

Abstract
The obligate intracellular bacterium Chlamydia pneumoniae is not only a causative agent of community-acquired pneumonia but is also associated with a more serious chronic disease, asthma, which might be exacerbated by air pollution containing carbon nanoparticles. Although a detailed mechanism of exacerbation remains unknown, the proinflammatory cytokine interleukin-1β (IL-1β) is a critical player in the pathogenesis of asthma. C. pneumoniae induces IL-1β in macrophages via NACHT, LRR, and PYD domain-containing protein 3 (NLRP3) inflammasome activation and Toll-like receptor 2/4 (TLR2/4) stimulation. Carbon nanoparticles, such as carbon nanotubes (CNTs), can also evoke the NLRP3 inflammasome to trigger IL-1β secretion from lipopolysaccharide-primed macrophages. This study assessed whether costimulation of C. pneumoniae with CNTs synergistically enhanced IL-1β secretion from macrophages, and determined the molecular mechanism involved. Enhanced IL-1β secretion from C. pneumoniae-infected macrophages by CNTs was dose and time dependent. Transmission electron microscopy revealed that C. pneumoniae and CNTs were engulfed concurrently by macrophages. Inhibitors of actin polymerization or caspase-1, a component of the inflammasome, significantly blocked IL-1β secretion. Gene silencing using small interfering RNA (siRNA) targeting the NLRP3 gene also abolished IL-1β secretion. Other inhibitors (K+ efflux inhibitor, cathepsin B inhibitor, and reactive oxygen species-generating inhibitor) also blocked IL-1β secretion. Taken together, these findings demonstrated that CNTs synergistically enhanced IL-1β secretion from C. pneumoniae-infected macrophages via the NLRP3 inflammasome and caspase-1 activation, providing novel insight into our understanding of how C. pneumoniae infection can exacerbate asthma.
INTRODUCTION
Chlamydia pneumoniae is an obligate intracellular bacterium and a causative agent of respiratory tract infections, including community-acquired pneumonia (1, 2). The seroprevalence rates of C. pneumoniae infection, which start to rise relatively early in childhood, are increased by 50% at 20 years of age and subsequently reach 70 to 80% by 60 to 70 years of age (3, 4), suggesting most individuals will have had some exposure to the bacterium in their lifetime. Therefore, C. pneumoniae is likely a ubiquitous pathogen in individuals worldwide (4). The symptoms of pulmonary infection vary considerably according to age from asymptomatic or mild illness to serious pneumonia, especially in pediatric infection (1–3). Regarding this, several studies indicate that bacterial infection might exacerbate pulmonary inflammation and thus is associated with a more serious chronic disease, asthma (3). The proinflammatory cytokine interleukin-1β (IL-1β), which is induced by C. pneumoniae (5–8), is thought to play a critical role in the development of chronic inflammatory disease, although detailed mechanisms remain unclear. Air pollution containing carbon nanoparticles, environmental air contaminants that might be easily inhaled with C. pneumoniae, also appears to be a critical factor in the pathogenesis of asthma (9–11). Recent studies indicated that carbon black or carbon nanotubes (CNTs), belonging to carbon nanoparticles, stimulate IL-1β secretion from lipopolysaccharide (LPS)-primed macrophages (12–14).
IL-1β secretion is strictly controlled by two different steps: (i) pro-IL-1β transcription and (ii) the cleavage of pro-IL-1β by a caspase-1-containing protein complex, the inflammasome (15–17). First, pro-IL-1β is transcribed via NF-κB activation following signaling from pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) (signal 1). TLR signaling is initiated by binding to ligands, known as pathogen-associated molecular patterns (PAMPs), such as LPS, flagella, or lipoprotein (18), and then pro-IL-1β is cleaved by the activated inflammasome, which consists of caspase-1, nucleotide-binding oligomerization domain (NOD)-like receptor (NLR), and an adapter molecule, ASC, which is the designation for apoptosis-associated speck-like protein containing a carboxy-terminal caspase recruitment domain (CARD) (signal 2). NLRs recognize PAMPs as well as danger-associated molecular patterns (DAMPs), including nuclear DNA or RNA, and cytosolic proteins (18). Interestingly, while IL-1β secretion from C. pneumoniae-infected cells requires both NF-κB activation via TLR2/4 recognition (signal 1) and NLRP3 inflammasome activation (signal 2) (5, 19, 20), LPS-primed macrophages stimulated with carbon nanoparticles activate the NLRP3 inflammasome (signal 2) (12–14). It is likely that while C. pneumoniae activates both signal 1 and 2, CNTs stimulate signal 2 alone. Thus, carbon nanoparticles are likely to be a precipitating cause of NLRP3 inflammasome activation during C. pneumoniae infection.
In the present study, we assessed whether costimulation of C. pneumoniae with CNTs synergistically enhanced IL-1β secretion from macrophages and determined the molecular mechanism involved.
MATERIALS AND METHODS
Chemical reagents.
Phorbol 12-myristate 13-acetate (PMA), Escherichia coli LPS (O55:B5), ATP, and N-acetyl-l-cysteine (NAC) were purchased from Sigma-Aldrich (St. Louis, MO). The pan-caspase inhibitor Z-VAD-FMK (Z-Val-Ala-Asp-fluoromethylketone) and caspase-1 inhibitor Z-WHED-FMK (Z-Trp-His-Glu-Asp-fluoromethylketone) were also purchased from the Peptide Institute (Osaka, Japan) and Enzo Life Sciences (Farmingdale, NY), respectively. Other inhibitors, such as cytochalasin D and CA-074 methyl ester (Me), bafilomycin A1, and diphenyleniodonium chloride (DPI), were also obtained from Enzo Life Sciences (Farmingdale, NY), LC Laboratories (Woburn, MA), and Cayman Chemical (Ann Arbor, MI), respectively.
Cells.
HEp-2 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Sigma) containing 10% heat-inactivated fetal calf serum (FCS) and antibiotics (gentamicin sulfate, 10 μg/ml; vancomycin, 10 μg/ml; amphotericin B, 1 μg/ml) (Sigma) at 37°C in 5% CO2. Cells of the human acute monocytic leukemia cell line THP-1 were also cultured in RPMI 1640 (Sigma) containing 10% heat-inactivated FCS and antibiotics at 37°C in 5% CO2.
Bacteria.
C. pneumoniae TW183 was used in this study. The bacteria were propagated in HEp-2 cells as described previously (21). Infected cells were collected after 3 days of incubation and then disrupted by freeze-thawing. After brief centrifugation at 180 × g for 5 min to remove cell debris, bacteria were concentrated at 9,000 × g for 10 min at 4°C. The bacterial pellets were resuspended in sucrose-phosphate-glutamic acid buffer and stored at −80°C until use. The numbers of C. pneumoniae infectious progeny cells were determined as inclusion-forming units (IFU) by counting chlamydial inclusions formed in HEp-2 cells using fluorescein isothiocyanate (FITC)-conjugated monoclonal anti-Chlamydia antibody specific for Chlamydia LPS (Denka Seiken, Tokyo, Japan) as described previously (21).
CNTs.
Single-wall CNTs 1.1 nm in diameter and 2,000 to 5,000 nm in length (Carbon Nanotechnologies, Inc., Houston, TX), were used as carbon nanoparticles in this study. CNTs were suspended in distilled water according to the method described previously (22) and stored at 4°C until use.
Costimulation of C. pneumoniae with CNTs.
