

Abstract
Infection of C3H mice with Borrelia burgdorferi, the causative agent of Lyme disease, reliably produces an infectious arthritis and carditis that peak around 3 weeks postinfection and then spontaneously resolve. Macrophage polarization has been suggested to drive inflammation, the clearance of bacteria, and tissue repair and resolution in a variety of infectious disease models. During Lyme disease it is clear that macrophages are capable of clearing Borrelia spirochetes and exhausted neutrophils; however, the role of macrophage phenotype in disease development or resolution has not been studied. Using classical (NOS2) and alternative (CD206) macrophage subset-specific markers, we determined the phenotype of F4/80+ macrophages within the joints and heart throughout the infection time course. Within the joint, CD206+ macrophages dominated throughout the course of infection, and NOS2+ macrophage numbers became elevated only during the peak of inflammation. We also found dual NOS2+ CD206+ macrophages which increased during resolution. In contrast to findings for the ankle joints, numbers of NOS2+ and CD206+ macrophages in the heart were similar at the peak of inflammation. 5-Lipoxygenase-deficient (5-LOX−/−) mice, which display a failure of Lyme arthritis resolution, recruited fewer F4/80+ cells to the infected joints and heart, but macrophage subset populations were unchanged. These results highlight differences in the inflammatory infiltrates during Lyme arthritis and carditis and demonstrate the coexistence of multiple macrophage subsets within a single inflammatory site.
INTRODUCTION
Macrophages play a dynamic role in the immune system as both inflammatory and anti-inflammatory mediators. Their phenotype is highly flexible and dependent upon the cytokine stimulus of the microenvironment they infiltrate (1). They are routinely classified as proinflammatory and immunomodulatory (M1) or anti-inflammatory and remodeling (M2) macrophages, but their phenotype actually lies within a spectrum of inflammatory to anti-inflammatory polarization (2). The language within the field is currently being modified to ensure that the macrophages being studied have a clear phenotypic description, and we use the nomenclature recently suggested (3). Within the last 15 years, much work has been done to phenotypically characterize macrophage subsets. Identifying unique surface and intracellular markers, as well as understanding transcriptional regulation, has allowed the importance of specific macrophage phenotypes in infectious and autoimmune models to be uncovered.
Classically activated M1 macrophages are proinflammatory cells capable of destroying pathogens, tumor cells, and various foreign compounds. Stimulation of undifferentiated macrophages with lipopolysaccharide (LPS) and gamma interferon (IFN-γ), or tumor necrosis factor alpha (TNF-α), induces polarization to the M1 phenotype (4, 5). They can be recognized both in vitro and in vivo by the production of nitric oxide species (NOS), as well as other proinflammatory molecules, including TNF-α, interleukin 1 (IL-1), IL-6, and CCL2 (6). They are routinely identified by flow cytometry using antibodies against F4/80, NOS2, Marco, and IL-12b (7). In contrast, alternatively activated M2 macrophages are responsible for promoting wound healing, tissue regeneration, and antagonizing M1 macrophage responses (8). Polarization of undifferentiated macrophages into the M2 subset in vitro can occur by stimulating them with IL-4, with or without IL-13 (9). M2 macrophages display elevated arginase activity and release IL-10, transforming growth factor β (TGF-β), and platelet-derived growth factor (PDGF), and their identification using flow cytometry relies on expression of F4/80, CD206, and CD163 (10, 11). Transcriptionally, IRF5 promotes M1 differentiation and actively inhibits M2 differentiation (12), while M2 polarization is regulated by both IRF3 and IRF4 (13, 14). Subclasses of the M2 phenotype have been described in vitro, including M2a, M2b, and M2c; however, the existence or relevance of these subtypes has yet to be explored in vivo (15).
Macrophage phenotype is thought to play an important role in models of infection, with M1 responses considered to be protective and M2 conditions to be associated with bacterial persistence. Thus, during acute infection, M1 macrophages are often considered the main component of protection and survival. Mice deficient in IFN-γ or TNF-α succumb to infection with Listeria monocytogenes due to their inability to induce an M1 phenotype, critical for the engulfment and destruction of the bacteria before their phagosomal escape (16, 17). Similar mechanisms have been reported for Salmonella and Chlamydia infections (18, 19). M1 macrophages, while critical, cannot persist in the environment because the potential for tissue damage and chronic inflammation increases the longer they remain. To circumvent this, macrophage phenotype is flexible, and once the infection is cleared and a more anti-inflammatory environment is created, these inflammatory cells may switch to a proresolution M2 phenotype (1). Manipulation of macrophage polarization to the M2 phenotype has been shown to aid in tissue remodeling in a murine model of hindlimb ischemia (20). Failure to make the switch, such as in patients with chronic venous ulcers or severe burns, prolongs inflammation (21, 22).
Early work suggested that M1 macrophages would be present in greater numbers during the initiation of inflammation and that M2 macrophages would be recruited later, or converted from M1 macrophages, to aid in resolution. It now appears that this is an oversimplified model and macrophage polarization in vivo is actually comprised of a spectrum of phenotypes and not that of simply two extremes. In vivo studies have defined a subset of macrophages possessing phenotypic similarities of both M1 and M2 macrophages, the resolution-phase macrophage (rM) (23, 24). These cells may be critical in resolution of inflammation and a return of the tissue to homeostasis after insult or injury. Resolution-phase macrophages generally appear to be more M2-like because they express mannose receptor (CD206) and produce IL-10 and arginase, but they also produce nitric oxide like an M1 cell. Interestingly, genetic control of the rM phenotype appears to be different from that of both M1 and M2 macrophages (24).
While there is increasing interest in macrophage polarization in infectious models, macrophage phenotype has not been fully explored within the Lyme field. Lyme disease is caused by infection with the bacterial spirochete Borrelia burgdorferi, and susceptible mouse strains develop inflammatory arthritis and carditis that peak around 3 weeks postinfection, followed by spontaneous resolution. While the inflammatory infiltrate in the infected joints is primarily comprised of neutrophils, macrophages appear to predominate during Lyme carditis (25). Understanding the polarization status of macrophages throughout a Lyme infection may help to define the mechanisms that drive both inflammation and disease resolution.
MATERIALS AND METHODS
Animals.
Female wild-type (WT) C3H/HeJ mice, age 4 to 6 weeks, were purchased from The Jackson Laboratory (Bar Harbor, ME). 5-LOX+/− mice (B6;129S2-Alox5tm1fun/J) were purchased as breeders from The Jackson Laboratory and were backcrossed for 10 generations onto the C3H/HeJ background. Heterozygous mice were then intercrossed to produce C3H 5-lipoxygenase-deficient (5-LOX−/−) mice. Animals were given sterile food and water ad libitum and housed in a specific-pathogen-free facility. All work was done in accordance with the Animal Care and Use Committee of the University of Missouri.
