

Abstract
Chronic periodontitis is a local inflammatory disease induced by a dysbiotic microbiota and leading to destruction of the tooth-supporting structures. Microbial nucleic acids are abundantly present in the periodontium, derived through release after phagocytic uptake of microbes and/or from biofilm-associated extracellular DNA. Binding of microbial DNA to its cognate receptors, such as Toll-like receptor 9 (TLR9), can trigger inflammation. In this study, we utilized TLR9 knockout (TLR9−/−) mice and wild-type (WT) controls in a murine model of Porphyromonas gingivalis-induced periodontitis and report the first in vivo evidence that TLR9 signaling mediates the induction of periodontal bone loss. P. gingivalis-infected WT mice exhibited significantly increased bone loss compared to that in sham-infected WT mice or P. gingivalis-infected TLR9−/− mice, which were resistant to bone loss. Consistent with this, the expression levels of interleukin 6 (IL-6), tumor necrosis factor (TNF), and receptor-activator of nuclear factor kappa B ligand (RANKL) were significantly elevated in the gingival tissues of the infected WT mice but not in infected TLR9−/− mice compared to their levels in controls. Ex vivo studies using splenocytes and bone marrow-derived macrophages revealed significantly diminished cytokine production in TLR9−/− cells relative to the cytokine production in WT cells in response to P. gingivalis, thereby implicating TLR9 in inflammatory responses to this organism. Intriguingly, compared to the cytokine production in WT cells, TLR9−/− cells exhibited significantly decreased proinflammatory cytokine production upon challenge with lipopolysaccharide (LPS) (TLR4 agonist) or Pam3Cys (TLR2 agonist), suggesting possible cross talk between TLR9, TLR4, and TLR2. Collectively, our results provide the first proof-of-concept evidence implicating TLR9-triggered inflammation in periodontal disease pathogenesis, thereby identifying a new potential therapeutic target to control periodontal inflammation.
INTRODUCTION
Periodontitis is one of the most common inflammatory diseases worldwide, affecting more than 47% of the U.S. adult population and leading to destruction of tooth-supporting structures (1). Although a dysbiotic biofilm structure initiates the disease, the periodontal tissue destruction occurs as a result of a dysregulated immune response to the microbial insult (2). Chronic, persistent immune responses to this complex microbiome not only can lead to tooth loss but also are associated with increased risk for several systemic complications, including atherosclerosis, diabetes, adverse pregnancy outcomes, pulmonary diseases, rheumatoid arthritis, and cancer (3–8). Periodontal research currently focuses primarily on identifying disease biomarkers, understanding mechanisms of pathogenesis, and identifying therapeutic targets to prevent the deleterious effects of periodontal inflammation on both local and systemic tissues.
The host immune response is initiated upon sensing of microbe-associated molecular patterns (MAMPs) by families of receptors collectively called pattern recognition receptors (PRRs) (9). The binding of MAMPs to PRRs promotes the release of inflammatory mediators that promote the induction of innate immune mechanisms aiming to eliminate pathogens and also orchestrate the development of the adaptive immune response. Toll-like receptors (TLRs) are the most widely studied PRRs that play a critical role as the first line of defense against microbial insults and also serve as a bridge between innate and adaptive immunity. Several types of cells can express TLRs, and each receptor is involved in the sensing of distinct microbial products. TLRs that are located on the plasma membrane (TLR1, -2, -4, -5, -6, and -10) recognize structural components of pathogens, such as proteins, lipoproteins, and polysaccharides. The receptors that reside in the endosomes (TLR3, -7, -8, and -9) are specific for nucleic acids, including microbial or self RNA and DNA (9). TLRs are type I transmembrane proteins composed of an extracellular leucine-rich repeat (LRR) domain that is involved in ligand recognition, a transmembrane domain, and a Toll-interleukin 1 (IL-1) receptor (TIR) domain that is involved in signaling. The signaling pathways activated by TLRs engage adaptor molecules that are recruited by TIR/TIR domain interactions and include myeloid differentiation primary response 88 (MyD88), TIR domain-containing adaptor protein (TIRAP, also known as MAL for MyD88 adaptor-like), TIR domain-containing adaptor inducing interferon (IFN) (TRIF), and TRIF-related adaptor molecule (TRAM). MyD88 is essential for signaling through all TLRs except TLR3 and is involved in early nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) activation and proinflammatory gene expression. Although the innate immune response, including TLR-mediated inflammatory responses, is potentially protective against infectious challenge, excessive or dysregulated activation of these sensors may lead to persistent, chronic inflammation in many conditions, including periodontal disease. Hence, a thorough understanding of the molecular mechanisms involved in TLR activation is considered to be crucial in the development of therapeutics in several diseases, including periodontitis (10, 11).
Nucleic acids represent one of the key components that are sensed by the innate immune system (12). During infection, DNA and RNA that are sequestered within bacteria, viruses, or host cells can be released and subsequently detected by specific receptors, activating inflammatory signaling cascades (13). Toll like receptor 9 (TLR9) is the major sensor for microbial unmethylated/hypomethylated CpG (cytosine-phosphate-guanosine) DNA motifs. It activates nuclear factor kappa B (NF-κB), activator protein 1 (AP-1), and mitogen-activated protein (MAP) kinase signaling pathways, which stimulate proinflammatory activities, but also the IFN regulatory factor (IRF) pathway, which can induce type I interferon and anti-inflammatory activities (9). Periodontal tissues are constantly exposed to microbes, and tissue homeostasis is mediated by a balance between continual turnover of microbial and host cells. The premise of this study was that the constant cell turnover provides a platform for the continuous presence of nucleic acids in the periodontium which could thereby exacerbate inflammatory responses through recognition by TLR9. Despite having been fairly well studied in relation to various diseases of infectious or immunological origin, the role of nucleic acid sensing by intracellular PRRs in the pathogenesis of periodontitis has received little attention. Previous clinical studies from our group and others have demonstrated increased TLR9 gene and protein expression in gingival tissues associated with chronic periodontitis (14–16). TLR9 showed the highest gene expression among all innate receptors, and the increased expression was predominantly localized in the connective tissue and basal epithelial layers (14). It is also well established that the host genetic background affects susceptibility to periodontitis (17). Recently, both our group and others revealed differential expression of specific single-nucleotide polymorphisms (SNPs) in the TLR9 gene in individuals with chronic periodontitis, providing further evidence for the involvement of TLR9 in periodontal disease (18, 19). Additionally, in vitro investigations demonstrated that periodontitis-associated bacterial DNA upregulates several genes of the innate immune response and induces the production of proinflammatory cytokines through TLR9 in macrophages and epithelial cells (20–22). In fact, a recent study has identified the TLR9 gene as one of the most promising candidate genes involved in the pathogenesis of periodontitis by using an integrative gene prioritization method (23). Collectively, emerging evidence suggests that TLR9-triggered immune responses may constitute a novel inflammatory pathway in periodontitis (24).
The importance of specific PRRs in mediating pathological inflammation requires the use of in vivo models, which, unlike in vitro models, can replicate the complexity of the interactions between the immune response, the microbiome, and the host tissue (25). One of the most widely used in vivo models of periodontitis involves inoculation of the murine oral cavity with Porphyromonas gingivalis by oral gavage (25–27). P. gingivalis is a keystone pathogen in periodontitis and orchestrates inflammatory bone loss upon its colonization of the murine oral cavity (2). As alluded to above, we hypothesize that TLR9 activation contributes to the pathogenesis of periodontitis by promoting enhanced inflammation. To test this hypothesis, we utilized the oral gavage model with P. gingivalis and TLR9 knockout (KO) mice to characterize the role of TLR9 in periodontal disease pathogenesis. We further determined whether lack of TLR9 signaling affects inflammatory responses to P. gingivalis and various TLR ligands by using ex vivo models. To our knowledge, this is the first in vivo study to investigate the role of an intracellular nucleic acid sensor in the pathogenesis of periodontitis.
