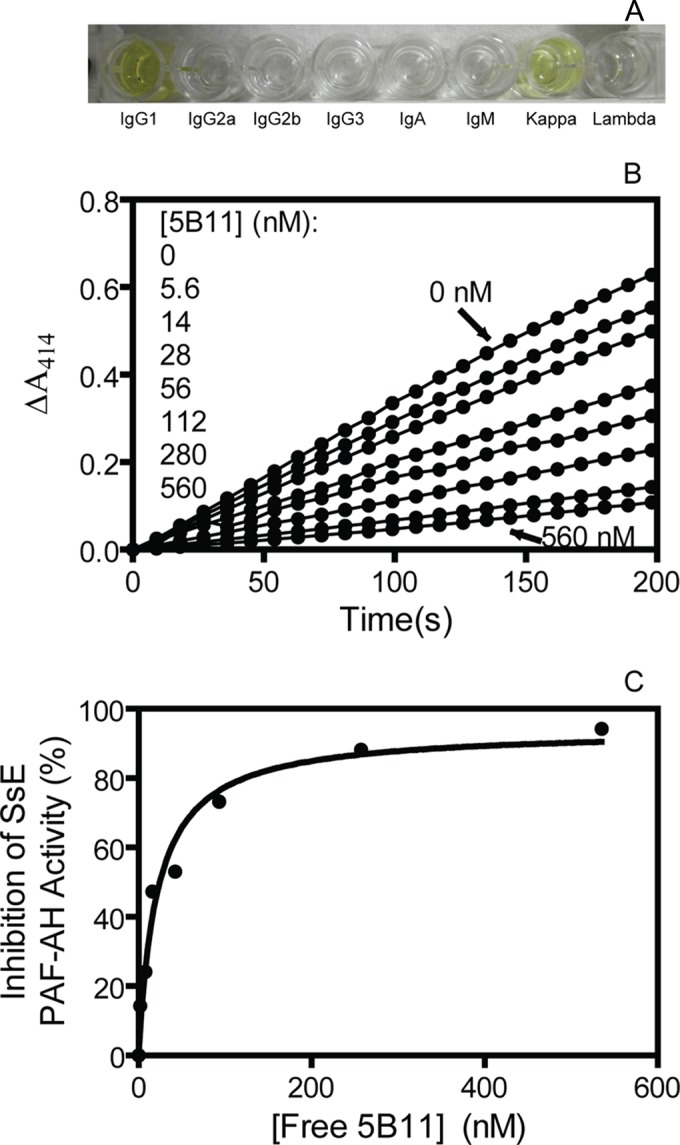
Anti-SsE IgG1 MAb 2B11 neutralizes the PAF acetylhydrolase activity of SsE. (A) ELISA for the antibody isotype. (B) Plots of ΔA414 versus time for the measurement of the SsE activity to hydrolyze 2-thio-PAF in the presence of 2B11 at the indicated concentrations. In the assay, 26 nM SsE was used. The lines from top to bottom correspond to descending 5B11 concentrations as indicated on the figure. (C) Inhibition of the SsE activity as a function of the concentration of free 2B11. Inhibition percentages were calculated from the slopes of the lines in panel B, and free 5B11 concentrations were obtained by subtracting the concentrations of SsE-5B11 complex that were estimated according to the inhibition extent of the SsE activity.
Anti-SsE MAb 2B11 protects mice against MGAS5005 infection in passive immunization.
Whether MAb 2B11 is protective was tested in a passive immunization and challenge experiment. Nine of 10 mice that received MAb 2B11 through the intraperitoneal route 6 h prior to subcutaneous MGAS5005 inoculation survived, whereas 8 of 10 mice that received DPBS did not survive (Fig. 4A). All the nonsurviving mice had high GAS burdens in the liver, whereas no GAS was detected in the liver of surviving mice at day 12 after MGAS5005 inoculation (Fig. 4B). The infected skin of surviving mice fell off during resolution of infection, and the area without fur of nine surviving anti-SsE MAb-treated mice had a mean size ± standard deviation of 0.98 ± 46 cm2. The inflammation size for eight nonsurviving mice was 9.60 ± 2.68 cm2. These data indicate that passive immunization with anti-SsE MAb 2B11 protects mice and reduces skin invasion in MGAS5005 infection.