THP-1 cells (4 × 106 cells/well) were seeded into a 6-well plate and allowed to differentiate into adherent macrophages in 10% FCS–RPMI 1640 supplemented with 50 nM PMA for 3 days. After being washed with serum-free RPMI, PMA-stimulated THP-1 cells were suspended in serum-free Opti-MEM (Invitrogen, Carlsbad, CA) with antibiotics and then incubated with C. pneumoniae at a multiplicity of infection (MOI) of 5. After 4 h, the cells were further incubated with 30 μg/ml CNTs for 24 h. Cells stimulated with 100 ng/ml E. coli LPS followed by 5 mM ATP were used as a positive control. IL-1β production from the supernatants and cell lysates was then determined by Western blotting. In some experiments, cells were preincubated with 2 μM cytochalasin D (actin polymerization inhibitor), 100 nM bafilomycin A1 (vacuolar H-ATPase inhibitor), 10 μM Z-VAD-FMK (pan-caspase inhibitor), 10 μM Z-WHED-FMK (caspase-1 inhibitor), 70 mM KCl (K+ efflux inhibitor), 10 μM CA-074 Me (cathepsin B inhibitor), 5 mM NAC (reactive oxygen species [ROS] inhibitor [antioxidant]), or 10 μM DPI (ROS inhibitor [ROS generating pathway inhibitor]) 1 h prior to the inoculation of CNTs.
Western blotting.
Culture supernatants were obtained from PMA-stimulated THP-1 cells. The supernatants were precipitated with 10% trichloroacetic acid (Sigma) for concentration by desalinization, washed with acetone, and then dried at room temperature. The dried pellets and the remaining cells were suspended in a reducing sample buffer containing 2-mercaptoethanol. Samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Separated proteins were electrotransferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA) with a semidry electroblotter (Bio-Rad, Richmond, CA). Membranes were blocked with 1% (wt/vol) skimmed milk in Tris-buffered saline (TBS, pH 7.5) containing 0.05% Tween 20 (TBS-T) and incubated with mouse anti-IL-1β antibody (Cell Signaling Technology, Beverly, MA) or mouse anti-α-tubulin antibody (Cedarlane, Ontario, Canada) in Immuno-enhancer reagent A (Wako Pure Chemical Industries) overnight at 4°C. After being washed with TBS-T, membranes were incubated with a horseradish peroxidase-conjugated goat anti-mouse IgG antibody (Sigma) in Immuno-enhancer reagent B for 1 h at room temperature. Labeled proteins were visualized with ImmunoStar LD Western blotting substrate (Wako Pure Chemical Industries) or Western blotting substrate (Thermo Scientific, Waltham, MA).
qPCR.
Total RNA was extracted from stimulated or unstimulated cells using the High Pure RNA isolation kit (Roche, Indianapolis, IN) according to the manufacturer's instructions. Extracted RNA was treated with DNA-free (Ambion, Austin, TX). cDNA was synthesized with random primers in ReverTra Ace quantitative PCR reverse transcription (qPCR RT) master mix (Toyobo, Osaka, Japan). qPCR was performed with primers specific for IL-1β (forward, 5′-ACA GAT GAA GTG CTC CTT CCA-3′; reverse, 5′-GTC GGA GAT TCG TAG CTG GAT-3′), for IL-8 (forward, 5′-CTG CGC CAA CAC AGA AAT TA-3′; reverse, 5′-ATT GCA TCT GGC AAC CCT AC-3′), for tumor necrosis factor alpha (TNF-α) (forward, 5′-CCC CAG GGA CCT CTC TCT AA-3′; reverse, 5′-TGA GGT ACA GGC CCT CTG AT-3′) (23), and for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (forward, 5′-AAC GGG AAG CTC ACT GGC ATG-3′; reverse, 5′-TCC ACC ACC CTG TTG CTG TAG-3′) (24). The PCR conditions consisted of 5 min of denaturation at 95°C, followed by 40 cycles, each of 30 s of denaturation at 95°C, 30 s of annealing at 60°C, and 45 s of extension at 72°C. The amount of mRNA expression of IL-1β, IL-8, or TNF-α was normalized to that of GAPDH. Whole DNA was also extracted from stimulated or unstimulated cells using a High Pure PCR template preparation kit (Roche) according to the manufacturer's instructions. Extracted DNA was used for qPCR with pairs of primers specific for C. pneumoniae 16S rRNA (forward, 5′-GGT CTC AAC CCC ATC CGT GTC GG-3′; reverse, 5′-TGC GGA AAG CTG TAT TTC TAC AGT T-3′) (25) and host cellular GAPDH gene (24). The thermal cycling conditions were 95°C for 5 min, followed by 40 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 45 s. The amount of amplified DNA of the Chlamydia 16S rRNA gene was normalized to that of the GAPDH gene.
TEM.
PMA-stimulated THP-1 cells incubated with C. pneumoniae and CNTs were fixed with 3% glutaraldehyde (Sigma) in 0.1 M phosphate-buffered saline (PBS, pH 7.4) at 4°C. After being washed with PBS, the cells were processed for alcohol dehydration and embedding in Epon 813, as described previously (21). Ultrathin sections of the cells were stained with lead citrate and uranium acetate before being viewed by transmission electron microscopy (TEM).
ELISA.
IL-18 in culture supernatants from PMA-stimulated THP-1 cells was also measured with the human IL-18 enzyme-linked immunosorbent assay (ELISA) kit (MBL, Nagoya, Japan) according to the manufacturer's protocol.
RNA interference.
Small interfering RNA (siRNA) targeting the NLRP3 gene (sense, 5′-GGU GUU GGA AUU AGA CAA CdTdT-3′; antisense, 5′-GUU GUC UAA UUC CAA CAC CdTdT-3′) or control scrambled siRNA (sense, 5′-UUC UCC GAA CGU GUC ACG UdTdT-3′; antisense, 5′-ACG UGA CAC GUU CGG AGA AdTdT-3′) was used as described previously (19). One day before incubation with C. pneumoniae, siRNA or scrambled RNA was incubated with RNAiMAX transfection reagent (Invitrogen) according to the manufacturer's instructions. Gene silencing of the NLRP3 gene was confirmed by reverse transcription (RT)-PCR using primers specific for the NLRP3 gene (forward, 5′-AGC CAC GCT AAT GAT CGA CT-3′; reverse, 5′-CAG GCT CAG AAT GCT CAT CA-3′) (26) and the GAPDH gene (24).
Statistical analysis.
Statistical analysis was performed using an unpaired Student's t test. A P value of <0.05 was considered significant.
RESULTS
CNT treatment enhanced IL-1β secretion from Chlamydia-stimulated macrophages.
To assess our hypothesis, we first investigated whether CNT treatment could enhance IL-1β secretion from Chlamydia-stimulated macrophages using Western blotting. Mature IL-1β was clearly detected in supernatants from PMA-stimulated THP-1 cells incubated with C. pneumoniae for 24 h, confirming previous reports (5–8). However, mature IL-1β was not detected in supernatants from cells stimulated with CNTs alone (Fig. 1A, Cpn and CNT). IL-1β secretion was significantly enhanced when cells were incubated with combined C. pneumoniae and CNTs (Fig. 1A, Cpn plus CNT). IL-1β secretion also increased dependent upon time and dose (Fig. 1B and C, respectively). Thus, the results indicated a synergistic effect, whereby CNT treatment enhanced IL-1β secretion in C. pneumoniae-stimulated macrophages.
FIG 1.
