

Abstract
Vibrio cholerae is a genetically diverse species, and pathogenic strains can encode different virulence factors that mediate colonization and secretory diarrhea. Although the toxin-coregulated pilus (TCP) is the primary colonization factor in epidemic-causing V. cholerae strains, other strains do not encode the TCP and instead promote colonization via the activity of a type 3 secretion system (T3SS). Using the infant mouse model and T3SS-positive O39 serogroup strain AM-19226, we sought to determine which of 12 previously identified, T3SS-translocated proteins (Vops) are important for host colonization. We constructed in-frame deletions in each of the 12 loci in strain AM-19226 and identified five Vop deletion strains, including ΔVopM, which were severely attenuated for colonization. Interestingly, a subset of deletion strains was also incompetent for effector protein transport. Our collective data therefore suggest that several translocated proteins may also function as components of the structural apparatus or translocation machinery and indicate that while VopM is critical for establishing an infection, the combined activities of other effectors may also contribute to the ability of T3SS-positive strains to colonize host epithelial cell surfaces.
INTRODUCTION
The toxin-coregulated pilus (TCP) is the major colonization factor encoded by all pathogenic O1 and O139 serogroup Vibrio cholerae strains, which cause epidemic cholera. In contrast, most clinically isolated non-O1/non-O139 serogroup strains do not encode TCP and thus must employ other mechanisms to effectively colonize the human intestinal epithelium and cause sporadic, cholera-like disease (1–3). Genome sequence analysis of a clinically isolated O39 serogroup strain, named AM-19226, identified a pathogenicity island on the large chromosome that encodes the structural proteins for a type 3 secretion system (T3SS) (4). T3SSs function as principal virulence mechanisms in many Gram-negative bacterial pathogens (e.g., Escherichia, Salmonella, Pseudomonas, Shigella, and Yersinia spp.), and in vivo studies using different animal models confirmed that the V. cholerae T3SS is essential for causing disease (5–7). In addition, numerous groups have identified T3SS-positive V. cholerae strains in laboratory collections, from patients, and from endemic environments, suggesting that a subset of non-O1/non-O139 serogroup strains depends on T3SS activity for virulence (1, 3, 4, 8–10).
The V. cholerae T3SS is most closely related to the Vibrio parahaemolyticus T3SS2. T3SS2 is associated with pandemic V. parahaemolyticus strains, whereas T3SS1 is present in all strains (11). Comparison of the T3SS genomic islands in V. cholerae strain AM-19226 and V. parahaemolyticus strain RIMD2210633 reveals synteny within a conserved, central “core” region, flanked by 5′ and 3′ regions of greater coding diversity between clades and species (4, 10, 12–14). The core region encodes proteins that form the T3SS structural apparatus and is transcriptionally organized into four main operons in V. cholerae (Fig. 1) (15, 16). In vitro, operon expression can be activated in a bile-dependent manner by VttRA and VttRB, two ToxR-like transmembrane transcriptional regulatory proteins that are also encoded by the T3SS island (15, 16). Embedded among the structural genes are 5 of 12 loci whose gene products are translocated from strain AM-19226 into mammalian cells during in vitro coculture: VopH, -A, -M, -I, and -W (12). The functions of VopH, -A, and -I have not previously been defined. The V. parahaemolyticus VopW protein was also detected in mammalian cells by using translocation assays but was additionally shown to function as the hydrophilic translocator protein for T3SS2 (17). By analogy, VopW likely has a similar function in V. cholerae. The V. parahaemolyticus vopV and V. cholerae vopM loci lie within conserved regions of synteny in the T3SS island of each species and encode homologous proteins. VopV and VopM mediate actin bundling and the formation of stress fibers in cultured mammalian cells and contribute to enterotoxigenicity and effacement of the intestinal brush border in a rabbit model of infection (18, 19).
FIG 1.

Schematic of the AM-19226 T3SS genomic island. The AM-19226 T3SS genomic island is composed of a conserved “core” region that is similar among T3SS-positive Vibrio strains and mosaic 5′- and 3′-flanking regions that differ among clades and species. Genes encoding the T3SS structural proteins (VcsRTCNS2, VcsVUQ2, VcsJ2, and VspD) are shown as dark gray arrows, genes encoding the translocated proteins (Vops) are shown as checkered arrows, genes encoding the T3SS transcriptional regulators (VttRA and VttRB) are shown as black arrows, and genes encoding predicted virulence-related proteins (Acfs and TDH-related hemolysin [TRH]) are shown as light gray arrows. Note that genes for 5 of the 12 translocated proteins are interspersed within the four structural gene operons in the conserved core region, which also includes VopF. Arrows indicate the major transcriptional initiation sites based on transcriptome sequencing data (16).
Remodeling of the host actin cytoskeleton to promote bacterial attachment or invasion is a common theme among T3SS effectors (20), and in addition to VopM, VopF has been assigned a role in cytoskeletal rearrangement. Tam et al. reported that VopF binds and nucleates actin in vitro, influences the integrity of tight junctions in cultured mammalian monolayers, and contributes to colonization of the infant mouse intestine (5, 7). Similarly, the V. parahaemolyticus VopF paralog, VopL, has been shown to induce the formation of actin stress fibers in HeLa cells (7). VopF is conserved in all T3SS-positive Vibrio strains belonging to the alpha-clade, although it is not encoded in the structural gene operons but rather at the 3′ end of the conserved, core region (4, 7, 10).
