

Abstract
The metabotropic glutamate (mGlu) receptors are a group of eight family C G protein–coupled receptors that are expressed throughout the central nervous system (CNS) and periphery. Within the CNS the different subtypes are found in neurons, both pre- and/or postsynaptically, where they mediate modulatory roles and in glial cells. The mGlu receptor family provides attractive targets for numerous psychiatric and neurologic disorders, with the majority of discovery programs focused on targeting allosteric sites, with allosteric ligands now available for all mGlu receptor subtypes. However, the development of allosteric ligands remains challenging. Biased modulation, probe dependence, and molecular switches all contribute to the complex molecular pharmacology exhibited by mGlu receptor allosteric ligands. In recent years we have made significant progress in our understanding of this molecular complexity coupled with an increased understanding of the structural basis of mGlu allosteric modulation.
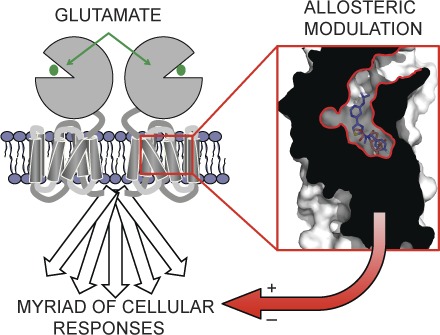
Introduction
Glutamate is the major excitatory neurotransmitter in the central nervous system (CNS), eliciting its effects via two distinct receptor classes. The ionotropic glutamate receptors are ligand-gated ion channels that mediate fast synaptic responses. The metabotropic glutamate (mGlu) receptors are G protein–coupled receptors (GPCRs) that play a modulatory role in the CNS. Ubiquitously expressed throughout the CNS in glia and neurons, found both pre- and postsynaptically, the mGlu receptors are involved in a broad range of physiologic functions in the brain. Comprised of eight members, the mGlu receptor family is often subclassified into three groups based on homology, pharmacology, and G protein coupling. Group I includes mGlu1 and mGlu5 that preferentially couple to Gq/11 and are typically found postsynaptically. Group II, mGlu2 and mGlu3; and group III, mGlu4, mGlu6, mGlu7, and mGlu8, members couple to Gi/o and with the exception of mGlu6, which is exclusively found in the retina, are predominantly located presynaptically. Multiple recent reviews have extensively discussed the roles of mGlu receptors in different neuronal circuits, modulating synaptic transmission, neuronal excitability, and other cellular functions within the CNS (Conn and Pin, 1997; Anwyl, 1999; Valenti et al., 2002; Coutinho and Knopfel, 2002; Bellone et al., 2008; Pinheiro and Mulle, 2008) as well as physiology in the periphery (Julio-Pieper et al., 2011). Based on this understanding, targeting activation or inhibition of specific mGlu receptor subtypes may provide a therapeutic benefit for a range of psychiatric and neurologic disorders. For neuropathic pain, inhibition of mGlu1 is being pursued as a therapeutic intervention with success reported in preclinical models (Mabire et al., 2005; Bennett et al., 2012). Agonists of group II receptors have reached clinical trials for schizophrenia and anxiety with varying success (Schoepp et al., 2003; Patil et al., 2007; Dunayevich et al., 2008; Kinon et al., 2011). Group II mGlu receptors are also attractive targets for addiction and depression (Ma et al., 2007; Moussawi and Kalivas, 2010; Chaki et al., 2013). Inhibitors of mGlu5 have demonstrated efficacy in preclinical studies for anxiety, depression, and Parkinson’s disease Levodopa-induced dyskinesias (Porter et al., 2005; Li et al., 2006; Belozertseva et al., 2007; Rylander et al., 2010; Hughes et al., 2013). Promisingly, multiple mGlu5 inhibitors have reached (Berry-Kravis et al., 2009; Keywood et al., 2009; Zerbib et al., 2010) or are currently in phase II clinical trials (www.clinicalTrials.gov) with varying successes and failures for fragile X syndrome, Parkinson’s disease Levodopa-induced dyskinesias, gastroesophogeal reflux disorder, and migraine. On the other hand, activators and/or potentiators of mGlu5 have efficacy in preclinical models for schizophrenia and cognition (Moghaddam, 2004; Noetzel et al., 2012a). Activation or potentiation of group III mGlu receptors, in particular mGlu4, has therapeutic potential for Parkinson’s disease (Marino et al., 2003; Jones et al., 2012), with mGlu4 also a potential target for pain (Vilar et al., 2013) and autism (Becker et al., 2014).