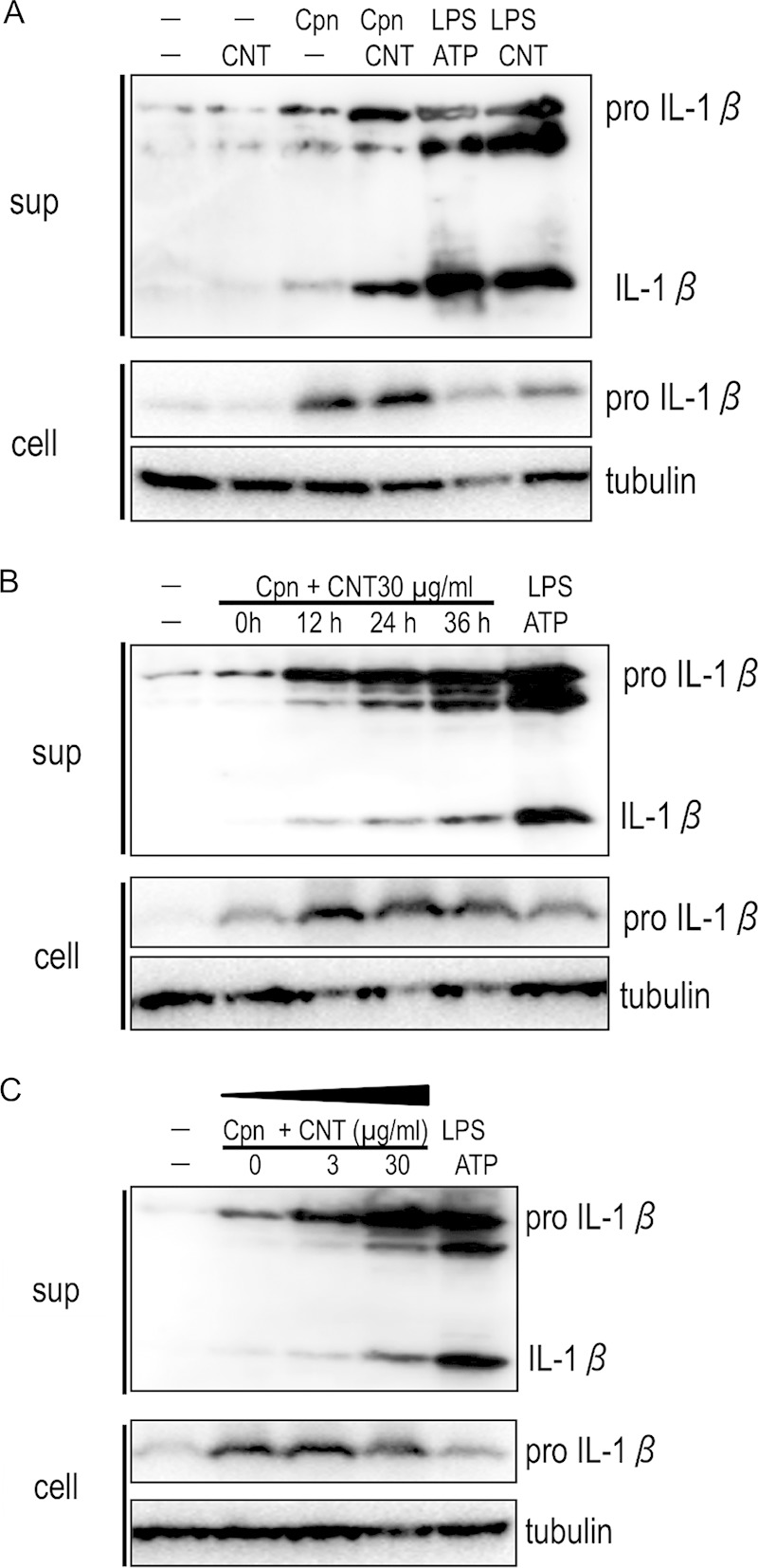
Representative Western blotting images show synergistic effects of Chlamydia pneumoniae with CNTs on IL-1β secretion from macrophages. (A) Mature IL-1β secretion from macrophages is significantly increased by costimulation of C. pneumoniae with CNTs at 24 h after incubation compared with CNTs or C. pneumoniae alone. (B) A time course experiment shows the synergistic increase of IL-1β secretion was time dependent. (C) A dose-response experiment revealed that the synergistic increase of IL-1β secretion also occurred in a dose-dependent manner, dependent upon the CNT concentration. The results are representative of three independent experiments. Tubulin was used as an internal control. sup, desalinized culture supernatants of THP-1 cells; cell, the remaining cells; Cpn, C. pneumoniae; CNT, carbon nanotube; LPS, lipopolysaccharide; IL-1β, interleukin-1β.
CNT treatment did not activate pro-IL-1β transcription in macrophages.
As mentioned above, IL-1β secretion is critically controlled by pro-IL-1β transcription via NF-κB activation and pro-IL-1β cleavage with inflammasome activation (15–17). We next investigated using qRT-PCR whether the synergistic effect of IL-1β maturation occurred at the mRNA expression level in C. pneumoniae-stimulated macrophages treated with CNTs. As shown in Fig. 2 (upper), while, IL-1β mRNA expression was significantly increased in THP-1 cells stimulated with either C. pneumoniae or C. pneumoniae plus CNTs, CNT treatment alone did not stimulate IL-1β mRNA expression. The transcription of mRNAs for other cytokines (IL-8 and TNF-α), which are also regulated through NF-κB, were also increased by C. pneumoniae or C. pneumoniae plus CNTs (Fig. 2, middle and lower). Thus, CNT treatment induced IL-1β secretion from C. pneumoniae-stimulated THP-1 cells at the maturation level via NLRP3 inflammasome activation, rather than by IL-1β mRNA transcription.
FIG 2.
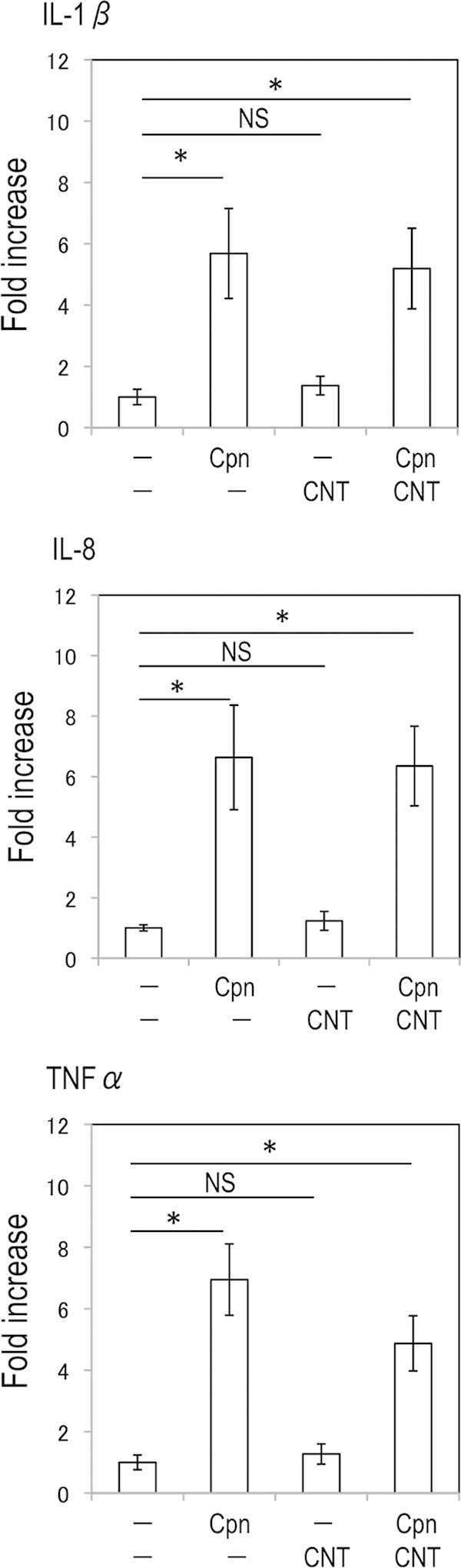
Increase of proinflammatory cytokine mRNA expression requires stimulation with either Chlamydia pneumoniae or C. pneumoniae plus CNTs, but not CNTs alone. Transcription of IL-1β, IL-8, or TNF-α mRNA was determined by qRT-PCR, and the data (means ± standard deviations) are shown as a ratio (fold change) of each of the target cytokine genes to the GAPDH gene. *, statistically significant difference (P < 0.05 versus control, Student's t test; n = 3). NS, not significant; Cpn, C. pneumoniae; CNT, carbon nanotube; IL-1β, interleukin-1β; TNF-α, tumor necrosis factor alpha.