Bacteria and infections.
A virulent clonal isolate of B. burgdorferi N40 strain was used for all infections. Frozen stocks were added to 7 ml of C-BSK-H medium (Sigma-Aldrich, St. Louis, MO) and grown to log phase at 32°C. Spirochetes were counted using dark-field microscopy in a Petroff-Hausser counting chamber (Hausser Scientific, Horsham, PA). Spirochete dilutions were made in sterile BSK-H medium, and mice were inoculated in each hind footpad with 50 μl of medium containing 5 × 104 spirochetes. Arthritis progression was monitored by measuring ankle swelling at the thickest craniocaudal portion of the joint using a metric caliper. The ankle diameter increase was determined by measuring the joint thickness prior to infection and then subtracting this value from the weekly measurements.
Reagents.
Antibodies for use in flow cytometry were obtained from eBioscience (CD45.2-peridinin chlorophyll protein [PerCP]-Cy5.5, CD16 CD32, NOS2-phycoerythrin [PE]), and BioLegend (F4/80-allophycocyanin [APC]-eF1.780, CD206-APC). Collagenase/dispase version 16 was purchased from Roche and resuspended according to the manufacturer's directions. DNase I DN-25 was purchased from Sigma and stored at −20°C in 2 mg/ml of 50% glycerol and 75 mM NaCl.
In vitro isolation of bone marrow derived macrophages (BMDM).
Bone marrow cells were isolated from C3H and C3H 5-LOX−/− mouse femurs and tibias and differentiated on 100- by 30-mm plastic petri dishes in medium containing RPMI 1640 supplemented with 30% L929 cell-conditioned medium, 10% fetal bovine serum (FBS), and 2% penicillin-streptomycin at 37°C in 5% CO2 for 6 days. Adherent cells were removed, washed, and plated in a 12-well plate at 1 × 106 per well overnight. For coculture experiments, B. burgdorferi was then added at a multiplicity of infection (MOI) of 10:1, and cultures were incubated for an additional 24 h. The wells were then washed and the cells removed by gentle scraping. Staining for the M1 or M2 phenotype was then completed as described below. For macrophage polarization experiments, the cells were then stimulated with 1 μg/ml of IL-4 (Peprotech) or LPS (Sigma) and IFN-γ (Peprotech) for 24 h to induce M2 and M1 subsets, respectively. Supernatant nitrite concentrations were determined by the Greiss reaction. Arginase activity was determined by incubating cell lysates with l-arginine for 1 h and measuring the amount of urea produced.
In vivo cell isolation.
Three infected WT C3H or C3H 5-LOX−/− mice were sacrificed at each time point. Ankles were removed from each mouse by removing the toes and carefully disarticulating the knee joint, particularly to avoid bone marrow contamination. Excess muscle tissue was trimmed to reduce blood contamination. Hearts were perfused with 10 ml of 1× phosphate-buffered saline (PBS), removed, and cut into fine pieces. Samples were placed in 15-ml conical tubes containing 5 ml of 1× PBS plus 4% FBS, 75 μl of diluted DNase I (0.03 mg; Sigma), and 50 μl of stock collagenase/dispase (Roche) and rocked at room temperature (RT) for 1 h. Samples were then placed in sterile petri dishes containing 5 ml of additional RPMI 1640 supplemented with 10% FBS, and ankles were carefully flayed apart using sterile rat tooth forceps. Cells were strained through a 70 μm filter (BD Falcon) into a 50-ml conical tube. Cells were washed with 5 ml of 1× PBS plus 4% FBS three times, and live cells were enumerated using trypan blue exclusion.
Cell staining.
A total of 1 × 106 cells were stained in a 96-well U-bottom plate (Corning, Inc.). All cells were treated with Fc block (anti-CD16/CD32; eBioscience) for 15 min at 4°C. Cells were stained on ice in the dark using stains specific for the following cell types: CD45.2-PerCP-Cy5.5 (hematopoietic cells), F4/80-APC-efluor780 (macrophages), intracellular NOS2-PE (M1 macrophages), and CD206-APC (M2 macrophages). Cells were then washed and fixed in 1% paraformaldehyde for 15 min and analyzed using a Dako CyAn flow cytometer and Summit V5.0 software.
Statistical analysis.
Statistical significance was assessed for most data using analysis of variance (ANOVA) followed by Dunnett's test for comparing multiple groups with a single control. For other data consisting of two groups we used an unpaired, two-tailed Student t test. Significance levels were set at a P value of <0.05. All experiments were completed at least twice, and figures are representative of a single experiment. For in vivo experiments, 3 animals were sacrificed per group per day.
RESULTS
Characterization of BMDM from C3H mice.
BMDM are commonly used in Lyme disease research as a representative macrophage for investigating in vitro cellular responses to B. burgdorferi infection (26–29). Following the culture and differentiation of BM precursors into BMDM in our laboratory, these cells appear to take on an uncommitted or M2 phenotype, as determined by flow cytometry expression of NOS2 or CD206 (Fig. 1A). Upon coculture with B. burgdorferi for 24 h, the cells become primarily M1-like cells, downregulating their expression of CD206 and increasing their expression of NOS2. They also acquire the M1-like ability to produce NO (Fig. 1B). Interestingly, a small population of cells appears to express markers of both M1 and M2 cells (NOS2+ CD206+). Whether these cells are in the process of transitioning from the M2 to M1 phenotype or represent a separate macrophage state, such as the resolution macrophage (rM) described by Bystrum et al. (23), is unclear at this time. However, it is clear that BMDM adopt an M1-like phenotype when cultured with B. burgdorferi in vitro.
FIG 1.