As mentioned above, six additional genes that lie outside the core region appear to be species and/or strain specific, based on comparative sequence analyses (10). For example, VopG in V. cholerae strain AM-19226 is encoded in the 3′-flanking region of the T3SS genomic island, but a similar protein is encoded in the 5′-flanking region of alpha-clade strains of V. parahaemolyticus (4, 9, 14). On the other hand, AM-19226 VopE, -X, -K, -Y, and -Z appear to be restricted to V. cholerae strains. VopE possesses a conserved Rho-GTPase-activating (GAP) motif that is also present in YopE of Yersinia spp. Based on the conserved domain and results suggesting a role in tight junction disruption, it was hypothesized that VopE may also modulate actin dynamics (7, 21). However, recent data indicate that the GAP activity is targeted to the mitochondrial Miro Rho-GTPases (22). The function(s) of VopX, -G, -K, -Y, and -Z is currently unknown.
However, as described previously, T3SS activity is essential for colonization, and an AM-19226 vcsN2 deletion strain (missing the ATPase component of the T3SS structural apparatus) is unable to establish infection in the infant mouse or rabbit intestinal tract model (5, 6). To determine which translocated proteins function during the initial stages of infection to colonize host tissues, we constructed in-frame deletions in each of the 12 genes encoding translocated proteins and determined their competitive index (CI) using the infant mouse model.
MATERIALS AND METHODS
Strains and culture conditions.
Bacterial strains used in this study are listed in Table 1. All strains were maintained at −80°C in Luria-Bertani (LB) broth containing 20% glycerol. For infant mouse competition and growth curve assays, parental and deletion strains were grown overnight in LB broth supplemented with streptomycin (100 μg/ml) on a roller drum at 37°C. For Escherichia coli and V. cholerae strains, ampicillin was used at 100 μg/ml.
TABLE 1.
Strains and plasmids
| Strain or plasmid | Genotype and/or descriptiona | Source or reference |
|---|---|---|
| Strains | ||
| V. cholerae | ||
| MD992 | AM-19226 R− M+ Strr | Laboratory stock |
| MD996 | AM-19226 R− M+ ΔlacZ Strr | Laboratory stock |
| AAC155 | MD992 Δhap ΔhlyA ΔrtxA | 12 |
| AAC330 | MD992 Δhap ΔhlyA ΔrtxA ΔvcsN2 | 12 |
| MC50 | MD992 ΔvopG | This study |
| MC96 | MD992 ΔvopZ | This study |
| MC150 | MD992 ΔvopF | This study |
| MC180 | MD992 ΔvopH | This study |
| MC188 | MD992 ΔvopA | This study |
| MC195 | MD992 ΔvopM | This study |
| KM121 | MD992 ΔvopY | This study |
| KM223 | MD992 ΔvopK | This study |
| JW6 | MD992 ΔvopW | This study |
| JW16 | MD992 ΔvopI | This study |
| VS4 | MD992 ΔvopE | This study |
| JC11 | MD992 vopX′ | This study |
| MC264 | MD992 ΔvopA (pBR322-vopA) | This study |
| CS11 | MD992 ΔvopH (pBR322-vopH) | This study |
| CS12 | MD992 ΔvopM (pBR322-vopM) | This study |
| CS13 | MD992 ΔvopW (pBR322-vopW) | This study |
| CS20 | MD992 ΔvopI (pBR322-vopI) | This study |
| CS27 | MD992 ΔvopG (pBR322-vopG) | This study |
| MC207 | AAC155 ΔvopH (pVTM30) | This study |
| MC210 | AAC155 ΔvopA (pVTM30) | This study |
| MC213 | AAC155 ΔvopH (pVTM30-VopM-Bla) | This study |
| MC214 | AAC155 ΔvopA (pVTM30-VopM-Bla) | This study |
| MC215 | AAC155 ΔvopM (pVTM30-VopG-Bla) | This study |
| MC217 | AAC155 ΔvopM (pVTM30) | This study |
| MC266 | AAC155 ΔvopF (pVTM30) | This study |
| MC268 | AAC155 ΔvopF (pVTM30-VopM-Bla) | This study |
| MS9 | AAC155 ΔvopI (pVTM30) | This study |
| MS10 | AAC155 ΔvopI (pVTM30-VopX-Bla) | This study |
| MS13 | AAC155 ΔvopW (pVTM30) | This study |
| MS15 | AAC155 ΔvopW (pVTM30-VopM-Bla) | This study |
| E. coli | ||
| DH5αλpir | F− (ϕ80 dlacΔM15) (lacZYA argF ΔU169)endA1 recA1 hsdR17 deoR thi-1 supE44 gyrA96 (Nalr) relA1 λpir | Laboratory stock |
| SM10λpir | thi thr leu tonA lacY supE recA RP4-2-Tc::M(λpir) Kanr | Laboratory stock |
| Plasmids | ||
| pCVD442 | Suicide vector; Ampr | Laboratory stock |
| pVTM30 | pDSW204-Bla; to construct Bla fusions; IPTG-inducible Ptrc; Ampr | 5 |
| pBR322 | Cloning vector; Ampr Tetr | 40 |
| pBR322-GW | pBR322 with Gateway cassette RfC.1; destination vector; Ampr Cmr | This study |
IPTG, isopropyl-β-d-thiogalactopyranoside.
Strain construction.
V. cholerae R− M+ lacZ+ strain AM-19226 (MD992) was used as the parental strain to construct nonpolar, in-frame deletions in genes encoding translocated proteins, using overlapping PCR and standard allelic exchange methods as previously described (15, 23). Deletions were confirmed by colony PCR and Southern blot analysis. The VopX truncation mutant (VopX′) was constructed by introducing a point mutation that resulted in a stop codon at residue 54 (K. A. Miller and M. Dziejman, unpublished data). For complementation studies, the vopA coding sequence was cloned into pBR322 by using standard restriction enzyme methods. For complementation of other deletions, the protein open reading frame was amplified by PCR, using primers that included att sites to facilitate recombination-mediated cloning into a pBR322 vector that was modified to serve as a Gateway destination vector (24, 25). Primer sequences are available upon request.
Infant mouse competition assays.