FIG 4.

Passive immunization with MAb 2B11 protects mice in subcutaneous MGAS5005 infection. Mice were injected i.p. with 250 mg of 2B11, DPBS buffer control, or control MAb MOPC-21 at 6 h prior to subcutaneous MGAS5005 inoculation. (A and D) Survival curves in tests with DPBS and MOPC-2 controls. (B and C) GAS loads in liver and lesion sizes at the endpoint of nonsurviving mice or at 12 days after GAS inoculation in the test with the DPBS control. The lesion sizes at 12 days after GAS inoculation correspond to the areas of holes in the skin.
It is possible that the antibody in the passive immunization nonspecifically enhanced innate immune responses. To rule out this possibility, passive immunization and challenge were performed using a murine IgG1 MAb, MOPC-21, as an isotype MAb control. Passive immunization with anti-SsE MAb 2B11 significantly protected mice against MGAS5005 infection compared with the treatment with MOPC-21, confirming the protective effect of anti-SsE MAb in passive immunization (Fig. 4D).
Reduction in skin invasion and systemic dissemination and enhancement of neutrophil infiltration by passive immunization with MAb 2B11.
SsE is important for GAS skin invasion and systemic dissemination (18) and for GAS inhibition of neutrophil infiltration (19). To determine whether 2B11 protects mice against MGAS5005 infection by reducing skin invasion and systemic dissemination and increasing neutrophil recruitment, we measured lesion sizes, GAS burdens in the liver and spleen, and neutrophil levels at skin infection sites at day 1 after subcutaneous MGAS5005 inoculation of mice that had been passively immunized with anti-SsE MAb 2B11 or control MAb MOPC-21. Representative images of inside-out infection sites of mice show that the mice passively immunized with anti-SsE MAb 2B11 (Fig. 5A) had smaller lesions and more neutrophils than the mice treated with MOPC-21 (Fig. 5B). Compared with the MOPC-21 treatment, the passive immunization with MAb 2B11 reduced lesion sizes by 60% (from 4.6 ± 0.7 cm2 to 1.8 ± 0.5 cm2) (Fig. 5C), GAS load in liver by 15-fold from (8.3 ± 4.8) × 105 CFU/g to (5.4 ± 5.1) × 104 CFU/g (Fig. 5D), and GAS load in spleen by 103-fold from (1.5 ± 1.5) × 107 CFU/g to (1.5 ± 2.2) × 104 CFU/g (Fig. 5E) but enhanced neutrophil recruitment by 6.8-fold from (2.26 ± 1.15) × 105 neutrophils/mm2 to (3.33 ± 1.07) × 104 neutrophils/mm2 (Fig. 5F). These effects of MAb 2B11 are equivalent to those of the sse deletion previously reported (19), supporting the idea that the neutralization of the enzymatic activity of SsE by MAb 2B11 diminishes the function of SsE in GAS skin invasion, systemic dissemination, and inhibition of neutrophil recruitment.
FIG 5.

Passive immunization with 2B11 reduces MGAS5005 skin invasion and systemic dissemination and increases neutrophil recruitment. The immunization and challenge were done as described in the legend of Fig. 4. Presented are inside-out images of skin infection sites (A and B), lesion sizes (C), GAS loads in liver (D) and spleen (E), and neutrophil recruitment in mice immunized with 2B11 and control MOPC-21 (F) at 24 h after GAS inoculation. The dashed circles in blue and solid traces in black in panel B indicate the inoculation site and spread area, respectively.
The effects of 2B11 on MGAS5005 skin invasion and neutrophil recruitment were further confirmed by histological analyses of the skin infection sites in mice passively treated with 2B11 and MOPC-21 by Gram and H&E stains. As shown in Fig. 6, the inoculation sites of mice treated with MOPC-21 had fewer inflammatory cells than those of mice treated with 2B11, and inflammatory cells in 2B11-treated mice directly engaged with GAS, especially at the edge of the GAS territory (Fig. 6E and F), but did not in MOPC-21-treated mice (Fig. 6A and B). The inflammatory region as indicated by the black traces in Fig. 5B had disseminated GAS in the interstice of adipose cells in the epidermis (Fig. 6C) and few inflammatory cells (Fig. 6D). Apparently, enhanced infiltration of inflammatory cells reduced GAS dissemination in 2B11-treated mice.