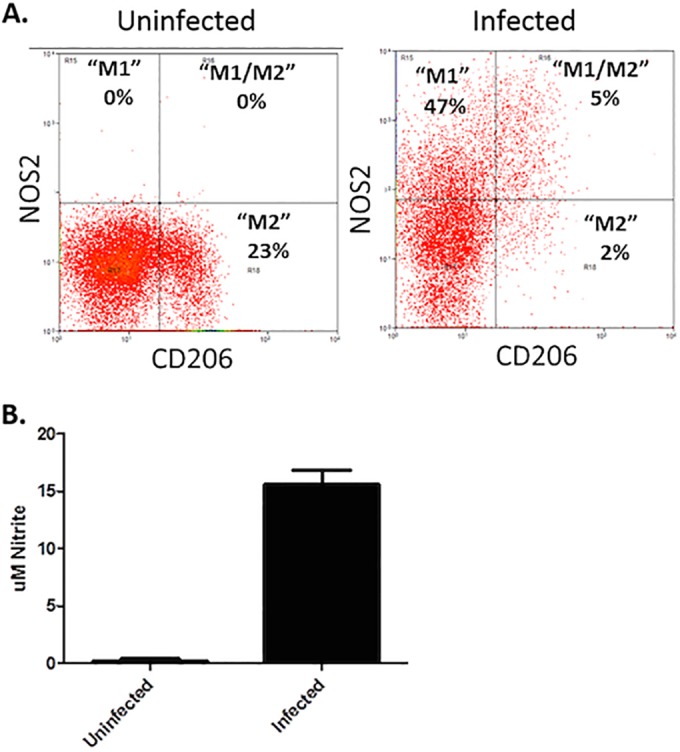
In vitro characterization of BMDM from C3H mice. BMDM were cultured with (infected) or without (uninfected) B. burgdorferi spirochetes (MOI, 10:1) for 24 h. Cells were gated on live cells and F4/80+ and then assessed for expression of CD206 and NOS2. Representative flow plots are shown, and percentages of F4/80+ cells expressing NOS2 (M1), CD206 (M2), or both NOS2 and CD206 (M1/M2) are presented (A). Levels of nitrite were determined in supernatants of uninfected (control) or infected BMDM (B).
Flow cytometric evaluation of macrophage subsets within experimental Lyme arthritis.
In order to study the development of macrophage subsets during experimental Lyme borreliosis, we infected susceptible C3H mice with 5 × 104 B. burgdorferi organisms per footpad and monitored arthritis progression. At various time points postinfection, groups of mice were sacrificed and joints and heart tissue were removed, homogenized, and stained for analysis by flow cytometry. Our gating strategy is shown in Fig. 2A. M1 macrophages were defined by their expression of NOS2, and M2 macrophages were defined by their expression of CD206 as reported by others (30–32). Following infection, arthritis development was monitored by measuring ankle swelling over the course of about 2 months (Fig. 2B). Ankle swelling is indicative of the underlying inflammatory response in WT C3H mice and is a convenient method to monitor both the development and resolution of experimental Lyme arthritis (33). As is typical for this infection, ankle swelling began during the second week of infection, peaked around the third week, and then resolved over the next few weeks (Fig. 2B). We found inflammatory hematopoietic cells (CD45.2+) beginning to infiltrate the ankle joint by day 7, increasing at day 14, peaking around day 21, and then resolving (Fig. 2C). Macrophage infiltration into the joint tissue (CD45.2+ F4/80+) was delayed slightly compared with all hematopoietic cells, and high levels were found during the day 14 and 21 time points (Fig. 2D). Macrophage levels were nearly returned to control levels by day 35 but then increased again at day 56; however, this increase was not statistically significant. Macrophage subsets, M1(F4/80+ NOS2+ CD206−) and M2 (F4/80+ NOS2− CD206+), were present in the joints with peaks at days 14 and 21 coinciding with total macrophages (Fig. 2E and F). However, at all time points the levels of M2 (F4/80+ NOS2− CD206+) macrophages were 100-fold greater than the levels of the M1 (F4/80+ NOS2+ CD206−) macrophages. This was unexpected, as this is the time of greatest inflammation in the infected joints (exemplified by the ankle swelling curve in Fig. 2B) and the proinflammatory M1 cell phenotype might be expected to dominate during this time. In addition, both cell subsets were near control levels on day 35 but then increased at day 56, again with M2 (F4/80+ NOS2− CD206+) macrophages dominating the cellular infiltrate, suggesting the M2-like cells may play two roles during Lyme arthritis: first limiting the overall inflammatory response during peak inflammation and then playing a reparative or healing role later. We also identified a population of macrophages expressing both NOS2 and CD206 (Fig. 2G). The numbers of these cells were similar to the numbers of M1 (F4/80+ NOS2+ CD206−) macrophages on days 14 and 21 but reached their highest levels on day 56. Others have described macrophage subsets displaying both M1- and M2-type markers from in vivo studies and suggested they are resolution macrophages (23, 24). While these cells may be simply transitioning from one cellular subset or the other, their high numbers at day 56 suggests they may represent an intermediate “resolution-type” cell and may play a role in returning the tissue to homeostasis.
FIG 2.

Flow cytometric characterization of macrophage subsets from ankle joints of mice infected with B. burgdorferi. Representative flow cytometry plots of cells isolated from mouse ankle joints were generated (A). Arthritis development was monitored by measuring ankle swelling over time (B). Cell number plots from joints of control (uninfected) or mice infected for various days with B. burgdorferi and gated for CD45.2+ hematopoietic cells (C), F4/80+ macrophages (D), M1 (NOS2+ CD206−) (E), M2 (NOS2− CD206+) (F), or rM (NOS2+ CD206+) (G) were generated. n = 4 for each time point and the experiment was performed twice. Asterisks indicate data are significantly different from control data at a P value of <0.05 as determined by ANOVA followed by Dunnett's test. Swelling curve differences were determined by comparing infected versus uninfected animals at each time point via Student's t test.
Macrophage phenotype over the course of murine Lyme carditis.
In Lyme carditis, macrophages are the primary inflammatory cell, whereas in Lyme arthritis, neutrophils predominate (25). It was therefore of interest to define macrophage polarization in the heart during Borrelia infection. Mice were infected as described above and groups of mice were sacrificed at specific time points. The hearts were perfused to remove blood and circulating cells, and were then removed. As with the joints, single cell suspensions were made, stained, and analyzed by flow cytometry. Infiltrating hematopoietic cells in the heart tissue peaked at day 21 postinfection and then resolved (Fig. 3A). There were fewer total inflammatory cells in B. burgdorferi-infected hearts than in the joint tissue. Higher numbers of F4/80+ macrophages were seen at days 14 and 21 postinfection, similar to the findings for the infected joint (Fig. 3B). Unlike the joint, M1 (F4/80+ NOS2+ CD206−) macrophages and M2 (F4/80+ NOS− CD206+) macrophages were present in the heart tissue at the peak of inflammation in roughly equal numbers (Fig. 3C and D), although M1-like macrophages peaked at day 14 postinfection, while the M2-like macrophages peaked around day 21 postinfection. The M2 (F4/80+ NOS− CD206+) macrophage population increased again at day 56 postinfection, perhaps signaling a role in tissue repair and resolution. The M1 (F4/80+ NOS2+ CD206−) macrophage population did not expand at this time point in the hearts. The rM (F4/80+ NOS2+ CD206+) expanded during the peak of infection, similar to the findings for the joints (Fig. 3E), and a small population was present during day 56 postinfection. These results demonstrate that different populations of macrophage subsets can exist within different infected tissues within the same animal.