MATERIALS AND METHODS
Bacteria.
P. gingivalis (strain ATCC 33277) was grown in an anaerobic chamber using brain heart infusion (BHI) broth (Difco Laboratories) supplemented with 0.5% yeast extract, 5 μg/ml hemin, 0.5 μg/ml vitamin K, and 0.1% cysteine as described previously (20).
Mice.
All the experiments involving studies with animals have been approved by the institutional animal care and use committee at Virginia Commonwealth University. TLR9−/− mice with the periodontitis-susceptible BALB/c background were obtained from Denis Klinmann (National Cancer Institute, Bethesda, MD) with the permission of Shizuo Akira (Osaka University). BALB/c wild-type (WT) mice were purchased from Jackson Laboratory (Bar Harbor, ME). All the animals were housed in a sterile, specific-pathogen-free room in individually ventilated cages. Age- and gender-matched groups of wild-type and knockout animals were used for each experiment.
P. gingivalis-induced murine periodontitis model.
Periodontal inflammation and bone loss were induced by oral inoculation with P. gingivalis following published protocols (27). Briefly, the mice were given kanamycin in drinking water (1 g/liter) for 7 days, followed by a 5-day antibiotic-free period. The mice were then infected with 2 × 109 CFU of live P. gingivalis (ATCC 33277) in 100 ml of vehicle comprised of phosphate-buffered saline (PBS) containing 2% carboxymethyl-cellulose (CMC) that was placed into the esophagus and oral cavity. The inoculations were performed three times within 48 h at the first and the second week (a total of 6 inoculations). Control mice from each group (wild type and knockout) received the antibiotic pretreatment and the CMC gavage without P. gingivalis. Seven weeks after the first gavage, the mice were euthanized by CO2 inhalation and cervical dislocation.
Oral microflora analysis.
The levels of P. gingivalis colonization were assessed using quantitative real-time PCR (qPCR) of the ISPg1 gene (P. gingivalis) (28). Briefly, bacterial samples were collected by paper point from the upper molars and incubated in BHI (Difco laboratories) supplemented with 0.5% yeast extract, 5 μg/ml hemin, 0.5 μg/ml vitamin K, and 0.1% cysteine anaerobically for 1 week (29, 30). The suspension was centrifuged, and the bacterial pellet was washed with PBS followed by isolation of genomic DNA (gDNA) using the DNeasy kit (Qiagen, Valencia, CA). qPCR was performed with the Applied Biosystems 7500 fast system using RT2 SYBR green ROX qPCR master mix (Qiagen) and the following primers (IDT): ISPg1 (P. gingivalis): 5′-CGCAGACGACAGAGAAGACA-3′ and 5′-ACGGACAACCTGTTTTTGATAATCCT-3′. A standard curve was obtained using 5-fold serial dilutions of 1 × 106 P. gingivalis DNA.
P. gingivalis antibody levels.
Blood samples were collected from each mouse strain before inoculation of bacteria and at the time of euthanasia at week 7 using cheek bleed or cardiac puncture, respectively. Sera were analyzed for P. gingivalis-specific-antibody levels using enzyme-linked immunosorbent assays (ELISAs) (31). Briefly, the wells of the 96-well plates (Costar; Fisher Scientific) were coated with formalin-fixed P. gingivalis (1 × 108/well) at 4°C overnight. The plates were washed using PBS with 0.05% Tween 20 (PBST) (wash buffer). To prevent nonspecific binding, plates were blocked with 1% bovine serum albumin (BSA) in PBS for 30 min at room temperature. The serum samples were added into each well and incubated for 2 h at 37°C. Serial dilutions of unlabeled mouse IgG (Southern Biotech) served as the standard. Following another wash step, P. gingivalis-specific IgG was incubated with horseradish peroxidase (HRP) conjugated to secondary antibody (goat anti-mouse IgG; Southern Biotech) for 1 h at 37°C. ELISA wells were color developed with 3,3′,5′5′-tetramethylbenzidine (TMB; SouthernBiotech) for 20 min at room temperature, and the enzyme reaction was stopped by adding 0.1 N H2SO4. The plates were read at 450 nm.
Determination of periodontal bone loss.
Alveolar bone loss around the maxillary molars was determined using microscopy as described previously (32). Briefly, at the end of each experiment, the animals were sacrificed, followed by defleshing of the skulls by autoclaving. The specimens were then immersed in 3% hydrogen peroxide overnight and stained with 1% methylene blue. The distance from the cemento-enamel junction (CEJ) to the alveolar bone crest (ABC) was measured at a total of 14 predetermined buccal sites per mouse using a stereoscopic zoom microscope (SMZ1000; Nikon) at a magnification of ×40. To calculate bone loss, the 14-site total CEJ-ABC distance for each mouse was subtracted from the mean CEJ-ABC distance of sham-infected mice. The results were expressed in millimeters, and negative values indicated bone loss relative to the results for sham controls. The specimens were imaged using a desktop micro-CT (computed tomography) system (Brüker Skyscan 1173 micro-CT scanner; Skyscan NV, Kontich, Belgium) at a resolution of 1,120 by 1,120 pixels in all three spatial dimensions by setting the sagittal plane parallel to the X-ray beam axis.
Determination of inflammatory mediator expression in gingival tissues.
The gingival tissues were harvested from around the maxillary molars at the time of euthanasia, and total RNA was isolated and cDNA was generated as described previously (14). The levels of IL-6, tumor necrosis factor (TNF), and receptor-activator of NF-κB ligand (RANKL) expression in gingival tissues of the mice in each group were determined by real-time PCR (Applied Biosystems 7500) using SYBR green master mix (SaBiosciences) and the following specific primer sets: for RANKL, forward, 5′ CAG CAT CGC TCT GTT CCT GTA 3′, and reverse, 5′ CTG CGT TTT CAT GGA GTC TCA 3′; for IL-6, forward, 5′ TCT ATA CCA CTT CAC AAG TCG GA 3′, and reverse, 5′ GAA TTG CCA TTG CAC AAC TCT TT 3′; for TNF, forward, 5′ CTG AAC TTC GGG GTG ATC GG 3′, and reverse, 5′ GGCTTG TCA CTC GAA TTT TGA GA 3′; and for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), forward, 5′ AGG TCG GTG TGA ACG GAT TTG 3′, and reverse, 5′ GGG GTC GTT GAT GGC AAC A 3′. The GAPDH gene was used as an internal control. The relative amount of each mRNA was calculated using the cycle threshold (2−ΔΔCT) method, where ΔCT = (CTmRNA − CTGAPDH).
Spleen harvesting and processing.
Uninfected naive mice (WT and TLR9−/−) were sacrificed, and spleens were removed, segmented, and placed in 10 ml serum-free RPMI 1640 medium (Invitrogen). Cells were dispersed through a 40-μm cell strainer and centrifuged at 1,500 rpm for 5 min. The pellet was suspended in 5 ml ACK lysis buffer (Quality Biological, Inc.) for 5 min to allow for the lysis of red blood cells. Serum-free medium was added to halt lysis, and the supernatant was collected and centrifuged at 1,500 rpm for 5 min to pellet the leukocytes. The total number of splenocytes was calculated by trypan blue exclusion assay, and further experiments were performed. The cells (2 × 106 cells/well) were cultured in RPMI 1640 medium (Invitrogen) supplemented with 2 mM l-glutamine, 10% fetal bovine serum (Invitrogen), 50 μM 2-mercaptoethanol, and 1% antibiotics (penicillin-streptomycin) at 37°C in a humidified chamber in the presence of 5% CO2 using 48-well culture plates for stimulation experiments.
Purification of bone marrow-derived macrophages.
