

Abstract
Cryptococcus neoformans is a fungal pathogen that causes pulmonary infections, which may progress into life-threatening meningitis. In commonly used mouse models of C. neoformans infections, fungal cells are not contained in the lungs, resulting in dissemination to the brain. We have previously reported the generation of an engineered C. neoformans strain (C. neoformans Δgcs1) which can be contained in lung granulomas in the mouse model and have shown that granuloma formation is dependent upon the enzyme sphingosine kinase 1 (SK1) and its product, sphingosine 1-phosphate (S1P). In this study, we have used four mouse models, CBA/J and C57BL6/J (both immunocompetent), Tgε26 (an isogenic strain of strain CBA/J lacking T and NK cells), and SK−/− (an isogenic strain of strain C57BL6/J lacking SK1), to investigate how the granulomatous response and SK1-S1P pathway are interrelated during C. neoformans infections. S1P and monocyte chemotactic protein-1 (MCP-1) levels were significantly elevated in the bronchoalveolar lavage fluid of all mice infected with C. neoformans Δgcs1 but not in mice infected with the C. neoformans wild type. SK1−/− mice did not show elevated levels of S1P or MCP-1. Primary neutrophils isolated from SK1−/− mice showed impaired antifungal activity that could be restored by the addition of extracellular S1P. In addition, high levels of tumor necrosis factor alpha were found in the mice infected with C. neoformans Δgcs1 in comparison to the levels found in mice infected with the C. neoformans wild type, and their levels were also dependent on the SK1-S1P pathway. Taken together, these results suggest that the SK1-S1P pathway promotes host defense against C. neoformans infections by regulating cytokine levels, promoting extracellular killing by phagocytes, and generating a granulomatous response.
INTRODUCTION
Cryptococcosis is a serious fungal infection in immunocompromised hosts that results in deadly meningitis once fungi disseminate to the central nervous system (CNS) (1–3). Cryptococcus neoformans is a common environmental fungus, and exposure is thought to be extremely prevalent but rarely progresses to disease in healthy individuals (4–6). Most human hosts are able to combat and contain C. neoformans in the lungs to prevent spread to the CNS. A successful immune response results in the killing of C. neoformans and the formation of a granuloma in the lungs, which is thought to prevent C. neoformans from accessing the vasculature and causing infection of the CNS. Under conditions of immune suppression, this response does not occur successfully and C. neoformans survives within macrophages, which are thought to transport C. neoformans across the blood-brain barrier and lead to life-threatening meningitis (3, 5). Although significant work has been done to elucidate the role of the host defense during early pulmonary infection, much work regarding the development of a granuloma in response to C. neoformans remains to be done (7, 8).
Despite the induction of proinflammatory cytokines in commonly used mouse models of C. neoformans infection, granuloma formation does not occur and immunocompetent mice succumb to infection with wild-type (WT) strain H99 of C. neoformans (2). In an effort to recapitulate the progression of the human disease, work has been done to study an attenuated mutant strain of C. neoformans, C. neoformans Δgcs1, that is unable to synthesize the sphingolipid glucosylceramide. The absence of this lipid prevents the fungi from replicating in the extracellular lung environment, which is neutral or alkaline in pH. Instead, C. neoformans Δgcs1 grows normally only under acidic conditions, such as those found in the phagolysosome (9). This mutation causes C. neoformans Δgcs1 to become an obligate intracellular pathogen, whereas wild-type C. neoformans is a facultative (intracellular) pathogen (10). Interestingly, infection of wild-type mice with the C. neoformans Δgcs1 strain results in granuloma formation and containment of the fungi in the lungs, whereas infection of immunocompromised Tgε26 mice with this strain results in fungal dissemination and death of the host (2, 9, 10). This phenomenon mimics the human physiopathology of C. neoformans infection in immunocompetent and immunocompromised subjects. Previous work has used this model to study the development of granuloma in the mouse model and found it to be dependent on the sphingosine 1-phosphate (S1P) pathway (2).
S1P is a bioactive sphingolipid that serves important functions in many biological processes. S1P is produced by the phosphorylation of sphingosine by one of two sphingosine kinases (SKs; SK1 and SK2) (11). S1P signals extracellularly through a family of G-protein-coupled receptors (S1PRs) and also functions intracellularly independently of S1PRs (12). S1P signaling has been shown to play a role in angiogenesis, vascular permeability, brain and cardiac development, and cancer growth and metastasis (13–15). In addition to these functions, S1P has been implicated as a key player in the immune response. Mast cells, platelets, and mononuclear phagocytes are the main sources of S1P in the immune system (16). Additionally, S1P serves important functions in both the innate and adaptive immune responses. S1P receptors are important for thymus-derived lymphocyte (T-cell) egress and trafficking and proinflammatory pathways (17, 18).
Evidence from the literature suggests that S1P plays an important role in the immune response to microbial pathogens. Garg and colleagues reported an increase in macrophage intracellular killing of Mycobacterium tuberculosis and Mycobacterium smegmatis in response to S1P addition and elucidated a protective role for S1P in lung fluids during Mycobacterium infections (19, 20). We previously showed that S1P promotes the phagocytosis of C. neoformans cells by macrophages (21). Importantly, we also found that SK1 and S1P are required for the formation of a granuloma in mice infected with the C. neoformans Δgcs1 strain (2, 21). Due to the established role of SK1 in the S1P signaling pathway and the role of S1P in immune signaling, in this study we sought to elucidate how S1P contributes to the granulomatous response and containment of C. neoformans infection using the C. neoformans Δgcs1 mouse model of cryptococcosis.
MATERIALS AND METHODS
Mouse strains.
Five- to 7-week-old mice were used for the experiments in this study. Four different mouse strains were used: CBA/J and C57BL6/J (The Harlan Laboratory and The Jackson Laboratory, respectively), Tgε26 (an isogenic strain of strain CBA/J lacking T cells), and SK1−/− (an isogenic strain of strain CBA/J lacking SK1). Tgε26 and SK1−/− mice were generated previously, and colonies were maintained as described previously (10, 22–24). All mice were available to us through the Medical University of South Carolina Center of Biomedical Research Excellence (MUSC COBRE) Animal Core Facility, directed by Toshihiko Kawamori, who provided breeding pairs for this study. For all experiments, mutant mice were age and sex matched with isogenic wild-type mice. All work was approved by the MUSC Institutional Animal Care and Use Committee.