Synergistic effect of C. pneumoniae with CNTs on IL-1β secretion from macrophages does not require bacterial growth or de novo bacterial protein synthesis.
To assess whether the synergistic effect on IL-1β secretion required bacterial growth or de novo bacterial protein synthesis, bacterial growth with IL-1β secretion was monitored over 3 days in C. pneumoniae-stimulated macrophages treated with CNTs in the presence or absence of chloramphenicol, a protein synthesis inhibitor. qPCR analysis revealed no significant change in bacteria numbers during the culture period, even in the absence or presence of CNTs (Fig. 3A). The addition of chloramphenicol to the cultures had no influence on the synergistic effect of C. pneumoniae and CNTs on IL-1β secretion from macrophages (Fig. 3B). Thus, the synergistic effect on IL-1β secretion from macrophages does not require either bacterial growth or de novo bacterial protein synthesis in cells.
FIG 3.
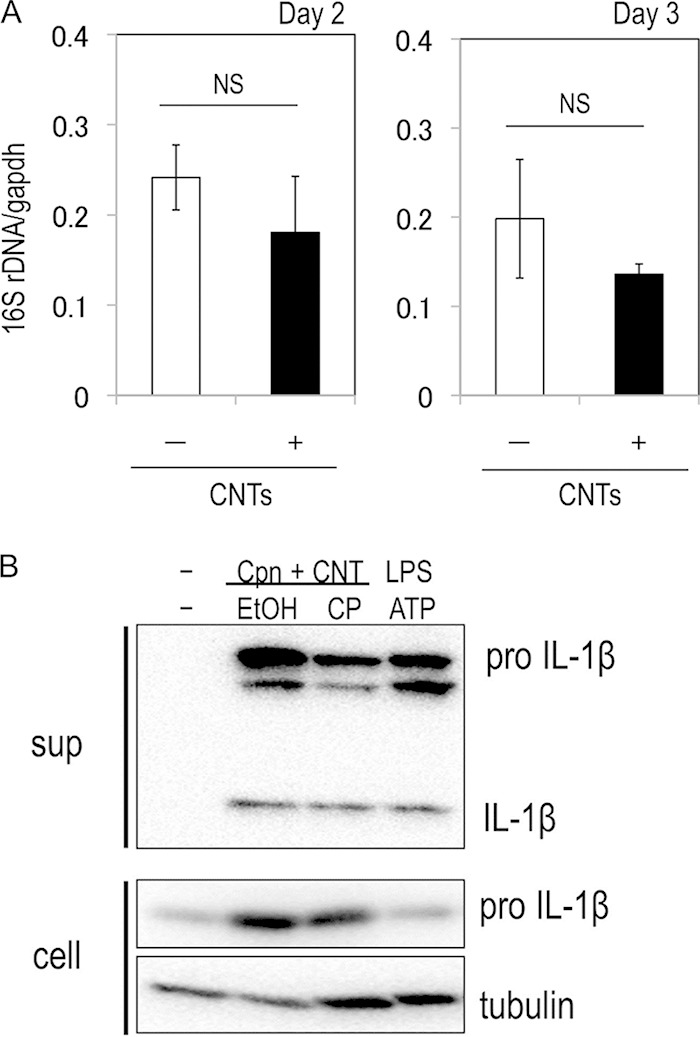
Synergistic effect of C. pneumoniae with CNTs on IL-1β secretion from macrophages does not require bacterial growth or de novo bacterial protein synthesis. (A) Chlamydial growth in macrophages treated with or without CNTs. The C. pneumoniae-stimulated THP-1 cells were cultured during 3 days in the presence or absence of CNTs. The amounts of chlamydial (16S rRNA genes [rDNA]) and host cell DNA (GAPDH gene) were monitored by qPCR, and the data (averages ± standard deviations) show a ratio of chlamydial 16S rRNA genes to the GAPDH gene. NS, not significant; Cpn; C. pneumoniae. (B) Effect of de novo protein synthesis by C. pneumoniae on IL-1β secretion from macrophages. The C. pneumoniae-stimulated THP-1 cells treated with CNTs were cultured for 24 h in the presence or absence of 50 μg/ml chloramphenicol (with ethyl acohol [EtOH] as the solvent control), and IL-1β secretion was detected by Western blotting. CP, chloramphenicol; sup, desalinized culture supernatants of THP-1 cells; cell, the remaining cells; Cpn, C. pneumoniae; CNT, carbon nanotube.
Synergistic effect of C. pneumoniae with CNTs on IL-1β secretion from macrophages requires uptake of bacteria and CNTs and is caspase-1 dependent.
Through experiments with the specific inhibitors cytochalasin D (an actin polymerization inhibitor) and bafilomycin A1 (a vacuolar H-ATPase inhibitor) and TEM observation, we assessed whether the synergistic effect required uptake of bacteria with CNTs or lysosomal maturation. In contrast to bafilomycin A1, treatment with cytochalasin D clearly blocked the synergistic effect on IL-1β secretion, likely via blocking C. pneumoniae infection (Fig. 4A). TEM observation revealed the coexistence of C. pneumoniae with CNTs in a macrophage (untreated with cytochalasin D) at 24 h after incubation (Fig. 4B). Thus, the synergistic effect required the uptake of bacteria with CNTs into cells regardless of lysosomal maturation. Furthermore, because bacteria with CNTs stimulate the NLRP3 inflammasome to activate caspase-1 followed by IL-1β maturation (signal 2) (5, 19, 20), we also performed experiments using caspase inhibitors (Z-VAD-FMK, a pan-caspase inhibitor, and Z-WHED-FMK, a caspase-1 inhibitor). Both inhibitors significantly blocked the synergistic effect on IL-1β secretion (Fig. 5A), and the inhibition occurred in a dose-dependent manner for the caspase-1 inhibitor (Fig. 5B), indicating the requirement of caspase-1 activation for the synergistic effect. Thus, the synergistic effect of C. pneumoniae with CNTs on IL-1β secretion from macrophages requires both the uptake of bacteria with CNTs into cells and caspase-1 activation.
FIG 4.

Synergistic effect of Chlamydia pneumoniae with CNTs on IL-1β secretion from macrophages requires uptake of bacteria into cells but not lysosomal maturation. (A) Cells stimulated with or without C. pneumoniae in the presence or absence of CNTs were incubated with either 2 μM cytochalasin D (an actin polymerization inhibitor) or 50 nM bafilomysin A1 (a vacuolar H-ATPase inhibitor) for 24 h, and then IL-1β secretion from macrophages was detected. Results are representative of three independent experiments. (B) A representative transmission electron microscopy image reveals uptake of C. pneumoniae with CNTs into a macrophage. The inset is enlarged in the right panel. Arrows, CNTs; arrowhead, C. pneumoniae. N, nucleus; sup, desalinized culture supernatants of THP-1 cells; cell, the remaining cells; CD, cytochalasin D; BAF, bafilomycin A1; Cpn, C. pneumoniae; CNT, carbon nanotube; DMSO, dimethyl sulfoxide; IL-1β, interleukin-1β.
