

Abstract
Recently, a number of studies have reported the presence of interleukin 17 (IL-17) in patients with Lyme disease, and several murine studies have suggested a role for this cytokine in the development of Lyme arthritis. However, the role of IL-17 has not been studied using the experimental Lyme borreliosis model of infection of C3H mice with Borrelia burgdorferi. In the current study, we investigated the role of IL-17 in the development of experimental Lyme borreliosis by infecting C3H mice devoid of the common IL-17 receptor A subunit (IL-17RA) and thus deficient in most IL-17 signaling. Infection of both C3H and C3H IL-17RA−/− mice led to the production of high levels of IL-17 in the serum, low levels in the heart tissue, and no detectable IL-17 in the joint tissue. The development and severity of arthritis and carditis in the C3H IL-17RA−/− mice were similar to what was seen in wild-type C3H mice. In addition, development of antiborrelia antibodies and clearance of spirochetes from tissues were similar for the two mouse strains. These results demonstrate a limited role for IL-17 signaling through IL-17RA in the development of disease following infection of C3H mice with B. burgdorferi.
INTRODUCTION
Despite much effort, the mechanisms responsible for the development of arthritis and carditis following infection with Borrelia burgdorferi remain unclear. According to the CDC, Lyme disease is the most commonly reported vector-borne disease in the United States with a reported 30,000 new cases each year (1). However, it is also estimated that the real incidence of Lyme disease may be much larger, with around 300,000 new cases per year (www.CDC.gov). Lyme disease is not endemic in every state and is present primarily in the upper midwest and northeastern states. B. burgdorferi is transmitted to humans through the bite of an infected Ixodes tick and if left untreated can lead to the development of arthritis, potentially life-threatening carditis, and neurological disease (2). Researchers studying the mechanisms of Lyme disease pathogenesis primarily use a murine model of experimental Lyme borreliosis in which disease-susceptible C3H mice are infected with B. burgdorferi and development of arthritis and carditis are followed over time (3). This model reproduces a portion of the disease seen in infected humans with Lyme disease, and the mechanisms of disease pathogenesis appear to correlate well between this experimental mouse model and human Lyme disease patients.
Interleukin 17 (IL-17) is a proinflammatory cytokine associated with chronic inflammatory conditions such as rheumatoid arthritis and multiple sclerosis (4, 5) and has been shown to play a role in collagen-induced arthritis in mice (6). Most IL-17 is produced by T cells (Th17), but innate immune cells such as γδ T cells, invariant natural killer T (iNKT) cells, and mast cells can also produce it (7, 8). The IL-17 family consists of 6 members, IL-17A to F, with IL-17A, IL-17E (also known as IL-25), and IL-17F as the most well-studied and understood members. These cytokines signal through a set of receptors (IL-17RA, IL-17RB, IL-17RC, IL-17RD, and IL-17RE), which most likely form heterodimers, although only a couple have been identified (9). IL-17A and IL-17F signal through a heterodimer of IL-17RA and IL-17RC, while IL-17E signals through a heterodimer of IL-17RA and IL-17RB (9). Binding partners for the other signaling chains have not yet been identified. Mice deficient in expression of the common IL-17RA receptor subunit have been reported and used to study the effects of IL-17 in a number of diseases. IL-17RA signaling stimulates production of neutrophilic chemokines, such as CXCL1, CXCL2, CXCL5, CXCL6, and CXCL8 (10). IL-17RA-deficient mice infected with Klebsiella pneumoniae quickly succumb to infection due to a failure to recruit a sufficient number of neutrophils (11). Similarly, IL-17RA-deficient mice infected with influenza also recruit significantly fewer neutrophils and suffer less-severe lung injury (10). In contrast, IL-17 was not required for neutrophil recruitment and clearance of Candida albicans (12). These results demonstrate the complex role of IL-17 during immune responses, especially to infectious agents.
B. burgdorferi can stimulate the production of IL-17 from various cell types and tissues, but its role in disease pathogenesis remains unclear. In mice, helper T cells primed in the presence of either B. burgdorferi or synthetic Borrelia outer surface proteins preferentially express IL-17 (13). Similarly, iNKT cells or spleen cells stimulated with B. burgdorferi produce low levels of IL-17 (14, 15), and bone marrow-derived dendritic cells (BMDC) produce IL-23, which can drive the production of IL-17 from T cells (16). These studies demonstrate that murine cells can be induced to produce IL-17 in response to exposure to Borrelial antigens. In humans, the correlation between B. burgdorferi exposure and IL-17 production is not as clear. Stimulation of peripheral blood mononuclear cells (PBMC) from normal donors did not induce IL-17 production (17), and no serum IL-17 could be detected in B. burgdorferi antibody-positive asymptomatic children (18). In contrast, IL-17 can be found in the cerebrospinal fluid of neuroborreliosis patients (19) and synovial T cells produce IL-17 in response to stimulation with the neutrophil-activating protein A (NapA) of B. burgdorferi (20). However, the incidence of prolonged Lyme disease symptoms such as arthritis was not altered in Lyme disease patients producing low levels of IL-17 due to the presence of an IL-23 genetic polymorphism (21). Thus, altered IL-17 signaling was not crucial for the pathogenesis of prolonged Lyme disease in humans.
In an elegant series of studies, Nardelli et al. have reported a critical role for IL-17 in the induction of murine Lyme arthritis. They first demonstrated that depletion of IL-17 could suppress the development of severe destructive arthritis in B. burgdorferi-vaccinated and -challenged gamma interferon (IFN-γ)-deficient mice (22) and that this was due to the induction of CD4+ CD25+ T regulatory cells (23, 24). Several cytokines important for the induction of IL-17 were also shown to have a role in the development of arthritis in this model (25, 26). However, these results have come from the use of a novel vaccination and challenge model of Lyme disease that relies upon the induction of T cell responses. In the C3H mouse infection model of Lyme disease, induction of T cells is not required for the development of Lyme arthritis or carditis (27), as disease pathogenesis is driven by innate immune cells. Indeed, while the production of IL-17 plays an important role in the development of Lyme arthritis in the vaccination and challenge model, IL-17 has been reported in the sera of B. burgdorferi-infected C3H mice in only a single study (28). Despite multiple attempts in our laboratory, we have not been able to demonstrate the production of IL-17 in the joints of infected C3H mice (C. Brown, unpublished observations), and similar results were recently reported by others (29). Thus, the importance of IL-17 in the pathogenesis of Lyme arthritis depends upon which model system is used, and how this relates to human disease has yet to be determined. In the current study, we infected C3H mice deficient in the IL-17RA subunit and followed arthritis and carditis development over time. We found no significant differences in the response to infection or development of disease between wild-type (WT) and IL-17RA-deficient mice. We conclude that IL-17 signaling through IL-17RA does not play any significant role in the development of Lyme borreliosis in the infection model of experimental Lyme disease.