Cryptococcus strains.
C. neoformans var. grubii serotype A strain H99 (wild type) (obtained from the Duke University Medical Center, Durham, NC, USA) and a mutant C. neoformans strain lacking GCS1 (C. neoformans Δgcs1) derived from H99 (9) were used in this study. Both strains were grown in yeast extract-peptone-dextrose (YPD) medium for 16 to 18 h at 30°C in a shaking cell culture incubator.
Infection of mice.
Prior to inoculation, the mice were anesthetized with an intraperitoneal injection of 60 μl of a xylazine-ketamine mixture containing 95 mg of ketamine per kilogram of body weight and 5 mg of xylazine per kilogram of body weight. Then, the C. neoformans strains were pipetted into the nasal cavity of anesthetized mice using 20 μl of phosphate-buffered saline (PBS; pH 7.0) containing 5 × 105 fungal cells.
Cell lines.
The MHS alveolar macrophage cell line was obtained from the American Type Culture Collection and maintained according to the culture conditions prescribed by the ATCC. The medium used was RPMI 1640 supplemented with 2-mercaptoethanol to a final concentration of 0.05 mM and fetal bovine serum to a final concentration of 10%.
Isolation of primary cell lines.
Murine primary alveolar macrophages were isolated from the lungs of mice as described previously (2). Briefly, cells collected from bronchoalveolar lavage (BAL) fluid were allowed to adhere for 30 min before the cell dishes were washed three times and fresh medium (RPMI 1640 supplemented with 0.1% penicillin-streptomycin) was added. Cells were incubated for an additional 90 min prior to experimentation. Murine primary neutrophils were obtained from CBA/J SK1+/+ and CBA/J SK1−/− mice using the Ficoll-Paque Premium (catalog no. 17-5442-02; GE Healthcare) method, which has been described previously (24).
Infection of primary alveolar macrophages.
C. neoformans cells were coincubated with primary macrophages in their medium for 4 h at a ratio of 1 macrophage to 10 C. neoformans cells in the absence of lipopolysaccharide, gamma interferon, or antibody so that the C. neoformans cells would remain extracellular.
Mass spectroscopy.
Total lipids were extracted as described previously (25). In brief, lipid extraction was performed according to the methods of Bligh and Dyer (26). A quarter of the samples were aliquoted for inorganic phosphate determination. The tubes were vacuum dried and used for mass spectrometry analysis. S1P levels were measured by liquid chromatography-mass spectrometry at the Medical University of South Carolina.
Cytokine analysis.
Cytokine levels were ascertained by enzyme-linked immunosorbent assay (ELISA). Many cytokines (∼16 or more) were tested using a Cytometric Bead Array system kit from BD Biosciences. The results for those cytokines that showed significant changes were then validated using ELISA kits directed against those specific cytokines. For monocyte chemoattractant protein-1 (MCP-1), a mouse MCP-1 ELISA set from BD Biosciences was used. For soluble tumor necrosis factor alpha receptor 1 (sTNA-αR1), a custom ELISA kit from BD Biosciences was used.
Neutrophil killing assay.
According to the methods of a previously described experiment (24), freshly isolated murine neutrophils were incubated with C. neoformans Δgcs1 (20:1 ratio of murine neutrophils/C. neoformans cells) in filtered PBS with 10% pooled mouse serum from the respective mouse strain for 4 h at 37°C in a 5% CO2 atmosphere. At the end of the incubation, the cultures were serially diluted, streaked onto YPD agar, and incubated for 48 h at 30°C. The numbers of CFU were counted to assess the killing of C. neoformans compared with that of control cultures of C. neoformans alone with no neutrophils (24). At 2 h after infection, extracellular S1P at nanomolar concentrations dissolved in methanol was added to the neutrophils. The results are reported as the percentage of killed cells compared to the number of C. neoformans cells not exposed to neutrophils or S1P.
Histology.
Organs were harvested and fixed overnight in 37% formaldehyde, paraffin embedded, sectioned, and stained with hematoxylin and eosin (H&E) or Movat. Visualization of host components was done using light microscopy. The Histology Core Facility at the Medical University of South Carolina performed all staining and tissue processing.
Statistical analysis.
All data are expressed as the mean ± standard deviation. No samples or animals were excluded from the analysis. For animal studies, group sizes were chosen so that they were sufficient to reach a statistical power of at least 80% (http://www.statisticalsolutions.net/pss_calc.php). Mice were randomly assigned to treatment groups. Statistical analysis was performed using unpaired t tests (for comparison of two groups). Data were compared for statistically significant differences. Statistical tests were carried out using GraphPad Prism (La Jolla, CA, USA) software for the Macintosh (v.400). The replicates used were biological replicates. Results were considered significant at P values of ≤0.05.
RESULTS
S1P levels in BAL fluid.
Since SK1 and S1P are regulators of host immunity (27–31) and antimicrobial activity (2, 20), we wondered whether the SK1-S1P pathway is stimulated by C. neoformans infection and if there is a difference in the stimulation of this pathway between wild-type (WT) C. neoformans strain H99 (which causes 100% mortality in mouse models) and C. neoformans Δgcs1 (which can be contained in the lungs of mice without causing mortality) (9).
To determine if the SK1-S1P pathway is stimulated by C. neoformans, we used mass spectrometry to measure the levels of S1P in the bronchoalveolar lavage (BAL) fluid of mice 4 days after intranasal infection with C. neoformans. Four different mouse models with the following four mouse strains were used to examine whether S1P generation is affected by immunodeficiency or SK1 activity: CBA/J and C57BL6/J (both immunocompetent), Tgε26 (an isogenic strain of strain CBA/J lacking T and NK cells), and SK1−/− (an isogenic strain of strain C57BL6/J lacking SK1). The levels of extracellular S1P were significantly increased after infection with C. neoformans Δgcs1 in all mice except SK1−/− mice, indicating that SK1 is the enzyme responsible for S1P production in the lungs upon infection (Fig. 1A to C). The significant difference in S1P levels was observed at the earliest time point of 4 days after infection (Fig. 1A) and continued throughout the experiment (Fig. 1B and C). Since macrophages cannot be activated through a gamma interferon-mediated mechanism in Tgε26 mice, the high level of extracellular S1P in Tgε26 mice suggests that macrophage activation is not required for S1P accumulation. Interestingly, the C. neoformans Δgcs1 strain stimulated S1P accumulation significantly more than the WT strain (e.g., 20.5 ± 2.5 pmol/ml of BAL fluid versus 5.8 ± 0.6 pmol/ml of BAL fluid in CBA/J mice at day 4 after infection), suggesting improved immunity against the mutant strain.