Bone marrow-derived macrophages were propagated and purified following the methods of previously published studies (33). Briefly, bone marrow was obtained from the femurs and tibias of 8-to-12-week-old wild-type and TLR9−/− mice. Red blood cells were osmotically lysed using ACK lysis buffer. Following lysis, cells were washed and filtered through a 70-μM nylon mesh filter to remove debris. Bone marrow cells were resuspended in bone marrow growth medium (BMM) (DMEM containing 4.5 g/liter glucose, 100 mg/liter sodium pyruvate, 10% fetal calf serum, glutamine, 5 × 10−5 M beta-mercaptoethanol, and 10% supernatant from colony-stimulating factor 1 (CSF-1)-transfected NIH 3T3 cells (courtesy of Marlena Westcott, Department of Microbiology and Immunology, Wake Forest School of Medicine). Cells were seeded at a density of 4 × 106 cells in 10 ml medium in a 125- by 50-mm Lab-Tek non-tissue culture-treated petri dish and fed on day 3 by gently adding an additional 10 ml of BMM. On day 6, the monolayer of macrophages was washed by aspirating the medium and gently adding PBS to remove any nonadherent or dead cells. Adherent macrophages were harvested by adding PBS to the monolayer, incubating at 4°C for 10 min, and gently pipetting to remove cells. Macrophages were further purified by CD11c-negative selection using magnetic bead isolation (Miltenyi Biotec) according to the manufacturer's protocol. Purified macrophages were seeded at a concentration of 4 × 105 cells in 160 μl of BMM without CSF-1-containing supernatant in a 48-well plate for 12 to 18 h prior to stimulation.
Stimulation of splenocytes and bone marrow-derived macrophages.
Heat-killed P. gingivalis was used to challenge the splenocytes and bone marrow-derived macrophages. P. gingivalis DNA was isolated using repeated phenol-chloroform extractions as described previously (20). The cells were stimulated with ODN 1668 (TLR9 agonist, 100 ng/μl; InviVogen), P. gingivalis (multiplicity of infection [MOI] of 1:100), P. gingivalis DNA (100 ng/μl), P. gingivalis lipopolysaccharide (LPS) (10 ng/μl; InviVogen), Escherichia coli LPS (10 ng/μl; InviVogen), and Pam3Cys (1 ng/μl; InviVogen) for 24 h. Inflammatory cytokine levels (IL-6 and TNF) were determined in cell-free culture supernatants using ELISAs (eBiosciences). Unstimulated cells were used as negative controls.
Surface staining and flow cytometry.
Individual cell populations of the splenocytes were determined in unstimulated and stimulated cells at baseline and at 24 and 48 h using flow cytometry. The following antibodies were used: anti-mouse Ly-6G–fluorescein isothiocyanate (FITC) antibody (clone 1A8), anti-mouse CD3-phycoerythrin (PE) antibody (clone 145-2C11), anti-mouse Gr-1–peridinin chlorophyll protein (PerCP) antibody (clone RB6-8C5), anti-mouse B220-allophycocyanin (APC) antibody (clone RA3-6B2), anti-mouse CD11c-APC-Cy7 antibody (clone N418), anti-mouse F4/80-PE-Cy7 antibody (clone BM8), anti-mouse/human CD11b-brilliant violet 711 antibody (clone M1/70), and rat anti-mouse Ly-6C–brilliant violet 421 antibody (clone AL-21). All antibodies were purchased from BioLegend except for the Ly-6C antibody, which was from BD Pharmingen. Surface staining of splenocytes for flow cytometry was performed by incubating cells for 30 min on ice in a 1:100 dilution of antibody in 2% fluorescence-activated cell sorting (FACS) buffer (PBS supplemented with 2% fetal calf serum). After being washed three times with FACS buffer, cells were fixed in 2% paraformaldehyde (Sigma-Aldrich). All samples were acquired on a BD LSRFortessa instrument, and the data analyzed using FlowJo software (TreeStar). Following gating on the viable single cell population, macrophages were defined as CD11bhi Gr-1− F4/80+, neutrophils as CD11b+ Gr-1+ Ly-6Clo, monocytes as CD11b+ Gr-1+ Ly-6Chi, conventional dendritic cells as CD11b+ B220−, B cells as B220+, and T cells as CD3+.
RNA isolation and real-time PCR.
Total RNA was isolated from splenocytes using the RNeasy kit (Qiagen) and gDNA eliminator spin columns. The amount of RNA was determined spectrophotometrically, and the RNA quality was assessed by performing 1% agarose gel electrophoresis. cDNA was synthesized from 1.0 μg total RNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems) according to the manufacturer's protocol. cDNA was kept on ice prior to gene expression analyses or stored at −20°C until use. The expression of the receptors was determined by real-time PCR (Applied Biosystems 7500) using specific primers (for TLR2, forward, 5′-CCT GGC CCT CTC TAC AAA CTT-3′, and reverse, 5′-ACT GTG TAT TCG TGT GCT GGA TA-3′, and for TLR4, forward, 5′-TGC TGC CGT TTT ATC ACG GA-3′, and reverse, 5′-CTA AAC TCT GGA TGG GGT TTC C-3′; IDT, Inc.) and SYBR green master mix (SaBiosciences). The GAPDH gene was used as an internal control. The relative amount of each gene was calculated using the 2−ΔΔCT method, where ΔCT = (CTmRNA − CTGAPDH).
Western blotting.
Murine splenocytes were stimulated with E. coli LPS or Pam3Cys or left unstimulated for 24 h in 24-well plates (6 × 105/well). After being washed with ice-cold PBS, the cells were collected and lysed in 100 μl of ice-cold NP-40 lysis buffer (25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 1 mM EDTA, pH 8), including 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM Na3VO4, 1 mM dithiothreitol (DTT), and 1× protease inhibitor cocktail (Sigma-Aldrich), by repeated pipetting and then homogenized twice at 50% pulse for 30 s each time using an ultrasonic homogenizer (BioLogics, Inc., Cary, NC). The protein concentration was determined using the bicinchoninic acid (BCA) protein assay kit (Thermo Scientific). Cell lysates (25 μg protein) were subjected to electrophoresis on 4-to-15% Mini-Protean TGX gels (Bio-Rad) and transferred to nitrocellulose membranes, followed by Western blotting according to standard techniques. Briefly, the membranes were incubated with primary antibody (rabbit monoclonal anti-mouse TLR-2 antibody, 1:1,000 dilution; Cell Signaling) or anti-mouse actin antibody (1:200 dilution; Santa Cruz Biotechnology) in 5% nonfat dry milk–Tris-buffered saline supplemented with 0.05% Tween 20 (TBST) solution at 4°C overnight and then with Immun-Star HRP-conjugated secondary antibody (goat anti-rabbit or goat anti-mouse IgG, 1:3,333) in 5% nonfat dry milk–1× TBST solution at room temperature for 1 h. Immunoreactive bands were visualized by using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) followed by exposure to X-ray film. Densitometric analyses were performed using the G:BOX Chem XX6 system and GeneTools image analysis software (Syngene). TLR-2 levels in control samples were determined as the percentages of total actin protein bands. To quantify TLR-2 levels following stimulation, the levels of actin and TLR-2 were normalized between samples. The TLR-2 signal was then normalized to actin protein levels.
Statistical analysis.
Data were analyzed by one-way analysis of variance (ANOVA) and the Tukey multiple-comparison test or unpaired t test using the InStat program (GraphPad Software, San Diego, CA). A P value of <0.05 was considered significant.