FIG 5.
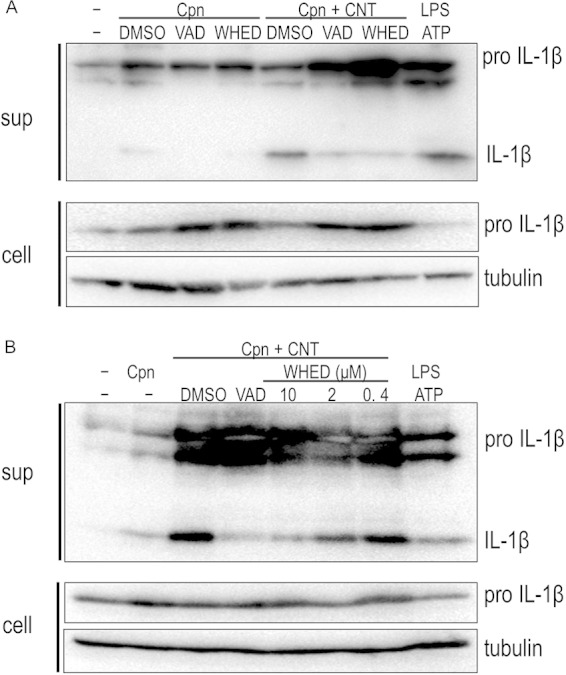
Synergistic effect of Chlamydia pneumoniae with CNTs on IL-1β secretion from macrophages is blocked by treatment with caspase inhibitors. (A) Cells stimulated with or without C. pneumoniae in the presence of absence of CNTs were incubated with either 10 μM Z-VAD-FMK (a pan-caspase inhibitor) or 10 μM Z-WHED-FMK (a caspase-1 inhibitor) for 24 h, and then IL-1β secretion from macrophages was detected. Results are representative of three independent experiments. (B) Dose-dependent inhibition of IL-1β secretion from macrophages by the treatment with Z-WHED-FMK. sup, desalinized culture supernatants of THP-1 cells; cell, the remaining cells; VAD, Z-VAD-FMK; WHED, Z-WHED-FMK; Cpn, C. pneumoniae; CNT, carbon nanotube; DMSO, dimethyl sulfoxide; IL-1β, interleukin-1β; LPS, lipopolysaccharide.
Synergistic effect of C. pneumoniae with CNTs on IL-18 secretion from macrophages.
To confirm caspase-1 activation, we quantitatively measured the amount of IL-18 in supernatants as well as IL-1β cleaved by caspase-1 secreted from macrophages stimulated with either C. pneumoniae or C. pneumoniae plus CNTs by an alternative method (not Western blotting) with a commercially available ELISA kit (see Materials and Methods). As expected, IL-18 secretion was significantly enhanced when incubated under the combination of C. pneumoniae with CNTs (Fig. 6A). Furthermore, it was also confirmed that the IL-18 secretion was increased in a time-dependent manner (Fig. 6B) and that the secretion as well as IL-1β maturation was significantly inhibited in a dose-dependent manner of the caspase-1 inhibitor (Fig. 6C). Thus, the results were supported by the evidence showing the synergistic effects of C. pneumoniae with CNTs into IL-1β maturation via caspase-1 activation.
FIG 6.
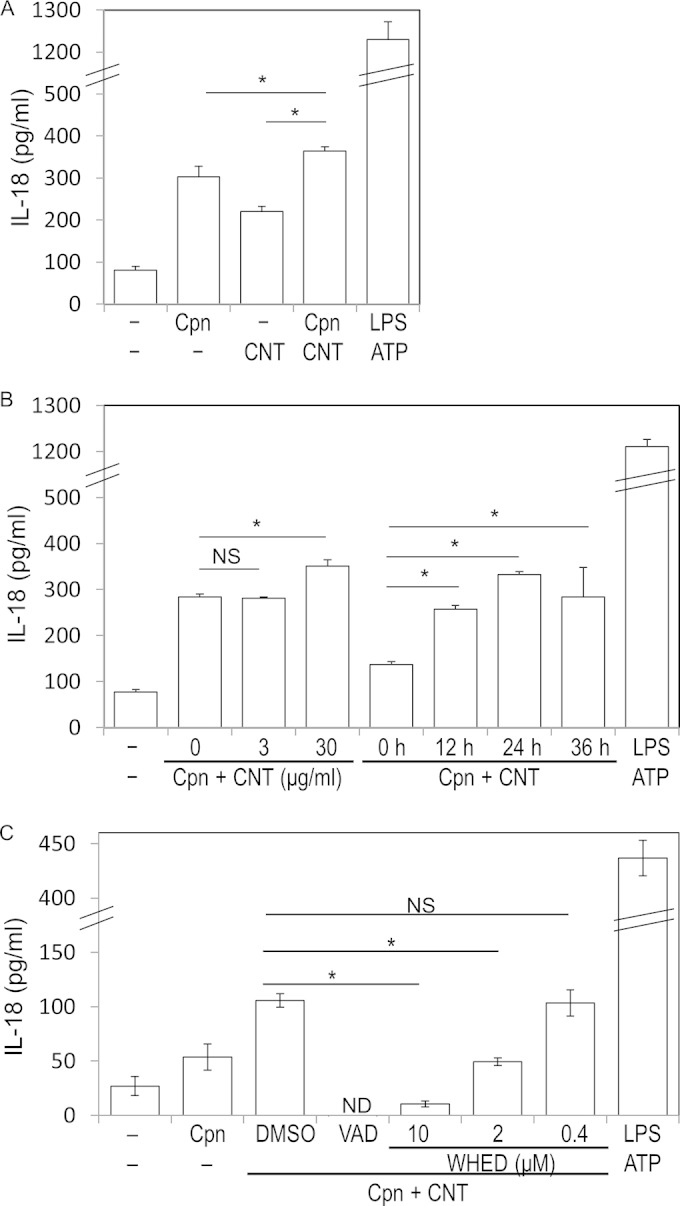
Synergistic effects of C. pneumoniae with CNTs on IL-18 secretion from macrophages. (A) IL-18 secretion was significantly increased by costimulation of C. pneumoniae with CNTs after incubation compared to that of either CNTs or C. pneumoniae alone. The data are shown as averages ± standard deviations. *, statistically significant difference (P < 0.05, Student's t test; n = 3). NS, not significant; Cpn, C. pneumoniae. (B) Dose and time course experiments reveal that the synergistic increase of IL-18 secretion occurred in a time-dependent manner for the incubation. (C) Dose-dependent inhibition of IL-18 secretion from macrophages by the treatment with Z-WHED-FMK. ND, not detected (described above).
Synergistic effect of C. pneumoniae with CNTs on IL-1β secretion from macrophages occurs via NLRP3 inflammasome activation.
IL-1β maturation absolutely requires pro-IL-1β cleavage with caspase-1, following the construction of a large protein complex, the inflammasome, comprising caspase-1, NLR, and ASC (15–17). The inflammasome is commonly activated by bacterial infections, including C. pneumoniae (5–8) or by stimulation with DAMPs (15, 16). Using siRNA to silence NLRP3 in cells (27), we assessed whether stimulation of C. pneumoniae with CNTs triggered the NLRP3 inflammasome, which is required for IL-1β secretion. The knockdown of NLRP3 gene mRNA expression was confirmed by RT-PCR (Fig. 7A). In contrast to scrambled siRNA, NLRP3 gene knockdown cells failed to show enhanced IL-1β secretion even under costimulation of C. pneumoniae with CNTs (Fig. 7B). Thus, stimulation with bacteria and CNTs enhanced NLRP3 inflammasome activation.