FIG 3.

Flow cytometric characterization of macrophage subsets from hearts of mice infected with B. burgdorferi. Hearts were perfused and digested, and single-cell suspensions were isolated as described in Materials and Methods. Cell number plots from hearts of control (uninfected) or mice infected for various days with B. burgdorferi and gated for CD45.2+ hematopoietic cells (A), F4/80+ macrophages (B), M1 (NOS2+ CD206−) (C), M2 (NOS2− CD206+) (D), or rM (NOS2+ CD206+) (E) were generated. n = 4 for each time point, and the experiment was performed twice. Asterisks indicate data are significantly different from control data at a P value of <0.05 as determined by ANOVA followed by Dunnett's test.
Macrophage subsets in 5-lipoxygenase-deficient BMDM.
Our laboratory has previously reported that 5-lipoxygenase-deficient (5-LOX−/−) mice develop arthritis similar to wild-type controls yet fail to resolve inflammation (34). Macrophages from 5-LOX−/− mice also displayed reduced ability to phagocytose opsonized spirochetes (34). Macrophage polarization and subset development in these mice have not been previously described. BMDM from 5-LOX−/− mice stimulated in vitro under M1 (IFN-γ, LPS) polarizing conditions make levels of nitrite comparable to those of wild-type mice (Fig. 4A). Under M2 (IL-4) polarizing conditions, 5-LOX−/− BMDM also displayed no difference in arginase activity compared to that of wild-type controls (Fig. 4B). Thus, the ability of BMDM from 5-LOX−/− mice to polarize under M1 or M2 conditions appears to be intact. Upon analysis of specific macrophage subset marker expression under in vitro M1 (IFN-γ, LPS) and M2 (IL-4) conditions, we noticed some variations. 5-LOX−/− BMDM polarized under M1 (IFN-γ, LPS) conditions displayed a slightly higher percentage of M1 (F4/80+ NOS2+ CD206−) cells and a slightly lower percentage of M2 (F4/80+ NOS− CD206+) cells (Fig. 4C and D). The production of rM (F4/80+ NOS2+ CD206+) cells was also decreased by the 5-LOX−/− BMDM (Fig. 4E). These results suggest that macrophage subset development in 5-LOX−/− mice during inflammatory responses is generally intact but may be skewed slightly toward the M1-like subset.
FIG 4.

In vitro polarization and characterization of BMDM from wild-type (WT) or 5-LOX−/− mice. Following treatment with polarizing cytokines M(LPS+IFN-γ) or M(IL-4) for 24 h, supernatant nitrite (A) and cell lysate arginase activity (B) were determined. Cells were gated on live cells and F4/80+ and then assessed for percentage of expression of NOS2 (C), CD206 (D), or both NOS2 and CD206 (E) by flow cytometry. Asterisks indicate statistically significant differences between 5-LOX cells compared with WT controls from the same group at a P value of >0.05.
Macrophage subsets in joints of 5-lipoxygenase-deficient animals.
To determine what effect the loss of 5-LOX might have on macrophage subset development during an inflammatory response in vivo, we infected C3H 5-LOX−/− mice with B. burgdorferi and sacrificed groups on days 21 and 56 postinfection. These time points are indicative of the peak and resolution of inflammation in WT mice. We have previously reported that 5-LOX−/− mice exhibit exacerbated ankle swelling and prolonged joint inflammation following B. burgdorferi infection compared to those in WT mice (34), and thus, it was of interest to determine if this correlated with altered macrophage subset development. We found that levels of F4/80+ macrophages were decreased in the joints of 5-LOX−/− mice at day 21 postinfection compared with those in WT mice (Fig. 5A). This difference appeared to result primarily from a decrease in M2 (F4/80+ NOS− CD206+) and rM (F4/80+ NOS2+ CD206+) cells, since M1 (F4/80+ NOS2+ CD206−) cells were similar between WT and 5-LOX−/− (Fig. 5B), while M2-like cells and rM-like cells were significantly decreased in the 5-LOX−/− mice (Fig. 5C and D). No differences were detected between WT and 5-LOX−/− mice in any macrophage population at day 56 postinfection (Fig. 5A to D); however, populations of M2 (F4/80+ NOS− CD206+) and rM (F4/80+ NOS2+ CD206+) cells persisted in the joints compared with day 21 levels. How these cells might contribute to the resolution of inflammation at this time point is currently unclear and under investigation.
FIG 5.
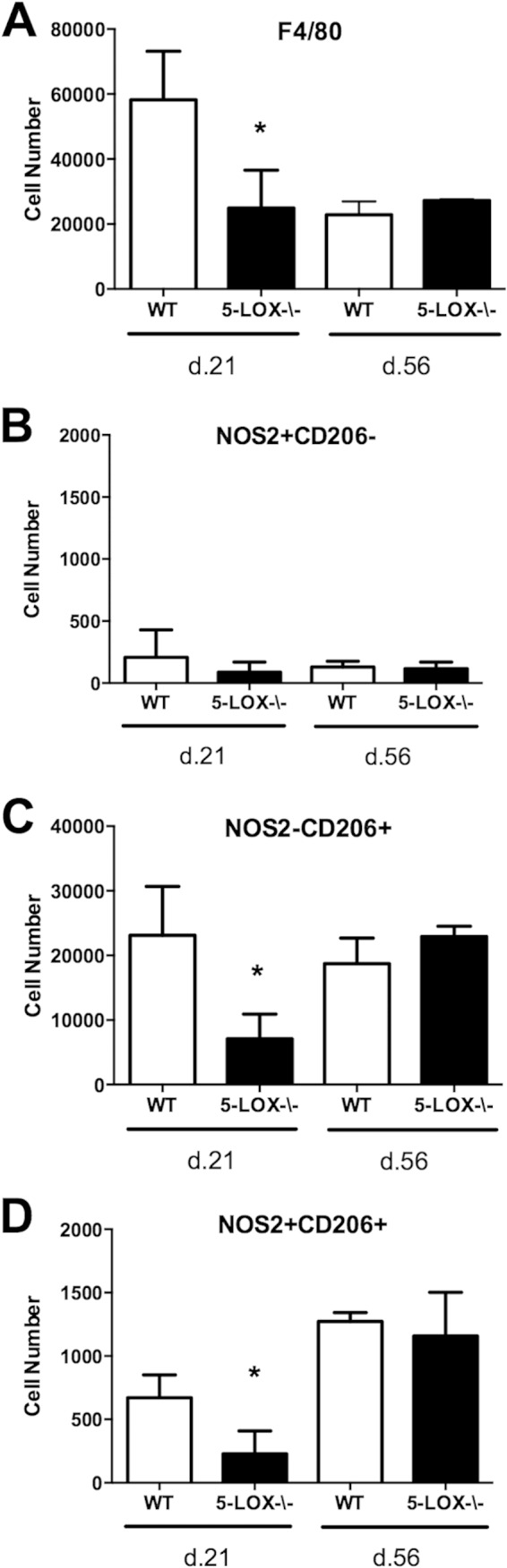
Macrophage phenotype and numbers were analyzed from the joints of Borrelia-infected WT or 5-LOX-deficient mice on days 21 and 56 postinfection. Numbers of F4/80+ macrophages (A), M1 (NOS2+ CD206−) (B), M2 (NOS2− CD206+) (C), or rM (NOS2+ CD206+) (D) were determined. n = 4 for each time point, and the experiment was performed twice. Asterisks indicate data that are significantly different from control data at a P value of <0.05 as determined by ANOVA followed by Dunnett's test.