FIG 1.

Measurement of S1P levels after infection of mice or an alveolar macrophage cell line with C. neoformans (Cn). (A to C) S1P levels in the BAL fluid of CBA/J, Tgε26, C57BL6/J, and SK1−/− mice uninfected or infected with the C. neoformans H99 WT or C. neoformans Δgcs1 strain. Data were collected from three mice per time point at 4 (A), 8 (B), and 16 (C) days postinfection. (D) S1P levels in the medium of the MHS macrophage cell line 2 h after infection with C. neoformans (MHS/C. neoformans ratio = 1:10). *, values significantly (P < 0.01) greater than those for both uninfected mice and mice infected with the C. neoformans H99 WT strain.
The mice infected with the wild-type strain do not survive the infection after 21 days (2), and this number is even lower for the Tgε26 mice, which have an average survival of 17.2 days (10). Thus, the measurements could not be continued for all models after this length of time. However, the measurements were continued in CBA/J and C57BL6/J mice until 60 days postinfection. The measurements showed that the S1P levels in mice infected with the C. neoformans Δgcs1 strain remained high up to 60 days after infection (34.2 ± 5.1 pmol/ml of BAL fluid in CBA/J mice and 39.6 ± 4.3 pmol/ml of BAL fluid in C57BL6/J mice). These values were significantly higher than the S1P values in uninfected mice at the same time (4.2 ± 0.7 pmol/ml of BAL fluid in CBA/J mice and 4.2 ± 0.6 pmol/ml of BAL fluid in C57BL6/J mice). It should be noted that reconstitution of strain C. neoformans Δgcs1 (the C. neoformans Δgcs1 GCS1 strain) restored the phenotypes observed for the wild-type strain and established virulence in all four mouse models. Thus, this strain was not included in these studies to reduce the number of animals required for the study.
Macrophages are known sources of S1P generation (16). Thus, one possible explanation for the differences in S1P levels observed in mice infected with various C. neoformans strains could be that the C. neoformans Δgcs1 strain was able to induce S1P generation by macrophages more efficiently. To test this hypothesis, an MHS macrophage cell line was infected with C. neoformans strains and examined for S1P generation (Fig. 1D). Macrophages showed a significant increase in S1P levels upon infection with C. neoformans Δgcs1 (26.5 ± 2.3 pmol/ml of medium versus 7.7 ± 1.1 pmol/ml of medium in noninfected cells), which was in agreement with the measurements in the BAL fluid. A slight accumulation of S1P was observed after infection of macrophages with the C. neoformans WT (12.3 ± 1.0 pmol/ml of medium), while the level of S1P detected from the fungal cells in the absence of mammalian cells was significantly lower (0.4 ± 0.2 pmol/ml of medium).
MCP-1 levels in BAL fluid.
The studies of S1P were followed by a screening of the level of cytokines in the BAL fluid following infection with the C. neoformans WT and Δgcs1 strains in the same mouse models. The cytokine that was most stimulated after infection was monocyte chemotactic protein-1 (MCP-1) (Fig. 2A), a chemokine that is responsible for the recruitment of monocytes and is a critical factor in the Th1-type immune response against pulmonary cryptococcosis (32–35). Similar to the results of the S1P measurement experiments, the levels of MCP-1 were significantly higher in the BAL fluid of animals infected with the C. neoformans Δgcs1 strain until 16 days postinfection (Fig. 2B and C). MCP-1 stimulation was totally abrogated in the SK1−/− mice but preserved in the Tgε26 mice, indicating that the SK1-S1P pathway and not the T (or NK) cells were responsible for the accumulation of MCP-1. Analogously to S1P, MCP-1 levels remained high until 60 days postinfection with C. neoformans Δgcs1 in CBA/J and C57BL6/J mice (27.9 ± 4.1 pg/ml of BAL fluid in CBA/J mice versus 2.1 ± 0.7 pg/ml of BAL fluid in uninfected mice; 29.7 ± 5.4 pg/ml of BAL fluid in C57BL6/J mice versus 1.7 ± 0.8 pg/ml of BAL fluid in uninfected mice).
FIG 2.

Measurement of MCP-1 levels after infection of mice or primary alveolar macrophages with C. neoformans. (A to C) MCP-1 levels in the BAL fluid of CBA/J, Tgε26, C57BL6/J, and SK1−/− mice uninfected or infected with the C. neoformans H99 or C. neoformans Δgcs1 strain. Data were collected from three mice per time point at 4 (A), 8 (B), and 16 (C) days postinfection. (D) MCP-1 levels in the medium of primary alveolar macrophages isolated from SK1−/− or SK1+/+ mice 2 h after infection with C. neoformans (MHS/C. neoformans ratio = 1:10). *, P < 0.01 versus the results for uninfected mice or mice infected with the C. neoformans H99 WT strain.
To understand whether the bronchoalveolar macrophages were involved in the process of in vivo MCP-1 generation following the infection, these cells were separated from the C57BL/6J mice (isogenic for SK1−/− mice) and SK1-knockout mice, and the levels of MCP-1 in the medium were measured 2 h after infection with the C. neoformans WT and Δgcs1 strains (Fig. 2D). A significant increase in the levels of MCP-1 in the medium was observed in the macrophages extracted from the C57BL/6J mice 2 h after infection with the C. neoformans Δgcs1 strain (310.3 ± 54.5 pg/ml of medium versus 10.3 ± 1.6 pg/ml of medium for the uninfected macrophages). A slight but significant increase in the MCP-1 level was also observed after infection with the WT strain. However, no changes in the MCP-1 levels of macrophages from SK1−/− mice were observed after infection with either strain. This finding was in agreement with the observations from the mouse experiments (Fig. 2A), further confirming the importance of the SK1-S1P pathway for MCP-1 production.
The effect of extracellular S1P on antifungal activity.