FIG 6.

Histological analyses of MGAS5005 infection sites in mice passively immunized with MOPC-21 (A to D) and 2B11 (E and F) MAbs. Three BALB/c mice treated with 2B11 or MOPC-21 were subcutaneously inoculated on the back with 1.0 × 108 CFU of MGAS5005, and the skin samples were collected at 24 h after inoculation. Shown are representative pictures of the Gram- and H&E-stained samples taken at a ×20 magnification. Images in panels A and B are of the inoculation site, and images in panels C and D are of the spread area in MOPC-21-treated mice. No extensive spreading was present in 2B11-treated mice.
Reduced protection of mice by 2B11 administration post-GAS infection.
To determine whether anti-SsE MAb has therapeutic value, mice were first infected with MGAS5005, and MAb 2B11 or MOPC-21 was injected into the intraperitoneal cavity 6 h after MGAS5005 inoculation. The majority of both anti-SsE MAb- and MPOC-21-treated mice did not survive (P = 0.6482) (Fig. 7A); however, the sizes of the inflammatory areas at infection sites of nonsurviving mice that received MAb 2B11 treatment (820 ± 67 mm2) were significantly smaller than those in nonsurviving MOPC-21-treated control mice (1,036 ± 57 mm2) (P = 0.0276) (Fig. 7B). Thus, when MAb 2B11 was administered after GAS inoculation, it had some protective effect but could not provide sufficient protection against GAS infections.
FIG 7.
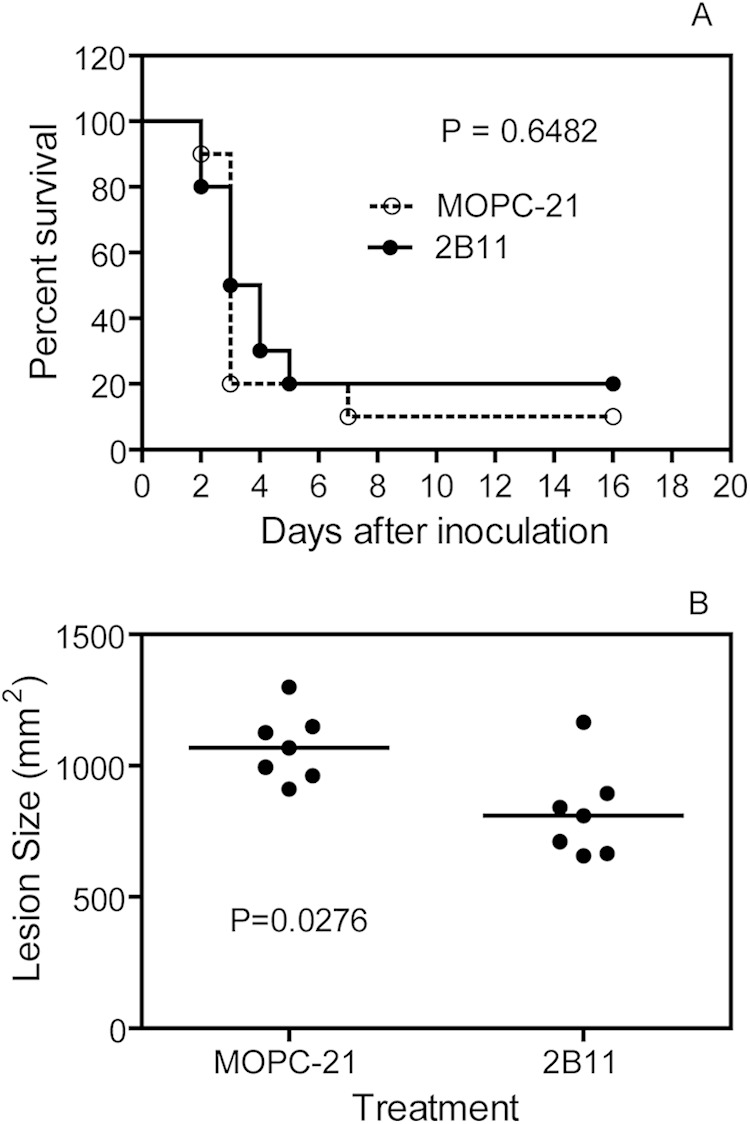
Reduced protection of mice by 2B11 administered i.p. at 6 h after MGAS5005 inoculation. Mice were first infected with MGAS5005, and, 6 h later, 250 mg of 2B11 or MOPC-21 was injected i.p. Presented are survival rates and the lesion sizes of nonsurviving mice.