Macrophage subsets during Lyme carditis in 5-lipoxygenase-deficient mice.
The role of 5-LOX in Lyme carditis has not been studied, and we were interested in the macrophage phenotype within the hearts of 5-LOX−/− mice compared to that in WT controls. During Lyme carditis, macrophages predominate over neutrophils in the cellular inflammatory infiltrate (25). At days 21 and 56 postinfection, hearts were perfused and cells were harvested for analysis by flow cytometry. Similar to what was found in the joint tissue, 5-LOX−/− mice displayed significantly fewer F4/80+ macrophages at day 21 than did WT controls (Fig. 6A). Although the levels of M1 (F4/80+ NOS2+ CD206−) and rM (F4/80+ NOS2+ CD206+) macrophages trended lower at this time point, they did not reach statistical significance (Fig. 6B and D). Only the 5-LOX−/− M2 (F4/80+ NOS2− CD206+) macrophages were significantly different from those in the WT animals (Fig. 6C). As with the joint tissues, there were no differences in macrophage subset numbers between WT and 5-LOX−/− mice in the hearts at day 56 postinfection. Numbers of M2 (F4/80+ NOS2− CD206+) macrophages remained elevated in hearts of both WT and 5-LOX−/− mice, suggesting a role for these cells in repair and resolution processes. rM (F4/80+ NOS2+ CD206+) macrophages were elevated in the hearts of the 5-LOX−/− mice at day 56 postinfection (Fig. 6D), but the values did not reach statistically significant difference from WT mice. However, this may suggest a role for these cells in the resolution of Lyme carditis, but further work is required to understand what this role might be.
FIG 6.
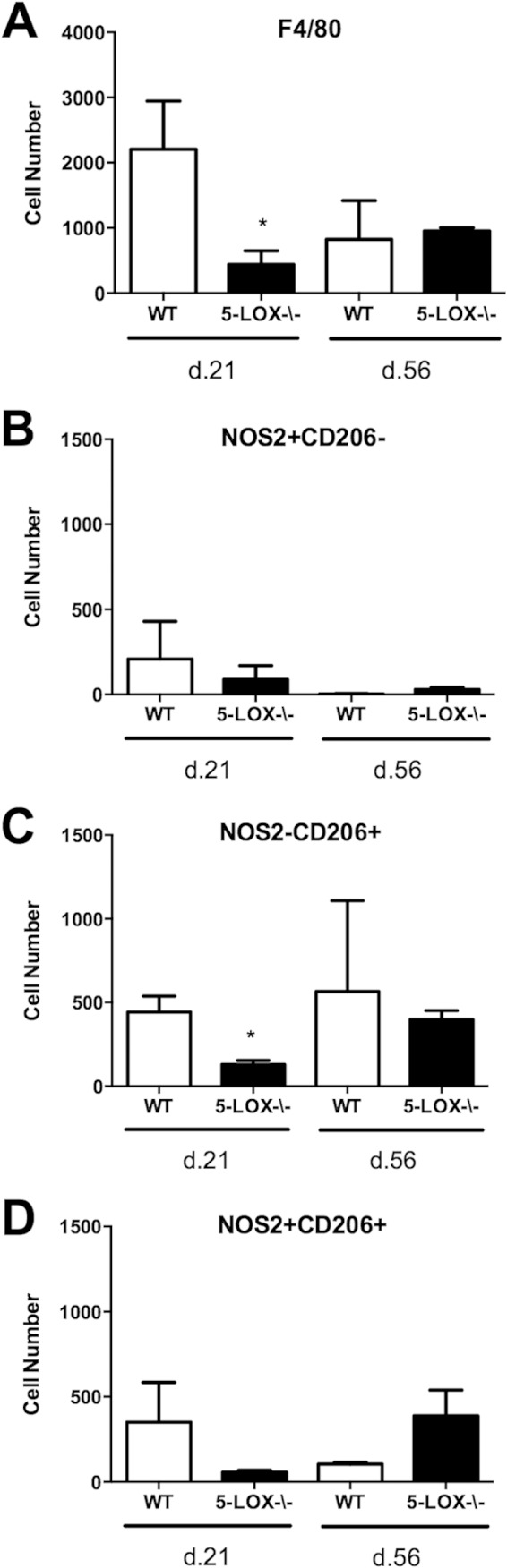
Macrophage phenotype and numbers were analyzed from the hearts of Borrelia-infected WT or 5-LOX-deficient mice on days 21 and 56 postinfection. Numbers of F4/80+ macrophages (A), M1 (NOS2+ CD206−) (B), M2 (NOS2− CD206+) (C), or rM (NOS2+ CD206+) (D) were determined. n = 4 for each time point, and the experiment was performed twice. Asterisks indicate data that are significantly different from control data at a P value of <0.05 as determined by ANOVA followed by Dunnett's test.