RESULTS
P. gingivalis infection.
We investigated the role of TLR9 in periodontal disease pathogenesis using the P. gingivalis-induced murine periodontitis model. P. gingivalis colonization, as well as P. gingivalis-specific antibody levels in the serum, were assessed to confirm that the mice were successfully infected with P. gingivalis. P. gingivalis was detected in WT and TLR9−/− mice at comparable levels 2 weeks postinoculation (Fig. 1A). Moreover, there was a significant increase in P. gingivalis-specific antibody levels in both WT and TLR9−/− mice inoculated with this organism compared to the levels in sham-infected controls (P < 0.0001) (Fig. 1B). Sera obtained prior to inoculation had no specific antibody activity against P. gingivalis, similar to sera from sham-infected mice (data not shown). No significant difference was observed between the antibody levels of P. gingivalis-infected WT and TLR9−/− mice (Fig. 1B). Therefore, TLR9 deficiency did not significantly affect P. gingivalis colonization or the induction of systemic antibody responses to this organism. This is important for the validity of the bone loss study (see below), since any potential differences in bone loss between WT and TLR9−/− mice would not be attributed to differences in P. gingivalis colonization.
FIG 1.

Comparison of P. gingivalis levels within the periodontal tissues (A) and P. gingivalis-specific antibody responses in serum (B) in TLR9−/− and WT mice. Each strain was orally inoculated with P. gingivalis or vehicle only (sham). The P. gingivalis levels were determined by qPCR of the ISPg1 gene (P. gingivalis) at 2 weeks postinfection. There was no statistically significant difference in P. gingivalis levels among groups. P. gingivalis-specific antibody responses were determined at the termination of the experiment. The antibody titers in TLR9−/− mice and WT mice infected with P. gingivalis were significantly higher than the titers in the uninfected mice (P < 0.0001). The data shown are the mean results ± standard deviations (SD; n = 8 mice per group) and were analyzed using either the unpaired t test or one-factor analysis of variance (ANOVA) and the Tukey test.
TLR9-mediated inflammation promotes P. gingivalis-induced periodontal bone loss.
Next, we determined the effect of TLR9 deficiency in periodontal disease progression. The disease phenotype was evaluated among 4 groups of mice, which included sham- or P. gingivalis-infected wild-type and TLR9 knockout animals. The distance from the cemento-enamel junction to the alveolar bone crest (CEJ-ABC distance) was measured at a total of 14 predetermined buccal sites per mouse using a microscope. The mean CEJ-ABC measurement for each mouse is shown in Fig. 2A. To calculate bone loss, the 14-site total CEJ-ABC distance for each mouse was subtracted from the mean CEJ-ABC distance of sham-infected mice. The results are expressed in millimeters, and negative values indicate bone loss relative to the results for sham controls (Fig. 2B). Analyses of the mean CEJ-ABC measurements and the change in maxillary alveolar bone levels among each group revealed significantly increased bone loss in the P. gingivalis-infected WT mice compared to the results for the sham-infected WT mice (P < 0.05) (Fig. 2A and B). In contrast, P. gingivalis-infected TLR9−/− mice exhibited no bone loss compared to the results for sham-infected controls (Fig. 2A and B). Moreover, P. gingivalis-infected WT mice exhibited significantly more bone loss than P. gingivalis-infected TLR9−/− mice (P < 0.01) (Fig. 2A and B). Alveolar bone levels were imaged using micro-CT, which displayed visible bone destruction in infected WT mice, while no bone loss was observed in infected TLR9 KO mice (Fig. 2C). Collectively, these results show for the first time that TLR9 signaling mediates the induction of alveolar bone loss.
FIG 2.
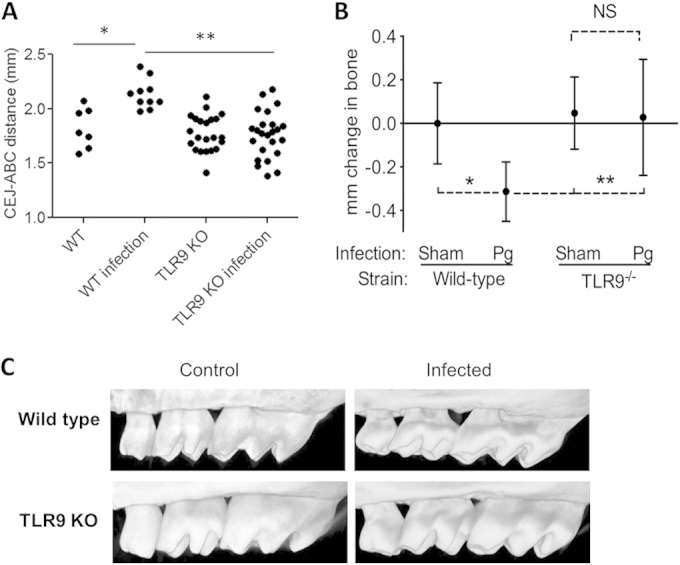
TLR9 KO (TLR9−/−) mice are resistant to P. gingivalis-instigated periodontal bone loss. Groups of mice (WT [n = 17] and TLR9−/− [n = 47]) were infected with P. gingivalis (Pg) or sham infected and euthanized 42 days later. Measurements were performed in defleshed maxillae. The data are represented as the mean results ± SD (n = 64 mice). (A) Distance (in millimeters) between the cemento-enamel junction (CEJ) and alveolar bone crest (ABC) in each group of animals. (B) Amount of bone change in WT and TLR9−/− mice. Negative values indicate bone loss in P. gingivalis-inoculated mice relative to the results for vehicle-inoculated (sham) controls. (C) Representative micro-CT images of maxillae from each group of mice. *, P < 0.05; **, P < 0.01.
Lack of TLR9 signaling suppresses inflammation in gingival tissues of P. gingivalis-infected mice.
Our results revealed significantly less bone loss in P. gingivalis-infected TLR9-deficient mice compared to the amount of bone loss in infected WT mice. We therefore hypothesized that TLR9 activation promotes inflammation and osteoclastogenesis. To investigate this, dissected gingival tissues from each group of mice were processed for qPCR to determine the mRNA expression of inflammatory molecules. In line with their susceptibility to bone loss, P. gingivalis-infected WT mice displayed significantly increased levels of expression of IL-6, TNF, and RANKL, a key osteoclastogenic factor, compared to the expression levels in sham-infected WT mice (P < 0.05) (Fig. 3). In stark contrast, no significant increase of any of the inflammatory markers was detected in infected TLR9−/− mice compared to their levels in sham-infected KO mice (Fig. 3). These findings confirm and expand the bone loss data and suggest that TLR9 signaling can modulate the progression of periodontal disease through upregulation of inflammatory and osteoclastogenic molecules in local tissues.
FIG 3.
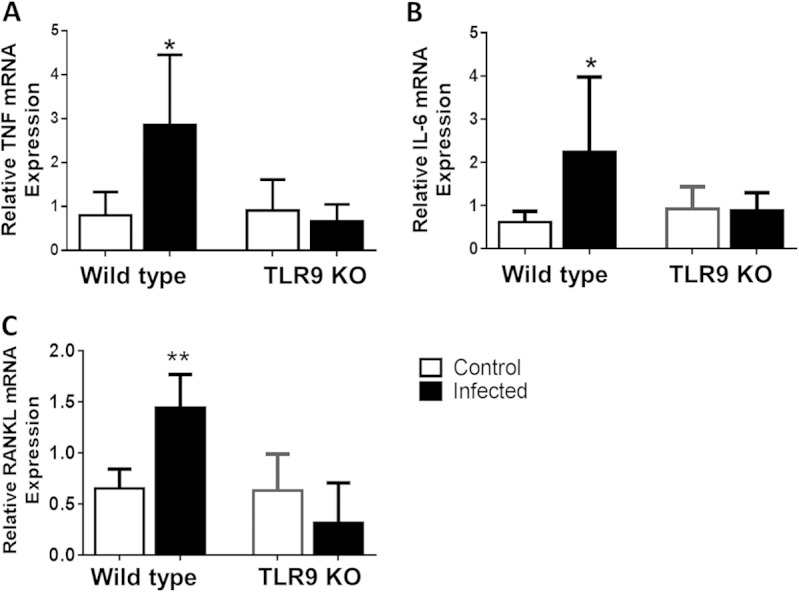
Inflammatory molecule expression in gingival tissues. WT and TLR9−/− mice were orally inoculated with P. gingivalis or vehicle only (sham) and euthanized 42 days later. Gingival tissues around maxillary molars were excised and processed for qPCR analyses to determine mRNA expression of TNF (A), IL-6 (B), and RANKL (C). Results are reported as fold induction after normalization to GAPDH. The data shown are the mean results ± SD (n = 5 or 6 mice per group) and were analyzed using the unpaired t test. *, P < 0.05; **, P < 0.01.