Competition assays were performed as previously described and in accordance with the University of Rochester Committee on Animal Resources-approved protocols (15, 26). Briefly, 3- to 5-day-old CD1 mice (n = 7 to 10) were intragastrically inoculated with a total of ∼105 bacteria at a 1:1 ratio of the deletion strain (lacZ+) and an isogenic parent strain (lacZ negative). After 16 to 18 h at 30°C, mice were euthanized, and the small intestines were surgically removed, homogenized, and plated onto LB agar plates containing 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal). The competitive index (CI) was calculated based on output and input ratios of the parental and the deletion strains, where CI = (mutant output/parental output)/(mutant input/parental input). The limit of detection for our protocol is 100 CFU/intestine.
Translocation assay.
Fluorescence resonance energy transfer (FRET)-based translocation assays were conducted as previously described (12). Briefly, V. cholerae strains containing pVTM30-based transmission electron microscopy (TEM) fusions to VopM and VopX (see Fig. 3) or VopG (data not shown) were used to infect HeLa cells at a multiplicity of infection (MOI) of ∼10 for 3 h and then washed three times with Hanks' balanced salt solution (HBSS) and incubated with coumarin-cephalosporin-fluorescein (2)-acetoxymethyl (CCF2-AM) (GeneBLAzer detection kits; Invitrogen) for 1 h. Fluorescence was observed following excitation at 409 nm by using an Olympus FV1000 confocal microscope at a ×20 magnification. Emission at 520 nm (green fluorescence) was scored as no translocation, whereas emission at 447 nm (blue fluorescence) was scored as positive for translocation.
FIG 3.
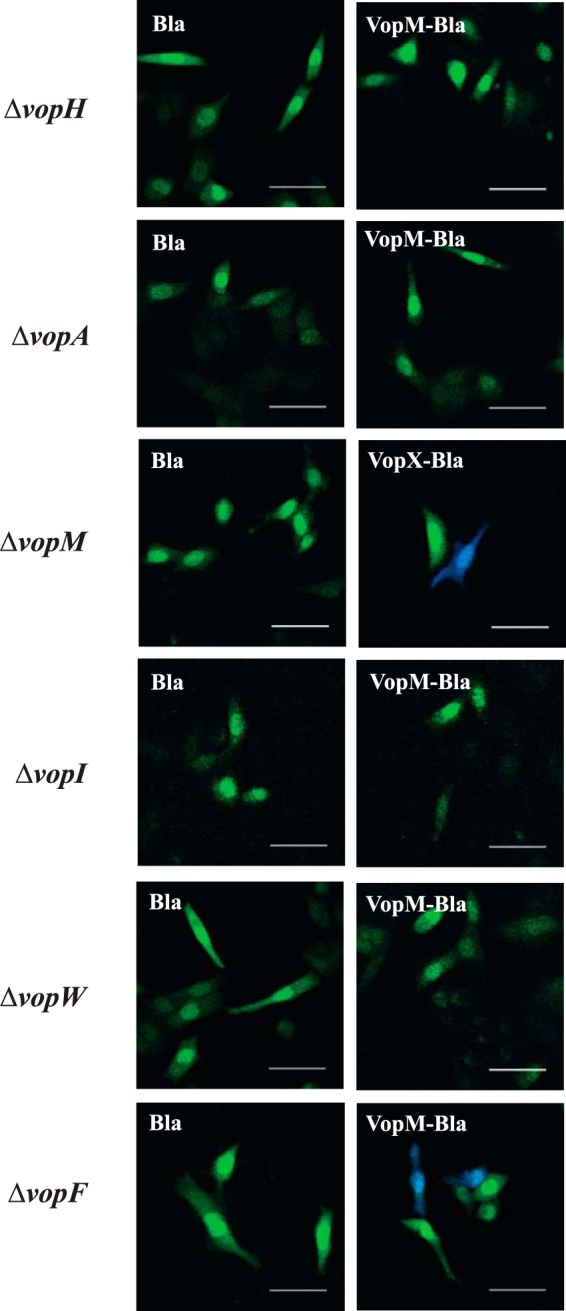
VopH, -A, -I, and -W are required for protein translocation. Shown are confocal microscopy images of FRET-based translocation assay results using Vop deletion strains carrying VopM-Bla or VopX-Bla constructs cocultured with HeLa cells. Green fluorescence indicates no translocation of effector-Bla fusion proteins, whereas blue fluorescence indicates translocation. Bars, 50 μm. Representative images from two or more experiments are shown.
Coverslip adherence assay.
Caco2-BBE cells were seeded onto collagen-coated coverslips (Neuvitro) in a 24-well plate, using 4 × 105 cells/well and Dulbecco's modified Eagle medium (DMEM) supplemented with 5% fetal bovine serum (FBS). After 48 h, confluent monolayers were washed once with phosphate-buffered saline (PBS) and provided fresh DMEM with 5% FBS and 0.2% bile (Sigma). The Caco2-BBE cells were then cocultured with bacterial strains at an MOI of ∼10 for 2 h. Bacterial cells were also added to wells containing the medium alone to calculate bacterial growth (output) under the same conditions as those used during coculture. After coculture, wells and coverslips were washed twice with PBS, and the coverslips were transferred to fresh wells and washed with PBS again. The Caco2-BBE cells were lysed with 0.1% Triton X-100 by gentle shaking for 1 h. LB broth was added to the lysate, and 10-fold serial dilutions were plated to determine the number of adherent bacteria. The ratio of output bacteria to adherent bacteria was used to calculate percent adherence to mammalian cells, where percent adherent bacteria = (coverslip-associated bacteria at 2 h/output at 2 h) × 100. Adherence assays were repeated at least twice with 3 to 5 colonies per strain.
Statistical analyses.
One-way analysis of variance (ANOVA) with a Dunnett post hoc test was performed to determine statistical significance, and P values of <0.05 were considered statistically significant.
RESULTS
Core-encoded, translocated proteins conserved between Vibrio species are required for in vivo colonization.