Vascular effusion at skin infection sites during early stages of infection.
Based on the protection results, we speculated that antibodies might be able to get to infection sites at early stages of infection but not later. We tested vascular effusion at 6 h and 20 h after MGAS5005 inoculation using Evans blue as an indicator for the effusion of albumin from blood vessels at infection sites. Evans blue was injected into the vein of the tail at 6 h and 20 h after MGAS5005 inoculation, and, 30 min later, the skin at infection sites was peeled off to observe leakage of Evans blue. Inoculation sites at 6 h after inoculation appeared intensely blue (Fig. 8A), indicating that there was vascular effusion. However, the infection site at 20 h after GAS inoculation did not appear blue (Fig. 8B). These results suggest that MAb 2B11 administered prior to MGAS5005 inoculation had a chance to enter infection sites through vascular effusion at the early hours of MGAS5005 infection but might not be able to enter the infection sites to neutralize the function of SsE at later time points.
FIG 8.
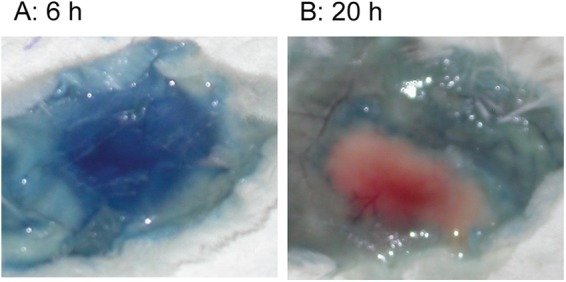
Images of the Miles assay showing vascular effusion to infection sites at 6 h, but not 20 h, after MGAS5005 inoculation of mice. The pictures are representative for each group of five mice.
DISCUSSION
The purpose of this study is to determine whether an antibody neutralizing the enzymatic activity of SsE inhibits the function of SsE and thus has the potential to be used as a therapeutic for the treatment of necrotizing fasciitis caused by group A Streptococcus. SsE is required for the skin invasion by hypervirulent serotype M1 GAS isolates in a mouse model of necrotizing fasciitis (18). The basis for the role of SsE in skin invasion has been proposed to be mediated by SsE inhibition of neutrophil recruitment in GAS skin infections (19, 24), and the SsE inhibition of the neutrophil response is partly mediated by SsE-catalyzed hydrolysis of PAF (19). In this study, we first demonstrated that the MGAS5005 derivative that produced an enzymatically inactive SsES178A mutant protein had the same phenotype as the reported phenotype of an sse deletion mutant of MGAS5005, with a reduction in skin invasion and enhancement in neutrophil recruitment during subcutaneous infection of mice (18, 19). Neither the sse deletion nor the SsE S178A substitution altered the resistance of MGAS5005 to phagocytosis by neutrophils and the survival ability of MGAS5005 in nonimmune human blood. These data indicate that the enzymatic activity of SsE is essential for its function to reduce neutrophil infiltration and to enhance skin invasion.
The second finding of this study is that an inhibitory IgG1 antibody of SsE can neutralize the function of SsE, conferring protection in skin infection. By enhancing neutrophil infiltration, passive immunization with the neutralizing MAb of SsE inhibited the function of SsE to enhance GAS skin invasion and systemic dissemination. The anti-SsE MAb enhances the recruitment of neutrophils, which in turn prevents the dissemination of GAS in the epidermis. These findings are consistent with our previous finding that active immunization with recombinant SsE protein and passive immunization with anti-SsE antiserum protect mice against subcutaneous GAS infection (23). Our new finding suggests that the protection conferred by the active immunization is mediated by antibodies that neutralize the enzymatic activity of SsE, supporting the concept that neutralization of the function of virulence factors by specific antibodies is part of the humoral immunity against GAS. The new data also support the idea that SsE can be included in a vaccine to prevent severe invasive GAS infections, such as necrotizing fasciitis.