MATERIALS AND METHODS
Animals.
Female WT C3H/HeJ mice, 4 to 6 weeks of age, were purchased from The Jackson Laboratory (Bar Harbor, ME). C57BL/6 IL-17RA-deficient mice were obtained from Immunex Corporation and backcrossed for 10 generations onto the C3H genetic background in our mouse colony. Heterozygous mice were then intercrossed to produce WT and C3H IL-17RA knockout mice for experiments. Animals were given sterile food and water ad libitum and housed in a specific-pathogen-free facility. All work was done in accordance with the Animal Care and Use Committee of the University of Missouri.
Bacteria and infections.
Frozen stocks of a virulent, passage 8, clonal isolate of B. burgdorferi strain N40 (a kind gift from Stephen Barthold) was used for all infections. Stocks were added to 7 ml C-BSKH medium (BSK-H medium containing 6% rabbit serum) (Sigma-Aldrich, St. Louis, MO) and grown to log phase at 32°C. Spirochetes were counted under dark-field microscopy using a Petroff-Hausser counting chamber (Hausser Scientific, Horsham, PA). Spirochete dilutions were made in sterile BSK-H medium, and the mice were inoculated in each hind footpad with 50 μl medium containing 5 × 104 B. burgdorferi organisms. Ankle swelling was measured weekly throughout the infection at the thickest craniocaudal portion of the joint using a metric caliper.
Assessment of pathology.
Infected WT C3H and C3H IL-17RA−/− mice were sacrificed on days 21 and 42 postinfection. One ankle joint and one-half of the heart were obtained for histological analysis. Tissues were fixed in 10% zinc–formalin and paraffin embedded, and sections were stained with hematoxylin and eosin (H&E). Ankle and heart sections were evaluated for disease severity and inflammatory cell type scores as previously described (30) on a scale of 0 to 4 with 0 representing no inflammation and 4 representing severe inflammation.
Cell isolation for flow cytometry.
At sacrifice, one ankle was removed from each mouse by removing the toes and carefully cutting through the knee joint to avoid bone marrow contamination. Excess muscle tissue was removed to reduce blood contamination. Ankles were placed in 15-ml conical tubes containing 5 ml 1× phosphate-buffered saline (PBS) and 4% fetal bovine serum (FBS), 75 μl (0.03 mg) DNase I (Sigma), and 50 μl of 100 mg/ml stock collagenase/dispase (Roche, Indianapolis, IN). Joints were placed on a rocker at room temperature for 1 h before being placed into sterile petri dishes with 5 ml of additional RPMI medium supplemented with 10% FBS. Ankles were carefully flayed apart using sterile rat tooth forceps. Cells were then strained through a 70-μm filter (BD Falcon) and washed with 5 ml 1× PBS with 4% FBS three times. Live cells were counted using trypan blue exclusion. A total of 1 × 106 cells were stained in a 96-well U bottom plate (Corning, Inc.). All wells were blocked using Fc Block (anti-CD16/CD32) for 15 min at 4°C. Cells were stained on ice in the dark using antibodies (eBioscience, San Diego, CA, and Leinco, St. Louis, MO) specific for the following cell types: CD45.2-PerCP-Cy5.5 (hematopoietic cells), F4/80-APC-eFluor780 (macrophages), CD3e-PE (T cells), B220-PECy7 (B cells), and Ly6g-APC (neutrophils). After staining, cells were washed and fixed in 1% paraformaldehyde for 15 min. Joint cellular infiltrate was analyzed using the Dako Cyan flow cytometer and Summit V5.0 software.
Quantification of IL-17.
Protein was isolated from tissue samples as previously described (31). Briefly, ankle joints and hearts were removed following sacrifice and immediately frozen in liquid nitrogen. The frozen samples were removed from liquid nitrogen, wrapped in aluminum foil, and pulverized using a hammer. The frozen tissue powder was then placed in 1 ml of Hanks balanced salt solution (HBSS) containing a protease inhibitor cocktail (Sigma). The samples were sonicated and then centrifuged at 8,000 rpm, 4°C, for 10 min. The supernatant was filtered through a 45-μm filter and the volume adjusted to 1.5 ml with HBSS. Total protein in each sample was quantified using a bicinchoninic acid (BCA) assay (Pierce Chemical Co., Rockford, IL), and IL-17 was measured using an IL-17A enzyme-linked immunosorbent assay (ELISA) from BioLegend (San Diego, CA). Results are presented in picograms of IL-17/milligram of protein. IL-17 levels in serum samples were also determined by ELISA with results reported in picograms/milliliter.
Quantification of B. burgdorferi in tissues.
Quantification of numbers of B. burgdorferi in tissue was completed using quantitative real-time PCR as described previously (31). Duplex PCR was performed on total DNA isolated from ear tissue, and quantification of spirochetes was determined by amplification of B. burgdorferi flaB and murine nidogen genes. Bacterial loads are expressed as copies of flagellin per 1,000 copies of nidogen as described previously (32).
Phagocytosis assay.
Neutrophil phagocytosis of B. burgdorferi was determined as described previously (33). Briefly, murine neutrophils were isolated from peripheral blood over a Percoll gradient. Isolated cells were washed and then incubated with green fluorescent protein (GFP)-expressing B. burgdorferi (a kind gift from James Carroll, NIH) for 2 h at a multiplicity of infection (MOI) of 10 (34). The cells were washed again and then stained for Ly6g. The percentages of Ly6g+ cells (neutrophils) expressing GFP (phagocytosed Borrelia) were determined using flow cytometry.
Borrelia-specific antibody production.