Although the addition of extracellular S1P increases the phagocytic index of macrophages in response to C. neoformans infection, the intracellular fungicidal activity of alveolar macrophages remains unchanged following S1P addition (21). Since C. neoformans Δgcs1 infection stimulates the SK1-S1P pathway, the effects of S1P on the extracellular fungicidal activity of primary neutrophils isolated from C57BL/6J and SK1−/− mice were examined. For these experiments, primary neutrophils were obtained from the SK1−/− and C57BL/6J (SK1+/+) mice, and their extracellular killing activity was monitored. Neutrophils isolated from the SK1−/− mice showed an almost 3-fold decrease in killing activity compared to that of neutrophils isolated from C57BL/6J (SK1+/+) mice (Fig. 3). Interestingly, addition of nanomolar concentrations of extracellular S1P restored the ability of primary neutrophils from SK1−/− mice to kill C. neoformans Δgcs1. This increase in killing ability was dose dependent and increased with increasing concentrations of S1P, indicating that extracellular S1P regulates the killing activity of neutrophils. It should be noted that S1P by itself did not show fungicidal activity on C. neoformans even at micromolar levels.
FIG 3.
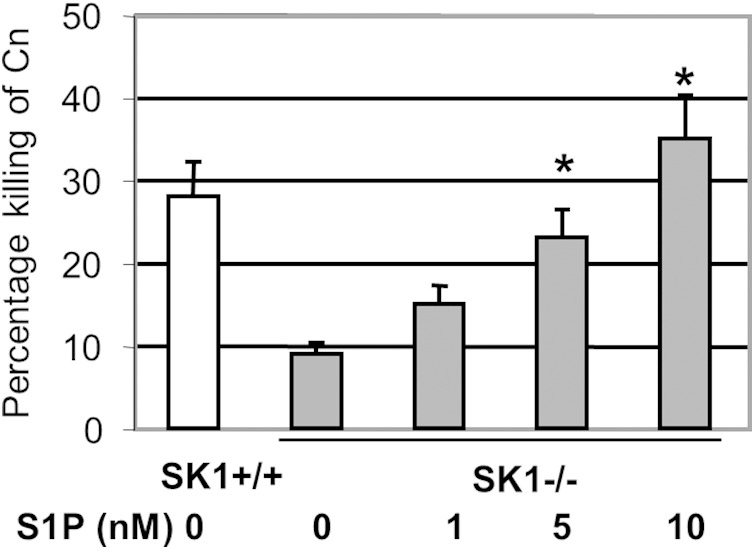
Killing activity of primary neutrophils isolated from SK1+/+ or SK1−/− mice untreated or treated with S1P. *, P < 0.01 versus the results for SK1−/− mice not treated with S1P.
Granulomatous response to C. neoformans infection.
Our previous survival studies in mouse models of cryptococcosis showed that mice succumb to intranasal infections with the C. neoformans WT but are able to survive infections with C. neoformans Δgcs1 (9). The observations regarding the stimulation of the SK1-S1P pathway can partly explain the differences in mouse survival following infection with each strain. However, analysis of cytokines in the BAL fluid of mice infected with these strains revealed another interesting difference: the level of soluble tumor necrosis factor alpha (TNF-α) receptor 1 (sTNF-αR1), which sequesters free TNF-α, was significantly higher in mice infected with C. neoformans WT than in uninfected mice and mice infected with C. neoformans Δgcs1 (Fig. 4A). This was observed in all mouse models regardless of T or NK cell levels and SK1 deficiency. The reduced amount of sTNF-αR1 in mice infected with C. neoformans Δgcs1 suggested an increased level of free TNF-α in these mice; thus, the levels of this cytokine were also measured. These measurements confirmed the increased levels of free TNF-α in C. neoformans Δgcs1-infected mice (Fig. 4B). Interestingly, a correlation between increased levels of sTNF-αR1 and decreased levels of TNF-α after infection was observed for all mouse models except the model with SK1−/− mice (Fig. 4B). The same trends were observed in all mouse models until 12 days postinfection (data not shown). Given that SK1−/− mice are unable to form granulomas, this suggests that TNF-α, the levels of which are at least partially regulated by sTNF-αR1 and the SK1-S1P pathway, can account for granuloma formation.
FIG 4.

Measurement of sTNF-αR1 (A) and TNF-α (B) levels in the BAL fluid of CBA/J, Tgε26, C57BL6/J, and SK1−/− mice uninfected or infected with the C. neoformans H99 or C. neoformans Δgcs1 strain. Data were collected from 3 mice at 4 days after infection. *, P < 0.001 versus the results for uninfected mice or C. neoformans Δgcs1-infected mice (A); ✪, P < 0.001 versus the results for uninfected mice or C. neoformans WT strain-infected mice (B).
In previous studies, we showed that a granuloma is observed in the lungs of C57BL/6J mice infected intranasally with C. neoformans Δgcs1, which was controlled by the SK1-S1P pathway (2). This granulomatous response was also observed in CBA/J mice infected with the same strain (Fig. 5A and B) but was absent from SK1−/− mice (not shown). The granuloma was marked by a central necrotic area, in which fungal cells were contained (Fig. 5A). This area was surrounded by a ring of giant multinuclear macrophages and a ring of fibrotic tissue infiltrated with lymphocytes and neutrophils (Fig. 5B). The structure of the granuloma that developed in C. neoformans Δgcs1-infected mice was very similar to that of the granuloma that developed in the lung of an immunocompetent human subject with cryptococcosis (Fig. 5C), both of which were characterized by a ring of fibrotic tissue with multinucleated macrophages.
FIG 5.
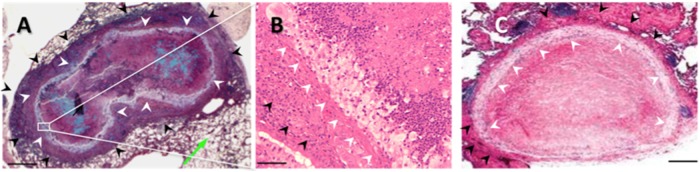
(A) Movat staining of a mouse (CBA/J) lung cryptococcal granuloma infected with C. neoformans Δgcs1. White arrowheads, the ring of multinucleated macrophages containing the C. neoformans cells (blue) in a necrotic center; green arrow, normal lung tissue; black arrowheads, ring of fibrotic tissue with a lymphocyte and neutrophil component. (B) H&E staining of the granuloma in panel A showing the macrophage ring (white arrowheads) and neutrophils and lymphocytes (black arrowheads). Histology sections were prepared at the end of the experiment (60 days postinfection). (C) H&E staining of a human lung cryptococcal granuloma showing a similar ring of multinucleated macrophages. (From the New England Journal of Medicine, R. M. Fairhurst and D. A. Pegues, Pulmonary cryptococcal granulomas, vol. 347, p. 497, Copyright 2002 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.) Bars = 200 μm (A and C) and 50 μm (B).
