

Abstract
Thioredoxins are highly conserved throughout a wide range of organisms, and they are essential for the isurvival of oxygen-sensitive cells. The gastric pathogen Helicobacter pylori uses the thioredoxin system to maintain its thiol/disulfide balance. There are two thioredoxins present in H. pylori, Trx1 and Trx2 (herein referred to as TrxA and TrxC). TrxA has been shown to be important as an electron donor for some antioxidant enzymes, but the function of TrxC remains unknown (L. M. Baker, A. Raudonikiene, P. S. Hoffman, and L. B. Poole, J Bacteriol 183:1961–1973, 2001; P. Alamuri and R. J. Maier, J Bacteriol 188:5839–5850, 2006). We demonstrate that both TrxA and TrxC are important in protecting H. pylori from oxidative stress. Individual ΔtrxA and ΔtrxC deletion mutant strains each show a greater abundance of lipid peroxides and suffer more DNA damage and more protein carbonylation than the parent. Both deletion mutants were much more sensitive to O2-mediated viability loss than the parent. Unexpectedly, the oxidative DNA damage and protein carbonylation was more severe in the ΔtrxC mutant than in the ΔtrxA mutant; it had 20-fold- and 4-fold-more carbonylated protein content than the wild type and the ΔtrxA strain, respectively, after 4 h of atmospheric O2 stress. trx transcript abundance was altered by the deletion of the heterologous trx gene. The ΔtrxC mutant lacked mouse colonization ability, while the ability to colonize mouse stomachs was significantly reduced in the ΔtrxA mutant.
INTRODUCTION
Oxidative stress is a problem encountered by most organisms, and anaerobic and microaerophilic microorganisms frequently have a limited tolerance for oxygen. Reactive oxygen species (ROS) are abundantly generated in the environment, which leads to every living organism, at least transiently, having contact with different reactive oxygen molecules. ROS are generated in the form of superoxide (O2–), hydroxyl radical (OH·), hydrogen peroxide (H2O2), and hypochlorous acid (HOCl). All of these can be generated by irradiation (X rays, gamma rays, and ultraviolent radiation) or by normal metabolic processes, including autoxidation of reduced electron carriers, lipid peroxidation, and metal-catalyzed reactions (1). These forms of ROS can be detrimental to organisms, as they react with and damage macromolecules (DNA, lipids, and proteins) of the cell if not scavenged by antioxidant proteins. Pathogens have to combat an additional source of ROS, the oxidative burst generated by host immune cells (2–6).
Helicobacter pylori is a Gram-negative microaerophilic organism that colonizes the gastric mucosa and is the causative agent of peptic ulcers and chronic gastritis. It is chronically in contact with an array of ROS during colonization due to the host inflammatory response (2, 7–10). The oxidative-stress response of H. pylori is made up of many enzymatic systems, including superoxide dismutase (SodB), catalase (KatA), methionine sulfoxide reductase (Msr), alkyl hydroperoxide reductase (AhpC), thiol-peroxidase (Tpx), and bacterioferritin comigratory protein (Bcp) (11–20). These enzymes function in concert as the primary means of combatting oxidative stress in the cell. These systems, combined with the Trx system as the reductant, provide H. pylori with the tools needed to maintain a reduced environment in times of oxygen stress. H. pylori lacks genes for oxidative-stress regulators and does not contain the glutathione-glutaredoxin reduction system (GSH) (21). The GSH reduction system is often used in addition to the Trx system in bacteria to maintain a reduced state inside the cell (22). Organisms that lack GSH, such as Lactobacillus casei, Bacillus subtilis, Bacteroides fragilis, Staphylococcus aureus, and H. pylori, presumably must rely on the Trx system to maintain thiol/disulfide balance in the cell (21, 23–26). The mechanisms for the maintenance of this balance in H. pylori have not been studied in great detail.
The Trx system is highly conserved throughout many organisms and is comprised of thioredoxin reductase (TrxR), NADPH, and Trx. H. pylori contains two genes that encode Trx, namely, TrxA (KEGG accession number HP0824) and TrxC (HP1458), but only a single gene that encodes the reductase TrxR (HP0825) (13). The first known function of TrxA was identified in Escherichia coli, and it was found to provide electrons to ribonucleotide reductase (RNR). RNR is an essential enzyme in the replication and repair of DNA, as it functions to catalyze the synthesis of deoxyribonucleotides from ribonucleotides. Evidence for this function in H. pylori has not been presented. However, H. pylori TrxA is expressed in greater amounts as a response to stress agents (27), and it is the electron donor for two important antioxidant enzymes, AhpC and Msr (13, 19). TrxA but not TrxC also acts as a chaperone in H. pylori, in which it renatures denatured arginase into its active form (28).
Notably, an H. pylori ΔtrxA mutant was shown to be more sensitive to oxidative stress agents (such as paraquat, S-nitrosoglutathione, and O2) than the wild type (strain 26695), while the ΔtrxC mutant was much less sensitive to these agents than the ΔtrxA strain (15). No specific roles (e.g., electron donor) for H. pylori TrxC in conferring resistance to oxidative stress or involvement in any cell process are known. We sought to further compare the antioxidant roles of TrxA and TrxC in H. pylori (strain 43504). We approached this by studying the effect of trx deletions on the subsequent state of macromolecules in the cell, with a focus on biomarker damage for oxidative stress. We determined the effects of oxygen on cell viability, total protein carbonyls, lipid hydroperoxides, and 8-oxoguanine (8-oxoG) content in the DNA of the strains and determined the colonization ability of the strains compared to that of the wild-type strain in a mouse model. The studies infer an important role for these reductant proteins in maintaining the macromolecule integrity and survival of H. pylori under oxidative-stress conditions, including in vivo.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
H. pylori 26695 (21) or ATCC 43504 was used for all in vitro studies, and strain X47 (29) was used for the mouse colonization assay. H. pylori cells were routinely grown on Brucella agar (BA; Oxoid) plates containing 10% defibrinated sheep blood (HemoStat Laboratories, Bogart, GA) and maintained in a 37°C humidified chamber under 5% CO2 and low levels of oxygen (4%), with N2 used as the balance. Chloramphenicol was added to the plates at a concentration of 30 μg/ml as needed.
Oxygen stress conditions.
H. pylori 43504, the ΔtrxA mutant, or the ΔtrxC mutant from BA plates after 2 days of growth were resuspended in phosphate-buffered saline (PBS), pH 7.4, and exposed to either atmospheric oxygen (20%) or HOCl. To expose cells to atmospheric oxygen (20%), cells were resuspended in PBS to an optical density at 600 nm (OD600) of 2 and placed at 37°C while being shaken at 60 rpm for up to 12 h. For viability determination, cells were taken at each 2-h time point; dilutions were made and plated on BA plates. All plates were then incubated at 4% O2 for 5 days, and the number of viable cells was determined. Cells exposed to HOCl were resuspended in PBS to an OD600 of 2, subjected to HOCl treatment (0 or 200 μM), and incubated at 37°C with shaking for 1 h at 200 rpm. Cells were then washed with PBS and harvested via centrifugation at 7,500 × g for 10 min at 4°C.
Construction of mutant strains. (i) Construction of the ΔtrxA mutant.
Primers trxA-F and trxA-R (Table 1) were used to PCR amplify an 864-bp fragment containing the H. pylori trxA gene (hp0824). The PCR fragment was directly cloned into the pGEM-T vector (Promega) according to the manufacturer's instructions to generate pGEM-trxA. The host strain used for cloning was DH5α. Subsequently, a chloramphenicol acetyltransferase (CAT) cassette was inserted at the unique NheI (nucleotide G130) site within the trxA sequence of pGEM-trxA. The allelic-exchange (chloramphenicol-resistant) transformants were isolated by selection of single colonies upon incubation under 4% O2 partial-pressure conditions. The balance of the atmosphere was N2 (91%) and CO2 (5%). The disruption of the gene in the genome of the mutant strain was confirmed by PCR using primers trxA-1 and trxA-4, which lie outside the mutagenized region. The PCR product indicated an increase to the expected size, indicating insertion of the CAT cassette.