Levels of Borrelia-specific IgM and IgG in the serum were determined as described previously (35).
Statistical analysis.
Each experiment was completed at least twice with at least 5 mice per experimental group. GraphPad Prism software was used to determine statistical differences between group means. Student's t test or the Mann-Whitney U test for nonparametric data was used with α set at 0.05.
RESULTS
IL-17RA deficiency does not alter ankle swelling or disease severity.
To determine if IL-17 played a role in the infection model of experimental Lyme borreliosis, we infected C3H WT and C3H IL-17RA−/− mice with B. burgdorferi. The IL-17RA−/− mice are devoid of the IL-17 common receptor A subunit and are thus unresponsive to IL-17A, IL-17F, and IL-17E (9). Ankle swelling was monitored throughout the infection time course, and joints and hearts were harvested at days 21 and 42 postinfection. Despite an inability to respond to IL-17, B. burgdorferi-infected IL-17RA−/− mice displayed no differences in ankle swelling compared to wild-type controls over the 42-day infection time course (Fig. 1). To determine if there were differences in disease development, we examined H&E-stained histology sections of ankles and joints. These sections were evaluated in a blinded manner and scored for their overall level of inflammation (severity score) and for the ratio of neutrophils to macrophages (type score) as described previously (30). Higher type scores are interpreted as acute inflammation and lower scores as resolving inflammation. Overall, there were no differences in the severity scores or the type scores in the ankles or hearts between the WT and IL-17RA−/− mice on day 21 or 42 postinfection (Table 1). All scores were higher at the day 21 postinfection time point, which is typically near the peak of inflammation (Fig. 1), and lower scores were associated with the resolution of inflammation at day 42 postinfection. These results indicate that the IL-17RA−/− mice develop Lyme arthritis and carditis and resolve their disease in a manner similar to that of WT mice.
FIG 1.
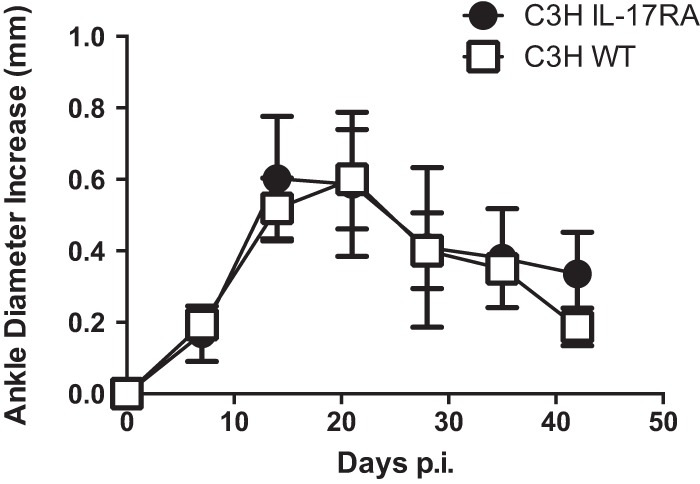
C3H WT and C3H IL-17RA-deficient mice were infected with B. burgdorferi, and ankle swelling was monitored throughout the infection time course. n = 5 per group.
TABLE 1.
Ankle and heart severity scoresa
| Mouse strain | Day postinfection | Arthritis score |
Carditis score |
||
|---|---|---|---|---|---|
| Severity | Type | Severity | Type | ||
| WT | 21 | 3.0 ± 0.0 | 3.0 ± 0.0 | 4.0 ± 0.0 | 4.0 ± 0.0 |
| IL-17RA | 21 | 3.6 ± 0.5 | 3.2 ± 1.2 | 3.3 ± 0.8 | 3.7 ± 0.5 |
| WT | 42 | 1.5 ± 0.7 | 1.0 ± 0.0 | 2.0 ± 0.0 | 2.0 ± 1.4 |
| IL-17RA | 42 | 2.2 ± 1.1 | 2.2 ± 1.1 | 1.7 ± 0.6 | 2.0 ± 1.7 |
Scores were determined from H&E-stained sections of tibiotarsal joints and hearts or B. burgdorferi-infected mice. Data are means ± standard deviations.
Cellular infiltration into joint tissue during Lyme arthritis.
IL-17RA−/− mice have been shown to have lowered levels of circulating neutrophils (36). We have previously shown that altering circulating neutrophil levels can have an effect on Lyme arthritis development (37). In addition, IL-17 can regulate neutrophil recruitment to sites of infection by inducing the production of the chemokine KC, which we have shown is critical for the development of Lyme arthritis (30). To examine the effect of IL-17RA deficiency on the recruitment of inflammatory cells into the infected joint, we used flow cytometry to analyze cellular infiltration into the joint tissue. Single-cell suspensions were made from joint tissue, and cells were first gated on live cells and doublets excluded. Next, the cells were gated on CD45.2+ hematopoietic (inflammatory) cells and analyzed for the infiltration of macrophages (F4/80), mature neutrophils (Ly6ghi), B cells (B220/IgM), and T cells (CD3e). Despite the reported neutropenia in IL-17RA−/− mice (36), we found no significant differences in the total number of recruited inflammatory cells (Fig. 2A) or in the numbers of neutrophils recruited to the joints (Fig. 2B) between WT and IL-17RA−/− mice. In addition, we found no differences in macrophage (Fig. 2C), B cell (Fig. 2D), or T cell (Fig. 2E) recruitment between the wild-type and knockout mice. Thus, recruitment of inflammatory cells to sites of infection did not appear to be compromised in the IL-17RA−/− mice.
FIG 2.

Cellular infiltrates into infected joints. Ankle joints of B. burgdorferi-infected C3H WT (white bars) and C3H IL-17RA-deficient (black bars) mice were analyzed for cellular infiltration using flow cytometry. Numbers of total inflammatory cells (A), neutrophils (B), macrophages (C), B cells (D), and T cells (E) were evaluated on days 21 and 42 postinfection using cell-specific markers. n = 5 per group.
IL-17RA-deficient mice efficiently clear B. burgdorferi.