Therapeutic Targeting of mGlus with Allosteric Modulators
The prevailing strategy for GPCR-based drug discovery has been to target the endogenous ligand binding site, referred to as an orthosteric site, to competitively block or mimic the actions of the endogenous ligand. However, multiple receptor families and transporters recognize glutamate, and as such, glutamate binding pockets are highly conserved such that orthosteric ligands often lack sufficient selectivity. Based on this and difficulties in optimizing glutamate analogs as drug candidates, mGlu receptor-based drug discovery programs have turned their attention to alternative strategies to develop more selective agents. One such approach is to target allosteric binding sites that are topographically distinct from the orthosteric site, such that the receptor can be simultaneously bound by more than one ligand. These ligands are referred to as allosteric modulators and have the capacity to perturb the affinity and/or efficacy of an orthosteric ligand. Because allosteric sites are often located in regions of the receptor that show greater diversity between subtypes, allosteric modulators offer the potential for greater subtype selectivity than their orthosteric counterparts. Allosteric modulators may potentiate or inhibit the affinity and/or efficacy of an orthosteric ligand, a property referred to as “cooperativity.” A potentiator that has positive cooperativity with the orthosteric ligand is a positive allosteric modulator (PAM), whereas an inhibitor is a negative allosteric modulator (NAM). Cooperativity is saturable, therefore allosteric modulators have the theoretical advantage of being safer in the case of overdose, as there is a limit to the modulatory effect. A third class of ligands that interact with allosteric sites but have neutral cooperativity with the orthosteric ligand are called neutral allosteric ligands and represent excellent pharmacological tools for dissecting allosteric modulator pharmacology (Christopoulos et al., 2014). A third advantage of allosteric modulators is the potential to maintain spatial and temporal aspects of receptor activation, i.e., modulation will only occur when and where the orthosteric agonist is present. However, it is important to note that allosteric ligands may possess intrinsic efficacy (either positive or inverse) in addition to, or exclusive of, cooperativity with orthosteric ligands (Annoura et al., 1996; Litschig et al., 1999; Niswender et al., 2008; Duvoisin et al., 2010; Gregory et al., 2012, 2013a; Noetzel et al., 2012b; Lavreysen et al., 2013; Rook et al., 2013). The first allosteric modulator discovered for the mGlu family was CPCCOEt [7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester], an mGlu1 NAM of multiple orthosteric agonists, including glutamate (Annoura et al., 1996; Litschig et al., 1999). The past 15 years has seen the discovery of selective allosteric modulators for most of the mGlu family, and for many subtypes multiple chemotypes and different classes of allosteric ligands have been disclosed (Tables 1, 2, and 3).
TABLE 1.
Molecular pharmacology of selected examples of allosteric ligands for group I metabotropic glutamate receptors
| Ligand/Selectivity | Allosteric Pharmacology | References | |
|---|---|---|---|
| CPCCOEt | 1 | NAM: glutamate, DHPG, quisqualate, and ACPD mediated inositol phosphate accumulation (IP1); glutamate mediated intracellular Ca2+ mobilization (iCa2+); quisqualate induced changes in cell impedance (xCelligence); extracellular Ca2+ induced changes in iCa2+. NAL: [3H]glutamate binding. | Annoura et al., 1996; Litschig et al., 1999; Scandroglio et al., 2010; Jiang et al., 2014 |
| JNJ-16259685 | 1 > >5 | NAM: glutamate mediated iCa2+ and IP1; quisqualate induced changes in cell impedance (xCelligence). Inverse agonist: IP1. NAL: [3H]quisqualate binding. | Lavreysen et al., 2004; Scandroglio et al., 2010; |
| R214127 | 1/5 | NAM: glutamate mediated iCa2+. Inverse agonist: IP1. | Lavreysen et al., 2003 |
| Ro 67-7476 | 1 | PAM: glutamate efficacy for mediating iCa2+ and cAMP accumulation. Agonist: pERK1/2. | Hemstapat et al., 2006; Sheffler et al., 2008 |
| Ro0711401 | 1 | Agonist: iCa2+. PAM: glutamate mediated K+ currents via GIRK activation. | Vieira et al., 2009 |
| YM-298198 | 1 | NAM: glutamate induced IP1; quisqualate induced changes in cell impedance (xCelligence). NAL: orthosteric agonists and affinity of radiolabeled ligand. | Kohara et al., 2005; Scandroglio et al., 2010 |
| 5MPEP | 5 | NAL: glutamate efficacy for iCa2+; glutamate and DHPG efficacy for IP1; quisqualate affinity. | Rodriguez et al., 2005; Chen et al., 2007, 2008; Ayala et al., 2009; Hammond et al., 2010; Bradley et al., 2011 |
| 5PAM523 | 5 | PAM: glutamate efficacy for iCa2+. NAL: [3H]quisqualate binding. | Parmentier-Batteur et al., 2014 |