We previously identified 12 loci encoding gene products that were translocated into eukaryotic cells during coculture and named them Vops (12). As shown in Fig. 1, all 12 vop genes lie within the T3SS genomic island, with the coding sequences for VopH, -A, -M, -I, and -W being interspersed among the genes encoding T3SS structural proteins. We investigated the role of each Vop in colonization by constructing individual unmarked, in-frame deletions and evaluated the ability of each resulting strain to colonize infant mouse intestines. Under standard in vitro growth conditions in LB broth, all deletion strains grew as well as the isogenic, parental strain (data not shown). The results from a representative in vivo experiment for each deletion strain are shown in Fig. 2. Table 2 lists the average competitive indices for two or more independent experiments.
FIG 2.

Colonization requires multiple T3SS island-encoded proteins. Competitive indices for AM-19226 strains deleted for 11 effector protein genes and a vopX point mutant are shown, and their position within the T3SS genomic island is indicated at the bottom. Each triangle represents the CI for a single mouse. The black bars denote the mean CI. Asterisks indicate that ΔvopA, ΔvopI, and ΔvopW strains were not recoverable from the infant mouse small intestine. Results from one of two or three experiments performed for each deletion strain are shown, with duplicate experiments producing similar results.
TABLE 2.
Summary of data from infant mouse competition assaysa
| Defect and strain | Mean CI |
|
|---|---|---|
| Deletion strain/WT | Complemented strain/WT | |
| 100–1,000-fold defect | ||
| ΔvopH | 0.0088 | 4.3700 |
| ΔvopA | 0.0029 | 0.5252 |
| ΔvopM | 0.0073 | 0.2612 |
| ΔvopI | 0.0090 | 0.0026 |
| ΔvopW | 0.0144 | 0.6715 |
| <20-fold defect | ||
| ΔvopF | 0.2776 | Previously reported (7) |
| ΔvopG | 0.0530 | 0.0308 |
| No defect | ||
| ΔvopE | 0.7007 | |
| vopX′ | 1.1009 | |
| ΔvopK | 1.1533 | |
| ΔvopY | 1.2108 | |
| ΔvopZ | 0.7902 | |
All strains were competed against an isogenic parental strain that was of the wild type (WT) for all T3SS loci (vop genes). A CI of ∼1 therefore represents a strain that colonized as well as the wild type, whereas a CI of <1 indicates a colonization defect.
Strains deleted for each of the five following individual vop genes were severely attenuated (>100- to 1,000-fold) for colonization compared to the wild-type strain: vopH, vopA, vopM, vopI, and vopW (Fig. 2 and Table 2). Strains deleted for genes encoding VopA, -I, and -W were not recoverable (≥1,000-fold defect) from infant mouse intestines, and in at least one experiment, very few colonies were recovered for a ΔvopH or ΔvopM strain. VopF and -G deletion strains had more moderate defects of <20-fold.
For each deletion strain having a defect, we constructed a pBR-based complementing plasmid that constitutively expressed the gene of interest from the tet promoter. The deletion strain carrying the complementing plasmid was competed against the wild-type strain by using the same in vivo assay. (Successful complementation should result in a competitive index of 1.) Using this method, we were able to complement the colonization defect of the ΔvopA, ΔvopH, and ΔvopW strains (Table 2). The ΔvopA, ΔvopH, and ΔvopW strains were also successfully complemented in vitro in a mammalian coculture assay, supporting the conclusion that the phenotype in vivo is due specifically to each gene deletion (Miller and Dziejman, unpublished). We observed moderate complementation with the construct expressing vopM, which likely reflects loss of the plasmid during infection. However, expressing vopI or vopG in the respective deletion strains did not restore the ability to colonize, suggesting that even though we had used an “in-frame” construction strategy, adjacent or downstream gene expression might be adversely affected (Table 2). A vopF deletion was previously complemented (7). Given the collective data at this time, we conclude that there is an absolute requirement for VopH, -A, and -W for colonization of the infant mouse small intestine.
The individual absence of other V. cholerae-specific effectors does not affect colonization.
Figure 2 shows that the ΔvopE, ΔvopK, ΔvopY, and ΔvopZ strains are each able to colonize as well as the wild-type strain. We previously reported that an in-frame deletion of vopX, where sequences coding for 3 N-terminal amino acids and 8 C-terminal amino acids remained after the deletion, resulted in a strain having a 5-fold colonization defect (12). However, preliminary data suggested that the deletion could not be complemented in trans with a wild-type copy of vopX in vitro (Miller and Dziejman, unpublished). Whole-genome transcript analyses performed by our laboratory revealed that the vttRA gene, which is adjacent and divergently transcribed, likely shares overlapping regulatory regions with vopX that also extend into the vopX coding sequence (data not shown). In fact, the vopX deletion could be complemented in other assays by providing VttRA (Miller and Dziejman, unpublished). Therefore, we tested the colonization properties of a strain that encodes a truncated protein, VopX′, resulting from an engineered point mutation that introduced a premature stop codon at amino acid 54 (Miller and Dziejman, unpublished). The VopX′ strain was able to colonize as well as the parental strain, and we therefore conclude that VopX does not contribute to colonization of the infant mouse intestine (Fig. 2 and Table 2).
VopH, -A, and -W are required for translocation of other T3SS effectors.
As described above, the genes encoding T3SS structural proteins are organized in the central, core region of the T3SS genomic island. However, genes that encode proteins for the needle filament, the needle length control protein, a component of the translocon, and the inner rod were not initially identified (4). VopW was subsequently described as the hydrophilic translocator protein in V. parahaemolyticus, raising the possibility that proteins previously identified as being translocated into host cells might also serve structural functions (17).