Clearance of B. burgdorferi from tissues by specific antibody production is thought to mediate disease resolution (38). We recently reported that mice deficient in cyclooxygenase 1 (COX-1) and infected with B. burgdorferi made a good Borrelia-specific IgM response but were defective in class switching to IgG (28). This defect correlated with low levels of IL-17 in the serum and could be rescued by exogenous delivery of IL-17. Thus, it was of interest to determine the production of Borrelia-specific antibody responses in the IL-17RA−/− mice. We found that the IL-17RA−/− mice had slightly higher levels of Borrelia-specific IgM and slightly lower levels of Borrelia-specific IgG on day 24 of infection, but these differences were not statistically significant (Fig. 3). In addition, spirochete loads and clearance from ear tissue in the IL-17RA−/− mice were no different than those in the WT mice (Fig. 4). IL-17 has also been shown to increase phagocytosis in neutrophils (39); therefore, we were curious to see if the lack of IL-17 signaling would impair neutrophil clearance of B. burgdorferi. Neutrophils were isolated from WT and IL-17RA−/− mice and cultured with GFP-B. burgdorferi for 2 h at an MOI of 10. The uptake of labeled spirochetes was then assessed by flow cytometry. Neutrophils from WT and IL-17RA−/− mice contained similar levels of GFP-Borrelia (Fig. 5) indicating that a lack of IL-17 signaling had no effect on neutrophil phagocytosis of spirochetes. Together these results demonstrate no functional defects in the host response to B. burgdorferi infection in IL-17 RA−/− mice.
FIG 3.
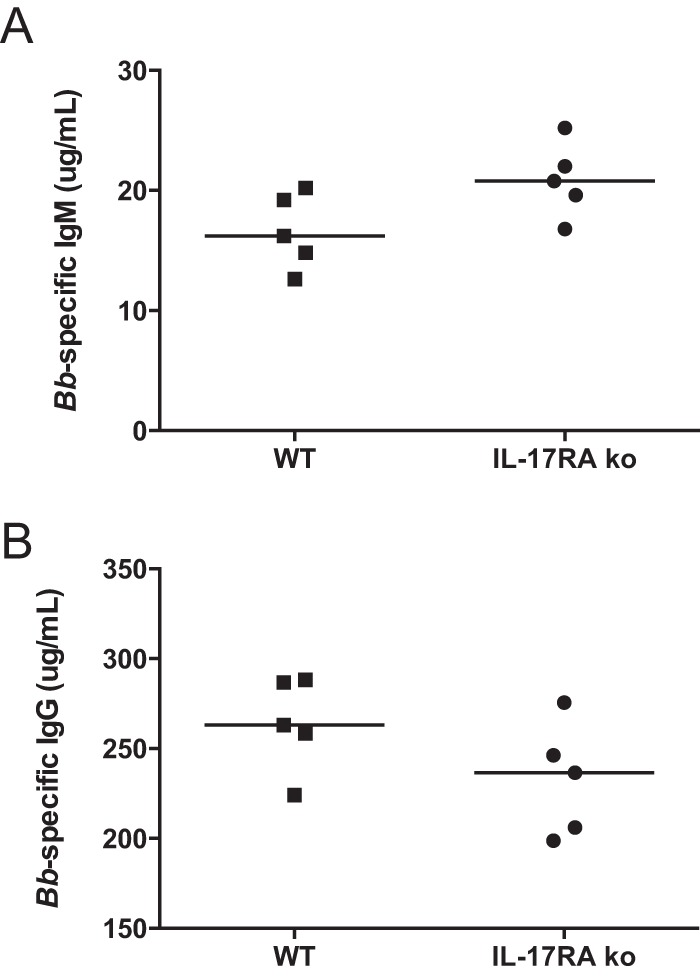
Quantification of Borrelia-specific antibody in sera of infected mice. Levels of B. burgdorferi-specific IgM (A) and IgG (B) were determined from sera of B. burgdorferi-infected C3H WT and C3H IL-17RA-deficient mice at day 24 postinfection. Each symbol represents an individual mouse. The bar represents the group median.
FIG 4.
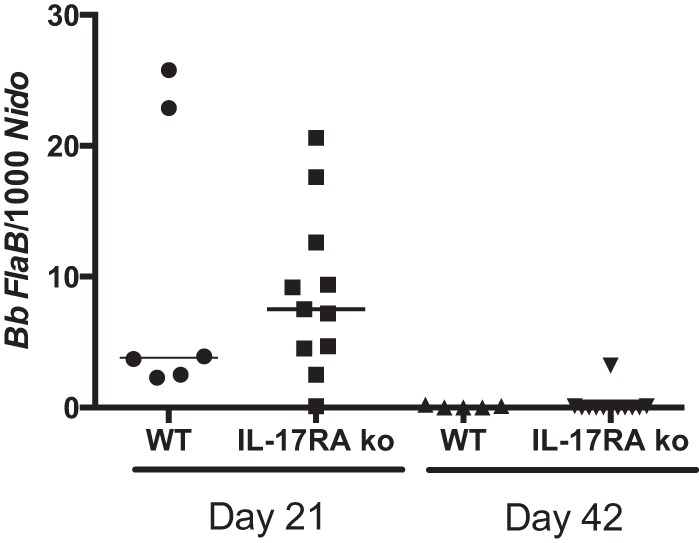
Effect of IL-17RA deficiency on levels of B. burgdorferi in tissue. DNA was isolated from ear tissue of B. burgdorferi-infected C3H WT and C3H IL-17RA-deficient mice at days 21 and 42 postinfection. Each symbol represents an individual mouse. The bar represents the group median.
FIG 5.