| ADX47273 | 5 | PAM: quisqualate affinity; quisqualate efficacy for IP1. | Bradley et al., 2011 |
| CDPPB | 5 | PAM: glutamate efficacy for iCa2+; quisqualate affinity; quisqualate efficacy for IP1. PAM-agonist: glutamate efficacy for iCa2+, pERK1/2 and p-Akt. | Chen et al., 2007; Bradley et al., 2011; Gregory et al., 2012; Doria et al., 2013; Parmentier-Batteur et al., 2014 |
| CPPHA | 5 > 1 > 4/8 | mGlu5 PAM: glutamate efficacy for iCa2+; quisqualate efficacy for IP1 and iCa2+. mGlu5 NAL: quisqualate and glutamate affinity. mGlu5 PAM-agonist: glutamate efficacy for pERK1/2. NAM-agonist: DHPG efficacy for pERK1/2 (cortical astrocytes). mGlu4 and mGlu8 NAM: glutamate-mediated Ca2+ signaling. | O’Brien et al., 2004; Zhang et al., 2005; Chen et al., 2008; Bradley et al., 2011; Gregory et al., 2012; |
| DPFE | 5 | PAM-agonist: glutamate efficacy for iCa2+and pERK1/2. | Gregory et al., 2013b |
| M-5MPEP | 5 | NAM (low cooperativity): glutamate efficacy for iCa2+; quisqualate and DHPG efficacy for IP1. NAM: glutamate, DHPG and quisqualate mediated Ca2+ oscillations; glutamate efficacy for pERK1/2. NAL: quisqualate affinity. | Rodriguez et al., 2005; Bradley et al., 2011; Gregory et al., 2012 |
| MPEP | 5 > >4 | mGlu5 NAM: glutamate efficacy for iCa2+and pERK1/2; quisqualate and DHPG stimulated IP1; glutamate, DHPG and quisqualate induced Ca2+ oscillations. mGlu5 inverse agonist: IP1. mGlu5 NAL: quisqualate and glutamate affinity. mGlu4 PAM-agonist: L-AP4 mediated iCa2+. mGlu4 PAM: L-AP4 induced changes in VFT. | Gasparini et al., 1999; Chen et al., 2007; Bradley et al., 2011; Gregory et al., 2012; Rovira et al., 2015 |
| VU0357121 | 5 | NAL: glutamate affinity. PAM: glutamate efficacy for iCa2+. PAM-agonist: glutamate efficacy for pERK1/2. | Hammond et al., 2010; Gregory et al., 2012 |
| VU0360172 | 5 | PAM: glutamate efficacy for iCa2+. NAL: glutamate affinity. PAM-agonist: glutamate efficacy for pERK1/2. | Gregory et al., 2012 |
ACPD, (1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid; ADX47273, (4-fluorophenyl)-[(3S)-3-[3-(4-fluorophenyl)-1,2,4-oxadiazol-5-yl]piperidin-1-yl]methanone; DPFE, 1-(4-(2,4-difluorophenyl)piperazin-1-yl)-2-((4-fluorobenzyl)oxy)ethanone; GIRK, G protein–coupled inwardly rectifying potassium channel; JNJ-16259685, 3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl-(4-methoxycyclohexyl)methanone; NAL, neutral allosteric ligand; 5PAM523, 5-fluoro-2-{3-[(3S,6R)-1-[(4-fluorophenyl)carbonyl]-6-methylpiperidin-3-yl]-1,2,4-oxadiazol-5-yl}pyridine; Ro0711401, N-[4-(trifluoromethyl)-1,3-oxazol-2-yl]-9H-xanthene-9-carboxamide; Ro 67-7476, (S)-2-(4-fluorophenyl)-1-(toluene-4-sulfonyl)pyrrolidine; VU0360172, N-cyclobutyl-6-[2-(3-fluorophenyl)ethynyl]pyridine-3-carboxamide; YM-298198, 6-amino-N-cyclohexyl-N,3-dimethylthiazolo[3,2-a]benzimidazole-2-carboxamide.
TABLE 2.
Molecular pharmacology of selected allosteric ligands for group II metabotropic glutamate receptors
| Ligand/Selectivity | Allosteric Pharmacology | References | |
|---|---|---|---|
| BINA | 2 | PAM-agonist: glutamate stimulated [35S]GTPγS binding at human mGlu2. PAM: glutamate stimulated [35S]GTPγS binding at rat mGlu2 and iCa2+ at human and rat mGlu2; [3H]DCG-IV binding to human mGlu2; LY541850 mediated decreases in Ca2+ oscillation frequency in primary neuronal cultures. NAL: [3H]LY341495 binding to human mGlu2, LY379268 mediated decreases in Ca2+ oscillation frequency in primary neuronal cultures. | Galici et al., 2006; Hemstapat et al., 2007; Lavreysen et al., 2013; Sanger et al., 2013 |
| CBiPES | PAM: glutamate mediated iCa2+; LY379268 and LY541850 mediated decreases in Ca2+ oscillation frequency in primary neuronal cultures. | Johnson et al., 2005; Sanger et al., 2013 | |
| JNJ-40068782 | 2 | PAM-agonist: glutamate stimulated [35S]GTPγS binding and iCa2+ at human mGlu2. PAM: glutamate stimulated [35S]GTPγS binding at rat mGlu2; [3H]DCG-IV binding to human mGlu2; glutamate affinity (displacement of [3H]DCG-IV). NAL: [3H]LY341495 binding. | Lavreysen et al., 2013 |
| LY2607540 (THIIC) | 2 | PAM-agonist: glutamate stimulation of iCa2+ and [35S]GTPγS binding at human receptor. PAM: glutamate stimulation of iCa2+ and [35S]GTPγS binding at rat receptor. | Fell et al., 2011; Lavreysen et al., 2013 |
| LY487379 | 2 | PAM-agonist: glutamate stimulation of [35S]GTPγS binding to rat cerebral cortical membranes. PAM: glutamate, DCG-IV, LCCG-I and LY379268 stimulated [35S]GTPγS binding and glutamate mediated iCa2+; [3H]DCG-IV binding to human mGlu2. NAL: [3H]LY341495 binding to human mGlu2. | Schaffhauser et al., 2003; Hemstapat et al., 2007; Odagaki et al., 2011, 2013; Lavreysen et al., 2013 |
| MNI-137 | 2/3 | mGlu2 NAM: glutamate and DCG-IV mediated iCa2+ and glutamate stimulated [35S]GTPγS binding. mGlu3 NAM: glutamate mediated iCa2+ and [35S]GTPγS binding. mGlu2 NAL: [3H]LY341495 binding and glutamate affinity. | Hemstapat et al., 2007; Yin et al., 2014; |