Lack of TLR9 signaling leads to decreased cytokine production in splenocytes and macrophages challenged with P. gingivalis.
Our in vivo studies demonstrated diminished inflammation and periodontal bone loss in P. gingivalis-infected TLR9−/− mice. Our subsequent experiments assessed whether cytokine responses to bacterial challenge differed between WT and TLR9−/− cells ex vivo. Briefly, splenocytes and bone marrow-derived macrophages from WT and TLR9−/− mice were challenged with heat-killed P. gingivalis (MOI of 1:100) for 24 h, and the levels of proinflammatory cytokines (IL-6 and TNF) in the cell culture supernatants were determined. Flow cytometry analyses showed that the ex vivo splenocyte cell populations consisted of 47.58% B cells, 38.52% T cells, and 13.9% other cells (monocytes, macrophages, neutrophils, and dendritic cells) for WT mice and 50.92% B cells, 37.99% T cells, and 11.09% other cells for TLR9−/− animals at baseline. The distribution of individual cell populations was also comparable between WT and knockout groups in unstimulated and stimulated cells at the termination of the experiments (data not shown). As expected, the results of the ex vivo studies were consistent with the in vivo observations and revealed significantly less IL-6 and TNF production (P < 0.05) in response to P. gingivalis challenge in TLR9−/− macrophages (Fig. 4A and B) and splenocytes (Fig. 4C and D) than in WT cells. ODN 1668 (synthetic TLR9 agonist) and P. gingivalis DNA were included as controls, and as expected, TLR9−/− cells failed to produce any of the cytokines when challenged with ODN 1668 and bacterial DNA.
FIG 4.

Comparison of proinflammatory cytokine production in WT versus TLR9−/− macrophages (A, B) and splenocytes (C, D) in response to P. gingivalis challenge after 24 h. The cells were stimulated with heat-killed P. gingivalis (MOI of 1:100), P. gingivalis DNA (100 ng/μl), and ODN 1668 (TLR9 agonist), and cell-free supernatants were analyzed for the presence of IL-6 and TNF using ELISA. Comparisons between the results for WT and TLR9 KO cells were performed using the unpaired student t test. The levels of IL-6 and TNF production were significantly reduced in TLR9−/− macrophages (A, B) and splenocytes (C, D) compared to the levels in WT cells. The results shown are representative of at least 3 independent experiments that were run in triplicates. The data shown are the mean results ± SD (n = 9). *, P < 0.05; ***, P < 0.001; ****, P < 0.0001.
Lack of TLR9 signaling affects cytokine production in splenocytes and macrophages challenged with TLR2 and TLR4 agonists.
Physiological responses require interaction of multiple components of the host and the microbiome, and cross talk among different innate immune signaling pathways plays a crucial role in determining the disease outcome (34). Therefore, the development of effective therapeutics ideally requires careful characterization of all possible interactions. Previously, we reported that inhibition of TLR9 signaling in human macrophage-like cells (THP-1) affects cytokine production in response to P. gingivalis LPS (21). This prompted us to think of a possible interaction of TLR9 in TLR2/TLR4-driven periodontal inflammation. Given the evidence regarding TLR9 cooperation with TLR2 and TLR4 in other disease models, we determined whether the immune responses vary in cells from WT and TLR9−/− mice upon challenge with TLR2 and TLR4 agonists. Splenocytes and bone marrow-derived macrophages from WT and TLR9−/− mice were challenged with different TLR agonists (Pam3Cys for TLR2, E. coli LPS for TLR4, P. gingivalis LPS for TLR4, and ODN 1668 for TLR9) for 24 h, and cytokine production was determined in cell culture supernatants using ELISA. There were significantly increased levels of both IL-6 and TNF in WT cells in response to all of the stimuli compared to the levels in unstimulated WT cells (Fig. 5). Accordingly, TLR9−/− cells also responded to each challenge except for the ODN 1668 challenge with significantly increased cytokine production compared to the levels in unstimulated TLR9−/− cells (Fig. 5). There were variations in the cytokine levels produced by macrophages and splenocytes depending on each stimulus. Upon challenge with Pam3Cys and E. coli LPS, TLR9−/− macrophages produced significantly less IL-6 than WT cells. In contrast, the IL-6 and TNF levels were similar in KO and WT macrophages in response to P. gingivalis and E. coli LPS and Pam3Cys (Fig. 5A and B). TLR9−/− splenocytes produced significantly less IL-6 in response to all of the ligands (Fig. 5C). TNF production was also significantly diminished in TLR9−/− splenocytes in response to each challenge except E. coli LPS (Fig. 5D). Our further analyses revealed equal levels of expression of TLR2 and TLR4 in WT and KO cells, indicating that the diminished cytokine levels in TLR9-deficient cells were not due to altered TLR2 and TLR4 expression (Fig. 6). The levels of TLR2 and TLR4 mRNA expression were comparable in wild-type and TLR9-deficient cells both at baseline and upon ligand challenge (Fig. 6A and B). Western blot analyses also revealed equal levels of expression of TLR2 in WT and TLR9−/− cells (Fig. 6C, D, and E). We were unable to detect TLR4 expression in either cell type by Western blot analyses (data not shown). Together, our ex vivo studies provide evidence that TLR9 modulates TLR2- and TLR4-triggered inflammation and suggest that TLR9 signaling possibly contributes to periodontal disease through cross talk with other innate signaling pathways also.
FIG 5.

Comparison of proinflammatory cytokine production in WT and TLR9−/− macrophages (A, B) and splenocytes (C, D) in response to different TLR agonists. The cells were stimulated with ODN 1668 (TLR9 agonist; 100 ng/μl), P. gingivalis LPS (TLR4 agonist; 10 ng/μl), E. coli LPS (TLR4 agonist; 10 ng/μl), or Pam3Cys (TLR2 agonist; 1 ng/μl) for 24 h. Cell-free supernatants were analyzed for the presence of IL-6 and TNF using ELISA. The results shown are representative of at least 3 independent experiments that were run in triplicates. The data shown are the mean results ± SD (n = 9). Comparisons between WT and KO cells were performed using the unpaired Student t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
FIG 6.

TLR2 and TLR4 expression in WT versus TLR9−/− splenocytes. The cells were stimulated with E. coli LPS (TLR4 agonist; 10 ng/μl) and Pam3Cys (TLR2 agonist; 1 ng/μl) for 24 h. The gene (A, B) and protein (C, D, E) expression levels in unstimulated and stimulated cells were determined using qPCR and Western blotting. TLR2 and TLR4 mRNA expression levels were equivalent in WT and TLR9−/− cells at baseline and following Pam3Cys (A) and LPS (B) challenge. The baseline and stimulated TLR2 protein levels were also equal in WT and TLR9−/− cells (C, D, E).