DISCUSSION
In the mouse model of cryptococcosis, the ability of animals to survive infection depends on the generation of a granulomatous response, which can contain the yeast cells in the lungs and avoid infection of the central nervous system. Our previous studies, using an obligate intracellular Cryptococcus strain (C. neoformans Δgcs1), revealed that granuloma formation in the mouse model is dependent on the SK1-S1P pathway and mice deficient in SK1 are unable to form a granuloma against the infection (2). The results of the current study show that mouse infection with C. neoformans Δgcs1 stimulates the accumulation of S1P, which is accompanied by increased levels of MCP-1 and TNF-α. Interestingly, these phenomena were observed only when mice or macrophages were infected with the Δgcs1 strain, and infection with the wild type significantly reduced or totally abrogated the response. The increased levels of MCP-1 and TNF-α in response to the C. neoformans Δgcs1 infection were dependent on the SK1-S1P pathway, and cytokine levels were significantly reduced in the SK1−/− mice.
A number of studies have focused on the role of cytokines and chemokines following intranasal infections with C. neoformans (33, 36, 37). However, despite the recent evidence that S1P affects phagocytosis and granuloma formation in mouse models of cryptococcosis (2, 21), the mechanisms by which this bioactive lipid affects the mouse immune response following infection remain poorly understood. Our studies reveal that S1P levels were increased after infection with C. neoformans Δgcs1 by more than 8-fold (Fig. 1). Interestingly, infection with the C. neoformans WT led to a less than 2-fold increase in S1P levels, suggesting that differences in S1P levels play an important role in the immune response of mice against these infections (all mice except for Tgε26 and SK1−/− mice survived the C. neoformans Δgcs1 infection, whereas all mice succumbed to the C. neoformans H99 infection). The increase in S1P levels was dependent on SK1 but not on mouse immunocompetence, as SK1−/− mice showed no change in S1P levels following infection. An increase in S1P levels in all other mice was observed as soon as 4 days after infection (Fig. 1), which was the earliest time point of evaluation in this study, and continued for the entire 60-day period of the mouse survival study. Since S1P is known to contribute to immune cell migration (29, 38, 39) and phagocytosis (21), these results indicate a sustainable immune response against C. neoformans Δgcs1 infection throughout the infection.
Macrophages, platelets, and mast cells are known intracellular sources of S1P (16). Of these, macrophages are the major effector cells in the alveoli and the first line of defense against pulmonary cryptococcosis. Stimulation of a macrophage cell line by C. neoformans cells resulted in a marked increase in S1P production (Fig. 1D), suggesting that macrophages are responsible for the spike in S1P levels in the BAL fluid following infection and are able to release S1P extracellularly. Extracellular S1P acts through S1P receptors 1 to 5, which display selective cellular expression (40). S1P receptors 1, 3, and 5 are involved in B- and T-cell migration and monocyte recruitment (27, 41, 42), while S1P receptor 4 plays a role in neutrophil trafficking (17). In addition, we have previously shown that S1P receptor 2 regulates the expression of the phagocytic Fcγ receptors, thereby playing an important role in the phagocytosis of C. neoformans (21). Thus, the extracellular release of S1P can significantly boost the immune response against C. neoformans infection by initiating the migration of immune cells or boosting the phagocytic activity of resident macrophages.
Examination of cytokine levels in the BAL fluid of mice revealed that MCP-1 was the cytokine that was the most stimulated by C. neoformans Δgcs1 infection (Fig. 2A). MCP-1 plays an important role in cell-mediated immunity against cryptococcosis (33). It has been reported that the neutralization of MCP-1 using an anti-MCP-1 antibody significantly reduces the recruitment of macrophages and CD4+ T cells, leading to inhibition of cryptococcal clearance (33). The increase in MCP-1 levels was not dependent on the immune cells but was controlled by SK1 and was totally abrogated in SK1-deficient mice, indicating the importance of the SK1-S1P pathway in the production of this cytokine. Huffnagle et al. (33) have reported an increase in MCP-1 levels following infection with C. neoformans strain 52D. This increase was time dependent and peaked at 4 weeks postinfection. In the current study, a similar time-dependent increase in MCP-1 levels was observed following infection with C. neoformans Δgcs1. MCP-1 levels in the BAL fluid increased at 4 days after infection and plateaued at 30 days postinfection. However, in contrast to the study of Huffnagle and colleagues (33), a very modest increase in MCP-1 levels was observed following infection with the wild-type strain. This difference might have been caused by two factors. (i) The increase in MCP-1 levels in both studies was time dependent; thus, it is possible that the mice infected with the wild-type strain did not survive long enough to demonstrate a spike in MCP-1 (the mice died in an average of 21 days, with the latest measurement being performed at 16 days postinfection). (ii) The strain used in the study of Huffnagle et al. (33), strain 52D, is less virulent than the strain used in the current study, strain H99 (43). It is possible that a stronger immune response is elicited against less virulent cryptococcal strains, and this notion is consistent with our observations that MCP-1 levels were significantly increased in response to the attenuated C. neoformans Δgcs1 strain but not in response to the wild type. Additional information is needed to characterize the cell types recruited to the site of infection following infection with the wild-type and attenuated strains, which can be the focus of future studies.
To understand whether the increase in MCP-1 levels in vivo was caused by resident alveolar macrophages, primary alveolar macrophages were isolated from C57BL6/J and SK1−/− mice (SK1−/− is an isogenic strain of C57BL6/J mice) and infected with C. neoformans. An increase in MCP-1 levels was observed only in mice that were not SK1 deficient (Fig. 2D). These observations suggest that MCP-1 production in macrophages is also controlled by the SK1-S1P pathway. This finding is in agreement with previous reports showing that incubation of human mammary endothelial cells and a neuroblastoma cell line with extracellular S1P increases MCP-1 expression in a time- and concentration-dependent manner (44, 45).