| RO4491533 | 2/3 | mGlu2 NAM: [3H]LY354740 affinity; ACPD inhibition of cAMP accumulation; glutamate stimulation of GIRK channels and [35S]GTPγS binding. mGlu2 NAL: [3H]LY341495 binding to human mGlu2. mGlu3 NAM: glutamate stimulation of GIRK channels. | Campo et al., 2011; Woltering et al., 2010 |
| RO4988546 | 2/3 | mGlu2 NAM: [3H]LY354740 binding; LY354740 stimulation of [35S]GTPγS binding and iCa2+. mGlu2 NAL: [3H]HYDIA binding. | Lundstrom et al., 2011 |
| RO5488608 | 2/3 | mGlu2 NAM: [3H]LY354740 binding; LY354740 stimulation of [35S]GTPγS binding and iCa2+. mGlu2 NAL: [3H]HYDIA binding. | Lundstrom et al., 2011 |
ACPD, (1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid; BINA, biphenyl-indanone A; CBiPES, N-(4′-cyano-biphenyl-3-yl)-N-(3-pyridinylmethyl)-ethanesulfonamide hydrochloride; DCG-IV, (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine; GIRK, G protein–coupled inwardly rectifying potassium channel; HYDIA, (1S,2R,3R,5R,6S)-2-amino-3-hydroxy-bicyclo[3.1.0]hexane-2,6-dicarboxylic acid; L-CCG-I, (2S,1′S,2′S)-2-(carboxycyclopropyl)glycine; LY2607540, THIIC, N-(4-((2-(trifluoromethyl)-3-hydroxy-4-(isobutyryl)phenoxy)methyl)benzyl)-1-methyl-1H-imidazole-4-carboxamide; LY354740, (1S,2R,5R,6S)-2-amino-bicyclo[3.1.0]hexane-2,6-dicarboxylic acid; LY487379, 2,2,2-trifluoro-N-[4-(2-methoxyphenoxy)phenyl]-N-(pyridin-3-ylmethyl)ethanesulfonamide; MNI-137, 4-(8-bromo-5-oxo-3,4,5,6-tetrahydro-1,6-benzodiazocin-2-yl)pyridine-2-carbonitrile; NAL, neutral allosteric ligand; RO4491533, 4-[3-(2,6-dimethylpyridin-4-yl)phenyl]-7-methyl-8-(trifluoromethyl)-1,3-dihydro-1,5-benzodiazepin-2-one; RO4988546, 5-[7-trifluoromethyl-5-(4-trifluoromethyl-phenyl)-pyrazolo[1,5-a]pyrimidin-3-ylethynyl]-pyridine-3-sulphonic acid; RO5488608, 3′-(8-methyl-4-oxo-7-trifluoromethyl-4,5-dihydro-3H-benzo[b][1,4]diazepin-2-yl)-biphenyl-3-sulphonic acid.
TABLE 3.
Molecular pharmacology of selected allosteric ligands for group III metabotropic glutamate receptors
| Ligand/Selectivity | Allosteric Pharmacology | References | |
|---|---|---|---|
| ADX71743 | 7 | NAM: L-AP4 efficacy for iCa2+ and inhibition of cAMP accumulation; glutamate in mediating inhibition of cAMP accumulation. | Kalinichev et al., 2013 |
| AMN082 | 7# | NAL-agonist: inhibition of cAMP accumulation, stimulates pERK1/2 and receptor internalization. NAL: MSOP (does not block AMN082 internalization). | Mitsukawa et al., 2005; Pelkey et al., 2007; Suzuki et al., 2007; Iacovelli et al., 2014 |
| AZ12216052 | 8 | PAM-agonist: glutamate stimulation of [35S]GTPγS binding. | Duvoisin et al., 2010 |
| MMPIP | 7 | NAM: L-AP4 efficacy for iCa2+ and changes in cell mass distribution; L-CCG-I and CPPG efficacy for iCa2+; glutamate stimulation of GIRK channels. Inverse/agonist: cAMP accumulation and Epic label-free assay. NAL: [3H]LY341495 binding. | Suzuki et al., 2007; Niswender et al., 2010 |
| PHCCC | 4/6 | PAM: glutamate for iCa2+ and GIRK channel activation; L-AP4 stimulated iCa2+ and changes in VFT. PAM-agonist: L-AP4 and glutamate stimulated [35S]GTPγS binding. mGlu1 NAM: glutamate mediated iCa2+. mGlu6 agonist: glutamate mediated calcium current inhibition. | Maj et al., 2003; Marino et al., 2003; Beqollari and Kammermeier, 2008; Niswender et al., 2008; Yin et al., 2014; Rovira et al., 2015 |
| VU0155041 | 4 | PAM-agonist: glutamate mediated iCa2+and GIRK channel activation. PAM: L-AP4 mediated iCa2+and changes in VFT. | Niswender et al., 2008; Yin et al., 2013; Rovira et al., 2015 |
| VU0155094 | 4-8 | PAM: glutamate apparent affinity (mGlu4, 7, 8); glutamate efficacy for stimulation of GIRK channels (mGlu8) and iCa2+ (mGlu4,7); L-AP4 apparent affinity (mGlu7, 8); L-AP4 efficacy for iCa2+ (mGlu4); LSP4-2022 apparent affinity (mGlu4, 7, 8); LSP4-2022 efficacy for iCa2+ (mGlu4). NAL/NAM: L-AP4 apparent affinity (mGlu4); L-AP4 and LSP4-2022 efficacy for iCa2+ (mGlu7). NAL: glutamate, L-AP4, LSP4-2022 efficacy for iCa2+ (mGlu8). | Jalan-Sakrikar et al., 2014 |
| VU0422288 | 4-8 | PAM: glutamate, L-AP4 and LSP4-2022 efficacy for iCa2+and GIRK channel activation (mGlu4,7,8); glutamate, L-AP4, LSP4-2022 apparent affinity (mGlu7,8). NAL: glutamate apparent affinity (mGlu4). NAM: L-AP4 and LSP4-2022 apparent affinity (mGlu4). | Jalan-Sakrikar et al., 2014 |
ADX71743, 6-(2,4-dimethylphenyl)-2-ethyl-4,5,6,7-tetrahydro-1,3-benzoxazol-4-one; AZ12216052, 2-{[(4-bromophenyl)methyl]sulfanyl}-N-[4-(butan-2-yl)phenyl]acetamide; CPPG, 2-amino-2-cyclopropyl-2-(4-phosphonophenyl)acetic acid; GIRK, G protein–coupled inwardly rectifying potassium channel; L-CCG-I, (2S,1′S,2′S)-2-(carboxycyclopropyl)glycine; MMPIP, 6-(4-methoxyphenyl)-5-methyl-3-(4-pyridinyl)-isoxazolo [4,5-c]pyridine-4(5H)-one hydrochloride; MSOP, 2-amino-2-methyl-3-phosphonooxypropanoic acid; NAL, neutral allosteric ligand.