TABLE 1.
List of primers used in this study
| Primera | Sequence (5′ to 3′) |
|---|---|
| trxA-1 | TTGAAATGGTGGAGAGGAAC |
| trxA-4 | CCGGTAGCGATAATCACGC |
| trxA-F | ATTTATGCGATCACGCCGAG |
| trxA-R | AAGTCTTGCCATCTTCTGCC |
| trxC-1 | CGCGATGCAAGAGGGCTTG |
| trxC-2 | ATCCACTTTTCAATCTATATCAATCATT |
| trxC-3 | CCCAGTTTGTCGCACTGATAACGTTAT |
| trxC-4 | CGCTTGAGCAACCCACC |
| trxC-F | ACACCATATGTCAGAAATGATTAACGG |
| trxC-R | ACACCTCGAGCAATAACGCTTTTAGAG |
| trxA-F(rt) | GTGGGCCTTGTAAGATG |
| trxA-R(rt) | CTGATACCAATTTTGCGCTC |
| trxC-F(rt) | GGATTGCAGAAAGATTGAGC |
| trxC-R(rt) | CTTGCGGATGCCTAAG |
| gyrA-F(rt) | GCTAGGATCGTGGGTGATGT |
| gyrA-R(rt) | TGGCTTCAGTGTAACGCATC |
All primers were obtained from Integrated DNA Technologies.
(ii) Construction of the ΔtrxC mutant.
The ΔtrxC mutant strain was constructed by overlapping PCR. Genomic DNA and primers trxC-1 and trxC-2cat were used to PCR amplify a 388-bp region that contains part of the hp1459 gene and part of the hp1458 and cat (Cmr) genes. Next, primers trxC-3 and trxC-4 were used to PCR amplify a 409-bp region containing part of the cat and hp1458 genes and the hp1457 gene. After each PCR, the products were gel purified and used for a final PCR step using primers trxC-1 and trxC-4. The final elongation step yielded a 1.5-kb product that was then used for (natural) transformation into strain 26695, 43504, or X47. The transformants (grown at 4% O2) were transferred onto BA plates supplemented with 30 μg/ml chloramphenicol, and individual colonies appeared after 3 to 5 days of incubation at 4% O2. The mutant strains grow at a rate similar to that of the wild type on BA at 4% O2. The deletion of the ΔtrxC mutant was confirmed by PCR using genomic DNA from each chosen clone. The PCR product was analyzed on a 0.7% Tris-acetate-EDTA (TAE) agarose gel. The clones were further confirmed via sequencing at the Georgia Genomics Facility (University of Georgia, Athens, GA). Construction of deletion mutants using overlapping PCR was also confirmed by reverse transcription-PCR (RT-PCR).
(iii) Construction of complemented mutant strains.
E. coli strain DH5α was used for all plasmid DNA manipulations. The genes trxA and trxC were PCR amplified from H. pylori genomic DNA using primers trxA-F and trxA-R or trxC-F and trxC-R. The PCR products were digested with NdeI and XhoI and ligated into the pPA plasmid digested with the same restriction enzymes. This created pPAtrxA or pPAtrxC, such that the genes were located downstream of the ureA promoter. Digestion of the plasmids with BglII and XhoI released the fragment. The fragments were ligated into pEM39kan, also digested with BglII and XhoI. The plasmid pEM39kan has been shown to efficiently insert DNA by homologous recombination into the region of the chromosome corresponding to the HP0405 site. Disruption of this gene has been shown to have no effect on the cell (30, 31). The plasmid was then naturally transformed into the trxA or trxC mutant strain and selected on BA containing 30 μg/ml kanamycin and 30 μg/ml chloramphenicol. This results in the insertion of trxA or trxC along with the ureA promoter at the HP0405 site in the chromosome. Successful recombination of trxA or trxC into the chromosome was confirmed via PCR and functional restoration by determining the viability of the complemented strains, the lipid peroxide level, the carbonylated protein content, and mouse colonization as described previously. Experiments in which the genetically complemented strains were used apply to cell survival (see Fig. 1), lipid peroxidation (see Fig. 3), protein carbonyl content (see Fig. 5), and mouse colonization (see Fig. 6).
FIG 1.
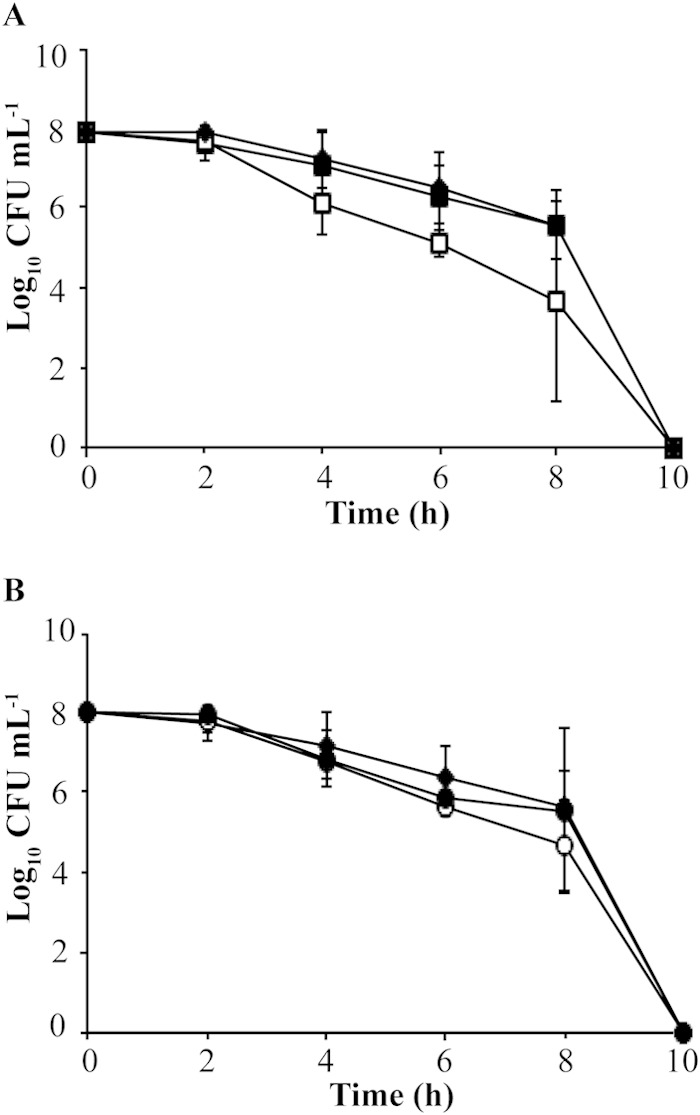
Survival of nongrowing cells incubated under atmospheric oxygen. (A) Filled diamonds, wild type; open squares, ΔtrxA mutant; filled squares, ΔtrxA complemented strain; (B) filled diamonds, wild type; open circles, ΔtrxC mutant; filled circles, ΔtrxC complemented strain. All strains were resuspended in sterile PBS and incubated under atmospheric oxygen for a 12-h period. Samples were taken at 2-h intervals, diluted, and plated onto BA plates. Dilutions were then incubated in a 4% partial-pressure O2 atmosphere and colony counts determined. The means and standard deviations from results of 4 independent observations are plotted.
FIG 3.
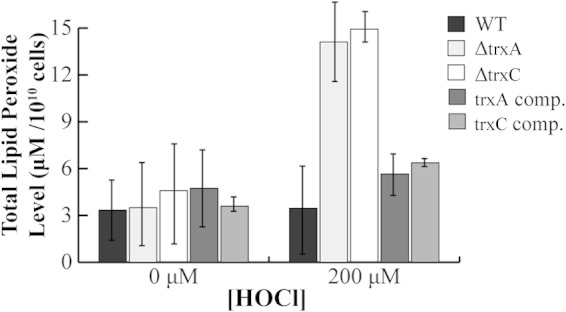
Lipid peroxide level in H. pylori trx mutants exposed to hypochlorous acid (HOCl) stress. H. pylori was grown on BA plates for 2 days and exposed to oxidant (or not), and cell extracts were obtained. The total amount of lipid peroxides was determined, and the hydroperoxide concentration in the sample was calculated. Data are presented as the averages from three independent experiments. Based on the Student t test, the ΔtrxA and ΔtrxC strains exposed to HOCl contain significantly greater amounts of hydroperoxide than the wild type at the 99% level of confidence. comp., complemented.