FIG 7.
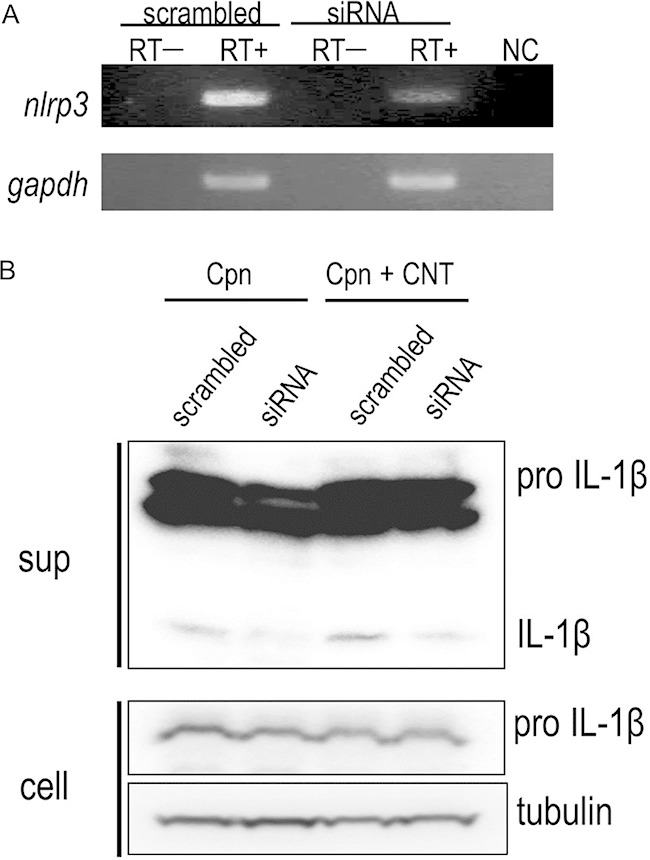
Synergistic effect of Chlamydia pneumoniae with CNTs on IL-1β secretion from macrophages requires NLRP3 inflammasome activation followed by caspase-1 activation. (A) Gene silencing of NLRP3 gene knockdown (KD) (siRNA) and control (scrambled) cells was confirmed by RT-PCR. (B) Representative Western blotting images show siRNA treatment diminished IL-1β secretion from NLRP3 KD cells stimulated with C. pneumoniae and/or CNTs. Results are representative of three independent experiments. sup, desalinized culture supernatants of THP-1 cells; cell, the remaining cells; RT, reverse transcription; Cpn, C. pneumoniae; CNT, carbon nanotube; IL-1β, interleukin-1β; NC, negative control.
Synergistic effect of C. pneumoniae with CNTs on IL-1β secretion from macrophages requires upstream signals, including K+ efflux, lysosomal degradation, and ROS production.
It is well known that the NLRP3 inflammasome is fully activated by distinct upstream signals, including K+ efflux (26), lysosomal degradation (28), and ROS production (29). We therefore investigated the upstream signal pathways required for NLRP3 inflammasome activation in cells stimulated by C. pneumoniae with CNTs, using specific inhibitors (KCl, a K+ efflux inhibitor, CA-074 Me, a cathepsin B inhibitor, NAC, an ROS production inhibitor as an antioxidant, and DPI, an ROS production inhibitor as the generating pathway inhibitor). IL-1β secretion was clearly inhibited by treatment with KCl compared with NaCl (control), indicating a requirement for K+ efflux (Fig. 8A). In addition, treatment with CA-074 Me obviously blocked IL-1β secretion, indicating that IL-1β secretion requires lysosomal degradation (Fig. 8B). Furthermore, treatment with DPI, but not NAC, inhibited IL-1β secretion, indicating IL-1β secretion partially requires ROS generation (Fig. 8C). Together, the results suggested that NLRP3 inflammasome activation coincidently requires three distinct upstream signals: K+ efflux, lysosomal degradation, and ROS production.
FIG 8.
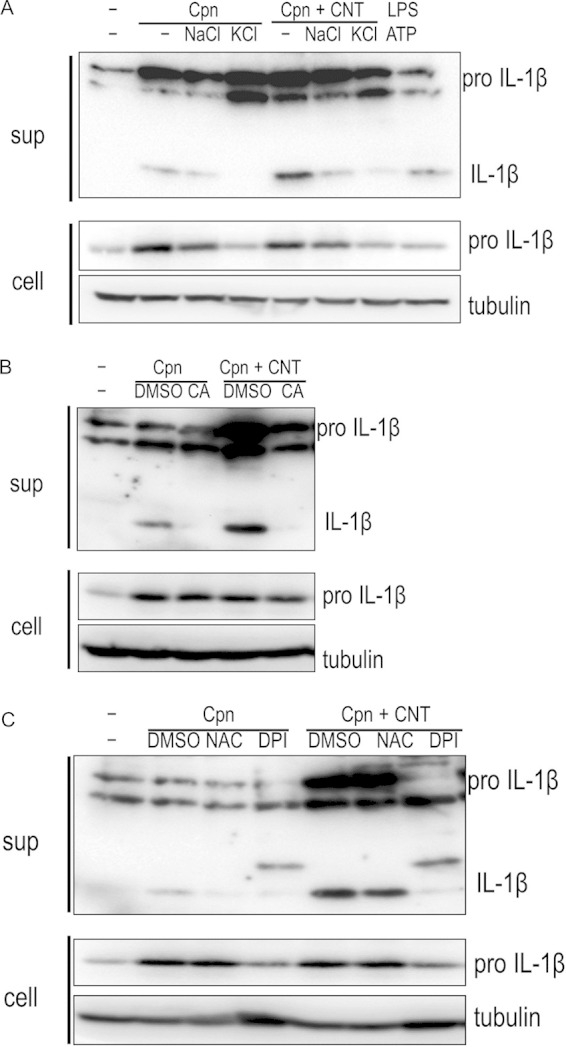
NLRP3 inflammasome activation requires three distinct upstream signals: K+ efflux, lysosomal degradation, and ROS production. Cells stimulated with or without Chlamydia pneumoniae in the presence of absence of CNTs were incubated with either 70 mM KCl, 10 μM CA-074 Me (a cathepsin B inhibitor), or ROS inhibitors (5 mM NAC or 10 μM DPI) for 24 h, and then IL-1β secretion from macrophages was detected. Treatment with KCl (A), CA-074 Me (B), or DPI (C), but not NAC, blocked IL-1β secretion. Results are representative of three independent experiments. Cpn, C. pneumoniae; CNT, carbon nanotube; IL-1β, interleukin-1β; LPS, lipopolysaccharide; DMSO, dimethyl sulfoxide; ROS, reactive oxygen species.
DISCUSSION
Seroepidemiological studies indicate C. pneumoniae infection has a high seroprevalence, indicating its ubiquitous distribution throughout the world (3, 4). While C. pneumoniae is a causative agent of pulmonary infection, symptoms of the infection vary from asymptomatic to serious pneumonia with high mortality (30). Furthermore, accumulated studies reveal an association of C. pneumoniae infection with chronic diseases, such as asthma (3), which is evoked by air pollution containing carbon nanoparticles (9–11). However, the detailed mechanisms by which C. pneumoniae is involved in the development of asthma remain unclear. We therefore studied the synergistic effect of C. pneumoniae with carbon nanoparticle derivatives, CNTs, on IL-1β secretion, to determine whether it exacerbated the mechanisms involved in C. pneumoniae infection. We found that CNTs synergistically enhanced IL-1β secretion from C. pneumoniae-infected macrophages.