Compound and/or its metabolite hits other non-mGlu targets (Ayala et al., 2008; Sukoff Rizzo et al., 2011).
Complexities of mGlu Receptor Allosteric Modulator Pharmacology
Stimulus-Bias.
Despite definite success in the discovery of mGlu allosteric ligands, the pharmacology of allosteric modulators is inherently more complex than their orthosteric counterparts. The overall pharmacological effect of an allosteric ligand includes affinity and efficacy as well as the cooperativity between the allosteric and orthosteric ligands. With respect to ligand efficacy it has become increasingly accepted that occupancy and efficacy are not necessarily linearly linked. The concept that orthosteric ligands may differentially impact the G protein coupling and other functional responses mediated by activation of the receptor has come to the fore in recent years (Fig. 1). This phenomenon referred to as “stimulus-bias” (although it has had many other monikers including “agonist-directed trafficking of receptor stimulus,” “functional selectivity,” and “biased agonism”) has now been observed for many GPCRs, including mGlu1, mGlu4, mGlu7, and mGlu8 (Emery et al., 2012; Gregory et al., 2012; Jalan-Sakrikar et al., 2014). For example, stimulus-bias is evident when comparing orthosteric agonists L-AP4 [l-2-amino-4-phosphonobutyrate] and LSP4-2022 [(2S)-2-amino-4-([[4-(carboxymethoxy)phenyl](hydroxy)methyl](hydroxy)phosphoryl)butanoic acid] with glutamate between Ca2+ mobilization and G protein–coupled inwardly rectifying potassium channel activation at a single group III subtype (Jalan-Sakrikar et al., 2014). Interestingly, when these orthosteric agonist bias profiles are compared between mGlu7 and either mGlu4 or mGlu8, there is a complete reversal in the direction of bias for the same ligands across the same two pathways. This finding highlights the important concept that the endogenous ligand itself will have a “natural bias,” revealed in this study by differences between different group III mGlu subtypes. Stimulus-bias is thought to arise from ligands stabilizing different complements of receptor conformations that favor one suite of receptor activation outcomes over others. It is perhaps not surprising then that there is preliminary evidence for stimulus-bias when comparing allosteric agonist activity to that with glutamate. For example, VU29 [N-(1,3-diphenyl-1H-pyrazolo-5-yl)-4-nitrobenzamide] and CDPPB [3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide] are PAM-agonists of mGlu5 for both intracellular Ca2+ mobilization and phosphorylated extracellular-signal regulated kinases 1 and 2 (pERK1/2) and notably have higher intrinsic efficacy for pERK1/2 relative to Ca2+ signaling (Gregory et al., 2012). Glutamate, however, shows the reverse, with higher efficacy for Ca2+ signaling over pERK1/2. Similar observations have been reported for mGlu1 PAM-agonists relative to glutamate between Ca2+ mobilization and cAMP accumulation (Sheffler and Conn, 2008). Moreover, many of the mGlu5 PAM-agonists for pERK1/2 are super-agonists, capable of achieving higher maximal responses than glutamate (Gregory et al., 2012). Although it should be noted that both ligand and system kinetics may contribute to these observations of biased agonism. Negative allosteric modulators are also often reported to be inverse agonists (Tables 1, 2, and 3); however, these observations are assay dependent, likely a result of receptor constitutive activity and assay resolution. Comprehensive investigation into stimulus-bias that factors in these contributing mechanisms and confounds is sorely needed.
Fig. 1.

Schematic depicting the concepts of biased agonism and modulation. (A) The endogenous ligand (blue triangle) activates the receptor, resulting in intracellular signal transduction. However, not all pathways will be activated to the same extent; this is the natural bias of the receptor/system. (B) A surrogate orthosteric agonist (red triangle) may engender a different suite of active receptor conformations, resulting in biased signaling compared with the endogenous ligand; in this case the strength of signaling to response 1 versus 2 is reversed. (C) The binding of an allosteric modulator (orange star) potentiates the response to the endogenous agonist for all signal pathways to the same extent. (D) In contrast, a biased allosteric modulator (purple star) may have different magnitude or direction on cooperativity depending on the response measured; in this case, response 1 is potentiated whereas response 2 is inhibited.