DISCUSSION
Macrophages have been shown to play a role in the development of both Lyme arthritis and carditis (35), and we were interested in determining whether macrophage subsets played a role in disease pathogenesis. Experimental Lyme borreliosis is an excellent model to study immune regulation since the mice develop an inflammatory arthritis and carditis that peak and then resolve as the spirochetes are cleared from the infected tissues (36). We used cell-specific markers NOS2 and CD206 as representatives of M1 and M2 cells, respectively. Others have used these markers to investigate M1 and M2 subsets in murine models of arthritis and carditis (30–32). Using these markers, we were able to phenotype M1 (F4/80+ NOS2+ CD206−) and M2 (F4/80+ NOS2− CD206+) macrophages within the joints and hearts of Borrelia-infected mice at a number of key time points. We tracked macrophage infiltration during the onset, peak, and resolution of disease.
Macrophages make up a significant proportion of the inflammatory infiltrate in infected joints and hearts of B. burgdorferi-infected mice (37, 38). However, the contribution of these cells to the development and resolution of Lyme arthritis and carditis has not been directly studied. In vitro stimulation of macrophages with B. burgdorferi or its outer surface lipoproteins generally results in the production of proinflammatory cytokines, such as IL-12, IL-6, IL-1β, and TNF-α, as well as NO production (39–43). Based upon these studies, in vitro stimulation of both human and mouse macrophages would be expected to push them into the M1 phenotype. This is confirmed in the current study, whereby murine BMDM from C3H mice expressed high levels of NOS2 and low levels of CD206 following in vitro coculture with B. burgdorferi spirochetes. This is in contrast to what we found in vivo, where we found M2 (F4/80+ NOS2− CD206+) cells to outnumber M1(F4/80+ NOS2+ CD206−) cells 100-fold throughout the disease time course in the infected joints. This M2 dominance may also explain why inhibition or genetic depletion of inducible NOS (iNOS) had little effect upon Lyme arthritis development in mice (42, 44). Since in vitro coculture with B. burgdorferi appears to drive macrophage M1 development, while in vivo infection drives development of a predominant M2 subset in the joint, caution should be used in interpreting macrophage responses to B. burgdorferi based upon in vitro data alone.
The resolution of Lyme arthritis occurs spontaneously and is thought to be primarily driven by antibody-mediated clearance of the spirochetes from the infected joint (45). However, under certain circumstances there is a failure or delay in arthritis resolution despite the seemingly appropriate production of anti-Borrelia antibodies (29, 34, 46, 47), suggesting a role for innate immune cells in arthritis resolution. We show that during resolution, at day 56 postinfection, there is an increase in hybrid NOS2+ CD206+ macrophages. The unique phenotype and time of increase indicate that these cells are likely resolution-phase macrophages. Resolution-phase macrophages, rM (F4/80+ NOS2+ CD206+), are neither classically nor alternatively activated; their phenotype is a combination of the two (23). Until now, resolution-phase macrophages have not been implicated in the resolution of Lyme arthritis. Further study may lead to the development of pharmaceuticals to target this unique cell type and help control chronic inflammation.
Lyme carditis is unique from arthritis in that macrophages appear to be the driving force in disease progression. Macrophage numbers within the heart predominate over those of neutrophils and peak around day 14 postinfection (48). Unlike for the joints, we found that M1 (F4/80+ NOS2+ CD206−) cells peaked early (day 14 postinfection) and their numbers then decreased significantly by day 21. The numbers of M2 (F4/80+ NOS2− CD206+) cells in the infected hearts peaked on day 21, were virtually absent on day 35, and then returned on day 56 postinfection. The majority of macrophages at this time point were M1 (F4/80+ NOS2+ CD206−) macrophages. This pattern of expression was similar for the rM (F4/80+ NOS2+ CD206+) macrophages. These results suggest that macrophages may have a unique role in disease development in the heart compared to the infected joint. M1 cells may be more involved in driving the initial inflammatory response in the heart, which may then be limited by M2 and/or rM cells at day 21, with further reparative/restoration processes occurring later (day 56). More work is required to define the roles of these cell subsets in mediating Lyme carditis development and resolution.
To better understand the role of macrophage polarization on disease development, we also studied a nonresolving mouse model. 5-Lipoxygenase (5-LOX)-deficient mice are susceptible to arthritis but fail to resolve normally (34). 5-Lipoxygenase converts arachidonic acid into leukotriene A4 (LTA4), which is then converted by downstream enzymes into LTB4 and the cysteinyl-LTs. 5-LOX products are also important for the transcellular biosynthesis of the proresolution lipoxins, which are important mediators of the resolution of inflammation (49). In vitro stimulation of BMDM from 5-LOX−/− mice resulted in shift toward the M1 subtype and slightly away from the M2 or rM phenotypes. Similarly, in vivo C3H 5-LOX−/− mice infected with B. burgdorferi had fewer F4/80+ macrophages in the joints and heart tissue than WT mice at day 21 postinfection. This defect was due to a deficit of M2 (F4/80+ NOS2− CD206+) and rM (F4/80+ NOS2+ CD206+) macrophages, while M1 (F4/80+ NOS2+ CD206−) cells were similar to those in WT mice. However, by the resolution of disease at day 56 postinfection, both M2 and rM cell numbers were comparable between the knockout and wild-type mice. Early production of lipid mediators has been shown to significantly impact resolution of inflammatory responses later during the immune response (50). Therefore, it is possible the lack of M2 or rM in the joints and hearts of B. burgdorferi-infected 5-LOX−/− mice may disrupt the mechanism of inflammatory resolution and lead to the chronic inflammation seen in these mice. Studies are currently ongoing to define these mechanisms.
In conclusion, macrophage subsets can be identified within the joints and hearts of B. burgdorferi-infected mice. In the joints, M2 (F4/80+ NOS2− CD206+) macrophages outnumber M1 (F4/80+ NOS2+ CD206−) macrophages 10:1, while in the heart tissue the numbers are similar, but with slightly different kinetics. At the time of disease resolution, a resolution macrophage rM (F4/80+ NOS2+ CD206+) appears to be present, but more work is necessary to elucidate its exact function. Nonresolving 5-LOX−/− mice show a defect in the production of both M2 and rM macrophages in both the joints and hearts at the peak of inflammation. This may drive chronic inflammation by exacerbating the strength and persistence of proinflammatory signals or by the failure of initiating a proresolution program. These possibilities are under investigation.
ACKNOWLEDGMENT
This work was supported by the University of Missouri, College of Veterinary Medicine Committee on Research.