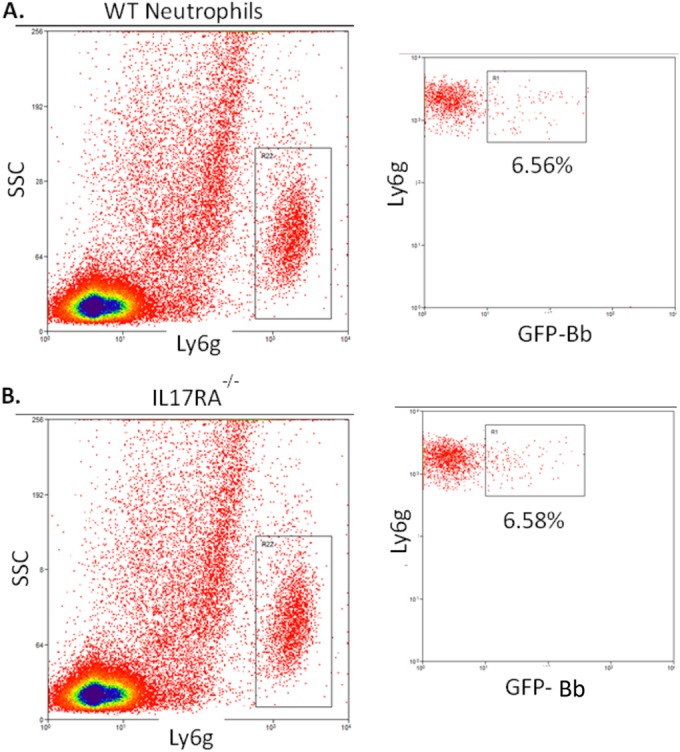
Quantification of neutrophil phagocytic capacity. Representative flow cytometry of neutrophils from B. burgdorferi-infected C3H WT (A) and C3H IL-17RA-deficient (B) mice. Neutrophils (Ly6g+) were gated and analyzed for their expression of GFP (internalized B. burgdorferi). The percentage of total neutrophils is shown.
IL-17 production in B. burgdorferi-infected mice.
IL-17 has been shown to be important in the vaccination and challenge model of Lyme disease (22). We have previously reported IL-17 in the serum of B. burgdorferi-infected mice (28) but have been unable to demonstrate it in infected joint tissue (Brown, unpublished), and similar results were reported by Nardelli et al. (29). As we previously reported, IL-17 was present in the sera of WT and IL-17RA−/− mice (Fig. 6A). The IL-17RA−/− mice tended to have higher levels than the WT mice, but this was not significant and may be due to their inability to use IL-17. We also found low levels of IL-17 in the heart tissue of both WT and IL-17RA−/− mice on days 21 and 42 postinfection (Fig. 6B). However, since the hearts were not perfused it is possible that this was due to residual blood contamination and not to the production of IL-17 directly within the heart tissue. Finally, we were unable to detect IL-17 in the joints of either WT or IL-17RA−/− mice (data not shown). These results demonstrate the systemic production of IL-17 in the sera of mice infected with B. burgdorferi but little or no IL-17 at sites of disease pathogenesis.
FIG 6.
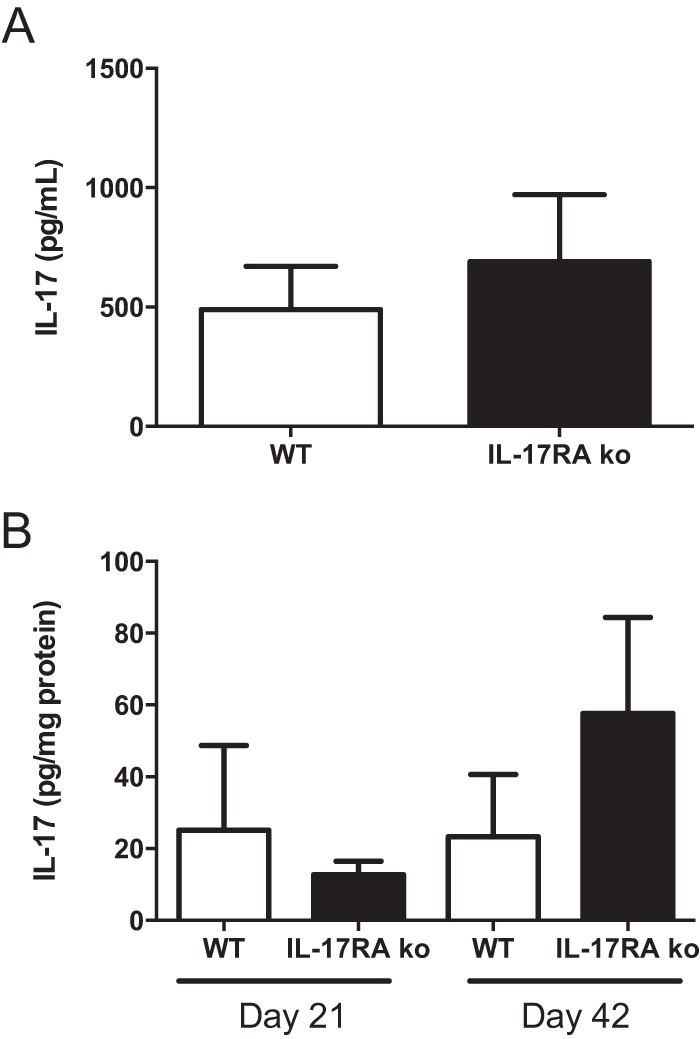
Quantification of IL-17 in sera and tissues from infected mice. Levels of IL-17A from B. burgdorferi-infected C3H WT (white bars) and C3H IL-17RA-deficient (black bars) mice were determined from day 21 serum (A) or heart tissue (B) at days 21 and 42 postinfection. IL-17A was not detectable in sera or tissues from uninfected mice or from ankle tissues of infected mice (data not shown). n = 5 per group.
DISCUSSION
IL-17 is an important proinflammatory cytokine produced by Th17 cells and other innate immune cells (8). It is thought to play an important role in the pathogenesis of rheumatoid and osteoarthritis (40); however, it is also an important regulator of host defense through granulopoiesis and neutrophil recruitment (41). Several studies have proposed that IL-17 may play a role in neuroborreliosis and Lyme arthritis in humans (19, 20), although this remains controversial (17, 18). Using a murine vaccination and challenge model system, others have found that IL-17 directly impacts Lyme arthritis development (22). IFN-γ-deficient B6 mice infected with strain 297 of B. burgdorferi and challenged with the C-1-11 strain developed severe destructive arthritis (22). When these mice were treated with IL-17 neutralizing antibodies, the development of arthritis was inhibited (22). When the treatment was combined with the administration of anti-transforming growth factor β (TGF-β) and anti-IL-6 antibodies, arthritis development and ankle swelling were completely eliminated (26). Treatment of B. burgdorferi-vaccinated and -challenged C3H/HeN mice with anti-IL-17 antibody decreased arthritis severity, but not to the same extent as in B6 mice (29). As stated above, the vaccination and challenge model of Lyme arthritis relies upon the induction of T cell responses and cross-reactive epitopes on antigens from two different strains of B. burgdorferi. Thus, disease induction in this model is mediated by adaptive immunity. In contrast, disease induction following infection of C3H mice with B. burgdorferi is induced by innate immunity and can occur independently of T cell involvement (42, 43). Infection of C3H mice with B. burgdorferi strain 297 did not induce the production of IL-17 in the sera of these mice (29). This is in contrast to what we have previously reported (28) and to results from the current study, in which we find systemic IL-17 production. Differences in the B. burgdorferi strains used (297 versus N40) or infectious doses may account for differences in reported results.