FIG 5.
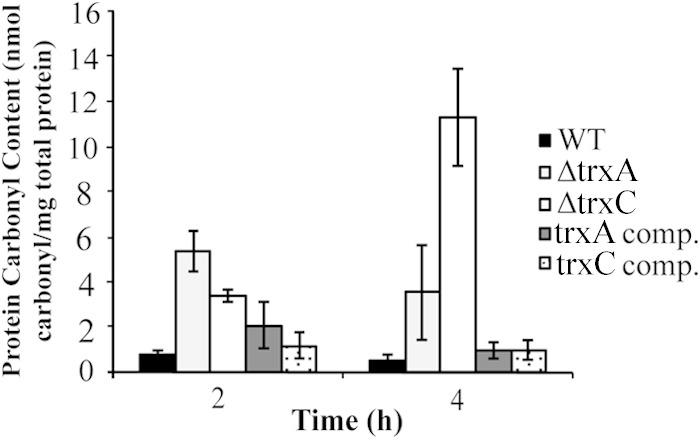
Protein carbonyl content in cells exposed to oxygen stress (20%) for 2 or 4 h. The wild-type, ΔtrxA mutant, ΔtrxC mutant, trxA complemented, or trxC complemented strain were exposed to 20% O2 for 2 or 4 h, and extracts were reacted with DNPH. The carbonyl content was determined by measuring the absorbance at 340 nm. Total carbonyl content is expressed as nanomoles of carbonyl per milligram of total protein. Results shown are the averages of results from three independent experiments. The carbonyl content of both deletion strains was significantly greater than that of the wild type at both the 2- and 4-h time points (P < 0.01). In low O2 (i.e., 4%), the carbonyl content of the mutant strains was not significantly greater than that of the wild type.
FIG 6.
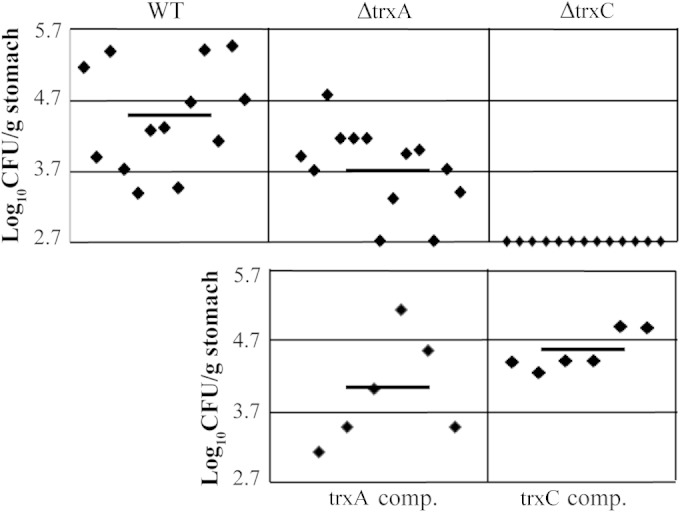
Mouse colonization assay of H. pylori X47 and trx strains. The mice were inoculated with a dose of 1.7 × 108 viable cells. Colonization of the mouse stomachs was determined 3 weeks postinoculation. Mouse stomachs were homogenized, and serial dilutions were plated. Data are presented as a scatter plot of numbers of CFU per gram of stomach as determined by plate counts. Each point represents a CFU count for one stomach, and the solid horizontal line represents the mean colonization for each strain. The baseline [log10(CFU g−1) = 2.7] is the detection limit of the assay, which represents a count below 500 CFU g−1 stomach. Horizontal bars represent the average log10 number of CFU/g stomach.
Preparation of cell extracts.
H. pylori was grown on BA plates for 2 days before being harvested. Cells were resuspended in sterile PBS, pH 7.4, to an optical density of 1 to 2 at 600 nm. Cells were then broken by sonication (Ultrasonics Heat Systems sonicator) in 3 sets of 10-s intervals (4-W output power and 40% duty cycle) and placed on ice. Cell debris was removed by centrifugation at 14,000 × g for 10 min at room temperature (25°C).
Real-time qPCR.
The Aurum total RNA minikit (Bio-Rad) was used to extract total RNA from H. pylori wild-type, ΔtrxA, and ΔtrxC cells grown under 4% O2. The Turbo DNA-free kit (Ambion) was used to degrade any remaining DNA. cDNA was then synthesized using the iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer's instructions. The iQ SYBR green supermix (Bio-Rad) kit was used for quantitative PCR (qPCR) according to the manufacturer's instructions. Primers specific for trxA [trxA-F/R(rt)] or trxC [trxC-F/R(rt)] were used along with primers specific for the housekeeping gene gyrA (Table 1). Relative transcript abundance was calculated using the formula 2−ΔCT (where CT is threshold cycle) (32).
Determination of lipid hydroperoxides.
Lipid hydroperoxides were extracted from the sample into chloroform as described in the work of Wang et al. (33), and the total amount of lipid hydroperoxides formed was quantified using a lipid hydroperoxide assay kit (Cayman Chemical, Ann Arbor, MI) by following the manufacturer's instructions. The chloroform extract was mixed with chloroform-methanol solvent and then combined with reagents 1 and 2 of the kit. Reagent 1 contained ferrous sulfate, which reacts with hydroperoxides to produce ferric ions. The resulting ferric ions react with ammonium thiocyanate, a chromogen that is detected at 500 nm with an extinction coefficient of 16,667 M−1 cm−1.
Fluorescent staining of cells and quantification of 8-oxoG.
H. pylori 43504, the ΔtrxA mutant, and the ΔtrxC mutant were analyzed for the presence of 8-oxoG by fluorescent staining of the cells as described by Wang et al. (33). 8-oxoG is structurally similar to avidin's natural substrate biotin, so avidin binding to 8-oxoG can be used for detection of 8-oxoG (34). Eight sets of the immunofluorescent images were examined for the luminosity of fluorescein isothiocyanate (FITC) and propidium iodide (PI) as well as for cell morphology, and a representative set of images is shown in Fig. 4. The average ratio of the luminosity of FITC to PI was calculated from 8 sets of images for the wild-type and the mutant cells.
FIG 4.

Detection of 8-oxoG by immunofluorescent staining. Wild-type H. pylori, ΔtrxA mutant, and ΔtrxC mutant cells were fixed on a glass slide and stained with 8-oxoG-specific avidin-FITC conjugate (lower panel) and propidium iodide (upper panel), followed by examination via fluorescence microscopy. The contrast adjustment was normalized for all the images, and a representative set of images is shown here. Blue arrows point to some examples of bacillary cells in the wild-type and ΔtrxA strains, and green arrows highlight some coccoid or broken cells in the ΔtrxA and ΔtrxC mutant strains. A 2-μm bar is given as a size scale.
Isolation of carbonylated proteins.
H. pylori 43504 cells grown for 2 days on BA plates were exposed to 20% O2 for 2 or 4 h (as described above) and then washed twice (centrifugation at 10,000 × g for 5 min at room temperature) in 50 mM potassium phosphate buffer, pH 6.7, with 1 mM EDTA. Cells were then resuspended in this buffer and cell extracts obtained, as described above. Samples were analyzed for contaminating nucleic acids by determining the ratio of the OD280 to the OD260. The carbonylated proteins can be quantified from cell extracts by reacting them with 2,4-dinitrophenylhydrazine (DNPH) (35). DNPH (Sigma) at 10 mM dissolved in 2 M HCl was added to the sample tubes, and 2 M HCl was added to the control tubes. All samples were incubated for 1 h in the dark with vortexing every 15 min. After incubation with DNPH, 20% trichloroacetic acid (TCA; Sigma) was added to each tube. Samples were centrifuged at 10,000 × g for 10 min, and the pellet was resuspended in 10% TCA. Samples were centrifuged again at 10,000 × g for 10 min, and the pellet was resuspended in a 1:1 ethanol-ethyl acetate mixture and washed twice with this mixture. The remaining pellet was resuspended in 6 M guanidine hydrochloride (Fisher) and centrifuged at 10,000 × g for 10 min, and the supernatant was transferred to the wells of a microtiter plate. Absorbance was then determined at 340 nm. The supernatant was also used to measure protein concentration of the samples by reading the absorbance at 280 nm and comparing this to a bovine serum albumin (BSA) standard curve.