VopM was translocated to ≈31% of the HeLa cells in our FRET-based translocation assay (12). Although other proteins encoded in the core region of the T3SS genomic island (Fig. 1) were detected in a smaller percentage of mammalian cells, their localization was T3SS dependent, and they were classified as translocated proteins and given Vop designations (12). Because VopW was subsequently identified as part of the translocon, we were interested in determining whether other proteins with positive results in the translocation assay might serve structural functions or serve dual roles, similar to those proposed for SipC of Salmonella and IpaC of Shigella (27–29). Taken together with the in vivo data implicating VopH, -A, and -M as essential for colonization, we investigated whether VopH, -A, and -M influenced protein translocation by testing whether a VopX-TEM1 or VopM-TEM1 fusion protein could be detected in mammalian host cells. Although we could not complement the severe colonization defect of the ΔvopI strain by expressing VopI in trans, we predicted that the deletion strain would also be defective for protein translocation and included it in the assay.
The TEM1 fusion partner alone was not detected in mammalian cells. Western blot analyses using a monoclonal antibody against β-lactamase (TEM1) confirmed the expression of fusion products (data not shown). We also tested a ΔvopF strain to determine if VopF, an effector known to modulate the actin cytoskeleton, is required for the translocation of other effectors (i.e., VopM) that have also been reported to interact with actin. We used a ΔVopW strain as a control, since data support a role for VopW as part of the T3SS translocon. The results are shown in Fig. 3. Consistent with the reported translocon function for VopW, we did not detect translocation of VopM (as shown) or VopX (data not shown) in the ΔvopW strain. Interestingly, we did not observe VopM (or VopX [data not shown]) translocation in the ΔvopH, ΔvopA, or ΔvopI strain. However, the ΔvopM and ΔvopF strains were competent for protein translocation, consistent with their described roles in actin reorganization, versus a role in secretion. Interestingly, Phyre2 modeling of VopH predicted a cytoplasmic N-terminal structure similar to that of YscD, with 77.1% confidence (data not shown). YscD is the inner ring protein of the Yersinia T3SS structural apparatus, further supporting the hypothesis that VopH constitutes a component of the T3SS structural apparatus (30).
Since the function of VopA is unknown, we used a coverslip-based adherence assay to test the ability of a ΔvopA strain to attach to a monolayer of Caco2-BBE intestinal epithelial cells. We observed a ∼60% reduction in adherence compared to the adherence of the parental strain, further supporting a role in colonization (Fig. 4). A vcsN2 deletion strain also showed similarly reduced levels of adherence (Fig. 4), and we predict that the ΔVopW strain also attenuates adherence since VopW is required for protein translocation. Similarly, our collective results provide evidence that like VopW, VopH and VopA may also function to promote the translocation of other T3SS-encoded proteins yet can also be detected in eukaryotic cells.
FIG 4.
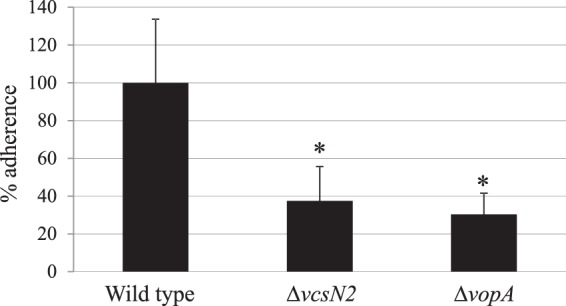
ΔvcsN2 and ΔvopA strains are deficient in adherence to Caco2-BBE cells. Caco2-BBE cells were grown to confluence on collagen-coated coverslips and cocultured for 2 h with either the AM-19226 parental strain or strains deleted for vcsN2 or vopA, after which nonadherent bacteria were washed away. After Triton X-100 treatment, the resulting lysates were serially diluted and plated onto selective medium to determine the number of adhered bacteria. *, P < 0.05.
DISCUSSION
Because V. cholerae strain AM-19226 does not express the TCP but does require T3SS function for colonization, we investigated the role of 12 vop genes that encode translocated proteins (12). We conducted our studies using the infant mouse model, which is a reliable method to assess the ability of strains to colonize the intestinal tract (31–33).
Testing of the roles of individual T3SS-translocated proteins in colonization using competition assays and subsequent complementation analyses allowed us to assign proteins to three broad categories: (i) those that do not contribute to colonization as individual proteins (VopE, -K, -X, -Y, and -Z), (ii) those that have a moderate (<20-fold) impact on colonization (VopF), and (iii) those that are absolutely required for colonization, having a 100- to 1,000-fold defect (VopH, -A, -M, and -W). In-frame deletions in individual genes encoding VopH, -A, -M, -I, or -W result in strains having a colonization defect of the same magnitude as that of the T3SS-null strain (ΔvcsN) (5, 6). However, because we could not complement the VopI deletion, we cannot at this time conclude that VopI is essential for colonization. Although VopF and VopE were previously shown to have effects on colonization and disease, the effects were not as dramatic as those of the T3SS-null deletion. Our results suggest that VopE is dispensable for colonization in mice and that VopF has a more moderate effect on the ability to establish an infection. Whereas the VopG deletion strain also is moderately attenuated, we were unable to complement the deletion, and therefore, in addition to VopI, the role of VopG is unclear at this time.
VopV (V. parahaemolyticus homolog of VopM) was shown previously to cause microvillus effacement, which promotes colonization by V. parahaemolyticus in infant rabbits, and our data provide evidence that this protein may have an analogous role in V. cholerae in the infant mouse model (18). Therefore, at this time, we conclude that 5 of 12 AM-19226 translocated proteins are required for colonization, prompting several interesting questions.