To assess the contribution of IL-17 to disease pathogenesis in the experimental Lyme borreliosis infection model, we generated IL-17RA−/− mice on the susceptible C3H genetic background (44).
Signaling through IL-17RA has been shown to be important in the development of collagen-induced arthritis (45); however, the role of this cytokine in the development of arthritis using an infectious model had not been tested. When IL-17RA−/− mice were infected with B. burgdorferi, we found no differences in ankle swelling or arthritis severity scores at any time point throughout the infection time course compared with WT mice. Similar results were found for Lyme carditis. Due to the importance of IL-17 in mediating neutrophil recruitment by regulating the production of chemokines (11) and the importance of KC/CXCR2 signaling for the development of Lyme arthritis (30, 31), we anticipated seeing less-severe inflammation. In an infectious model of acute lung injury, the absence of IL-17R signaling resulted in less lung destruction due to decreased production of reactive oxygen intermediates (10). Similar results were reported for Toxoplasma gondii (46) and Klebsiella pneumoniae (11) infections. In infection models, less inflammation is usually associated with significantly reduced neutrophil recruitment and higher bacterial loads. Because of the critical role of neutrophils in the development of Lyme arthritis, we set out to assess neutrophil recruitment in the B. burgdorferi-infected IL-17RA−/− mice. Surprisingly, we found that the numbers of infiltrating cells of IL-17RA−/− mice at days 21 and 42 postinfection were similar to those of WT controls. Thus, the development of Lyme arthritis and carditis in the infection model of Lyme disease was not impacted by the loss of IL-17 signaling.
While the presence of the spirochetes in tissues is required for the development of disease, their absolute numbers do not correlate with disease severity (47, 48). Thus, it is possible that while we did not find differences in disease severity, there could still be differences in spirochete clearance in the IL-17RA−/− mice. The production of specific antibody to B. burgdorferi is thought to be primarily responsible for spirochete clearance from tissues (38). IL-17 has been shown to play a role in antibody production, germinal center formation, and antibody class switching in autoimmune disease (49), and thus a defect in IL-17 signaling could impact spirochete clearance. We measured the levels of B. burgdorferi-specific IgM and IgG and found no significant differences between IL-17RA−/− mice and WT mice. In addition, spirochete loads in tissues were also similar between the two mouse strains, as was the ability of neutrophils to phagocytose GFP-labeled spirochetes. Taken together, we found no significant effects on the host response to B. burgdorferi infection in mice deficient in IL-17RA. We conclude that there is no role for IL-17 signaling through IL-17RA in the development of disease or host response in the infectious experimental model of Lyme borreliosis.
REFERENCES
- 1.CDC. 2010. Final 2009 reports of nationally notifiable infectious diseases. MMWR Morb Mortal Wkly Rep 59:1025–1039. [Google Scholar]
- 2.Bockenstedt LK, Wormser GP. 2014. Unraveling Lyme disease. Arthritis Rheumatol 66:2313–2323. doi: 10.1002/art.38756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wooten RM, Weis JJ. 2001. Host-pathogen interactions promoting inflammatory Lyme arthritis: use of mouse models for dissection of disease processes. Curr Opin Microbiol 4:274–279. doi: 10.1016/S1369-5274(00)00202-2. [DOI] [PubMed] [Google Scholar]
- 4.Leipe J, Grunke M, Dechant C, Reindl C, Kerzendorf U, Schulze-Koops H, Skapenko A. 2010. Role of Th17 cells in human autoimmune arthritis. Arthritis Rheum 62:2876–2885. doi: 10.1002/art.27622. [DOI] [PubMed] [Google Scholar]
- 5.Millward JM, Lobner M, Wheeler RD, Owens T. 2010. Inflammation in the central nervous system and Th17 responses are inhibited by IFN-gamma-induced IL-18 binding protein. J Immunol 185:2458–2466. doi: 10.4049/jimmunol.0902153. [DOI] [PubMed] [Google Scholar]
- 6.Mus AM, Cornelissen F, Asmawidjaja PS, van Hamburg JP, Boon L, Hendriks RW, Lubberts E. 2010. Interleukin-23 promotes Th17 differentiation by inhibiting T-bet and FoxP3 and is required for elevation of interleukin-22, but not interleukin-21, in autoimmune experimental arthritis. Arthritis Rheum 62:1043–1050. doi: 10.1002/art.27336. [DOI] [PubMed] [Google Scholar]
- 7.Jin W, Zhao Y, Yan W, Cao L, Zhang W, Wang M, Zhang T, Fu Q, Li Z. 2012. Elevated circulating interleukin-27 in patients with coronary artery disease is associated with dendritic cells, oxidized low-density lipoprotein, and severity of coronary artery stenosis. Mediators Inflamm 2012:506283. doi: 10.1155/2012/506283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cua DJ, Tato CM. 2010. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol 10:479–489. doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- 9.Zepp J, Wu L, Li X. 2011. IL-17 receptor signaling and T helper 17-mediated autoimmune demyelinating disease. Trends Immunol 32:232–239. doi: 10.1016/j.it.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crowe CR, Chen K, Pociask DA, Alcorn JF, Krivich C, Enelow RI, Ross TM, Witztum JL, Kolls JK. 2009. Critical role of IL-17RA in immunopathology of influenza infection. J Immunol 183:5301–5310. doi: 10.4049/jimmunol.0900995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. 2001. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 194:519–528. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trautwein-Weidner K, Gladiator A, Nur S, Diethelm P, LeibundGut-Landmann S. 2015. IL-17-mediated antifungal defense in the oral mucosa is independent of neutrophils. Mucosal Immunol 8:221–231. doi: 10.1038/mi.2014.57. [DOI] [PubMed] [Google Scholar]