Mouse colonization assay.
H. pylori X47, the ΔtrxA mutant, the ΔtrxC mutant, the trxA complemented strain, and the trxC complemented strain were resuspended in sterile PBS, pH 7.5, and inoculated into 5- to 6-week-old female C57BL/6NCr mice (NCI, Frederick, MD) via oral gavage with a 0.1-ml cell suspension. The 0.1 ml contained 1.7 × 108 viable cells. Three weeks after inoculation, mouse stomachs were removed and homogenized in PBS, pH 7.5 (36). The homogenized samples were then serially diluted, and 0.1 ml was plated onto BA plates containing 100 μg/ml bacitracin, 10 μg/ml vancomycin, and 10 μg/ml amphotericin. The plates were incubated in a partial-pressure O2 atmosphere (4%) for 5 to 7 days, the resulting colonies were counted, and numbers of CFU/g stomach were calculated. The data shown are the combined set from two separate experiments. The Institutional Animal Care and Use Committee gave prior approval for all animal work.
Statistical analysis.
Results were analyzed using the unpaired t test and are presented as means ± standard deviations (GraphPad). For the mouse colonization assay, the Wilcoxon signed-rank test was used to calculate statistical significance.
RESULTS
Oxygen stress tolerance of nongrowing cells.
To elucidate the roles of the trx genes on the ability of H. pylori to survive under oxygen stress, we determined the oxygen sensitivity (i.e., viability) of the trx deletion mutants constructed with strain 43504. We have experienced greater success in the construction of mutant strains with 43504 than with other strains, so this strain was chosen as the parent. Nongrowing cells were incubated under atmospheric oxygen (20%) for a 12-h period. This condition is considered a stress environment for H. pylori, as the bacterium grows optimally under low oxygen (2 to 10%). As shown in Fig. 1, the wild-type and the complemented strains are only slightly affected by exposure to oxygen for an 8-h period, but the ΔtrxA mutant shows a 100-fold loss in viability after 4 h of atmospheric oxygen exposure (Fig. 1A). The ΔtrxC mutant also shows a loss of viability as a result of oxygen stress, but its sensitivity was not as severe as the ΔtrxA mutant's. The ΔtrxC mutant is 100-fold decreased in viability compared to that of the wild type upon 6 h of atmospheric oxygen exposure (Fig. 1B). These results are similar to those shown with the trx deletion mutants constructed with strain 26695 (15). Growth rates were not compared among strains, as these experiments were performed in nongrowing cells. The goal was to assess/compare the viabilities of all trx strains while they were subjected to the same oxygen stress. Importantly, the viability of both strains was restored to wild-type levels upon complementation with the wild-type versions of both trxA and trxC. This makes conclusions about the roles of these enzymes from mutant strain analysis rigorous and indicates that both TrxA and TrxC play important roles in protecting the cell from oxidative stress. We conclude that trxA and trxC are essential for optimal growth under oxygen stress in H. pylori 43504.
Transcript abundance in the single trx gene deletion strains.
trx transcript abundance was analyzed using quantitative real-time PCR (Fig. 2). There was a 6-fold increase in transcript abundance of trxA in the ΔtrxC strain background compared to that in the wild type (Fig. 2A). However, a 4-fold decrease in the transcript abundance of trxC was observed in the ΔtrxA background compared to its abundance in the wild type (Fig. 2B). It is noteworthy that the overall abundance of trxC was 1,000-fold less than that of trxA in the wild type. Transcript abundance differences of trx have previously been shown in other organisms that possess multiple thioredoxins (25, 37–39).
FIG 2.
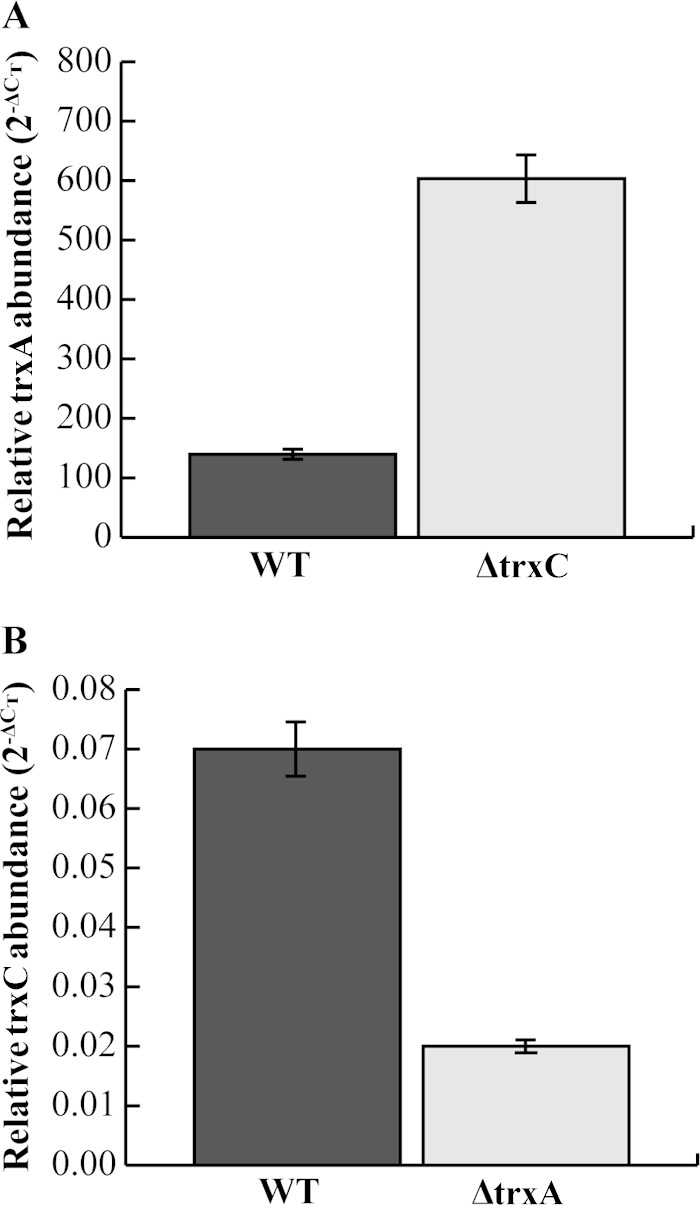
Relative trx transcript levels. (A) trxA abundance in the wild-type and ΔtrxC strains; (B) trxC abundance in the wild-type (WT) and ΔtrxA strains. Transcript abundance was determined by quantitative real-time PCR after cells were grown under conditions favorable for microaerophilic growth (4% O2). The gyrA housekeeping gene was used as an internal control. Results shown are from two separate experiments sampled in triplicate. The experiment was repeated again with similar results.
The amounts of lipid peroxides are greater in the Δtrx mutants than in the wild type under oxygen stress.
Unsaturated phospholipids, glycolipids, and cholesterol in the cell membrane are all targets of oxidant attack. Reactive oxygen-mediated oxidation of these molecules results in the initiation of lipid peroxidation and subsequent free radical chain reaction under times of oxidative stress (40). Thus, quantification of LOOH is often used to assess the level of oxidative membrane damage in cells under stress.