In contrast to treatment with a polymer such as dextran that enhances chlamydial growth in vitro (31), treatment with CNTs did not enhance C. pneumoniae growth in macrophages, excluding the possibility that the enhancement of infection by CNT treatment is a critical event in the synergistic effect. In addition, treatment with CNTs alone failed to induce IL-1β secretion (both mature and pro-IL-1β) from or in macrophages, and no increase of inflammatory cytokine mRNA transcription (IL-1β, IL-8, and TNF-α) was observed in cells stimulated with CNTs alone. IL-1β secretion is strictly controlled by IL-1β mRNA expression and IL-1β cleavage in macrophages (15–17); therefore, evidence from previous studies and the present study indicates CNTs alone have no effects on IL-1β transcription via signaling through TLRs (stimulation by signal 2). In contrast to CNTs, treatment with the bacterium alone was sufficient to stimulate IL-1β transcription, and caspase inhibitors clearly blocked IL-1β maturation. Furthermore, previous studies also indicated that IL-1β transcription occurred in a TLR2/4-dependent manner in macrophages stimulated with C. pneumoniae alone (5–8, 19, 20, 32, 33). Thus, taken together, while CNTs only stimulate signal 2, C. pneumoniae stimulation can induce both signals 1 and 2, and stimulation from CNTs and C. pneumoniae independently can activate the NLRP3 inflammasome (signal 2).
Because the NLRP3 inflammasome is activated by distinct upstream signals, including K+ efflux (26), lysosomal degradation (28), and ROS production (29), we attempted to determine the upstream signal pathway involved. We found that NLRP3 inflammasome activation required all three distinct upstream signals: K+ efflux, lysosomal degradation, and ROS production. Although antioxidant NAC failed to block the synergistic effect, ROS production may be partially involved in the effect. Further upstream signals, including PAMPs derived from C. pneumoniae or CNTs, require further study to determine their involvement. However, several studies have reported that type III secretion systems or their effectors are involved in NLRP3 inflammasome activation (34, 35); thus, these molecules might be attractive candidates for inflammasome activation. Moreover, recent studies indicated that Chlamydia induces cell death at late-stage infection with features of both apoptosis and necrosis (for a review, see 36). Oxidized mitochondrial DNA is also likely to be responsible for NLRP3 inflammasome activation and IL-1β secretion from C. pneumoniae-infected cells (37).
The present study indicates that full caspase-1 activation is important for C. pneumoniae-infected cell stimulation with CNTs. Although there was no direct evidence showing actual caspase-1 activation, our experiments using caspase inhibitors and significant increase of IL-18, which is another substrate of caspase-1 (15–17), support caspase-1 activation. Caspase-1 activation causes pyroptotic cell death by pore formation in plasma membranes (15, 38). We therefore confirmed whether cell death was induced by the stimulation of C. pneumoniae with CNTs using a pore-forming assay, as previously described (39, 40). However, no obvious increase in pyroptotic cell death was observed (data not shown). A recent report has demonstrated two distinct pathways for inflammasome activation either with or without ASC activation (41). Therefore, IL-1β secretion from the C. pneumoniae-infected cells with CNT treatment might occur in an ASC-dependent manner, although further study is required to clarify this.
In conclusion, this study showed that CNTs synergistically activate the NLRP3 inflammasome in C. pneumoniae-infected macrophages inducing caspase-1 activation that enhances IL-1β secretion, providing a novel insight into the mechanism of how CNTs exacerbate C. pneumoniae infection. Regarding this, we propose a possible mechanism for IL-1β secretion from C. pneumoniae-infected cells with CNT treatment (Fig. 9). While C. pneumoniae activates both signals 1 and 2, CNTs stimulate signal 2 alone, together enhancing the cleavage of pro-IL-1β cleavage to the mature form of IL-1β. To the best of our knowledge, this is the first study to show a synergistic effect of C. pneumoniae with carbon nanoparticles on IL-1β secretion from immortal human macrophages.
FIG 9.

Hypothetical model of synergistic effect of CNTs on IL-1β secretion from C. pneumoniae-infected macrophages. Cpn, C. pneumoniae; CNT, carbon nanotube; IL-1β, interleukin-1β.
ACKNOWLEDGMENTS
This study was supported by grants-in-aid for the Japan Society for the Promotion of Science (JSPS) KAKENHI, grant no. 25860824, 21590474, 24659194, and 24117501 “Innovation Areas (Matryoshka-Type Evolution).” K. Ishida is a Research Fellow of the Japan Society for the Promotion of Science.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Grayston JT. 1992. Chlamydia pneumoniae, strain TWAR pneumonia. Annu Rev Med 43:317–323. doi: 10.1146/annurev.me.43.020192.001533. [DOI] [PubMed] [Google Scholar]
- 2.Kuo CC, Jackson LA, Campbell LA, Grayston JT. 1995. Chlamydia pneumoniae (TWAR). Clin Microbiol Rev 8:451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blasi F, Tarsia P, Aliberti S. 2009. Chlamydophila pneumoniae. Clin Microbiol Infect 15:29–35. doi: 10.1111/j.1469-0691.2008.02130.x. [DOI] [PubMed] [Google Scholar]
- 4.Roulis E, Polkinghorne A, Timms P. 2013. Chlamydia pneumoniae: modern insights into an ancient pathogen. Trends Microbiol 21:120–128. doi: 10.1016/j.tim.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 5.He X, Mekasha S, Mavrogiorgos N, Fitzgerald KA, Lien E, Ingalls RR. 2010. Inflammation and fibrosis during Chlamydia pneumoniae infection is regulated by IL-1 and the NLRP3/ASC inflammasome. J Immunol 184:5743–5754. doi: 10.4049/jimmunol.0903937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Netea MG, Selzman CH, Kullberg BJ, Galama JM, Weinberg A, Stalenhoef AF, Van der Meer JW, Dinarello CA. 2000. Acellular components of Chlamydia pneumoniae stimulate cytokine production in human blood mononuclear cells. Eur J Immunol 30:541–549. doi:. [DOI] [PubMed] [Google Scholar]
- 7.Heinemann M, Susa M, Simnacher U, Marre R, Essig A. 1996. Growth of Chlamydia pneumoniae induces cytokine production and expression of CD14 in a human monocytic cell line. Infect Immun 64:4872–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaukoranta-Tolvanen SS, Teppo AM, Laitinen K, Saikku P, Linnavuori K, Leinonen M. 1996. Growth of Chlamydia pneumoniae in cultured human peripheral blood mononuclear cells and induction of a cytokine response. Microb Pathog 21:215–221. doi: 10.1006/mpat.1996.0056. [DOI] [PubMed] [Google Scholar]
- 9.Delzell JE., Jr 2013. Common lung conditions: environmental pollutants and lung disease. FP Essent 409:32–42. [PubMed] [Google Scholar]
- 10.Ganguly K, Upadhyay S, Irmler M, Takenaka S, Pukelsheim K, Beckers J, De Angelis MH, Hamelmann E, Stoeger T, Schulz H. 2011. Impaired resolution of inflammatory response in the lungs of JF1/Msf mice following carbon nanoparticle instillation. Respir Res 12:94. doi: 10.1186/1465-9921-12-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murr LE, Garza KM, Soto KF, Carrasco A, Powell TG, Ramirez DA, Guerrero PA, Lopez DA, Venzor J III. 2005. Cytotoxicity assessment of some carbon nanotubes and related carbon nanoparticle aggregates and the implications for anthropogenic carbon nanotube aggregates in the environment. Int J Environ Res Public Health 2:31–42. doi: 10.3390/ijerph2005010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palomäki J, Välimäki E, Sund J, Vippola M, Clausen PA, Jensen KA, Savolainen K, Matikainen S, Alenius H. 2011. Long, needle-like carbon nanotubes and asbestos activate the NLRP3 inflammasome through a similar mechanism. ACS Nano 5:6861–6870. doi: 10.1021/nn200595c. [DOI] [PubMed] [Google Scholar]