An additional mechanism that may also contribute to observations of stimulus-bias relates to receptor compartmentalization. For example, a large pool of intracellular mGlu5 is located on the nuclear membrane (Jong et al., 2005; Kumar et al., 2008). Activation of mGlu5 at the cell surface versus the nuclear membrane results in the activation of different signaling cascades, as demonstrated through the use of permeable/transported and impermeable/nontransported orthosteric and allosteric ligands of mGlu5 (Jong et al., 2009; Kumar et al., 2012). Furthermore, activation of cell surface mGlu5 leads to long-term potentiation and long-term depression in the hippocampus, whereas activation of intracellular mGlu5 only mediates long-term depression (Purgert et al., 2014). These findings highlight that stimulus-bias may also arise from a differential ability of different chemotypes to access distinct subcellular compartments where receptors may be located. Therefore the lipophilicity or transportability of an allosteric ligand may impact the overall functional outcome observed.
Biased Modulation.
A related concept to biased agonism is the phenomenon of biased modulation (Fig. 1), if we consider that the conformations engendered by the simultaneous occupation of a receptor with an orthosteric and allosteric ligand that give rise to cooperativity differ from those engendered by binding of orthosteric ligand alone. Therefore this raises the possibility that the “natural bias” of an endogenous ligand may be influenced by an allosteric modulator. Conceptually, this may manifest as a change in the magnitude and/or direction of the cooperativity factors that describe the allosteric interaction depending on the receptor activation outcome measured. Alternatively, allosteric modulators may also show differential apparent affinity estimates depending upon the receptor activation outcome measured. In recent years, there have been a growing number of reports of biased mGlu allosteric modulation. For example at mGlu5, CPPHA [N-(4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl)-2-hydroxybenzamide], and DFB [((3-fluorophenyl)methylene)hydrazone-3-fluorobenzaldehyde] are both PAMs for glutamate-mediated Ca2+ mobilization in rat cortical astrocytes; however, DFB potentiates, whereas CPPHA inhibits DHPG [3,5-dihydroxyphenylglycine]-stimulated pERK1/2 (Zhang et al., 2005). Furthermore, multiple mGlu5 modulators have lower positive cooperativity for glutamate-mediated pERK1/2 versus intracellular Ca2+ mobilization (Gregory et al., 2012). Recent studies suggest that biased modulation has relevance in more physiologic systems, raising the possibility that biased modulation may have clinical implications. For example, a novel mGlu5 selective modulator from the same chemotype as CPPHA, NCFP [N-(4-chloro-2-[(1,3-dioxoisoindolin-2-yl)methyl)phenyl)picolinamide], potentiated DHPG-induced depolarization of subthalamic nucleus neurons but lacked the ability to potentiate induction of long-term potentiation or long-term depression in the hippocampus (Noetzel et al., 2013). Importantly, another mGlu5 allosteric modulator, VU0092273 [3′-(8-methyl-4-oxo-7-trifluoromethyl-4,5-dihydro-3H-benzo[b][1,4]diazepin-2-yl)-biphenyl-3-sulfonic acid], that shared a similar pharmacological mGlu5 PAM profile in recombinant cells, potentiated all three slice electrophysiological responses (Noetzel et al., 2013), suggesting that NCFP is showing true biased modulation in a native environment. Another novel mGlu5 allosteric modulator, VU0409551 [5-[(4-fluorophenyl)carbonyl]-2-(phenoxymethyl)-4,5,6,7-tetrahydro[1,3]oxazolo[5,4-c]pyridine], was recently disclosed that potentiates multiple glutamate-induced mGlu5 signaling responses in cell-based assays, but has no efficacy in modulating hippocampal long-term potentiation or N-methyl-d-aspartate receptor currents (Rook et al., 2015). However, VU0409551 has efficacy in preclinical rodent models of psychosis and cognition (Rook et al., 2015), suggesting that biased allosteric modulators of mGlu5 can possess preclinical efficacy.
Biased modulation is not limited to mGlu5 PAMs, both positive and negative modulators of group III receptors have been suggested to induce biased receptor signaling (Niswender et al., 2010; Jalan-Sakrikar et al., 2014). The extent and prevalence of biased modulators is likely to be highly underappreciated. This is due to the fact that the vast majority of mGlu allosteric modulators are characterized in a single cell-based assay, generally Ca2+ mobilization, to classify and rank compounds on the basis of potency. Furthermore, the therapeutic relevance of biased pharmacology has yet to be realized; however, it is tempting to speculate that unappreciated bias may contribute to differential efficacy of allosteric modulators in behavioral models. For example, 1-(4-(2,4-difluorophenyl)piperazin-1-yl)-2-((4-fluorobenzyl)oxy)ethanone, an mGlu5 PAM of glutamate, requires far lower doses for efficacy in cognitive models compared with reversing amphetamine-induced hyperlocomotion, a model for antipsychotic efficacy (Gregory et al., 2013a). Moreover, there is the potential that biased modulation may be exploited to develop therapeutics that are not only subtype selective but also pathway selective, modulating receptor responses that yield therapeutic effects and avoiding those that give rise to on-target adverse effects.
Probe Dependence.