DISCUSSION
The oral mucosa is constantly exposed to microbes, and tissue homeostasis requires a balance between microbial cell turnover and host proinflammatory and anti-inflammatory responses. Disruption of this homeostatic balance by certain environmental exposures (e.g., smoking, obesity, or stress), host genetic and epigenetic defects in innate responses, or subversion of the host response by dysbiotic microflora can lead to destructive inflammation, precipitating periodontitis (35–41). Recently, TLR9 signaling has emerged as a potential inflammatory pathway in periodontitis pathogenesis on the basis of clinical and in vitro investigations. Confirming the role of specific genes and cells in a disease process requires in vivo investigations that better simulate the physiological environment (26). The results of the current study demonstrate that TLR9 signaling in vivo can mediate the induction of inflammatory and osteoclastogenic cytokines (IL-6, TNF, and RANKL), as well as bone loss, in a well-established and validated periodontitis model (26, 27). Complementing the in vivo results, our ex vivo studies also demonstrated diminished cytokine production in TLR9−/− macrophages and splenocytes in response to P. gingivalis challenge. Taken together, therefore, our preclinical data suggest that TLR9 activation may be involved in the induction and/or exacerbation of periodontal disease.
Intriguingly, our findings also revealed that TLR9 deficiency can affect the extent of inflammatory responses to TLR2 and TLR4 ligands despite similar levels of TLR2 and TLR4 expression in WT and TLR9 KO cells. The induction levels of the cytokines studied varied depending on the cell type being studied and the specificity of the ligand challenge. In this regard, TLR9 activation seemed to have a more pronounced effect on IL-6 levels than on TNF levels. It was reported previously that P. gingivalis can activate the MyD88-independent proinflammatory phosphatidylinositol 3-kinase (PI3K) pathway in neutrophils but not in macrophages (40), whereas P. gingivalis-induced TNF production is predominantly driven by the TLR2-MyD88 pathway in macrophages (41). The latter finding may possibly explain why TLR9 deficiency had a relatively modest effect on TNF production as opposed to its effect on IL-6. We also observed that TLR9−/− macrophages exhibited higher levels of IL-6 and TNF production in response to P. gingivalis DNA and whole bacteria than to ODN 1668. One possible explanation for this observation is that natural microbial DNA may have additional targets, whereas ODN 1668 may have exclusive specificity for TLR9. In this regard, there are other cytoplasmic receptors, such as absent in melanoma 2 (AIM2) and DNA-dependent activator of IFN regulatory factors (DAI), that can respond to microbial DNA (42). Although it is beyond the scope of this article, we can speculate that P. gingivalis DNA can be engaged by other nucleic acid sensors as well. In fact, our previous clinical studies also showed increased DAI expression in chronic periodontitis lesions compared to its expression in healthy tissues (14). Future studies are warranted to delineate the extent of involvement of other microbial DNA sensors in periodontitis pathogenesis.
The immune response to a particular antigen may also vary depending on the individual cell type, the tissue-specific localization of the antigen, and the receptors and signaling molecules involved. Our findings also revealed that immune responses to the same antigenic challenge vary in different cell types, e.g., macrophages versus splenocytes, which further supports this notion. Macrophages are thought to play important roles in periodontal lesions. As microbial DNA engages with TLR9 following phagocytosis, our current study primarily focused on an evaluation of inflammatory responses in macrophages ex vivo. However, it is important to note that periodontal tissues are composed of different cell types of both myeloid and nonmyeloid origin and TLR9 is expressed in multiple types of cells, including gingival keratinocytes, dendritic cells, and neutrophils. Additionally, our previous study revealed increased TLR9 expression within both the connective tissue and basal epithelial layers of the diseased periodontal tissues (14). Therefore, future studies utilizing more targeted approaches, such as cell lineage-specific conditional knockout mice, are warranted to determine the cell types that are driving TLR9-mediated inflammation in periodontal tissues.
The polymicrobial nature of periodontitis and the presence of various cells forming the periodontal structures create an environment for simultaneous interaction of distinct sensors and periodontal MAMPs. Hence, the activation of multiple receptors and cross talk among intracellular signaling proteins eventually determines the periodontal disease outcome. Although ligand recognition shows specificity for each receptor, the downstream signaling pathways activated by TLRs have some redundancy, generating the potential for signaling cross talk. In fact, the involvement of TLR2-mediated immune responses and their cooperation with other innate sensing molecules, such as complement receptors, in periodontal inflammation has been documented (43). Intriguingly, cross talk between TLR9, TLR2, and TLR4 has been reported in several other disease models (44–48). For example, a study using an in vivo model of hypersensitivity pneumonitis revealed that TLR2 and TLR9 cooperated in neutrophil recruitment and IL-17-associated cytokine production such that both neutrophil recruitment and cytokine production were significantly decreased in double-KO mice (TLR2/9−/−) compared to their levels in TLR2−/− mice (49). TLR9 has also been reported to be critical for the development of Th17-mediated granulomatous inflammation in the lung in response to Stachybotrys chartarum sensitization (50). The study demonstrated decreased expression of several proinflammatory cytokines, as well as IL-17 and IL-23, in TLR9 KO cells (50). Another in vivo investigation reported significant synergy between TLR2 and TLR9 for induction of the MyD88-dependent splenic cytokine and chemokine response to Streptococcus pneumoniae (46). Thus, it is plausible that TLR9 communicates with other receptors and signaling pathways in the course of periodontal inflammation as well. This notion is supported by our findings. First, our results revealed that the levels of IL-6 and TNF were significantly reduced in TLR9−/− cells challenged with TLR2 and TLR4 agonists compared to the levels of these cytokines in wild-type cells. These results imply that impaired TLR9 signaling can modulate TLR2- and TLR4-mediated immune responses, possibly communicating through downstream signaling pathways. However, it is also important to emphasize that although there were significant reductions in the cytokine levels in TLR9−/− cells compared to the levels in WT cells, the cytokine levels still remained significantly high in LPS- and Pam3Cys-treated TLR9−/− cells compared to the levels in unstimulated cells. Second, although TLR2 has been conclusively implicated in P. gingivalis-induced periodontal bone loss (2, 40, 41), TLR9−/− mice were completely resistant to P. gingivalis-induced bone loss in this study. Together, these findings suggest that TLR9 possibly interacts with and amplifies TLR2-mediated inflammation. The potential role of TLR9-mediated inflammation in periodontal disease pathogenesis is summarized in the model shown in Fig. 7. Our observations highlight a critical aspect of microbial DNA sensing in relation to other signaling pathways of importance for periodontitis and warrant further investigations utilizing in vivo models and transgenic animals to fully characterize the extent of communication and molecular pathways between TLR9 and other innate sensors in periodontitis pathogenesis.
FIG 7.

Model for TLR9-mediated periodontal inflammation. Polymicrobial challenge exposes periodontal tissues to various MAMPs associated with periodontal bacteria (e.g., LPS, fimbriae, lipoproteins, and nucleic acids, etc.), leading to activation of distinct host innate receptors, as indicated. Cross talk among different innate sensors can determine the periodontal disease clinical outcome. Microbial DNA, which is released following phagocytosis of microbial cells, can engage TLR9 within the endolysosomal compartment and start a cascade of signaling events that mediate inflammatory responses that contribute to periodontal tissue destruction, either alone or possibly through communication with other innate sensors, such as TLR2 and TLR4. MyD88, myeloid differentiation primary response 88; MAPK, mitogen-activated protein kinase; AP-1, activator protein 1; NF-κB, nuclear factor kappa B; IRF-7, interferon regulatory factor 7).
Conventional periodontal therapy is generally not effective to control the persistent forms of periodontal disease, and current research efforts focus on identifying alternative or adjunctive therapeutic options to prevent the progression of periodontal disease in local tissues and block its adverse effects on systemic tissues (51, 52). It is now well documented that communication among different receptors and downstream signaling molecules is important in the progression of periodontal inflammation. Therefore, controlling periodontal inflammation and pathological bone loss will require better understanding of the signaling pathways activated by multiple receptors. To our knowledge, this is the first in vivo demonstration that a nucleic acid sensor, TLR9, can mediate destructive periodontal inflammation and can therefore be considered a potential therapeutic target for periodontal disease. Small-molecule inhibitors of TLR9 signaling are already commercially available and are being evaluated for the treatment of various inflammatory disorders (53–55). Our findings provide for the first time a strong mechanistic rationale for investigating the efficacy of TLR9 small-molecule inhibitors in controlling periodontitis in a preclinical setting and, ultimately, in future clinical trials.