H. pylori possesses two peroxiredoxins that are known to reduce such hydroperoxide-derivative molecules to their corresponding alcohol, AhpC, and bacterioferritin comigratory protein (BCP) (33). Since TrxA is required for the function of AhpC, we reasoned that the ΔtrxA strain would contain a greater amount of lipid hydroperoxide than the wild type, especially under oxidative stress. Indeed, upon 200 μM HOCl exposure, we saw a 4-fold-greater lipid hydroperoxide level in the ΔtrxA mutant than in the wild type (Fig. 3). The ΔtrxC strain also had a 4-fold-greater level of lipid hydroperoxides than the parent strain (Fig. 3). The data from both mutants were shown to be statistically significantly greater than data for the wild type, with a 99% confidence level. This is similar to the lipid peroxide levels measured previously in the 43504 ΔahpC mutant (3-fold higher than in the wild type) (33), and a rigorous comparison of all the strains—the two types of ΔahpC strains plus the two Δtrx strains—with the parent strain and with multiple HOCl levels should reveal the genes most relevant to combating lipid peroxide formation. Importantly, complementation of either of the trx deletion strains with the wild-type version of the gene restored lipid peroxide levels to near wild-type levels (Fig. 3). The increase in lipid peroxides in the ΔtrxC mutant is interesting because Trx2 had previously been ruled out as an electron donor to AhpC (13). The results suggest that both Trxs are involved in preventing lipid peroxidation.
Oxidative stress affects cell morphology and the level of 8-oxoG in H. pylori Δtrx mutants.
Next, we examined the level of 8-oxoG DNA lesions in the mutant cells by using FITC-avidin/PI fluorescence microscopy. As FITC-avidin specifically labels DNA lesions while PI labels all DNA, the intensity ratio of stained FITC to PI reflects the level of 8-oxoG DNA lesions in the cells (41). This technique was successfully used in our lab to examine other oxidative-stress-related mutants of H. pylori (42, 43).
In this analysis, H. pylori wild-type or mutant cells were exposed to air for 4 h before subjecting them to immunofluorescent staining. Figure 4 shows that 4 h of exposure to air had no significant effect on the morphology of the wild-type cells, with ∼90% of the cells being in the bacillary form. However, when similarly treated, ∼45% of the ΔtrxA cells and ∼90% of the ΔtrxC cells transformed to the coccoid form or were lysed, suggesting that these cells suffer from persistent oxidative stress (44). The ratio of FITC-avidin to PI fluorescence intensity in the ΔtrxA and ΔtrxC mutant cells was determined to be 1.7- and 2.4-fold higher, respectively, than that in the wild-type cells (Table 2). These results demonstrated that the Δtrx mutant cells exposed to oxidative stress contain considerably higher levels of 8-oxoG DNA lesions and that the ΔtrxC mutant cells suffered more oxidative damage than the ΔtrxA mutant cells.
TABLE 2.
Cell morphology and the level of 8-oxo guanine in H. pylori cells
| Strain | % of damaged cellsa | 8-oxoG level (FITC/PI intensity ratio)b |
|---|---|---|
| Wild type | ∼10 | 0.86 + 0.05 |
| trxA mutant | ∼45 | 1.45 + 0.06 |
| trxC mutant | ∼85 | 2.05 + 0.13 |
Cell morphology was examined by fluorescence microscopy, and the numbers are estimated percentages of damaged cells, including coccoid and broken cells, in the whole-cell population.
The 8-oxoG level is expressed as the mean intensity ratio of FITC to PI, with the standard deviation. According to statistical analysis with Student's t test, the data for each mutant strain are significantly different from those for the wild type (P < 0.001).
In another experiment, H. pylori wild-type or Δtrx mutant cells were grown under optimum conditions with low O2 (4% partial pressure in the atmosphere) before immunofluorescent staining was conducted. No significant difference between wild-type and Δtrx cells was observed either in cell morphology or in the level of 8-oxoG DNA lesions generated (data not shown).
Oxidative damage to proteins.
To further assess the effect of the trx deletions on cells under oxidative stress, we estimated the overall oxidative modification of proteins by measuring the presence of carbonyl groups. Protein carbonyls are often used as a biomarker for severe oxidative stress, and the accumulation of carbonylated proteins is linked to E. coli cell death (33, 45–50). Although some amino acid residues are precursors to carbonylation and can be repaired, the addition of a carbonyl group is an irreversible posttranslational modification (45). Figure 5 shows that at 20% O2 exposure for 2 h, the ΔtrxA and ΔtrxC mutants contained 7- and 5-fold-greater amounts of carbonylated proteins, respectively, than the wild type. At 4 h, however, there was a greater amount of protein carbonyls in the ΔtrxC mutant than in the ΔtrxA mutant. A 7-fold-greater carbonyl level in the ΔtrxA mutant than in the wild type was observed, while that in the ΔtrxC mutant was increased 20-fold under 20% O2. Upon complementation, carbonyl levels were restored to wild-type levels. Taken together, these results indicate an increase in protein carbonylation in cells lacking the trx genes and indicate that these thioredoxins play roles in limiting protein damage.
The ΔtrxA and ΔtrxC mutants are decreased in mouse colonization ability.
To determine whether trxA or trxC had an effect on the ability of H. pylori to colonize the stomach, we inoculated mice with wild-type X47, the ΔtrxA mutant, the ΔtrxC mutant, or each complemented strain. Three weeks after inoculation, the mouse stomachs were harvested and colony counts determined. As shown in Fig. 6, we recovered H. pylori from all 13 mice inoculated with the wild-type strain at numbers ranging between 103 and 106 CFU per gram of stomach. Eleven of the 13 mice inoculated with the ΔtrxA mutant were H. pylori positive, but 2 of those 11 showed colonization numbers below 103 CFU/g stomach. All 13 mice inoculated with the ΔtrxC mutant were H. pylori negative. Importantly, both the trxA and trxC complemented strains colonized the stomachs at levels similar to those of the wild type. The colonization efficiency of the ΔtrxA mutant is significantly lower than that of the wild type at the 95% confidence level (P < 0.05), and that of the ΔtrxC strain is lower at a >99% level of confidence. These results suggest that both trxA and (particularly) trxC are important for host colonization by the pathogen.
DISCUSSION
Maintaining the redox balance within H. pylori is important for cell viability, growth, and metabolism (51). This renders the Trx system essential for survival under oxidative stress, as this is the only disulfide reductase system present in H. pylori. The glutathione-glutaredoxin system, present in many other organisms for maintaining the cellular thiol/disulfide balance, is lacking in H. pylori (21). In addition, the Trx system is important because it is the electron donor for all three peroxidases, BCP, Tpx, and AhpC (13, 43). H. pylori TrxA contains the typical Trx motif found in most Trx proteins (CGPC), but TrxC contains an unusual motif (CPDC) (27). It is not known how this affects its activity or its roles. Alignment of the protein sequences of TrxA and TrxC show that the two proteins are only 30% identical. TrxC has been ruled out as an electron donor for the known oxidative-stress-combating reductases tested (AhpC and Msr) (13, 19). The TrxC of H. pylori is, however, still a functional disulfide reductase, as it has been shown to successfully reduce insulin in vitro when used as a substrate (13).
The O2 exposure viability assay suggests that both Trxs have independent roles in protecting the cell from oxygen stress (Fig. 1). This is similar to what was concluded based on mutations in other organisms that contain more than one trx gene and no GSH reduction system. For example, L. casei is also a GSH-negative organism and has four trx genes. Disruption of either trxA or trxC resulted in moderate growth defects under aerobic conditions, and a trxA trxC double mutant of L. casei had severe growth defects (25). Still, the partial growth ability of the double mutant suggests that the other Trxs function to some extent. Similar results were shown for B. fragilis, which contains six trx genes. A single-mutation strain deleting one of the trx genes did not show any growth defect, but deletion of multiple genes in a single strain revealed a defective growth phenotype (52). A previous study using H. pylori 26695 as the parent strain showed defective growth associated with a ΔtrxA strain but a less severe defect in the ΔtrxC strain. A ΔtrxA trxC double mutant showed the largest growth defect (15). Our result indicates that TrxC is a major player in the protection of cells from oxygen stress and in their survival in vivo.