Mouse survival following intranasal C. neoformans infections depends on the ability of the animals to form a granuloma in the lungs and prevent the pathogen from invading the brain. Our cytokine analysis revealed a significant difference in the levels of sTNF-αR1 (Fig. 4A). sTNF-αR1 is ubiquitous, is expressed on almost all cell types, and sequesters free TNF-α (46, 47). Indeed, the increased levels of sTNF-αR1 were accompanied by a reduction in the amount of TNF-α in the BAL fluid (Fig. 4B). TNF-α is a key stimulator of the granulomatous response, and TNF-α-deficient mice have been shown to be unable to form tight granulomas in response to bacterial infections (48). In agreement with this observation, it has been reported that patients infected with M. tuberculosis who show a poor response to antimycobacterial treatment have higher levels of sTNF-αR1 in their serum than patients who respond well to the treatment (49). TNF-α also plays an important role in response to cryptococcosis, and elevated levels of TNF-α have been observed in SJL/J mice that are resistant to cryptococcal infections (50).
The increased levels of TNF-α might also provide a mechanism for the observed increase in MCP-1 generation and the subsequent strong immune response against C. neoformans Δgcs1 infection. TNF-α has been shown to induce S1P generation in murine and human cell lines (51). S1P generation has been shown to activate the nuclear factor kappa–light-chain enhancer of activated B cells (NF-κB) protein complex (52), which is believed to regulate MCP-1 gene expression (45). S1P is also able to increase MCP-1 expression (44, 45) and regulates TNF-α-induced MCP-1 expression (45). Thus, a TNF-α–S1P–NF-κB network might be in play in regulating MCP-1 generation.
Based on our observations in the current and previous studies, we propose the following model for the role of the SK1-S1P pathway in regulating the immune response against infection with the wild-type and C. neoformans Δgcs1 strains (Fig. 6). Upon infection with C. neoformans Δgcs1, resident macrophages are stimulated (either directly or through the action of TNF-α, the levels of which are regulated by sTNF-αR1) to convert sphingosine to S1P through the action of SK1. S1P production increases the phagocytosis of macrophages (21) and the extracellular killing of neutrophils (Fig. 3) and stimulates the generation of MCP-1. MCP-1 recruits macrophages/monocytes and CD4+ T cells to the site of the infection, resulting in a strong immune response against C. neoformans Δgcs1 (Fig. 6A). Upon infection with the wild type, sTNF-α R1 levels increase and reduce the levels of free TNF-α. This leads to lower levels of S1P generation and a weaker response by phagocytes in response to infection. MCP-1 levels are also reduced in this case, resulting in the recruitment of fewer immune cells or the delayed recruitment of immune cells to the site of the infection. This leads to the increased proliferation of fungal cells in the lungs, which, coupled with a lack of granuloma formation, can result in dissemination in the bloodstream and, eventually, to death of the host.
FIG 6.

Proposed model of the role of S1P during lung infection with C. neoformans Δgcs1 and C. neoformans H99. (A) In C. neoformans Δgcs1 infection, TNF-α stimulates resident macrophages to produce S1P from sphingosine (SPH) by the action of SK1. S1P contributes to phagocytosis and extracellular killing of C. neoformans cells and stimulates the production of MCP-1. MCP-1 recruits activated macrophages and other phagocytes, leading to a strong immune response. (B) In C. neoformans H99 infection, TNF-α and S1P levels are significantly reduced, leading to a reduction in the level of MCP-1 generation. This in turn hampers immune cell recruitment, phagocytosis, and extracellular killing and allows the fungus to proliferate and disseminate throughout the body.
The findings of the current study suggest that differences in the levels of S1P, MCP-1, and TNF-α are some of the factors contributing to the formation of lung granuloma around C. neoformans Δgcs1 but not the C. neoformans WT. The granuloma observed in mice following C. neoformans Δgcs1 infection is regulated by the SK1-S1P pathway and shows striking similarity to a human lung granuloma, characterized by multinucleated giant cells, fibrous tissue, and lymphocyte and neutrophil penetration, observed in an immunocompetent subject (53, 54). Thus, the C. neoformans Δgcs1 strain provides a model for studying the immunological basis of granuloma formation in cryptococcosis. Obviously, such a model does not perfectly recapitulate the physiological conditions of clinical relevance (i.e., a human host infected with the wild-type strain), and one should be cautious in drawing comparisons between granuloma formation in this model and granuloma formation in the human host.
ACKNOWLEDGMENTS
This work was supported by NIH grants AI56168, AI71142, AI87541, and AI100631 to M.D.P.
Maurizio Del Poeta is a Burroughs Wellcome investigator in infectious diseases.
We thank Alicja Bielawska and Jacek Bielawski from the Lipidomic Core at the Medical University of South Carolina for lipid measurements.