The protective effect of the SsE-neutralizing MAb diminishes if the antibody is administered after GAS is inoculated. Thus, the therapeutic value of neutralizing anti-SsE antibodies appears to be limited. However, these data coupled with the GAS-induced vascular effusion suggest some clues to the host responses to GAS infections and features of GAS skin infections. First, the host can mount a response through vascular effusion at early stages of skin infection, and protective antibodies in immune persons should enter infection sites to reduce or constrain GAS infection. Second, the data indicating that the anti-SsE MAb was protective when it was administered prior to, but not after, GAS inoculation could indicate that GAS in the mouse skin infection model disseminates into organs and blood at very early stages. Disseminated GAS at the early hours after GAS inoculation may be the primary cause for mouse mortality. This possibility is consistent with our previous finding that SsE is critical for virulence of MGAS5005 in subcutaneous inoculation but not in intraperitoneal infection of mice (18). The critical role of systemic GAS dissemination in GAS virulence is supported by the finding that the deletion of the covR gene enhances skin invasion by GAS but GAS fails to disseminate to organs and to cause lethal infection (31). Third, the diminished protection of the antibody 2B11 when it was administered after GAS inoculation might be due to the reduced access of 2B11 to the infection site after the early hours of the infection. The vascular effusion results suggest this possibility. This possible limitation in accessibility may also be linked to the basis for the ineffectiveness of antibiotic treatment of necrotizing fasciitis patients (4). The basis for the diminished vascular effusion to the skin infection site at 20 h after GAS inoculation is not clear and is the target of our follow-up studies.
The limited value of SsE-neutralizing MAb in treatment of severe invasive infection should not discourage efforts to develop MAb-based immunotherapy for severe invasive GAS infections. The finding that passive immunization with the antibody provides protection indicates that neutralizing antibodies can block the function of virulence factors. Although 2B11 MAb does not have a therapeutic value, an MAb-based immunotherapy for severe invasive infections may still be developed if the antigen target(s) is appropriate. SsE contributes to virulence in skin infection and appears not to be an ideal target for treating lethal systemic infection. GAS is notoriously good at resisting phagocytic killing in nonimmune blood but could not survive in immune blood. Monoclonal antibodies that can enhance opsonophagocytic killing of GAS are expected to have therapeutic values. Hasty et al. have generated anti-M24 protein MAbs that can enhance opsonophagocytosis of GAS and protect mice in passive immunization (32). It would be interesting to test whether these MAbs have therapeutic value against severe invasive infections. In addition, intravenous immunoglobulin treatment is actually an adjunctive treatment for severe invasive infections (6). Developing MAbs effective for enhancing GAS killing by the innate immune system may eventually lead to an efficacious immunotherapy for treating severe invasive GAS infections.
ACKNOWLEDGMENTS
This work was supported in part by grants AI097703, AI095704, and GM110732 from the National Institutes of Health, USDA Animal Formula Fund, equipment grants from the M. J. Murdock Charitable Trust, and the Montana State Agricultural Experimental Station.