The ΔtrxC strain contained more 8-oxoG than both the ΔtrxA strain and the wild type (Fig. 4). This effect on DNA is a novel finding and has not been demonstrated in any other organism. Trxs are known to have important roles in the reduction of ribonucleotide reductase, an enzyme required for DNA synthesis (53). The function of H. pylori Trx2 is not known, but it is possible that it is involved in DNA replication and/or DNA repair. One indication that Trx2 may be involved in DNA replication is based upon the location of trxC in the chromosome. The trxC gene is located near dnaE (825 bp upstream), encoding the DNA polymerase III alpha subunit. This is similar to what was observed regarding the location of the essential trxA gene in B. fragilis; trxA is located directly downstream of dnaE. Phylogenetic analysis of TrxA grouped it with Trxs of Campylobacter, Helicobacter, and Porphyromonas gingivalis, indicating that the roles of TrxA and Trxs in these organisms may overlap (52). Trx2 may be involved in DNA repair in addition to DNA synthesis. Under oxygen stress conditions, DNA can become damaged by guanine oxidation (54). Oxidized guanine can then be incorporated into the genome, resulting in mispaired bases. H. pylori contains several proteins that function to excise 8-oxoG: MutT, MutY, and MutS (55). MutY is an iron-sulfur-containing glycosylase that excises mispaired adenines from the 8-oxoG (56). The Trx reductase system has been shown to be essential for iron binding in the iron-sulfur cluster protein IscA in E. coli (57). It is possible that H. pylori Trx2 aids in the iron binding of MutY.
We demonstrated an association between individual Trxs and host colonization ability for the first time in a bacterial system. Previously, a B. fragilis Trx reductase (trxB) mutant was shown to be essential for survival in an in vivo mouse abscess model (24). This mutation renders the whole system incomplete (the reductase and Trx), so it does not provide insight as to the importance of Trx alone. We show that the H. pylori ΔtrxA mutant is decreased in its ability to colonize the mouse stomach (Fig. 6). This phenotype is not surprising in light of the known functions of TrxA as the reductant for oxidized Msr and AhpC and its function as a chaperone for proper protein folding (13, 19, 28). However, the complete lack of colonization by the ΔtrxC mutant (Fig. 6) was not expected. Based upon this result, it seems that H. pylori TrxC plays a large in vivo role. It may play a broad role like Trx proteins that function to provide electrons to DsbD, located in the cytoplasmic membrane (58). H. pylori contains several Dsb (disulfide bond) protein oxidases that allow for disulfide bond formation in the periplasm (59–62). This mechanism has been well characterized in E. coli but less so in H. pylori (63). In E. coli, DsbD functions as the acceptor of electrons from Trx and then transfers them to various periplasmic oxidoreductases and a cytochrome synthesis protein (DsbB, DsbA, DsbC, and CcmG). DsbD receives the electrons from cytoplasmic Trx proteins and has been shown to reduce electrons to proteins involved in disulfide bond isomerization, cytochrome c maturation, and possibly other processes (64). In H. pylori, homologues of DsbG, DsbC, DsbB (DsbI), and DsbD (CcdA) have been identified (60, 61). Both DscC and DsbG have the Trx CXXC motif, and DsbG has been shown to have reductase activity. The reductase activity combined with a Trx motif indicates that DsbG functions as an oxidoreductase in the periplasm (59).
There are many virulence proteins located in the periplasm of H. pylori that, if damaged, would likely result in a lack of host colonization. While Trx maintains the methionine repair enzyme Msr reduced in the H. pylori cytoplasm, the players carrying out this role in the periplasm (where Msr is also known to function) are not known (19, 65). A membrane-bound Trx-like protein, PilB, provides electrons to Msr in the Neisseria gonorrhoeae periplasm, and the N. gonorrhoeae and H. pylori Msrs are highly homologous to each other (18, 66). Catalase is also an important periplasmic virulence protein in H. pylori (67). Catalase has been shown to be susceptible to oxidative inactivation and subsequent repair by Msr. Failure to repair a methionine sulfoxide-containing catalase could certainly result in a decrease in host colonization ability (68). It is noteworthy that during long-term colonization, a Δmsr mutant could not be recovered from a mouse stomach, indicating the importance of Msr in host colonization (18).
A proteomic analysis of E. coli Trx-targeted proteins revealed Trx to be involved in at least 26 different cellular processes, including transcriptional regulation, cell division, energy transduction, and several biosynthetic pathways (69). Trx is likely to be involved in multiple processes in H. pylori as well. Further work is needed to ascertain if any new protein-protein interactions occur that would aid in revealing new Trx roles, ones perhaps aiding peroxidase activity or macromolecule repair processes. While the specific roles of TrxC are not known, the importance of this protein in survival is now revealed. Both Trxs studied herein aid macromolecule integrity and homeostasis.
ACKNOWLEDGMENT
This work was supported by National Institutes of Health grant RO1 AI077569.
REFERENCES
- 1.Stadtman ER, Levine RL. 2000. Protein oxidation. Ann N Y Acad Sci 899:191–208. doi: 10.1111/j.1749-6632.2000.tb06187.x. [DOI] [PubMed] [Google Scholar]
- 2.Bagchi D, Bhattacharya G, Stohs SJ. 1996. Production of reactive oxygen species by gastric cells in association with Helicobacter pylori. Free Radic Res 24:439–450. doi: 10.3109/10715769609088043. [DOI] [PubMed] [Google Scholar]
- 3.Ramarao N, Gray-Owen SD, Meyer TF. 2000. Helicobacter pylori induces but survives the extracellular release of oxygen radicals from professional phagocytes using its catalase activity. Mol Microbiol 38:103–113. doi: 10.1046/j.1365-2958.2000.02114.x. [DOI] [PubMed] [Google Scholar]
- 4.Davies GR, Simmonds NJ, Stevens TR, Sheaff MT, Banatvala N, Laurenson IF, Blake DR, Rampton DS. 1994. Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo. Gut 35:179–185. doi: 10.1136/gut.35.2.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baik SC, Youn HS, Chung MH, Lee WK, Cho MJ, Ko GH, Park CK, Kasai H, Rhee KH. 1996. Increased oxidative DNA damage in Helicobacter pylori-infected human gastric mucosa. Cancer Res 56:1279–1282. [PubMed] [Google Scholar]
- 6.Nardone G, Rocco A, Malfertheiner P. 2004. Review article: Helicobacter pylori and molecular events in precancerous gastric lesions. Aliment Pharmacol Ther 20:261–270. doi: 10.1111/j.1365-2036.2004.02075.x. [DOI] [PubMed] [Google Scholar]
- 7.Stent A, Every AL, Sutton P. 2012. Helicobacter pylori defense against oxidative attack. Am J Physiol Gastrointest Liver Physiol 302:G579–G587. doi: 10.1152/ajpgi.00495.2011. [DOI] [PubMed] [Google Scholar]
- 8.Bagchi D, McGinn TR, Ye X, Bagchi M, Krohn RL, Chatterjee A, Stohs SJ. 2002. Helicobacter pylori-induced oxidative stress and DNA damage in a primary culture of human gastric mucosal cells. Dig Dis Sci 47:1405–1412. doi: 10.1023/A:1015399204069. [DOI] [PubMed] [Google Scholar]