REFERENCES
- 1.Del Poeta M, Casadevall A. 2012. Ten challenges on Cryptococcus and cryptococcosis. Mycopathologia 173:303–310. doi: 10.1007/s11046-011-9473-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McQuiston T, Luberto C, Del Poeta M. 2010. Role of host sphingosine kinase 1 in the lung response against cryptococcosis. Infect Immun 78:2342–2352. doi: 10.1128/IAI.01140-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McQuiston TJ, Williamson PR. 2012. Paradoxical roles of alveolar macrophages in the host response to Cryptococcus neoformans. J Infect Chemother 18:1–9. doi: 10.1007/s10156-011-0306-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abadi J, Pirofski L. 1999. Antibodies reactive with the cryptococcal capsular polysaccharide glucuronoxylomannan are present in sera from children with and without human immunodeficiency virus infection. J Infect Dis 180:915–919. doi: 10.1086/314953. [DOI] [PubMed] [Google Scholar]
- 5.Coelho C, Bocca AL, Casadevall A. 2014. The intracellular life of Cryptococcus neoformans. Annu Rev Pathol 9:219–238. doi: 10.1146/annurev-pathol-012513-104653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeShaw M, Pirofski LA. 1995. Antibodies to the Cryptococcus neoformans capsular glucuronoxylomannan are ubiquitous in serum from HIV+ and HIV− individuals. Clin Exp Immunol 99:425–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osterholzer JJ, Surana R, Milam JE, Montano GT, Chen G-H, Sonstein J, Curtis JL, Huffnagle GB, Toews GB, Olszewski MA. 2009. Cryptococcal urease promotes the accumulation of immature dendritic cells and a non-protective T2 immune response within the lung. Am J Pathol 174:932–943. doi: 10.2353/ajpath.2009.080673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qiu Y, Davis MJ, Dayrit JK, Hadd Z, Meister DL, Osterholzer JJ, Williamson PR, Olszewski MA. 2012. Immune modulation mediated by cryptococcal laccase promotes pulmonary growth and brain dissemination of virulent Cryptococcus neoformans in mice. PLoS One 7:e47853. doi: 10.1371/journal.pone.0047853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rittershaus PC, Kechichian TB, Allegood JC, Merrill AH, Hennig M, Luberto C, Del Poeta M. 2006. Glucosylceramide synthase is an essential regulator of pathogenicity of Cryptococcus neoformans. J Clin Invest 116:1651–1659. doi: 10.1172/JCI27890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kechichian TB, Shea J, Del Poeta M. 2007. Depletion of alveolar macrophages decreases the dissemination of a glucosylceramide-deficient mutant of Cryptococcus neoformans in immunodeficient mice. Infect Immun 75:4792–4798. doi: 10.1128/IAI.00587-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chalfant C, Del Poeta M. 2010. Sphingolipids as signaling and regulatory molecules. Landes Bioscience, Austin, TX. [Google Scholar]
- 12.Strub GM, Maceyka M, Hait NC, Milstien S, Spiegel S. 2010. Extracellular and intracellular actions of sphingosine-1-phosphate. Adv Exp Med Biol 688:141–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fyrst H, Saba JD. 2010. An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat Chem Biol 6:489–497. doi: 10.1038/nchembio.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee M-J, Van Brocklyn JR, Thangada S, Liu CH, Hand AR, Menzeleev R, Spiegel S, Hla T. 1998. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science 279:1552–1555. doi: 10.1126/science.279.5356.1552. [DOI] [PubMed] [Google Scholar]
- 15.Ponnusamy S, Selvam SP, Mehrotra S, Kawamori T, Snider AJ, Obeid LM, Shao Y, Sabbadini R, Ogretmen B. 2012. Communication between host organism and cancer cells is transduced by systemic sphingosine kinase 1/sphingosine 1-phosphate signalling to regulate tumour metastasis. EMBO Mol Med 4:761–775. doi: 10.1002/emmm.201200244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosen H, Goetzl EJ. 2005. Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat Rev Immunol 5:560–570. doi: 10.1038/nri1650. [DOI] [PubMed] [Google Scholar]
- 17.Allende ML, Bektas M, Lee BG, Bonifacino E, Kang J, Tuymetova G, Chen W, Saba JD, Proia RL. 2011. Sphingosine-1-phosphate lyase deficiency produces a pro-inflammatory response while impairing neutrophil trafficking. J Biol Chem 286:7348–7358. doi: 10.1074/jbc.M110.171819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. 2004. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 427:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 19.Garg SK, Santucci MB, Panitti M, Pucillo L, Bocchino M, Okajima F, Bisen PS, Saltini C, Fraziano M. 2006. Does sphingosine 1-phosphate play a protective role in the course of pulmonary tuberculosis? Clin Immunol 121:260–264. doi: 10.1016/j.clim.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 20.Garg SK, Volpe E, Palmieri G, Mattei M, Galati D, Martino A, Piccioni MS, Valente E, Bonanno E, De Vito P. 2004. Sphingosine 1-phosphate induces antimicrobial activity both in vitro and in vivo. J Infect Dis 189:2129–2138. doi: 10.1086/386286. [DOI] [PubMed] [Google Scholar]
- 21.McQuiston T, Luberto C, Del Poeta M. 2011. Role of sphingosine-1-phosphate (S1P) and S1P receptor 2 in the phagocytosis of Cryptococcus neoformans by alveolar macrophages. Microbiology 157:1416–1427. doi: 10.1099/mic.0.045989-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allende ML, Dreier JL, Mandala S, Proia RL. 2004. Expression of the sphingosine 1-phosphate receptor, S1P1, on T-cells controls thymic emigration. J Biol Chem 279:15396–15401. doi: 10.1074/jbc.M314291200. [DOI] [PubMed] [Google Scholar]
- 23.Kono M, Mi Y, Liu Y, Sasaki T, Allende ML, Wu Y-P, Yamashita T, Proia RL. 2004. The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J Biol Chem 279:29367–29373. doi: 10.1074/jbc.M403937200. [DOI] [PubMed] [Google Scholar]
- 24.Qureshi A, Grey A, Rose KL, Schey KL, Del Poeta M. 2011. Cryptococcus neoformans modulates extracellular killing by neutrophils. Front Microbiol 2:193. doi: 10.3389/fmicb.2011.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh A, Wang H, Silva LC, Na C, Prieto M, Futerman AH, Luberto C, Del Poeta M. 2012. Methylation of glycosylated sphingolipid modulates membrane lipid topography and pathogenicity of Cryptococcus neoformans. Cell Microbiol 14:500–516. doi: 10.1111/j.1462-5822.2011.01735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 27.Cinamon G, Zachariah MA, Lam OM, Foss FW, Cyster JG. 2008. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat Immunol 9:54–62. doi: 10.1038/ni1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jenne CN, Enders A, Rivera R, Watson SR, Bankovich AJ, Pereira JP, Xu Y, Roots CM, Beilke JN, Banerjee A. 2009. T-bet-dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J Exp Med 206:2469–2481. doi: 10.1084/jem.20090525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rathinasamy A, Czeloth N, Pabst O, Förster R, Bernhardt G. 2010. The origin and maturity of dendritic cells determine the pattern of sphingosine 1-phosphate receptors expressed and required for efficient migration. J Immunol 185:4072–4081. doi: 10.4049/jimmunol.1000568. [DOI] [PubMed] [Google Scholar]