ACKNOWLEDGMENTS
The study was supported by U.S. Public Health Service grants DE022836, KL2TR000057-03, and UL1TR000058 to S.E.S., grants DE015254 and AI068730 to G.H. from NIH, and grant K12GM093857 for support of K.E.C.
We declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
REFERENCES
- 1.Eke PI, Thornton-Evans GO, Wei L, Borgnakke WS, Dye BA. 2010. Accuracy of NHANES periodontal examination protocols. J Dent Res 89:1208–1213. doi: 10.1177/0022034510377793. [DOI] [PubMed] [Google Scholar]
- 2.Hajishengallis G. 2014. Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response. Trends Immunol 35:3–11. doi: 10.1016/j.it.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schenkein HA, Loos BG. 2013. Inflammatory mechanisms linking periodontal diseases to cardiovascular diseases. J Periodontol 84:S51–S69. doi: 10.1902/jop.2013.134006. [DOI] [PubMed] [Google Scholar]
- 4.Lalla E, Papapanou PN. 2011. Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat Rev Endocrinol 7:738–748. doi: 10.1038/nrendo.2011.106. [DOI] [PubMed] [Google Scholar]
- 5.Linden GJ, Lyons A, Scannapieco FA. 2013. Periodontal systemic associations: review of the evidence. J Periodontol 84:S8–S19. doi: 10.1902/jop.2013.1340010. [DOI] [PubMed] [Google Scholar]
- 6.Ide M, Papapanou PN. 2013. Epidemiology of association between maternal periodontal disease and adverse pregnancy outcomes—systematic review. J Periodontol 84:S181–S194. doi: 10.1902/jop.2013.134009. [DOI] [PubMed] [Google Scholar]
- 7.Han YW, Houcken W, Loos BG, Schenkein HA, Tezal M. 2014. Periodontal disease, atherosclerosis, adverse pregnancy outcomes, and head-and-neck cancer. Adv Dent Res 26:47–55. doi: 10.1177/0022034514528334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hajishengallis G. 2015. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol 15:30–44. doi: 10.1038/nri3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akira S. 2009. Innate immunity to pathogens: diversity in receptors for microbial recognition. Immunol Rev 227:5–8. doi: 10.1111/j.1600-065X.2008.00739.x. [DOI] [PubMed] [Google Scholar]
- 10.Brown J, Wang H, Hajishengallis GN, Martin M. 2011. TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J Dent Res 90:417–427. doi: 10.1177/0022034510381264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Savva A, Roger T. 2013. Targeting Toll-like receptors: promising therapeutic strategies for the management of sepsis-associated pathology and infectious diseases. Front Immunol 4:387. doi: 10.3389/fimmu.2013.00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawasaki T, Kawai T, Akira S. 2011. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol Rev 243:61–73. doi: 10.1111/j.1600-065X.2011.01048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holm CK, Paludan SR, Fitzgerald KA. 2013. DNA recognition in immunity and disease. Curr Opin Immunol 25:13–18. doi: 10.1016/j.coi.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sahingur SE, Xia XJ, Voth SC, Yeudall WA, Gunsolley JC. 2013. Increased nucleic acid receptor expression in chronic periodontitis. J Periodontol 84:e48–e57. doi: 10.1902/jop.2013.120739. [DOI] [PubMed] [Google Scholar]
- 15.Kajita K, Honda T, Amanuma R, Domon H, Okui T, Ito H, Yoshie H, Tabeta K, Nakajima T, Yamazaki K. 2007. Quantitative messenger RNA expression of Toll-like receptors and interferon-alpha1 in gingivitis and periodontitis. Oral Microbiol Immunol 22:398–402. doi: 10.1111/j.1399-302X.2007.00377.x. [DOI] [PubMed] [Google Scholar]
- 16.Rojo-Botello NR, Garcia-Hernandez AL, Moreno-Fierros L. 2012. Expression of toll-like receptors 2, 4 and 9 is increased in gingival tissue from patients with type 2 diabetes and chronic periodontitis. J Periodont Res 47:62–73. doi: 10.1111/j.1600-0765.2011.01405.x. [DOI] [PubMed] [Google Scholar]
- 17.Kinane DF, Shiba H, Hart TC. 2005. The genetic basis of periodontitis. Periodontol 2000 39:91–117. doi: 10.1111/j.1600-0757.2005.00118.x. [DOI] [PubMed] [Google Scholar]
- 18.Sahingur SE, Xia XJ, Gunsolley J, Schenkein HA, Genco RJ, De Nardin E. 2011. Single nucleotide polymorphisms of pattern recognition receptors and chronic periodontitis. J Periodont Res 46:184–192. doi: 10.1111/j.1600-0765.2010.01327.x. [DOI] [PubMed] [Google Scholar]
- 19.Holla LI, Vokurka J, Hrdlickova B, Augustin P, Fassmann A. 2010. Association of Toll-like receptor 9 haplotypes with chronic periodontitis in Czech population. J Clin Periodontol 37:152–159. doi: 10.1111/j.1600-051X.2009.01523.x. [DOI] [PubMed] [Google Scholar]
- 20.Sahingur SE, Xia XJ, Alamgir S, Honma K, Sharma A, Schenkein HA. 2010. DNA from Porphyromonas gingivalis and Tannerella forsythia induce cytokine production in human monocytic cell lines. Mol Oral Microbiol 25:123–135. doi: 10.1111/j.2041-1014.2009.00551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sahingur SE, Xia XJ, Schifferle RE. 2012. Oral bacterial DNA differ in their ability to induce inflammatory responses in human monocytic cell lines. J Periodontol 83:1069–1077. doi: 10.1902/jop.2011.110522. [DOI] [PubMed] [Google Scholar]
- 22.Wara-aswapati N, Chayasadom A, Surarit R, Pitiphat W, Boch JA, Nagasawa T, Ishikawa I, Izumi Y. 2013. Induction of Toll-like receptor expression by Porphyromonas gingivalis. J Periodontol 84:1010–1018. doi: 10.1902/jop.2012.120362. [DOI] [PubMed] [Google Scholar]
- 23.Zhan Y, Zhang R, Lv H, Song X, Xu X, Chai L, Lv W, Shang Z, Jiang Y. 2014. Prioritization of candidate genes for periodontitis using multiple computational tools. J Periodontol 85:1059–1069. doi: 10.1902/jop.2014.130523. [DOI] [PubMed] [Google Scholar]
- 24.Hajishengallis G, Sahingur SE. 2014. Novel inflammatory pathways in periodontitis. Adv Dent Res 26:23–29. doi: 10.1177/0022034514526240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hajishengallis G, Lamont RJ, Graves DT. 2015. The enduring importance of animal models in understanding periodontal disease. Virulence 6:229–235. doi: 10.4161/21505594.2014.990806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graves DT, Fine D, Teng YT, Van Dyke TE, Hajishengallis G. 2008. The use of rodent models to investigate host-bacteria interactions related to periodontal diseases. J Clin Periodontol 35:89–105. doi: 10.1111/j.1600-051X.2007.01172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baker PJ, Dixon M, Evans RT, Roopenian DC. 2000. Heterogeneity of Porphyromonas gingivalis strains in the induction of alveolar bone loss in mice. Oral Microbiol Immunol 15:27–32. doi: 10.1034/j.1399-302x.2000.150105.x. [DOI] [PubMed] [Google Scholar]