The basic paradigm of O1 and O139 serogroup disease assigns specific protein functions for colonization (TCP) and secretory diarrhea (cholera toxin). The simplest hypothetical categorization for Vops that mediate T3SS-associated disease would also separate proteins into two categories. However, it is possible that translocated proteins have overlapping roles, that different effectors are required at different time points during colonization, or that effector interactions with eukaryotic host proteins resulting in colonization inadvertently disrupt homeostasis and cause a diarrheal response. Additional complexity arises if some translocated proteins function to counteract the activity of others, in order to minimize damage to host epithelial cells and promote a productive infection. The latter scenario is likely the case for VopF and VopM (VopV in V. parahaemolyticus), which have been reported to modulate the actin cytoskeleton in opposing manners. While VopF nucleates actin, VopM is speculated to depolymerize actin (5, 18, 19). In addition, redundancy is a common theme for T3SS effector activity in many pathogens and is likely to exist in V. cholerae as well (20).
Our data reveal that the proteins essential for colonization are encoded adjacent to T3SS structural apparatus genes in operons found in the central region of the AM-19226 T3SS genomic island. Previous data suggested that operon gene expression is regulated by VttRA/B (Fig. 1) (15, 16). Importantly, the central region is conserved among different Vibrio T3SS clades and species, whereas the 5′- and 3′-flanking regions carry genes that are less well conserved. We previously reported that five of the six proteins encoded in the central region of the T3SS genomic island (VopH, -A, -I, -W, and -F) are translocated into host cells at low frequencies in vitro (12). Based on our observations and the identification of VopW as an effector and a structural component (17), we explored the possibility that deletions in other Vops could also inhibit protein translocation. To this end, we tested the translocation of other effectors in strains deleted for VopH, -A, -M, -I, or -W to determine whether one or more of the proteins is required for the translocation of other effectors. We also tested translocation in a ΔvopF strain to determine whether the absence of a protein modulating actin dynamics can prevent the translocation of other effectors (VopM/VopV) that are also known to interact with actin. However, we observed translocation of other effectors in only the ΔvopM and ΔvopF strains, suggesting that actin reorganization activities are not required in order for other proteins to be translocated. Consistent with the reported function of VopW, we did not observe translocation of other Vops in the ΔvopW strain. We also did not observe translocation in the ΔvopH, ΔvopA, and ΔvopI strains. Combined with data from the colonization studies, our results further support the conclusion that VopW functions as a hydrophilic translocator component of the T3SS structural apparatus (17) and suggest that VopH and VopA could have structural or translocation functions as well. Because we were unable to complement the vopI and vopG deletions, the role of the encoded proteins and the transcriptional effects of the deletions must be further explored. The genes that encode the AM-19226 T3SS needle filament, the needle length control protein, and the inner ring protein have not been identified based on sequence homology to other T3SS proteins, although modeling data suggest that VopH may constitute the inner ring component. We therefore favor the interpretation that other translocated proteins function as T3SS structural proteins and may have additional, currently unidentified roles in the eukaryotic cell cytosol (17).
Notably, a strain deleted for vopA, within the vcsVUQ2 operon, was unrecoverable from the infant mouse intestine (Fig. 2). The addition of an episomal wild-type copy of vopA during infection restored the ability of the deletion strain to colonize (Table 2), suggesting that the vopA deletion did not have polar effects on downstream genes, which include the gene encoding VopH. Similarly, we were also able to complement deletions in the vopH and vopW genes. Our observations of in vivo complementation are consistent with those seen in vitro in our cell culture model that measures AM-19226-induced cytotoxicity, where we were able to complement VopA, -H, and -W but not VopI, which is encoded by an operon downstream of vspD, along with VopM (Miller and Dziejman, unpublished). Because the regulation of gene expression in T3SS operons is complex, we are currently investigating the transcriptional organization of the T3SS island to better understand the impact of individual gene deletions on the expression of neighboring genes. For example, the vopX coding region, which is present in all T3SS-positive V. cholerae strains but only in beta-clade T3SS-positive V. parahaemolyticus strains, overlaps the regulatory region governing the expression of vttRA (12, 34; Miller and Dziejman, unpublished).
A recently identified effector in V. parahaemolyticus, named VopZ (distinct from the VopZ protein described here), was shown to play a role in colonization (13). We have identified a homologous protein encoded by the AM-19226 T3SS genomic island at the junction of the 5′-flanking sequences and the central region, but its role in strain AM-19226 colonization remains to be tested. In addition, genes encoding accessory colonization factors, such as mshA, gbpA, and acfABCD, have been identified in different V. cholerae strains, and it is possible that similar gene products contribute to AM-19226 colonization by coordinating functions early in infection, providing activities that can sustain robust colonization, or fine-tuning localization in the host niche (35). For example, while V. cholerae mshA expression is repressed during human infection, the V. parahaemolyticus MshA pilus apparently enables T3SS-mediated pathogenicity in vitro (36, 37). In addition, GbpA contributes to the ability of O1 serogroup strains to successfully colonize mammalian host tissues and zooplankton, but substrate binding (chitin versus mucus) is specified by different domains within the modular structure of the protein (38, 39).
Nonetheless, the T3SS is clearly essential for V. cholerae to colonize. As previously demonstrated, a VopF deletion strain has moderate defects. The VopM deletion strain is severely attenuated in the infant mouse, further supporting data from V. parahaemolyticus studies indicating that the VopV counterpart plays an essential role during disease. Perhaps most interestingly, we found that other severely attenuated deletion strains are also incompetent for T3SS protein translocation. Our results therefore identified additional proteins required for T3SS secretion, highlighted the critical role for VopM in colonization, and suggest collaborative roles for other effectors, such as VopF, in mediating events in the host cell that lead to bacterial adherence and the establishment of an infection.
ACKNOWLEDGMENTS
We thank Stacy Gregoire, Natalie Liles, and Jennifer Colquhoun for excellent assistance with mouse experiments and strain and plasmid construction. We also appreciate the efforts of Martin Pavelka and members of the Dziejman laboratory who participated in helpful discussions and critical reading of the manuscript.
This work was supported by NIH/NIAID grant AI073785 (M.D.), NIH/NIAID grant T32 AI007362 (K.A.M.), NIH/NICHD grant T32 HD057821 (V.S.), and NIH/NIGM R25 GM064133 (C.S.).