- 30.Olivera A, Mizugishi K, Tikhonova A, Ciaccia L, Odom S, Proia RL, Rivera J. 2007. The sphingosine kinase-sphingosine-1-phosphate axis is a determinant of mast cell function and anaphylaxis. Immunity 26:287–297. doi: 10.1016/j.immuni.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 31.Oskeritzian CA, Alvarez SE, Hait NC, Price MM, Milstien S, Spiegel S. 2008. Distinct roles of sphingosine kinases 1 and 2 in human mast-cell functions. Blood 111:4193–4200. doi: 10.1182/blood-2007-09-115451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deshmane SL, Kremlev S, Amini S, Sawaya BE. 2009. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 29:313–326. doi: 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huffnagle GB, Strieter RM, Standiford TJ, McDonald RA, Burdick MD, Kunkel SL, Toews GB. 1995. The role of monocyte chemotactic protein-1 (MCP-1) in the recruitment of monocytes and CD4+ T cells during a pulmonary Cryptococcus neoformans infection. J Immunol 155:4790–4797. [PubMed] [Google Scholar]
- 34.Traynor TR, Kuziel WA, Toews GB, Huffnagle GB. 2000. CCR2 expression determines T1 versus T2 polarization during pulmonary Cryptococcus neoformans infection. J Immunol 164:2021–2027. doi: 10.4049/jimmunol.164.4.2021. [DOI] [PubMed] [Google Scholar]
- 35.Olszewski MA, Huffnagle GB, Traynor TR, McDonald RA, Cook DN, Toews GB. 2001. Regulatory effects of macrophage inflammatory protein 1α/CCL3 on the development of immunity to Cryptococcus neoformans depend on expression of early inflammatory cytokines. Infect Immun 69:6256–6263. doi: 10.1128/IAI.69.10.6256-6263.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huffnagle GB, Strieter RM, McNeil LK, McDonald RA, Burdick MD, Kunkel SL, Toews GB. 1997. Macrophage inflammatory protein-1alpha (MIP-1alpha) is required for the efferent phase of pulmonary cell-mediated immunity to a Cryptococcus neoformans infection. J Immunol 159:318–327. [PubMed] [Google Scholar]
- 37.Wozniak KL, Ravi S, Macias S, Young ML, Olszewski MA, Steele C, Wormley FL Jr. 2009. Insights into the mechanisms of protective immunity against Cryptococcus neoformans infection using a mouse model of pulmonary cryptococcosis. PLoS One 4:e6854. doi: 10.1371/journal.pone.0006854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, Barbour SE, Milstien S, Spiegel S. 2008. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J 22:2629–2638. doi: 10.1096/fj.08-107169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Z, Zhang Z-Y, Fauser U, Schluesener HJ. 2008. FTY720 ameliorates experimental autoimmune neuritis by inhibition of lymphocyte and monocyte infiltration into peripheral nerves. Exp Neurol 210:681–690. doi: 10.1016/j.expneurol.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 40.Adada M, Canals D, Hannun YA, Obeid LM. 2013. Sphingosine-1-phosphate receptor 2. FEBS J 280:6354–6366. doi: 10.1111/febs.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwab SR, Cyster JG. 2007. Finding a way out: lymphocyte egress from lymphoid organs. Nat Immunol 8:1295–1301. doi: 10.1038/ni1545. [DOI] [PubMed] [Google Scholar]
- 42.Keul P, Lucke S, von Wnuck Lipinski K, Bode C, Gräler M, Heusch G, Levkau B. 2011. Sphingosine-1-phosphate receptor 3 promotes recruitment of monocyte/macrophages in inflammation and atherosclerosis. Circ Res 108:314–323. doi: 10.1161/CIRCRESAHA.110.235028. [DOI] [PubMed] [Google Scholar]
- 43.Szymczak WA, Davis MJ, Lundy SK, Dufaud C, Olszewski M, Pirofski L. 2013. X-linked immunodeficient mice exhibit enhanced susceptibility to Cryptococcus neoformans infection. mBio 4(4):e00265-13. doi: 10.1128/mBio.00265-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li M-H, Harel M, Hla T, Ferrer F. 2014. Induction of chemokine (CC motif) ligand 2 by sphingosine-1-phosphate signaling in neuroblastoma. J Pediatr Surg 49:1286–1291. doi: 10.1016/j.jpedsurg.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen X-L, Grey JY, Thomas S, Qiu F-H, Medford RM, Wasserman MA, Kunsch C. 2004. Sphingosine kinase-1 mediates TNF-α-induced MCP-1 gene expression in endothelial cells: upregulation by oscillatory flow. Am J Physiol Heart Circ Physiol 287:H1452–H1458. doi: 10.1152/ajpheart.01101.2003. [DOI] [PubMed] [Google Scholar]
- 46.Fiers W. 1991. Tumor necrosis factor characterization at the molecular, cellular and in vivo level. FEBS Lett 285:199–212. doi: 10.1016/0014-5793(91)80803-B. [DOI] [PubMed] [Google Scholar]
- 47.Naudé PJ, den Boer JA, Luiten PG, Eisel UL. 2011. Tumor necrosis factor receptor cross-talk. FEBS J 278:888–898. doi: 10.1111/j.1742-4658.2011.08017.x. [DOI] [PubMed] [Google Scholar]
- 48.Roach DR, Bean AG, Demangel C, France MP, Briscoe H, Britton WJ. 2002. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol 168:4620–4627. doi: 10.4049/jimmunol.168.9.4620. [DOI] [PubMed] [Google Scholar]
- 49.Brahmbhatt S, Black G, Carroll N, Beyers N, Salker F, Kidd M, Lukey P, Duncan K, Van Helden P, Walzl G. 2006. Immune markers measured before treatment predict outcome of intensive phase tuberculosis therapy. Clin Exp Immunol 146:243–252. doi: 10.1111/j.1365-2249.2006.03211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guillot L, Carroll SF, Homer R, Qureshi ST. 2008. Enhanced innate immune responsiveness to pulmonary Cryptococcus neoformans infection is associated with resistance to progressive infection. Infect Immun 76:4745–4756. doi: 10.1128/IAI.00341-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pettus BJ, Bielawski J, Porcelli AM, Reames DL, Johnson KR, Morrow J, Chalfant CE, Obeid LM, Hannun YA. 2003. The sphingosine kinase 1/sphingosine-1-phosphate pathway mediates COX-2 induction and PGE2 production in response to TNF-α. FASEB J 17:1411–1421. doi: 10.1096/fj.02-1038com. [DOI] [PubMed] [Google Scholar]
- 52.Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, Maceyka M, Jiang H, Luo C, Kordula T. 2010. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465:1084–1088. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fairhurst RM, Pegues DA. 2002. Pulmonary cryptococcal granulomas. N Engl J Med 347:497. doi: 10.1056/NEJMicm010063. [DOI] [PubMed] [Google Scholar]
- 54.Shibuya K, Hirata A, Omuta J, Sugamata M, Katori S, Saito N, Murata N, Morita A, Takahashi K, Hasegawa C. 2005. Granuloma and cryptococcosis. J Infect Chemother 11:115–122. doi: 10.1007/s10156-005-0387-X. [DOI] [PubMed] [Google Scholar]