- 9.Chaturvedi R, Asim M, Romero-Gallo J, Barry DP, Hoge S, de Sablet T, Delgado AG, Wroblewski LE, Piazuelo MB, Yan F, Israel DA, Casero RA Jr, Correa P, Gobert AP, Polk DB, Peek RM Jr, Wilson KT. 2011. Spermine oxidase mediates the gastric cancer risk associated with Helicobacter pylori CagA. Gastroenterology 141:1696–1708.e2. doi: 10.1053/j.gastro.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Briggs RT, Drath DB, Karnovsky ML, Karnovsky MJ. 1975. Localization of NADH oxidase on the surface of human polymorphonuclear leukocytes by a new cytochemical method. J Cell Biol 67:566–586. doi: 10.1083/jcb.67.3.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spiegelhalder C, Gerstenecker B, Kersten A, Schiltz E, Kist M. 1993. Purification of Helicobacter pylori superoxide dismutase and cloning and sequencing of the gene. Infect Immun 61:5315–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hazell SL, Evans DJ Jr, Graham DY. 1991. Helicobacter pylori catalase. J Gen Microbiol 137:57–61. doi: 10.1099/00221287-137-1-57. [DOI] [PubMed] [Google Scholar]
- 13.Baker LM, Raudonikiene A, Hoffman PS, Poole LB. 2001. Essential thioredoxin-dependent peroxiredoxin system from Helicobacter pylori: genetic and kinetic characterization. J Bacteriol 183:1961–1973. doi: 10.1128/JB.183.6.1961-1973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou Y, Wan XY, Wang HL, Yan ZY, Hou YD, Jin DY. 1997. Bacterial scavengase p20 is structurally and functionally related to peroxiredoxins. Biochem Biophys Res Commun 233:848–852. doi: 10.1006/bbrc.1997.6564. [DOI] [PubMed] [Google Scholar]
- 15.Comtois SL, Gidley MD, Kelly DJ. 2003. Role of the thioredoxin system and the thiol-peroxidases Tpx and Bcp in mediating resistance to oxidative and nitrosative stress in Helicobacter pylori. Microbiology 149:121–129. doi: 10.1099/mic.0.25896-0. [DOI] [PubMed] [Google Scholar]
- 16.Olczak AA, Wang G, Maier RJ. 2005. Up-expression of NapA and other oxidative stress proteins is a compensatory response to loss of major Helicobacter pylori stress resistance factors. Free Radic Res 39:1173–1182. doi: 10.1080/10715760500306729. [DOI] [PubMed] [Google Scholar]
- 17.Wang G, Olczak AA, Walton JP, Maier RJ. 2005. Contribution of the Helicobacter pylori thiol peroxidase bacterioferritin comigratory protein to oxidative stress resistance and host colonization. Infect Immun 73:378–384. doi: 10.1128/IAI.73.1.378-384.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alamuri P, Maier RJ. 2004. Methionine sulphoxide reductase is an important antioxidant enzyme in the gastric pathogen Helicobacter pylori. Mol Microbiol 53:1397–1406. doi: 10.1111/j.1365-2958.2004.04190.x. [DOI] [PubMed] [Google Scholar]
- 19.Alamuri P, Maier RJ. 2006. Methionine sulfoxide reductase in Helicobacter pylori: interaction with methionine-rich proteins and stress-induced expression. J Bacteriol 188:5839–5850. doi: 10.1128/JB.00430-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pesci EC, Pickett CL. 1994. Genetic organization and enzymatic activity of a superoxide dismutase from the microaerophilic human pathogen, Helicobacter pylori. Gene 143:111–116. doi: 10.1016/0378-1119(94)90614-9. [DOI] [PubMed] [Google Scholar]
- 21.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 22.Lu J, Holmgren A. 2014. The thioredoxin antioxidant system. Free Radic Biol Med 66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 23.Newton GL, Arnold K, Price MS, Sherrill C, Delcardayre SB, Aharonowitz Y, Cohen G, Davies J, Fahey RC, Davis C. 1996. Distribution of thiols in microorganisms: mycothiol is a major thiol in most actinomycetes. J Bacteriol 178:1990–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rocha ER, Tzianabos AO, Smith CJ. 2007. Thioredoxin reductase is essential for thiol/disulfide redox control and oxidative stress survival of the anaerobe Bacteroides fragilis. J Bacteriol 189:8015–8023. doi: 10.1128/JB.00714-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serata M, Iino T, Yasuda E, Sako T. 2012. Roles of thioredoxin and thioredoxin reductase in the resistance to oxidative stress in Lactobacillus casei. Microbiology 158:953–962. doi: 10.1099/mic.0.053942-0. [DOI] [PubMed] [Google Scholar]
- 26.Fahey RC, Brown WC, Adams WB, Worsham MB. 1978. Occurrence of glutathione in bacteria. J Bacteriol 133:1126–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Windle HJ, Fox A, Ni Eidhin D, Kelleher D. 2000. The thioredoxin system of Helicobacter pylori. J Biol Chem 275:5081–5089. doi: 10.1074/jbc.275.7.5081. [DOI] [PubMed] [Google Scholar]
- 28.McGee DJ, Kumar S, Viator RJ, Bolland JR, Ruiz J, Spadafora D, Testerman TL, Kelly DJ, Pannell LK, Windle HJ. 2006. Helicobacter pylori thioredoxin is an arginase chaperone and guardian against oxidative and nitrosative stresses. J Biol Chem 281:3290–3296. doi: 10.1074/jbc.M506139200. [DOI] [PubMed] [Google Scholar]
- 29.Veyrier FJ, Ecobichon C, Boneca IG. 2013. Draft genome sequence of strain X47-2AL, a feline Helicobacter pylori isolate. Genome Announc 1:e01095-13. doi: 10.1128/genomeA.01095-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olson JW, Mehta NS, Maier RJ. 2001. Requirement of nickel metabolism proteins HypA and HypB for full activity of both hydrogenase and urease in Helicobacter pylori. Mol Microbiol 39:176–182. doi: 10.1046/j.1365-2958.2001.02244.x. [DOI] [PubMed] [Google Scholar]
- 31.Olson JW, Agar JN, Johnson MK, Maier RJ. 2000. Characterization of the NifU and NifS Fe-S cluster formation proteins essential for viability in Helicobacter pylori. Biochemistry 39:16213–16219. doi: 10.1021/bi001744s. [DOI] [PubMed] [Google Scholar]
- 32.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.Wang G, Hong Y, Johnson MK, Maier RJ. 2006. Lipid peroxidation as a source of oxidative damage in Helicobacter pylori: protective roles of peroxiredoxins. Biochim Biophys Acta 1760:1596–1603. doi: 10.1016/j.bbagen.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 34.Struthers L, Patel R, Clark J, Thomas S. 1998. Direct detection of 8-oxodeoxyguanosine and 8-oxoguanine by avidin and its analogues. Anal Biochem 255:20–31. doi: 10.1006/abio.1997.2354. [DOI] [PubMed] [Google Scholar]
- 35.Levine RL, Garland D, Oliver CN, Amici A, Climent I, Lenz AG, Ahn BW, Shaltiel S, Stadtman ER. 1990. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol 186:464–478. doi: 10.1016/0076-6879(90)86141-H. [DOI] [PubMed] [Google Scholar]
- 36.Seyler RW Jr, Olson JW, Maier RJ. 2001. Superoxide dismutase-deficient mutants of Helicobacter pylori are hypersensitive to oxidative stress and defective in host colonization. Infect Immun 69:4034–4040. doi: 10.1128/IAI.69.6.4034-4040.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gustafsson TN, Sahlin M, Lu J, Sjoberg BM, Holmgren A. 2012. Bacillus anthracis thioredoxin systems, characterization and role as electron donors for ribonucleotide reductase. J Biol Chem 287:39686–39697. doi: 10.1074/jbc.M112.413427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lillig CH, Prior A, Schwenn JD, Aslund F, Ritz D, Vlamis-Gardikas A, Holmgren A. 1999. New thioredoxins and glutaredoxins as electron donors of 3′-phosphoadenylylsulfate reductase. J Biol Chem 274:7695–7698. doi: 10.1074/jbc.274.12.7695. [DOI] [PubMed] [Google Scholar]