- 13.Infante-Duarte C, Horton HF, Byrne MC, Kamradt T. 2000. Microbial lipopeptides induce the production of IL-17 in Th cells. J Immunol 165:6107–6115. doi: 10.4049/jimmunol.165.11.6107. [DOI] [PubMed] [Google Scholar]
- 14.Michel ML, Keller AC, Paget C, Fujio M, Trottein F, Savage PB, Wong CH, Schneider E, Dy M, Leite-de-Moraes MC. 2007. Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J Exp Med 204:995–1001. doi: 10.1084/jem.20061551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oosting M, van de Veerdonk FL, Kanneganti TD, Sturm P, Verschueren I, Berende A, van der Meer JW, Kullberg BJ, Netea MG, Joosten LA. 2011. Borrelia species induce inflammasome activation and IL-17 production through a caspase-1-dependent mechanism. Eur J Immunol 41:172–181. doi: 10.1002/eji.201040385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knauer J, Siegemund S, Muller U, Al-Robaiy S, Kastelein RA, Alber G, Straubinger RK. 2007. Borrelia burgdorferi potently activates bone marrow-derived conventional dendritic cells for production of IL-23 required for IL-17 release by T cells. FEMS Immunol Med Microbiol 49:353–363. doi: 10.1111/j.1574-695X.2006.00210.x. [DOI] [PubMed] [Google Scholar]
- 17.Bachmann M, Horn K, Rudloff I, Goren I, Holdener M, Christen U, Darsow N, Hunfeld K-P, Koehl U, Kind P, Pfeilschifter J, Kraiczy P, Mühl H. 2010. Early production of IL-22 but not IL-17 by peripheral blood mononuclear cells exposed to live Borrelia burgdorferi: the role of monocytes and interleukin-1. PLoS Pathog 6:e1001144. doi: 10.1371/journal.ppat.1001144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skogman BH, Hellberg S, Ekerfelt C, Jenmalm MC, Forsberg P, Ludvigsson J, Bergstrom S, Ernerudh J. 2012. Adaptive and innate immune responsiveness to Borrelia burgdorferi sensu lato in exposed asymptomatic children and children with previous clinical Lyme borreliosis. Clin Dev Immunol 2012:294587. doi: 10.1155/2012/294587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henningsson AJ, Tjernberg I, Malmvall BE, Forsberg P, Ernerudh J. 2011. Indications of Th1 and Th17 responses in cerebrospinal fluid from patients with Lyme neuroborreliosis: a large retrospective study. J Neuroinflammation 8:36. doi: 10.1186/1742-2094-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Codolo G, Amedei A, Steere AC, Papinutto E, Cappon A, Polenghi A, Benagiano M, Paccani SR, Sambri V, Del PG, Baldari CT, Zanotti G, Montecucco C, D'Elios MM, de Bernard M. 2008. Borrelia burgdorferi NapA-driven Th17 cell inflammation in Lyme arthritis. Arthritis Rheum 58:3609–3617. doi: 10.1002/art.23972. [DOI] [PubMed] [Google Scholar]
- 21.Oosting M, ter Hofstede H, van de Veerdonk FL, Sturm P, Kullberg BJ, van der Meer JWM, Netea MG, Joosten LAB. 2011. Role of interleukin-23 (IL-23) receptor signaling for IL-17 responses in human Lyme disease. Infect Immun 79:4681–4687. doi: 10.1128/IAI.05242-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burchill MA, Nardelli DT, England DM, DeCoster DJ, Christopherson JA, Callister SM, Schell RF. 2003. Inhibition of interleukin-17 prevents the development of arthritis in vaccinated mice challenged with Borrelia burgdorferi. Infect Immun 71:3437. doi: 10.1128/IAI.71.6.3437-3442.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nardelli DT, Burchill MA, England DM, Torrealba J, Callister SM, Schell RF. 2004. Association of CD4+ CD25+ T cells with prevention of severe destructive arthritis in Borrelia burgdorferi-vaccinated and challenged gamma interferon-deficient mice treated with anti-interleukin-17 antibody. Clin Diagn Lab Immunol 11:1075–1084. doi: 10.1128/CDLI.11.6.1075-1084.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nardelli DT, Cloute JP, Luk KHK, Torrealba J, Warner TF, Callister SM, Schell RF. 2005. CD4+ CD25+ T cells prevent arthritis associated with Borrelia vaccination and infection. Clin Diagn Lab Immunol 12:786–792. doi: 10.1128/CDLI.12.6.786-792.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kotloski NJ, Nardelli DT, Peterson SH, Torrealba JR, Warner TF, Callister SM, Schell RF. 2008. Interleukin-23 is required for development of arthritis in mice vaccinated and challenged with Borrelia species. Clin Vaccine Immunol 15:1199–1207. doi: 10.1128/CVI.00129-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nardelli DT, Luk KHK, Kotloski NJ, Warner TF, Torrealba JR, Callister SM, Schell RF. 2008. Role of IL-17, transforming growth factor-beta, and IL-6 in the development of arthritis and production of anti-outer surface protein A borreliacidal antibodies in Borrelia-vaccinated and -challenged mice. FEMS Immunol Med Microbiol 53:265–274. doi: 10.1111/j.1574-695X.2008.00431.x. [DOI] [PubMed] [Google Scholar]
- 27.Brown CR, Reiner SL. 1999. Genetic control of experimental Lyme arthritis in the absence of specific immunity. Infect Immun 67:1967–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blaho VA, Buczynski MW, Dennis EA, Brown CR. 2009. Cyclooxygenase-1 orchestrates germinal center formation and antibody class-switch via regulation of IL-17. J Immunol 183:5644–5653. doi: 10.4049/jimmunol.0901499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nardelli DT, Luedtke JO, Munson EL, Warner TF, Callister SM, Schell RF. 2010. Significant differences between the Borrelia-infection and Borrelia-vaccination and -infection models of Lyme arthritis in C3H/HeN mice. FEMS Immunol Med Microbiol 60:78–89. doi: 10.1111/j.1574-695X.2010.00721.x. [DOI] [PubMed] [Google Scholar]