REFERENCES
- 1.Faruque SM, Chowdhury N, Kamruzzaman M, Dziejman M, Rahman MH, Sack DA, Nair GB, Mekalanos JJ. 2004. Genetic diversity and virulence potential of environmental Vibrio cholerae population in a cholera-endemic area. Proc Natl Acad Sci U S A 101:2123–2128. doi: 10.1073/pnas.0308485100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sack DA, Sack RB, Nair GB, Siddique AK. 2004. Cholera. Lancet 363:223–233. doi: 10.1016/S0140-6736(03)15328-7. [DOI] [PubMed] [Google Scholar]
- 3.Rahman MH, Biswas K, Hossain MA, Sack RB, Mekalanos JJ, Faruque SM. 2008. Distribution of genes for virulence and ecological fitness among diverse Vibrio cholerae population in a cholera endemic area: tracking the evolution of pathogenic strains. DNA Cell Biol 27:347–355. doi: 10.1089/dna.2008.0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dziejman M, Serruto D, Tam VC, Sturtevant D, Diraphat P, Faruque SM, Rahman MH, Heidelberg JF, Decker J, Li L, Montgomery KT, Grills G, Kucherlapati R, Mekalanos JJ. 2005. Genomic characterization of non-O1, non-O139 Vibrio cholerae reveals genes for a type III secretion system. Proc Natl Acad Sci U S A 102:3465–3470. doi: 10.1073/pnas.0409918102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tam VC, Serruto D, Dziejman M, Brieher W, Mekalanos JJ. 2007. A type III secretion system in Vibrio cholerae translocates a formin/spire hybrid-like actin nucleator to promote intestinal colonization. Cell Host Microbe 1:95–107. doi: 10.1016/j.chom.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 6.Shin OS, Tam VC, Suzuki M, Ritchie JM, Bronson RT, Waldor MK, Mekalanos JJ. 2011. Type III secretion is essential for the rapidly fatal diarrheal disease caused by non-O1, non-O139 Vibrio cholerae. mBio 2(3):e00106-11. doi: 10.1128/mBio.00106-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tam VC, Suzuki M, Coughlin M, Saslowsky D, Biswas K, Lencer WI, Faruque SM, Mekalanos JJ. 2010. Functional analysis of VopF activity required for colonization in Vibrio cholerae. mBio 1(5):e00289-10. doi: 10.1128/mBio.00289-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chatterjee S, Ghosh K, Raychoudhuri A, Chowdhury G, Bhattacharya MK, Mukhopadhyay AK, Ramamurthy T, Bhattacharya SK, Klose KE, Nandy RK. 2009. Incidence, virulence factors, and clonality among clinical strains of non-O1, non-O139 Vibrio cholerae isolates from hospitalized diarrheal patients in Kolkata, India. J Clin Microbiol 47:1087–1095. doi: 10.1128/JCM.02026-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y, Johnson JA, Pusch GD, Morris JG Jr, Stine OC. 2007. The genome of non-O1 Vibrio cholerae NRT36S demonstrates the presence of pathogenic mechanisms that are distinct from those of O1 Vibrio cholerae. Infect Immun 75:2645–2647. doi: 10.1128/IAI.01317-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okada N, Iida T, Park KS, Goto N, Yasunaga T, Hiyoshi H, Matsuda S, Kodama T, Honda T. 2009. Identification and characterization of a novel type III secretion system in trh-positive Vibrio parahaemolyticus strain TH3996 reveal genetic lineage and diversity of pathogenic machinery beyond the species level. Infect Immun 77:904–913. doi: 10.1128/IAI.01184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park KS, Ono T, Rokuda M, Jang MH, Okada K, Iida T, Honda T. 2004. Functional characterization of two type III secretion systems of Vibrio parahaemolyticus. Infect Immun 72:6659–6665. doi: 10.1128/IAI.72.11.6659-6665.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alam A, Miller KA, Chaand M, Butler JS, Dziejman M. 2011. Identification of Vibrio cholerae type III secretion system effector proteins. Infect Immun 79:1728–1740. doi: 10.1128/IAI.01194-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou X, Gewurz BE, Ritchie JM, Takasaki K, Greenfeld H, Kieff E, Davis BM, Waldor MK. 2013. A Vibrio parahaemolyticus T3SS effector mediates pathogenesis by independently enabling intestinal colonization and inhibiting Tak1 activation. Cell Rep 3:1690–1702. doi: 10.1016/j.celrep.2013.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, Tagomori K, Iijima Y, Najima M, Nakano M, Yamashita A, Kubota Y, Kimura S, Yasunaga T, Honda T, Shinagawa H, Hattori M, Iida T. 2003. Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V cholerae. Lancet 361:743–749. doi: 10.1016/S0140-6736(03)12659-1. [DOI] [PubMed] [Google Scholar]
- 15.Alam A, Tam V, Hamilton E, Dziejman M. 2010. VttRA and VttRB encode ToxR family proteins that mediate bile-induced expression of type three secretion system genes in a non-O1/non-O139 Vibrio cholerae strain. Infect Immun 78:2554–2570. doi: 10.1128/IAI.01073-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaand M, Dziejman M. 2013. Vibrio cholerae VttRA and VttRB regulatory influences extend beyond the type 3 secretion system genomic island. J Bacteriol 195:2424–2436. doi: 10.1128/JB.02151-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou X, Ritchie JM, Hiyoshi H, Iida T, Davis BM, Waldor MK, Kodama T. 2012. The hydrophilic translocator for Vibrio parahaemolyticus, T3SS2, is also translocated. Infect Immun 80:2940–2947. doi: 10.1128/IAI.00402-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou X, Massol RH, Nakamura F, Chen X, Gewurz BE, Davis BM, Lencer WI, Waldor MK. 2014. Remodeling of the intestinal brush border underlies adhesion and virulence of an enteric pathogen. mBio 5(4):e01639-14. doi: 10.1128/mBio.01639-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hiyoshi H, Kodama T, Saito K, Gotoh K, Matsuda S, Akeda Y, Honda T, Iida T. 2011. VopV, an F-actin-binding type III secretion effector, is required for Vibrio parahaemolyticus-induced enterotoxicity. Cell Host Microbe 10:401–409. doi: 10.1016/j.chom.