A related phenomenon to biased modulation is that of probe dependence, where allosteric interactions are dependent upon the chemical nature of the two ligands under investigation. The result being that the direction and/or magnitude of cooperativity may be significantly different. There are multiple examples of probe dependence at the group II mGlu receptors. Biphenyl-indanone A potentiates Ca2+ oscillations in cortical neurons induced by the mGlu2 selective orthosteric agonist LY541850 [(1S,2S,4R,5R,6S)-2-amino-4-methylbicyclo[3.1.0]hexane2,6-dicarboxylic acid] but not the mixed group II orthosteric agonist LY379268 [(−)-2-oxa-4-minobicyclo[3.1.0]hexane-4,6-dicarboxylate] (Sanger et al., 2013). JNJ-40068782 [3-cyano-1-cyclo-propylmethyl-4-(4-phenyl-piperidin-1-yl)-pyridine-2(1H)-one], a PAM-agonist of glutamate-stimulation of [35S]GTPγS binding and Ca2+ mobilization at mGlu2, has neutral cooperativity with the orthosteric antagonist radioligand [3H]LY341495 [(2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid] and positive cooperativity with the orthosteric agonist radioligand [3H]DCG-IV [(2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine] (Lavreysen et al., 2013). For group I and III mGlu receptors, there are also select examples of probe dependence. Affinity modulation occurs between the orthosteric agonist quisqualate and mGlu5 PAMs (Bradley et al., 2011); however, a separate study found no evidence for affinity modulation (i.e., neutral cooperativity) between glutamate and mGlu5 PAMs of glutamate in functional assays (Gregory et al., 2012). At mGlu4 and mGlu7, VU0155094 [methyl 4-(3-(2-((4-acetamidophenyl)thio)acetyl)-2,5-dimethyl-1H-pyrrol-1-yl)benzoate] and VU0422288 [N-(3-chloro-4-((5-chloropyridin-2-yl)oxy)phenyl)picolinamide] have different degrees of cooperativity depending upon both the orthosteric agonist and the assay endpoint used (Jalan-Sakrikar et al., 2014). For example, at mGlu7, VU0155094 is neutral with respect to the apparent affinity of LSP4-2022 and L-AP4 but a positive modulator of glutamate apparent affinity (Jalan-Sakrikar et al., 2014). Clearly, probe dependence is evident for the mGlu family and should be taken into consideration when designing screening paradigms and characterizing mGlu allosteric modulators. The choice of assay and/or probe can result in a very different pharmacological profile. This is of particular importance for mGlu7 where L-AP4 is most commonly used as the orthosteric agonist given the low affinity of this subtype for glutamate (in the millimolar range). Moreover, extracellular Ca2+ ions can activate mGlu1, influencing the responses to orthosteric agonists, indicating that two endogenous orthosteric agonists need to be considered with respect to probe dependence but also that different endogenous ligands can allosterically interact at mGlu1 (Jiang et al., 2014). Importantly, extracellular Ca2+ activation of mGlu1 can be modulated by small molecule positive and negative allosteric modulators of glutamate (Ro67-4853 [butyl (9H-xanthene-9-carbonyl)carbamate] and CPCCOEt) (Jiang et al., 2014). Probe dependence can also be an important consideration when assessing allosteric modulators in native systems (brain slice electrophysiology or primary neuronal cultures) where surrogate agonists are typically used to selectively activate a particular mGlu subtype and avoid the confounds of glutamate activating ionotropic glutamate receptors and being subject to endogenous transporters.
Protein-Protein Interactions.
Another important consideration in assessing allosteric modulator pharmacology in native systems is the fact that the mGlu family are obligate dimers (El-Moustaine et al., 2012) that also have the capacity to form heteromers with other subtypes (Doumazane et al., 2011; Kammermeier, 2012; Sevastyanova and Kammermeier, 2014; Yin et al., 2014) as well as with other unrelated GPCRs (Gama et al., 2001; Gonzalez-Maeso et al., 2008; Cabello et al., 2009). Unsurprisingly, such complexity in receptor configurations can influence the pharmacology of small molecules. Heterodimerization of mGlu4 and mGlu2 has differential effects on the pharmacology of different allosteric ligands (Kammermeier, 2012; Yin et al., 2014). For example, at the mGlu2/4 heteromer PHCCC [N-phenyl-7-(hydroxyimino)cyclopropa[b] chromen-1a-carboxamide] has lower cooperativity with L-AP4, whereas Lu AF21934 [(1S,2R)-2-[(aminooxy)methyl]-N-(3,4-dichlorophenyl)cyclohexane-1-carboxamide] and VU0155041 [(1R,2S)-2-[(3,5-dichlorophenyl)carbamoyl]cyclohexane-1-carboxylic acid] have decreased affinity but higher cooperativity compared with mGlu4 homodimers (Yin et al., 2014). This perturbation in the pharmacology of allosteric ligands has consequences in vivo at synapses where mGlu2 and mGlu4 are coexpressed. VU0155041 and Lu AF21934 are both effective at potentiating the actions of L-AP4 at corticostriatal synapses (Gubellini et al., 2001; Yin et al., 2014). However, PHCCC does not potentiate the actions of L-AP4 at corticostriatal synapses (Yin et al., 2014), despite being an efficacious mGlu4 PAM of L-AP4–induced responses at other synapses (Marino et al., 2003; Valenti et al., 2005; Jones et al., 2008). Direct protein-protein interactions are not necessarily required for a coexpressed receptor to impact the pharmacological effect of an mGlu ligand. Coexpression and coactivation of Gq-coupled receptors enhances mGlu4 stimulus-response coupling to orthosteric and allosteric agonists in intracellular Ca2+ mobilization but not cAMP accumulation assays (Yin et al., 2013). It is apparent that cellular context, measure of receptor function and coexpression of other GPCRs can impact the pharmacology of small molecule allosteric ligands, leading to unanticipated consequences in native systems. However, this complexity extends further to the structure-activity relationships of allosteric modulators themselves.