- 39.Miranda-Vizuete A, Damdimopoulos AE, Gustafsson J, Spyrou G. 1997. Cloning, expression, and characterization of a novel Escherichia coli thioredoxin. J Biol Chem 272:30841–30847. doi: 10.1074/jbc.272.49.30841. [DOI] [PubMed] [Google Scholar]
- 40.Girotti AW. 1998. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J Lipid Res 39:1529–1542. [PubMed] [Google Scholar]
- 41.Chen SK, Tsai MH, Hwang JJ, Chang WP. 2001. Determination of 8-oxoguanine in individual cell nucleus of gamma-irradiated mammalian cells. Radiat Res 155:832–836. doi: 10.1667/0033-7587(2001)155[0832:DOOIIC]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 42.Wang G, Alamuri P, Humayun MZ, Taylor DE, Maier RJ. 2005. The Helicobacter pylori MutS protein confers protection from oxidative DNA damage. Mol Microbiol 58:166–176. doi: 10.1111/j.1365-2958.2005.04833.x. [DOI] [PubMed] [Google Scholar]
- 43.Wang G, Conover RC, Olczak AA, Alamuri P, Johnson MK, Maier RJ. 2005. Oxidative stress defense mechanisms to counter iron-promoted DNA damage in Helicobacter pylori. Free Radic Res 39:1183–1191. doi: 10.1080/10715760500194018. [DOI] [PubMed] [Google Scholar]
- 44.Park AM, Li Q, Nagata K, Tamura T, Shimono K, Sato EF, Inoue M. 2004. Oxygen tension regulates reactive oxygen generation and mutation of Helicobacter pylori. Free Radic Biol Med 36:1126–1133. doi: 10.1016/j.freeradbiomed.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 45.Madian AG, Regnier FE. 2010. Proteomic identification of carbonylated proteins and their oxidation sites. J Proteome Res 9:3766–3780. doi: 10.1021/pr1002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dalle-Donne I, Rossi R, Giustarini D, Milzani A, Colombo R. 2003. Protein carbonyl groups as biomarkers of oxidative stress. Clin Chim Acta 329:23–38. doi: 10.1016/S0009-8981(03)00003-2. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki YJ, Carini M, Butterfield DA. 2010. Protein carbonylation. Antioxid Redox Signal 12:323–325. doi: 10.1089/ars.2009.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nystrom T. 2005. Role of oxidative carbonylation in protein quality control and senescence. EMBO J 24:1311–1317. doi: 10.1038/sj.emboj.7600599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gonidakis S, Finkel SE, Longo VD. 2010. E. coli hypoxia-inducible factor ArcA mediates lifespan extension in a lipoic acid synthase mutant by suppressing acetyl-CoA synthetase. Biol Chem 391:1139–1147. doi: 10.1515/BC.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dukan S, Nystrom T. 1999. Oxidative stress defense and deterioration of growth-arrested Escherichia coli cells. J Biol Chem 274:26027–26032. doi: 10.1074/jbc.274.37.26027. [DOI] [PubMed] [Google Scholar]
- 51.Kaakoush NO, Sterzenbach T, Miller WG, Suerbaum S, Mendz GL. 2007. Identification of disulfide reductases in Campylobacterales: a bioinformatics investigation. Antonie Van Leeuwenhoek 92:429–441. doi: 10.1007/s10482-007-9171-5. [DOI] [PubMed] [Google Scholar]
- 52.Reott MA, Parker AC, Rocha ER, Smith CJ. 2009. Thioredoxins in redox maintenance and survival during oxidative stress of Bacteroides fragilis. J Bacteriol 191:3384–3391. doi: 10.1128/JB.01665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holmgren A. 1985. Thioredoxin. Annu Rev Biochem 54:237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 54.Fowler RG, White SJ, Koyama C, Moore SC, Dunn RL, Schaaper RM. 2003. Interactions among the Escherichia coli mutT, mutM, and mutY damage prevention pathways. DNA Repair (Amst) 2:159–173. doi: 10.1016/S1568-7864(02)00193-3. [DOI] [PubMed] [Google Scholar]
- 55.Hsieh P. 2001. Molecular mechanisms of DNA mismatch repair. Mutat Res 486:71–87. doi: 10.1016/S0921-8777(01)00088-X. [DOI] [PubMed] [Google Scholar]
- 56.Au KG, Clark S, Miller JH, Modrich P. 1989. Escherichia coli mutY gene encodes an adenine glycosylase active on G-A mispairs. Proc Natl Acad Sci U S A 86:8877–8881. doi: 10.1073/pnas.86.22.8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ding H, Harrison K, Lu J. 2005. Thioredoxin reductase system mediates iron binding in IscA and iron delivery for the iron-sulfur cluster assembly in IscU. J Biol Chem 280:30432–30437. doi: 10.1074/jbc.M504638200. [DOI] [PubMed] [Google Scholar]
- 58.Rietsch A, Bessette P, Georgiou G, Beckwith J. 1997. Reduction of the periplasmic disulfide bond isomerase, DsbC, occurs by passage of electrons from cytoplasmic thioredoxin. J Bacteriol 179:6602–6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoon JY, Kim J, Lee SJ, Kim HS, Im HN, Yoon HJ, Kim KH, Kim SJ, Han BW, Suh SW. 2011. Structural and functional characterization of Helicobacter pylori DsbG. FEBS Lett 585:3862–3867. doi: 10.1016/j.febslet.2011.10.042. [DOI] [PubMed] [Google Scholar]
- 60.Kaakoush NO, Kovach Z, Mendz GL. 2007. Potential role of thiol:disulfide oxidoreductases in the pathogenesis of Helicobacter pylori. FEMS Immunol Med Microbiol 50:177–183. doi: 10.1111/j.1574-695X.2007.00259.x. [DOI] [PubMed] [Google Scholar]
- 61.Raczko AM, Bujnicki JM, Pawlowski M, Godlewska R, Lewandowska M, Jagusztyn-Krynicka EK. 2005. Characterization of new DsbB-like thiol-oxidoreductases of Campylobacter jejuni and Helicobacter pylori and classification of the DsbB family based on phylogenomic, structural and functional criteria. Microbiology 151:219–231. doi: 10.1099/mic.0.27483-0. [DOI] [PubMed] [Google Scholar]
- 62.Godlewska R, Dzwonek A, Mikula M, Ostrowski J, Pawlowski M, Bujnicki JM, Jagusztyn-Krynicka EK. 2006. Helicobacter pylori protein oxidation influences the colonization process. Int J Med Microbiol 296:321–324. doi: 10.1016/j.ijmm.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 63.Kadokura H, Katzen F, Beckwith J. 2003. Protein disulfide bond formation in prokaryotes. Annu Rev Biochem 72:111–135. doi: 10.1146/annurev.biochem.72.121801.161459. [DOI] [PubMed] [Google Scholar]
- 64.Ritz D, Beckwith J. 2001. Roles of thiol-redox pathways in bacteria. Annu Rev Microbiol 55:21–48. doi: 10.1146/annurev.micro.55.1.21. [DOI] [PubMed] [Google Scholar]
- 65.Gonzalez Porqué P, Baldesten A, Reichard P. 1970. The involvement of the thioredoxin system in the reduction of methionine sulfoxide and sulfate. J Biol Chem 245:2371–2374. [PubMed] [Google Scholar]
- 66.Brot N, Collet JF, Johnson LC, Jonsson TJ, Weissbach H, Lowther WT. 2006. The thioredoxin domain of Neisseria gonorrhoeae PilB can use electrons from DsbD to reduce downstream methionine sulfoxide reductases. J Biol Chem 281:32668–32675. doi: 10.1074/jbc.M604971200. [DOI] [PubMed] [Google Scholar]
- 67.Harris AG, Hazell SL. 2003. Localisation of Helicobacter pylori catalase in both the periplasm and cytoplasm, and its dependence on the twin-arginine target protein, KapA, for activity. FEMS Microbiol Lett 229:283–289. doi: 10.1016/S0378-1097(03)00850-4. [DOI] [PubMed] [Google Scholar]
- 68.Mahawar M, Tran V, Sharp JS, Maier RJ. 2011. Synergistic roles of Helicobacter pylori methionine sulfoxide reductase and GroEL in repairing oxidant-damaged catalase. J Biol Chem 286:19159–19169. doi: 10.1074/jbc.M111.223677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kumar JK, Tabor S, Richardson CC. 2004. Proteomic analysis of thioredoxin-targeted proteins in Escherichia coli. Proc Natl Acad Sci U S A 101:3759–3764. doi: 10.1073/pnas.0308701101. [DOI] [PMC free article] [PubMed] [Google Scholar]