REFERENCES
- 1.Davis MJ, Tsang TM, Qiu Y, Dayrit JK, Freij JB, Huffnagle GB, Olszewski MA. 2013. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. mBio 4(3):e00264-13. doi: 10.1128/mBio.00264-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gordon S. 2007. The macrophage: Past, present and future. Eur J Immunol 37:S9–S17. doi: 10.1002/eji.200737638. [DOI] [PubMed] [Google Scholar]
- 3.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, Wynn TA. 2014. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erwig L-P, Kluth DC, Walsh GM, Rees AJ. 1998. Initial cytokine exposure determines function of macrophages and renders them unresponsive to other cytokines. J Immunol 161:1983–1988. [PubMed] [Google Scholar]
- 5.Wallet MA, Wallet SM, Guiulfo G, Sleasman JW, Goodenow MM. 2010. IFNγ primes macrophages for inflammatory activation by high molecular weight hyaluronan. Cell Immunol 262:84–88. doi: 10.1016/j.cellimm.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laskin DL. 2009. Macrophages and inflammatory mediators in chemical toxicity: a battle of forces. Chem Res Toxicol 22:1376–1385. doi: 10.1021/tx900086v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murray PJ, Wynn TA. 2011. Obstacles and opportunities for understanding macrophage polarization. J Leukoc Biol 89:557–563. doi: 10.1189/jlb.0710409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murray PJ, Wynn TA. 2011. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. 2004. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 10.Misson P, van den Brûle S, Barbarin V, Lison D, Huaux F. 2004. Markers of macrophage differentiation in experimental silicosis. J Leukoc Biol 76:926–932. doi: 10.1189/jlb.0104019. [DOI] [PubMed] [Google Scholar]
- 11.Lolmede K, Campana L, Vezzoli M, Bosurgi L, Tonlorenzi R, Clementi E, Bianchi ME, Cossu G, Manfredi AA, Brunelli S, Rovere-Querini P. 2009. Inflammatory and alternatively activated human macrophages attract vessel-associated stem cells, relying on separate HMGB1- and MMP-9-dependent pathways. J Leukoc Biol 85:779–787. doi: 10.1189/jlb.0908579. [DOI] [PubMed] [Google Scholar]
- 12.Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, Hussell T, Feldmann M, Udalova IA. 2011. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol 12:231–238. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- 13.Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, Bottazzi B, Doni A, Vincenzo B, Pasqualini F, Vago L, Nebuloni M, Mantovani A, Sica A. 2006. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-κB and enhanced IRF-3/STAT1 activation). Blood 107:2112–2122. doi: 10.1182/blood-2005-01-0428. [DOI] [PubMed] [Google Scholar]
- 14.Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, Miyake T, Matsushita K, Okazaki T, Saitoh T, Honma K, Matsuyama T, Yui K, Tsujimura T, Standley DM, Nakanishi K, Nakai K, Akira S. 2010. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol 11:936–944. doi: 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- 15.Martinez FO, Gordon S. 2014. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep 6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pfeffer KD, Huecksteadt TP, Hoidal JR. 1993. Expression and regulation of tumor necrosis factor in macrophages from cystic fibrosis patients. Am J Respir Cell Mol Biol 9:511–519. doi: 10.1165/ajrcmb/9.5.511. [DOI] [PubMed] [Google Scholar]
- 17.Shaughnessy LM, Swanson JA. 2007. The role of the activated macrophage in clearing Listeria monocytogenes infection. Front Biosci 12:2683–2692. doi: 10.2741/2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Igietseme JU, Perry LL, Ananaba GA, Uriri IM, Ojior OO, Kumar SN, Caldwell HD. 1998. Chlamydial infection in inducible nitric oxide synthase knockout mice. Infect Immun 66:1282–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rottenberg MnE, Gigliotti-Rothfuchs A, Wigzell H. 2002. The role of IFN-γ in the outcome of chlamydial infection. Curr Opin Immunol 14:444–451. doi: 10.1016/S0952-7915(02)00361-8. [DOI] [PubMed] [Google Scholar]
- 20.Takeda Y, Costa S, Delamarre E, Roncal C, Leite de Oliveira R, Squadrito ML, Finisguerra V, Deschoemaeker S, Bruyere F, Wenes M, Hamm A, Serneels J, Magat J, Bhattacharyya T, Anisimov A, Jordan BF, Alitalo K, Maxwell P, Gallez B, Zhuang ZW, Saito Y, Simons M, De Palma M, Mazzone M. 2011. Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature 479:122–126. doi: 10.1038/nature10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H, Hainzl A, Schatz S, Qi Y, Schlecht A, Weiss JM, Wlaschek M, Sunderkotter C, Scharffetter-Kochanek K. 2011. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest 121:985–997. doi: 10.1172/JCI44490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi M, Jeschke MG, Shigematsu K, Asai A, Yoshida S, Herndon DN, Suzuki F. 2010. M2b monocytes predominated in peripheral blood of severely burned patients. J Immunol 185:7174–7179. doi: 10.4049/jimmunol.0903935. [DOI] [PubMed] [Google Scholar]
- 23.Bystrom J, Evans I, Newson J, Stables M, Toor I, van Rooijen N, Crawford M, Colville-Nash P, Farrow S, Gilroy DW. 2008. Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood 112:4117–4127. doi: 10.1182/blood-2007-12-129767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stables M, Shah S, Camon EB, Lovering RC, Newson J, Bystrom J, Farrow S, Gilroy DW. 2011. Transcriptomic analyses of murine resolution-phase macrophages. Blood 118:e192–e208. doi: 10.1182/blood-2011-04-345330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montgomery RR, Booth CJ, Wang X, Blaho VA, Malawista SE, Brown CR. 2007. Recruitment of macrophages and polymorphonuclear leukocytes in Lyme carditis. Infect Immun 75:613–620. doi: 10.1128/IAI.00685-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chung Y, Zhang N, Wooten RM. 2013. Borrelia burgdorferi elicited-IL-10 suppresses the production of inflammatory mediators, phagocytosis, and expression of co-stimulatory receptors by murine macrophages and/or dendritic cells. PLoS One 8:e84980. doi: 10.1371/journal.pone.0084980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hawley KL, Olson CM, Iglesias-Pedraz JM, Navasa N, Cervantes JL, Caimano MJ, Izadi H, Ingalls RR, Pal U, Salazar JC, Radolf JD, Anguita J. 2012. CD14 cooperates with complement receptor 3 to mediate MyD88-independent phagocytosis of Borrelia burgdorferi. Proc Natl Acad Sci U S A 109:1228–1232. doi: 10.1073/pnas.1112078109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sahay B, Singh A, Gnanamani A, Patsey RL, Blalock JE, Sellati TJ. 2011. CD14 signaling reciprocally controls collagen deposition and turnover to regulate the development of Lyme arthritis. Am J Pathol 178:724–734. doi: 10.1016/j.ajpath.