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 20.Galan JE. 2009. Common themes in the design and function of bacterial effectors. Cell Host Microbe 5:571–579. doi: 10.1016/j.chom.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aili M, Telepnev M, Hallberg B, Wolf-Watz H, Rosqvist R. 2003. In vitro GAP activity towards RhoA, Rac1 and Cdc42 is not a prerequisite for YopE induced HeLa cell cytotoxicity. Microb Pathog 34:297–308. doi: 10.1016/S0882-4010(03)00063-9. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki M, Danilchanka O, Mekalanos JJ. 2014. Vibrio cholerae T3SS effector VopE modulates mitochondrial dynamics and innate immune signaling by targeting Miro GTPases. Cell Host Microbe 16:581–591. doi: 10.1016/j.chom.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donnenberg MS, Kaper JB. 1991. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect Immun 59:4310–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walhout AJ, Temple GF, Brasch MA, Hartley JL, Lorson MA, van den Heuvel S, Vidal M. 2000. Gateway recombinational cloning: application to the cloning of large numbers of open reading frames or ORFeomes. Methods Enzymol 328:575–592. doi: 10.1016/S0076-6879(00)28419-X. [DOI] [PubMed] [Google Scholar]
- 25.Hartley JL, Temple GF, Brasch MA. 2000. DNA cloning using in vitro site-specific recombination. Genome Res 10:1788–1795. doi: 10.1101/gr.143000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gardel CL, Mekalanos JJ. 1996. Alterations in Vibrio cholerae motility phenotypes correlate with changes in virulence factor expression. Infect Immun 64:2246–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dean P. 2011. Functional domains and motifs of bacterial type III effector proteins and their roles in infection. FEMS Microbiol Rev 35:1100–1125. doi: 10.1111/j.1574-6976.2011.00271.x. [DOI] [PubMed] [Google Scholar]
- 28.Reis RS, Horn F. 2010. Enteropathogenic Escherichia coli, Salmonella, Shigella and Yersinia: cellular aspects of host-bacteria interactions in enteric diseases. Gut Pathog 2:8. doi: 10.1186/1757-4749-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mounier J, Popoff MR, Enninga J, Frame MC, Sansonetti PJ, Van Nhieu GT. 2009. The IpaC carboxyterminal effector domain mediates Src-dependent actin polymerization during Shigella invasion of epithelial cells. PLoS Pathog 5:e1000271. doi: 10.1371/journal.ppat.1000271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the PHYRE server. Nat Protoc 4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 31.Millet YA, Alvarez D, Ringgaard S, von Andrian UH, Davis BM, Waldor MK. 2014. Insights into Vibrio cholerae intestinal colonization from monitoring fluorescently labeled bacteria. PLoS Pathog 10:e1004405. doi: 10.1371/journal.ppat.1004405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ritchie JM, Waldor MK. 2009. Vibrio cholerae interactions with the gastrointestinal tract: lessons from animal studies. Curr Top Microbiol Immunol 337:37–59. doi: 10.1007/978-3-642-01846-6_2. [DOI] [PubMed] [Google Scholar]
- 33.Klose KE. 2000. The suckling mouse model of cholera. Trends Microbiol 8:189–191. doi: 10.1016/S0966-842X(00)01721-2. [DOI] [PubMed] [Google Scholar]
- 34.Morita M, Yamamoto S, Hiyoshi H, Kodama T, Okura M, Arakawa E, Alam M, Ohnishi M, Izumiya H, Watanabe H. 2013. Horizontal gene transfer of a genetic island encoding a type III secretion system distributed in Vibrio cholerae. Microbiol Immunol 57:334–339. doi: 10.1111/1348-0421.12039. [DOI] [PubMed] [Google Scholar]
- 35.Peterson KM, Mekalanos JJ. 1988. Characterization of the Vibrio cholerae ToxR regulon: identification of novel genes involved in intestinal colonization. Infect Immun 56:2822–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Boyle N, Houeix B, Kilcoyne M, Joshi L, Boyd A. 2013. The MshA pilus of Vibrio parahaemolyticus has lectin functionality and enables TTSS-mediated pathogenicity. Int J Med Microbiol 303:563–573. doi: 10.1016/j.ijmm.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 37.Hsiao A, Liu Z, Joelsson A, Zhu J. 2006. Vibrio cholerae virulence regulator-coordinated evasion of host immunity. Proc Natl Acad Sci U S A 103:14542–14547. doi: 10.1073/pnas.0604650103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirn TJ, Jude BA, Taylor RK. 2005. A colonization factor links Vibrio cholerae environmental survival and human infection. Nature 438:863–866. doi: 10.1038/nature04249. [DOI] [PubMed] [Google Scholar]
- 39.Wong E, Vaaje-Kolstad G, Ghosh A, Hurtado-Guerrero R, Konarev PV, Ibrahim AF, Svergun DI, Eijsink VG, Chatterjee NS, van Aalten DM. 2012. The Vibrio cholerae colonization factor GbpA possesses a modular structure that governs binding to different host surfaces. PLoS Pathog 8:e1002373. doi: 10.1371/journal.ppat.1002373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bolivar F, Rodriguez RL, Greene PJ, Betlach MC, Heyneker HL, Boyer HW, Crosa JH, Falkow S. 1997. Construction and characterization of new cloning vehicles. II. A multipurpose cloning system. Gene 2:95–113. [PubMed] [Google Scholar]