REFERENCES
- 1.Carapetis JR, Steer AC, Mulholland EK, Weber M. 2005. The global burden of group A streptococcal diseases. Lancet Infect Dis 5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 2.Olsen RJ, Musser JM. 2010. Molecular pathogenesis of necrotizing fasciitis. Annu Rev Pathol 5:1–31. doi: 10.1146/annurev-pathol-121808-102135. [DOI] [PubMed] [Google Scholar]
- 3.O'Loughlin RE, Roberson A, Cieslak PR, Lynfield R, Gershman K, Craig A, Albanese BA, Farley MM, Barrett NL, Spina NL, Beall B, Harrison LH, Reingold A, Van Beneden C, Active Bacterial Core Surveillance Team . 2007. The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000-2004. Clin Infect Dis 45:853–862. doi: 10.1086/521264. [DOI] [PubMed] [Google Scholar]
- 4.Stevens DL, Gibbons AE, Bergstrom R, Winn V. 1988. The Eagle effect revisited: efficacy of clindamycin, erythromycin, and penicillin in the treatment of streptococcal myositis. J Infect Dis 158:23–28. doi: 10.1093/infdis/158.1.23. [DOI] [PubMed] [Google Scholar]
- 5.Wong CH, Yam AK, Tan AB, Song C. 2008. Approach to debridement in necrotizing fasciitis. Am J Surg 196:e19–24. doi: 10.1016/j.amjsurg.2007.08.076. [DOI] [PubMed] [Google Scholar]
- 6.Cawley MJ, Briggs M, Haith LR Jr, Reilly KJ, Guilday RE, Braxton GR, Patton ML. 1999. Intravenous immunoglobulin as adjunctive treatment for streptococcal toxic shock syndrome associated with necrotizing fasciitis: case report and review. Pharmacotherapy 19:1094–1098. doi: 10.1592/phco.19.13.1094.31589. [DOI] [PubMed] [Google Scholar]
- 7.Young MH, Engleberg NC, Mulla ZD, Aronoff DM. 2006. Therapies for necrotizing fasciitis. Expert Opin Biol Ther 6:155–165. doi: 10.1517/14712598.6.2.155. [DOI] [PubMed] [Google Scholar]
- 8.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. 2006. Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog 2:e5. doi: 10.1371/journal.ppat.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, Zhu H, Feng W, Liu M, Song Y, Zhang X, Zhou Y, Bei W, Lei B. 2013. Regulation of inhibition of neutrophil infiltration by the two-component regulatory system CovRS in subcutaneous murine infection with group A streptococcus. Infect Immun 81:974–983. doi: 10.1128/IAI.01218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kansal RG, Datta V, Aziz RK, Abdeltawab NF, Rowe S, Kotb M. 2010. Dissection of the molecular basis for hypervirulence of an in vivo-selected phenotype of the widely disseminated M1T1 strain of group A Streptococcus bacteria. J Infect Dis 201:855–865. doi: 10.1086/651019. [DOI] [PubMed] [Google Scholar]
- 11.Li J, Liu G, Feng W, Zhou Y, Liu M, Wiley JA, Lei B. 2014. Neutrophils select hypervirulent CovRS mutants of M1T1 group A Streptococcus during subcutaneous infection of mice. Infect Immun 82:1579–1590. doi: 10.1128/IAI.01458-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levin JC, Wessels MR. 1998. Identification of csrR/csrS, a genetic locus that regulates hyaluronic acid capsule synthesis in group A Streptococcus. Mol Microbiol 30:209–219. doi: 10.1046/j.1365-2958.1998.01057.x. [DOI] [PubMed] [Google Scholar]
- 13.Heath A, DiRita VJ, Barg NL, Engleberg NC. 1999. A two-component regulatory system, CsrR-CsrS, represses expression of three Streptococcus pyogenes virulence factors, hyaluronic acid capsule, streptolysin S, and pyrogenic exotoxin B. Infect Immun 67:5298–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Federle MJ, McIver KS, Scott JR. 1999. A response regulator that represses transcription of several virulence operons in the group A Streptococcus. J Bacteriol 181:3649–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Treviño J, Perez N, Ramirez-Peña E, Liu Z, Shelburne SA, Musser JM, Sumby P. 2009. CovS simultaneously activates and inhibits the CovR-mediated repression of distinct subsets of group A Streptococcus virulence factor-encoding genes. Infect Immun 77:3141–3149. doi: 10.1128/IAI.01560-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walker MJ, Hollands A, Sanderson-Smith ML, Cole JN, Kirk JK, Henningham A, McArthur JD, Dinkla K, Aziz RK, Kansal RG, Simpson AJ, Buchanan JT, Chhatwal GS, Kotb M, Nizet V. 2007. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med 13:981–985. doi: 10.1038/nm1612. [DOI] [PubMed] [Google Scholar]