- 28.Liang S, Krauss JL, Domon H, McIntosh ML, Hosur KB, Qu H, Li F, Tzekou A, Lambris JD, Hajishengallis G. 2011. The C5a receptor impairs IL-12-dependent clearance of Porphyromonas gingivalis and is required for induction of periodontal bone loss. J Immunol 186:869–877. doi: 10.4049/jimmunol.1003252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA. 2011. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10:497–506. doi: 10.1016/j.chom.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abe T, Shin J, Hosur K, Udey MC, Chavakis T, Hajishengallis G. 2014. Regulation of osteoclast homeostasis and inflammatory bone loss by MFG-E8. J Immunol 193:1383–1391. doi: 10.4049/jimmunol.1400970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Polak D, Naddaf R, Shapira L, Weiss EI, Houri-Haddad Y. 2013. Protective potential of non-dialyzable material fraction of cranberry juice on the virulence of P. gingivalis and F. nucleatum mixed infection. J Periodontol 84:1019–1025. doi: 10.1902/jop.2012.120331. [DOI] [PubMed] [Google Scholar]
- 32.Abe T, Hosur KB, Hajishengallis E, Reis ES, Ricklin D, Lambris JD, Hajishengallis G. 2012. Local complement-targeted intervention in periodontitis: proof-of-concept using a C5a receptor (CD88) antagonist. J Immunol 189:5442–5448. doi: 10.4049/jimmunol.1202339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Westcott MM, Henry CJ, Cook AS, Grant KW, Hiltbold EM. 2007. Differential susceptibility of bone marrow-derived dendritic cells and macrophages to productive infection with Listeria monocytogenes. Cell Microbiol 9:1397–1411. doi: 10.1111/j.1462-5822.2006.00880.x. [DOI] [PubMed] [Google Scholar]
- 34.Kawai T, Akira S. 2011. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 35.Johannsen A, Susin C, Gustafsson A. 2014. Smoking and inflammation: evidence for a synergistic role in chronic disease. Periodontol 2000 64:111–126. doi: 10.1111/j.1600-0757.2012.00456.x. [DOI] [PubMed] [Google Scholar]
- 36.Bullon P, Newman HN, Battino M. 2014. Obesity, diabetes mellitus, atherosclerosis and chronic periodontitis: a shared pathology via oxidative stress and mitochondrial dysfunction? Periodontol 2000 64:139–153. doi: 10.1111/j.1600-0757.2012.00455.x. [DOI] [PubMed] [Google Scholar]
- 37.Doyle CJ, Bartold PM. 2012. How does stress influence periodontitis? J Int Acad Periodontol 14:42–49. [PubMed] [Google Scholar]
- 38.Rhodin K, Divaris K, North KE, Barros SP, Moss K, Beck JD, Offenbacher S. 2014. Chronic periodontitis genome-wide association studies: gene-centric and gene set enrichment analyses. J Dent Res 93:882–890. doi: 10.1177/0022034514544506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang S, Barros SP, Moretti AJ, Yu N, Zhou J, Preisser JS, Niculescu MD, Offenbacher S. 2013. Epigenetic regulation of TNFA expression in periodontal disease. J Periodontol 84:1606–1616. doi: 10.1902/jop.2013.120294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benakanakere M, Abdolhosseini M, Hosur K, Finoti LS, Kinane DF. 2015. TLR2 promoter hypermethylation creates innate immune dysbiosis. J Dent Res 94:183–191. doi: 10.1177/0022034514557545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hajishengallis G, Lamont RJ. 2014. Breaking bad: manipulation of the host response by Porphyromonas gingivalis. Eur J Immunol 44:328–338. doi: 10.1002/eji.201344202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. 2011. Pattern recognition receptors and the innate immune response to viral infection. Viruses 3:920–940. doi: 10.3390/v3060920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maekawa T, Abe T, Hajishengallis E, Hosur KB, DeAngelis RA, Ricklin D, Lambris JD, Hajishengallis G. 2014. Genetic and intervention studies implicating complement C3 as a major target for the treatment of periodontitis. J Immunol 192:6020–6027. doi: 10.4049/jimmunol.1400569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gravina HD, Antonelli L, Gazzinelli RT, Ropert C. 2013. Differential use of TLR2 and TLR9 in the regulation of immune responses during the infection with Trypanosoma cruzi. PLoS One 8:e63100. doi: 10.1371/journal.pone.0063100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bafica A, Santiago HC, Goldszmid R, Ropert C, Gazzinelli RT, Sher A. 2006. Cutting edge: TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J Immunol 177:3515–3519. doi: 10.4049/jimmunol.177.6.3515. [DOI] [PubMed] [Google Scholar]
- 46.Lee KS, Scanga CA, Bachelder EM, Chen Q, Snapper CM. 2007. TLR2 synergizes with both TLR4 and TLR9 for induction of the MyD88-dependent splenic cytokine and chemokine response to Streptococcus pneumoniae. Cell Immunol 245:103–110. doi: 10.1016/j.cellimm.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhan U, Ballinger MN, Zeng X, Newstead MJ, Cornicelli MD, Standiford TJ. 2010. Cooperative interactions between TLR4 and TLR9 regulate interleukin 23 and 17 production in a murine model of gram negative bacterial pneumonia. PLoS One 5:e9896. doi: 10.1371/journal.pone.0009896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Nardo D, De Nardo CM, Nguyen T, Hamilton JA, Scholz GM. 2009. Signaling crosstalk during sequential TLR4 and TLR9 activation amplifies the inflammatory response of mouse macrophages. J Immunol 183:8110–8118. doi: 10.4049/jimmunol.0901031. [DOI] [PubMed] [Google Scholar]
- 49.Andrews K, Abdelsamed H, Yi AK, Miller MA, Fitzpatrick EA. 2013. TLR2 regulates neutrophil recruitment and cytokine production with minor contributions from TLR9 during hypersensitivity pneumonitis. PLoS One 8:e73143. doi: 10.1371/journal.pone.0073143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhan U, Newstead MJ, Zeng X, Podsaid A, Goswami M, Ballinger MN, Kunkel SL, Standiford TJ. 2013. TLR9-dependent IL-23/IL-17 is required for the generation of Stachybotrys chartarum-induced hypersensitivity pneumonitis. J Immunol 190:349–356. doi: 10.4049/jimmunol.1202225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Colombo AP, Boches SK, Cotton SL, Goodson JM, Kent R, Haffajee AD, Socransky SS, Hasturk H, Van Dyke TE, Dewhirst F, Paster BJ. 2009. Comparisons of subgingival microbial profiles of refractory periodontitis, severe periodontitis, and periodontal health using the human oral microbe identification microarray. J Periodontol 80:1421–1432. doi: 10.1902/jop.2009.090185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Armitage GC. 2002. Classifying periodontal diseases—a long-standing dilemma. Periodontol 2000 30:9–23. doi: 10.1034/j.1600-0757.2002.03002.x. [DOI] [PubMed] [Google Scholar]
- 53.Hoque R, Farooq A, Malik A, Trawick BN, Berberich DW, McClurg JP, Galen KP, Mehal W. 2013. A novel small-molecule enantiomeric analogue of traditional (-)-morphinans has specific TLR9 antagonist properties and reduces sterile inflammation-induced organ damage. J Immunol 190:4297–4304. doi: 10.4049/jimmunol.1202184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suarez-Farinas M, Arbeit R, Jiang W, Ortenzio FS, Sullivan T, Krueger JG. 2013. Suppression of molecular inflammatory pathways by Toll-like receptor 7, 8, and 9 antagonists in a model of IL-23-induced skin inflammation. PLoS One 8:e84634. doi: 10.1371/journal.pone.0084634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kandimalla ER, Bhagat L, Wang D, Yu D, Sullivan T, La Monica N, Agrawal S. 2013. Design, synthesis and biological evaluation of novel antagonist compounds of Toll-like receptors 7, 8 and 9. Nucleic Acids Res 41:3947–3961. doi: 10.1093/nar/gkt078. [DOI] [PMC free article] [PubMed] [Google Scholar]