- 13.Reisetter AC, Stebounova LV, Baltrusaitis J, Powers L, Gupta A, Grassian VH, Monick MM. 2011. Induction of inflammasome-dependent pyroptosis by carbon black nanoparticles. J Biol Chem 286:21844–21852. doi: 10.1074/jbc.M111.238519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meunier E, Coste A, Olagnier D, Authier H, Lefèvre L, Dardenne C, Bernad J, Béraud M, Flahaut E, Pipy B. 2012. Double-walled carbon nanotubes trigger IL-1beta release in human monocytes through Nlrp3 inflammasome activation. Nanomedicine 8:987–995. doi: 10.1016/j.nano.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 15.Horvath GL, Schrum JE, De Nardo CM, Latz E. 2011. Intracellular sensing of microbes and danger signals by the inflammasomes. Immunol Rev 243:119–135. doi: 10.1111/j.1600-065X.2011.01050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gross O, Thomas CJ, Guarda G, Tschopp J. 2011. The inflammasome: an integrated view. Immunol Rev 243:136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- 17.Martinon F, Tschopp J. 2004. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell 117:561–574. doi: 10.1016/j.cell.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 18.Saïd-Sadier N, Ojcius DM. 2012. Alarmins, inflammasomes and immunity. Biomed J 35:437–439. doi: 10.4103/2319-4170.104408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eitel J, Meixenberger K, van Laak C, Orlovski C, Hocke A, Schmeck B, Hippenstiel S, N′Guessan PD, Suttorp N, Opitz B. 2012. Rac1 regulates the NLRP3 inflammasome which mediates IL-1beta production in Chlamydophila pneumoniae infected human mononuclear cells. PLoS One 7:e30379. doi: 10.1371/journal.pone.0030379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimada K, Crother TR, Arditi M. 2012. Innate immune responses to Chlamydia pneumoniae infection: role of TLRs, NLRs, and the inflammasome. Microbes Infect 14:1301–1307. doi: 10.1016/j.micinf.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishida K, Kubo T, Saeki A, Yamane C, Matsuo J, Yimin Nakamura S, Hayashi Y, Kunichika M, Yoshida M, Takahashi K, Hirai I, Yamamoto Y, Shibata K, Yamaguchi H. 2013. Chlamydophila pneumoniae in human immortal Jurkat cells and primary lymphocytes uncontrolled by interferon-gamma. Microbes Infect 15:192–200. doi: 10.1016/j.micinf.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 22.Takeda S, Hui SP, Fuda H, Jin S, Sakurai T, Ishii A, Mukasa K, Sueoka K, Chiba H. 2013. Evaluation of various electrode materials for detection of oxidized low-density lipoproteins. J Biomed Nanotechnol 9:303–306. doi: 10.1166/jbn.2013.1518. [DOI] [PubMed] [Google Scholar]
- 23.Miura K, Matsuo J, Rahman MA, Kumagai Y, Li X, Rikihisa Y. 2011. Ehrlichia chaffeensis induces monocyte inflammatory responses through MyD88, ERK, and NF-kappaB but not through TRIF, interleukin-1 receptor 1 (IL-1R1)/IL-18R1, or Toll-like receptors. Infect Immun 79:4947–4956. doi: 10.1128/IAI.05640-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gnoatto N, Lotufo RF, Matsuda M, Penna V, Marquezini MV. 2007. Expression of cell-surface heparan sulfate proteoglycans in human cyclosporin-induced gingival overgrowth. J Periodont Res 42:553–558. doi: 10.1111/j.1600-0765.2007.00981.x. [DOI] [PubMed] [Google Scholar]
- 25.Madico G, Quinn TC, Boman J, Gaydos CA. 2000. Touchdown enzyme time release-PCR for detection and identification of Chlamydia trachomatis, C. pneumoniae, and C. psittaci using the 16S and 16S-23S spacer rRNA genes. J Clin Microbiol 38:1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. 2007. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 14:1583–1589. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 27.Meixenberger K, Pache F, Eitel J, Schmeck B, Hippenstiel S, Slevogt H, N′Guessan P, Witzenrath M, Netea MG, Chakraborty T, Suttorp N, Opitz B. 2010. Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1beta, depending on listeriolysin O and NLRP3. J Immunol 184:922–930. doi: 10.4049/jimmunol.0901346. [DOI] [PubMed] [Google Scholar]
- 28.Hornung V, Latz E. 2010. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur J Immunol 40:620–623. doi: 10.1002/eji.200940185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tschopp J, Schroder K. 2010. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 30.Brown JS. 2012. Community-acquired pneumonia. Clin Med 12:538–543. doi: 10.7861/clinmedicine.12-6-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sabet SF, Simmons J, Caldwell HD. 1984. Enhancement of Chlamydia trachomatis infectious progeny by cultivation of HeLa 229 cells treated with DEAE-dextran and cycloheximide. J Clin Microbiol 20:217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prebeck S, Kirschning C, Dürr S, da Costa C, Donath B, Brand K, Redecke V, Wagner H, Miethke T. 2001. Predominant role of Toll-like receptor 2 versus 4 in Chlamydia pneumoniae-induced activation of dendritic cells. J Immunol 167:3316–3323. doi: 10.4049/jimmunol.167.6.3316. [DOI] [PubMed] [Google Scholar]
- 33.Jiang SJ, Kuo CC, Berry MW, Lee AW, Campbell LA. 2008. Identification and characterization of Chlamydia pneumoniae-specific proteins that activate tumor necrosis factor alpha production in RAW 264.7 murine macrophages. Infect Immun 76:1558–1564. doi: 10.1128/IAI.01331-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abdul-Sater AA, Said-Sadier N, Padilla EV, Ojcius DM. 2010. Chlamydial infection of monocytes stimulates IL-1beta secretion through activation of the NLRP3 inflammasome. Microbes Infect 12:652–661. doi: 10.1016/j.micinf.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abdul-Sater AA, Koo E, Hacker G, Ojcius DM. 2009. Inflammasome-dependent caspase-1 activation in cervical epithelial cells stimulates growth of the intracellular pathogen Chlamydia trachomatis. J Biol Chem 284:26789–26796. doi: 10.1074/jbc.M109.026823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ying S, Pettengill M, Ojcius DM, Häcker G. 2007. Host-cell survival and death during Chlamydia infection. Curr Immunol Rev 3:31–40. doi: 10.2174/157339507779802179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M. 2012. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duprez L, Wirawan E, Vanden Berghe T, Vandenabeele P. 2009. Major cell death pathways at a glance. Microbes Infect 11:1050–1062. doi: 10.1016/j.micinf.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 39.Silveira TN, Zamboni DS. 2010. Pore formation triggered by Legionella spp. is an Nlrc4 inflammasome-dependent host cell response that precedes pyroptosis. Infect Immun 78:1403–1413. doi: 10.1128/IAI.00905-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lage SL, Amarante-Mendes GP, Bortoluci KR. 2013. Evaluation of pyroptosis in macrophages using cytosolic delivery of purified flagellin. Methods 61:110–116. doi: 10.1016/j.ymeth.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 41.Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. 2010. Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8:471–483. doi: 10.1016/j.chom.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]