- 17.Edwards RJ, Taylor GW, Ferguson M, Murray S, Rendell N, Wrigley A, Bai Z, Boyle J, Finney SJ, Jones A, Russell HH, Turner C, Cohen J, Faulkner L, Sriskandan S. 2005. Specific C-terminal cleavage and inactivation of interleukin-8 by invasive disease isolates of Streptococcus pyogenes. J Infect Dis 192:783–790. doi: 10.1086/432485. [DOI] [PubMed] [Google Scholar]
- 18.Zhu H, Liu M, Sumby P, Lei B. 2009. The secreted esterase of group A Streptococcus is important for invasive skin infection and dissemination in mice. Infect Immun 77:5225–5232tk;3. doi: 10.1128/IAI.00636-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu M, Zhu H, Li J, Garcia CC, Feng W, Kirpotina LN, Hilmer J, Tavares LP, Layton AW, Quinn MT, Bothner B, Teixeira MM, Lei B. 2012. Group A Streptococcus secreted esterase hydrolyzes platelet-activating factor to impede neutrophil recruitment and facilitate innate immune evasion. PLoS Pathog 8:e1002624. doi: 10.1371/journal.ppat.1002624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aziz RK, Pabst MJ, Jeng A, Kansal R, Low DE, Nizet V, Kotb M. 2004. Invasive M1T1 group A Streptococcus undergoes a phase-shift in vivo to prevent proteolytic degradation of multiple virulence factors by SpeB. Mol Microbiol 51:123–134. doi: 10.1046/j.1365-2958.2003.03797.x. [DOI] [PubMed] [Google Scholar]
- 21.Cole JN, McArthur JD, McKay FC, Sanderson-Smith ML, Cork AJ, Ranson M, Rohde M, Itzek A, Sun H, Ginsburg D, Kotb M, Nizet V, Chhatwal GS, Walker MJ. 2006. Trigger for group A streptococcal M1T1 invasive disease. FASEB J 20:1745–1747. doi: 10.1096/fj.06-5804fje. [DOI] [PubMed] [Google Scholar]
- 22.Cole JN, Pence MA, von Köckritz-Blickwede M, Hollands A, Gallo RL, Walker MJ, Nizet V. 2010. M protein and hyaluronic acid capsule are essential for in vivo selection of covRS mutations characteristic of invasive serotype M1T1 group A Streptococcus. mBio 1(4):e00191-10. doi: 10.1128/mBio.00191-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu M, Zhu H, Zhang J, Lei B. 2007. Active and passive immunizations with the streptococcal esterase Sse protect mice against subcutaneous infection with group A streptococci. Infect Immun 75:3651–3657. doi: 10.1128/IAI.00038-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu G, Liu M, Xie G, Lei B. 2013. Characterization of streptococcal platelet-activating factor acetylhydrolase variants that are involved in innate immune evasion. Infect Immun 81:3128–3138. doi: 10.1128/IAI.00398-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 26.Hanks TS, Liu M, McClure MJ, Lei B. 2005. ABC transporter FtsABCD of Streptococcus pyogenes mediates uptake of ferric ferrichrome. BMC Microbiol 5:62. doi: 10.1186/1471-2180-5-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.White-Owen C, Alexander JW, Sramkoski RM, Babcock GF. 1992. Rapid whole-blood microassay using flow cytometry for measuring neutrophil phagocytosis. J Clin Microbiol 30:2071–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu M, Hanks TS, Zhang J, McClure MJ, Siemsen DW, Elser JL, Quinn MT, Lei B. 2006. Defects in ex vivo and in vivo growth and sensitivity to osmotic stress of group A Streptococcus caused by interruption of response regulator gene vicR. Microbiology 152:967–978. doi: 10.1099/mic.0.28706-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gozes Y, Moayeri M, Wiggins JF, Leppla SH. 2006. Anthrax lethal toxin induces ketotifen-sensitive intradermal vascular leakage in certain inbred mice. Infect Immun 74:1266–1272. doi: 10.1128/IAI.74.2.1266-1272.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou Y, Hanks TS, Feng W, Li J, Liu G, Liu M, Lei B. 2013. The sagA/pel locus does not regulate the expression of the M protein of the M1T1 lineage of group A Streptococcus. Virulence 4:698–706. doi: 10.4161/viru.26413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dalton TL, Hobb RI, Scott JR. 2006. Analysis of the role of CovR and CovS in the dissemination of Streptococcus pyogenes in invasive skin disease. Microb Pathog 40:221–227. doi: 10.1016/j.micpath.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 32.Hasty DL, Beachey EH, Simpson WA, Dale JB. 1982. Hybridoma antibodies against protective and nonprotective antigenic determinants of a structurally defined polypeptide fragment of streptococcal M protein. J Exp Med 155:1010–1018. doi: 10.1084/jem.155.4.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]