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wooten RM, Ma Y, Yoder RA, Brown JP, Weis JH, Zachary JF, Kirschning CJ, Weis JJ. 2002. Toll-like receptor 2 is required for innate, but not acquired, host defense to Borrelia burgdorferi. J Immunol 168:348–355. doi: 10.4049/jimmunol.168.1.348. [DOI] [PubMed] [Google Scholar]
- 30.Antonios JK, Yao Z, Li C, Rao AJ, Goodman SB. 2013. Macrophage polarization in response to wear particles in vitro. Cell Mol Immunol 10:471–482. doi: 10.1038/cmi.2013.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ye L, Wen Z, Li Y, Chen B, Yu T, Liu L, Zhang J, Ma Y, Xiao S, Ding L, Li L, Huang Z. 2014. Interleukin-10 attenuation of collagen-induced arthritis is associated with suppression of interleukin-17 and retinoid-related orphan receptor γt production in macrophages and repression of classically activated macrophages. Arthritis Res Ther 16:R96. doi: 10.1186/ar4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li K, Xu W, Guo Q, Jiang Z, Wang P, Yue Y, Xiong S. 2009. Differential macrophage polarization in male and female BALB/c mice infected with coxsackievirus B3 defines susceptibility to viral myocarditis. Circ Res 105:353–364. doi: 10.1161/CIRCRESAHA.109.195230. [DOI] [PubMed] [Google Scholar]
- 33.Barthold SW, Beck DS, Hansen GM, Terwilliger GA, Moody KD. 1990. Lyme borreliosis in selected strains and ages of laboratory mice. J Infect Dis 162:133–138. doi: 10.1093/infdis/162.1.133. [DOI] [PubMed] [Google Scholar]
- 34.Blaho VA, Zhang Y, Hughes-Hanks JM, Brown CR. 2011. 5-Lipoxygenase-deficient mice infected with Borrelia burgdorferi develop persistent arthritis. J Immunol 186:3076–3084. doi: 10.4049/jimmunol.1003473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wooten RM, Weis JJ. 2001. Host-pathogen interactions promoting inflammatory Lyme arthritis: use of mouse models for dissection of disease processes. Curr Opin Microbiol 4:274–279. doi: 10.1016/S1369-5274(00)00202-2. [DOI] [PubMed] [Google Scholar]
- 36.Barthold SW, Janotka JL, Smith AL, Persing DH. 1993. Chronic Lyme borreliosis in the laboratory mouse. Am J Pathol 143:959–971. [PMC free article] [PubMed] [Google Scholar]
- 37.Schaible UE, Kramer MD, Museteanu C, Zimmer G, Mossmann H, Simon MM. 1989. The severe combined immunodeficiency (scid) mouse. A laboratory model for the analysis of Lyme arthritis and carditis. J Exp Med 170:1427–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruderman EM, Kerr JS, Telford SR, Spielman A III, Glimcher LH, Gravallese EM. 1995. Early murine Lyme carditis has a macrophage predominance and is independent of major histocompatibility complex class II-CD4+ T cell interactions. J Infect Dis 171:362–370. doi: 10.1093/infdis/171.2.362. [DOI] [PubMed] [Google Scholar]
- 39.Habicht GS, Beck G, Benach JL, Coleman JL, Leichtling KD. 1985. Lyme disease spirochetes induce human and murine interleukin 1 production. J Immunol 134:3147–3154. [PubMed] [Google Scholar]
- 40.Ma Y, Weis JJ. 1993. Borrelia burgdorferi outer surface lipoproteins OspA and OspB possess B-cell mitogenic and cytokine-stimulatory properties. Infect Immun 61:3843–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Radolf JD, Arndt LL, Akins DR, Curetty LL, Levi ME, Shen Y, Davis LS, Norgard MV. 1995. Treponema pallidum and Borrelia burgdorferi lipoproteins and synthetic lipopeptides activate monocytes/macrophages. J Immunol 154:2866–2877. [PubMed] [Google Scholar]
- 42.Seiler KP, Vavrin Z, Eichwald E, Hibbs JB Jr, Weis JJ. 1995. Nitric oxide production during murine Lyme disease: lack of involvement in host resistance or pathology. Infect Immun 63:3886–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown JP, Zachary JF, Teuscher C, Weis JJ, Wooten RM. 1999. Dual role of interleukin-10 in murine Lyme disease: regulation of arthritis severity and host defense. Infect Immun 67:5142–5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown CR, Reiner SL. 1999. Development of Lyme arthritis in mice deficient in inducible nitric oxide synthase. J Infect Dis 179:1573–1576. doi: 10.1086/314774. [DOI] [PubMed] [Google Scholar]
- 45.Barthold SW, deSouza M, Feng S. 1996. Serum-mediated resolution of Lyme arthritis in mice. Lab Invest 74:57–67. [PubMed] [Google Scholar]
- 46.Blaho VA, Mitchell WJ, Brown CR. 2008. Arthritis develops but fails to resolve during inhibition of cyclooxygenase 2 in a murine model of Lyme disease. Arthritis Rheum 58:1485–1495. doi: 10.1002/art.23371. [DOI] [PubMed] [Google Scholar]
- 47.Bolz DD, Sundsbak RS, Ma Y, Akira S, Kirschning CJ, Zachary JF, Weis JH, Weis JJ. 2004. MyD88 plays a unique role in host defense but not arthritis development in Lyme disease. J Immunol 173:2003–2010. doi: 10.4049/jimmunol.173.3.2003. [DOI] [PubMed] [Google Scholar]
- 48.Armstrong AL, Barthold SW, Persing DH, Beck DS. 1992. Carditis in Lyme disease susceptible and resistant strains of laboratory mice infected with Borrelia burgdorferi. Am J Trop Med Hyg 47:249–258. [DOI] [PubMed] [Google Scholar]
- 49.Bannenberg G, Moussignac RL, Gronert K, Devchand PR, Schmidt BA, Guilford WJ, Bauman JG, Subramanyam B, Perez HD, Parkinson JF, Serhan CN. 2004. Lipoxins and novel 15-epi-lipoxin analogs display potent anti-inflammatory actions after oral administration. Br J Pharmacol 143:43–52. doi: 10.1038/sj.bjp.0705912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Serhan CN, Savill J. 2005. Resolution of inflammation: the beginning programs the end. Nat Immunol 6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]