- 30.Ritzman AM, Hughes-Hanks JM, Blaho VA, Wax LE, Mitchell WJ, Brown CR. 2010. The chemokine receptor CXCR2 ligand KC (CXCL1) mediates neutrophil recruitment and is critical for development of experimental Lyme arthritis and carditis. Infect Immun 78:4593–4600. doi: 10.1128/IAI.00798-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown CR, Blaho VA, Loiacono CM. 2003. Susceptibility to experimental Lyme arthritis correlates with KC and monocyte chemoattractant protein-1 production in joints and requires neutrophil recruitment via CXCR2. J Immunol 171:893–901. doi: 10.4049/jimmunol.171.2.893. [DOI] [PubMed] [Google Scholar]
- 32.Morrison TB, Ma Y, Weis JH, Weis JJ. 1999. Rapid and sensitive quantification of Borrelia burgdorferi-infected mouse tissues by continuous fluorescent monitoring of PCR. J Clin Microbiol 37:987–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blaho VA, Zhang Y, Hughes-Hanks JM, Brown CR. 2011. 5-Lipoxygenase-deficient mice infected with Borrelia burgdorferi develop persistent arthritis. J Immunol 186:3076–3084. doi: 10.4049/jimmunol.1003473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carroll JA, Stewart PE, Rosa P, Elias AF, Garon CF. 2003. An enhanced GFP reporter system to monitor gene expression in Borrelia burgdorferi. Microbiology 149:1819–1828. doi: 10.1099/mic.0.26165-0. [DOI] [PubMed] [Google Scholar]
- 35.Blaho VA, Mitchell WJ, Brown CR. 2008. Arthritis develops but fails to resolve during inhibition of cyclooxygenase 2 in a murine model of Lyme disease. Arthritis Rheum 58:1485–1495. doi: 10.1002/art.23371. [DOI] [PubMed] [Google Scholar]
- 36.von Vietinghoff S, Ley K. 2009. IL-17A controls IL-17F production and maintains blood neutrophil counts in mice. J Immunol 183:865–873. doi: 10.4049/jimmunol.0804080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown CR, Blaho VA, Loiacono CM. 2004. Treatment of mice with the neutrophil-depleting antibody RB6-8C5 results in early development of experimental Lyme arthritis via the recruitment of Gr-1- polymorphonuclear leukocyte-like cells. Infect Immun 72:4956–4965. doi: 10.1128/IAI.72.9.4956-4965.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barthold SW, Hodzic E, Tunev S, Feng S. 2006. Antibody-mediated disease remission in the mouse model of Lyme borreliosis. Infect Immun 74:4817–4825. doi: 10.1128/IAI.00469-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsu SC, Wang LT, Yao CL, Lai HY, Chan KY, Liu BS, Chong P, Lee OK, Chen HW. 2013. Mesenchymal stem cells promote neutrophil activation by inducing IL-17 production in CD4+ CD45RO+ T cells. Immunobiology 218:90–95. doi: 10.1016/j.imbio.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 40.van Baarsen LG, Lebre MC, van der Coelen D, Aarrass S, Tang MW, Ramwadhdoebe TH, Gerlag DM, Tak PP. 2014. Heterogeneous expression pattern of interleukin 17A (IL-17A), IL-17F and their receptors in synovium of rheumatoid arthritis, psoriatic arthritis and osteoarthritis: possible explanation for nonresponse to anti-IL-17 therapy? Arthritis Res Ther 16:426. doi: 10.1186/s13075-014-0426-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu JJ, Ruddy MJ, Wong GC, Sfintescu C, Baker PJ, Smith JB, Evans RT, Gaffen SL. 2007. An essential role for IL-17 in preventing pathogen-initiated bone destruction: recruitment of neutrophils to inflamed bone requires IL-17 receptor-dependent signals. Blood 109:3794–3802. doi: 10.1182/blood-2005-09-010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaible UE, Kramer MD, Museteanu C, Zimmer G, Mossmann H, Simon MM. 1989. The severe combined immunodeficiency (scid) mouse. A laboratory model for the analysis of Lyme arthritis and carditis. J Exp Med 170:1427–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barthold SW, Sidman CL, Smith AL. 1992. Lyme borreliosis in genetically resistant and susceptible mice with severe combined immunodeficiency. Am J Trop Med Hyg 47:605–613. [DOI] [PubMed] [Google Scholar]
- 44.Barthold SW, Beck DS, Hansen GM, Terwilliger GA, Moody KD. 1990. Lyme borreliosis in selected strains and ages of laboratory mice. J Infect Dis 162:133–138. doi: 10.1093/infdis/162.1.133. [DOI] [PubMed] [Google Scholar]
- 45.Corneth OBJ, Mus AMC, Asmawidjaja PS, Klein Wolterink RGJ, van Nimwegen M, Maarten DB, Hofman Y, Hendriks RW, Lubberts E. 2014. Absence of interleukin-17 receptor A signaling prevents autoimmune inflammation of the joint and leads to a Th2-like phenotype in collagen-induced arthritis. Arthritis Rheumatol 66:340–349. doi: 10.1002/art.38229. [DOI] [PubMed] [Google Scholar]
- 46.Guiton R, Zagani R, Dimier-Poisson I. 2009. Major role for CD8 T cells in the protection against Toxoplasma gondii following dendritic cell vaccination. Parasite Immunol 31:631–640. doi: 10.1111/j.1365-3024.2009.01146.x. [DOI] [PubMed] [Google Scholar]
- 47.Ma Y, Seiler KP, Eichwald EJ, Weis JH, Teuscher C, Weis JJ. 1998. Distinct characteristics of resistance to Borrelia burgdorferi-induced arthritis in C57BL/6N mice. Infect Immun 66:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown CR, Reiner SL. 1998. Clearance of Borrelia burgdorferi may not be required for resistance to experimental Lyme arthritis. Infect Immun 66:2065–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, Yi J, Guentert T, Tousson A, Stanus AL, Le Tv Lorenz RG, Xu H, Kolls JK, Carter RH, Chaplin DD, Williams RW, Mountz JD. 2008. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol 9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]