Molecular Switches.
Given the complexities of allosteric modulation surrounding stimulus-bias and probe dependence it is perhaps not surprising that structure-activity relationships (SAR) for mGlu receptor allosteric modulators are notoriously challenging. SAR is often reported to be “steep,” with minimal changes resulting in a complete loss of activity (Conn et al., 2014; Lindsley, 2014). In addition, mGlu allosteric chemotypes often show “molecular switches” where the same scaffold can give rise to modulators with negative, positive, and neutral cooperativity with the same orthosteric agonist (Wood et al., 2011). The phenomenon was first observed during the development of DFB, an mGlu5 PAM for glutamate-mediated intracellular Ca2+ mobilization (O'Brien et al., 2003). Recently, this propensity for molecular switching was exploited with the development of a photoswitchable allosteric modulator for mGlu5, where negative cooperativity with glutamate could be controlled by exposure to different light wavelengths (Pittolo et al., 2014). However, unexpected changes in cooperativity continue to be a challenge for medicinal chemistry programs and interpretation of SAR (Hammer et al., 1980; Sharma et al., 2009; Rodriguez et al., 2010; Schann et al., 2010; Zhou et al., 2010; Lamb et al., 2011; Sams et al., 2011; Sheffler et al., 2012). These difficulties in SAR interpretation can in part be attributed to a paucity in our understanding of the structural basis of allosterism. The majority of discovery programs rely on modulator potency curves in the presence of a single concentration of agonist to classify allosteric ligands as active/inactive and ascribe either positive or negative cooperativity. However, potency represents a composite measure of efficacy, affinity, and cooperativity (Gregory et al., 2012). As such, modifications to a chemotype may influence any one of these three parameters. Therefore, more quantitative analysis of allosteric chemotypes is required to truly understand how SAR relates to efficacy, affinity, and cooperativity. Going forward a key challenge will be coupling SAR interpretation as we improve our understanding of the structural basis of allosterism.
Structural Basis of Allosteric Modulation
The mGlus are family C GPCRs typified by their large extracellular N-terminal Venus flytrap domain (VFT), linked via a cysteine-rich domain to the seven transmembrane (TM) spanning α-helical domains that are the hallmark of the GPCR superfamily. The orthosteric site is located in the N-terminal domain and to date all small molecule allosteric modulators are thought to bind within the 7TMs. Historically, receptor chimeras proved valuable to (1) definitively demonstrate an allosteric mechanism of action for small molecule ligands and (2) to localize allosteric binding sites to the 7TM domains. Chimeric constructs swapping the VFT of one subtype onto another or even exchanging with non-mGlu family C GPCRs have been used. Chimeras between mGlu1 and mGlu5 or mGlu4 and the calcium-sensing receptor were used to validate the allosteric mechanism of action of mGlu1 selective allosteric modulator CPCCOEt (Brauner-Osborne et al., 1999; Litschig et al., 1999; Gasparini et al., 2001). Subsequent studies used mGlu1/5 chimeras to demonstrate MPEP (2-methyl-6-(phenylethynyl)pyridine; an mGlu5 selective NAM), and multiple mGlu1 PAMs recognized an overlapping binding site with that of CPCCOEt (Pagano et al., 2000; Knoflach et al., 2001). Chimeric constructs were also used to validate an allosteric mechanism of action for PHCCC, AMN082 [N,N′-dibenzhydrylethane-1,2-diamine dihydrochloride], and BAY36-7620 [(3aS,6aS)-hexahydro-5-methylene-6a-(2-naphthalenylmethyl)-1H-cyclopenta[c]furan-1-one] at mGlu4, mGlu7, and mGlu1, respectively (Carroll et al., 2001; Maj et al., 2003; Mitsukawa et al., 2005). Another approach to achieve gross localization of allosteric binding sites is through the generation of N-terminal truncated receptors. The extracellular VFT can be removed to generate a “headless” mGlu, yielding a receptor construct that does not respond to glutamate but can couple to intracellular signaling pathways (Goudet et al., 2004). Headless mGlu1 and mGlu5 are constitutively active such that negative allosteric modulators behave as inverse agonists (Goudet et al., 2004; Chen et al., 2007; Suzuki et al., 2007). Furthermore, headless mGlu5 can be stimulated by small molecule positive allosteric modulators that do not necessarily have intrinsic efficacy at the full-length receptor (Chen et al., 2007; Goudet et al., 2004; Noetzel et al., 2013). Interestingly, NCFP had little or no efficacy for stimulating headless mGlu5, despite having a similar magnitude of positive cooperativity as VU0092273, which can activate headless mGlu5 (Noetzel et al., 2013). The headless mGlu 7TM is thought to behave as a monomer (Goudet et al., 2004), so perhaps this discrepancy relates to receptor dimerization in the full-length receptor being important either for NCFP binding or cooperativity.
Two recent X-ray crystal structures were solved for the 7TMs of group I mGlus with allosteric modulators bound [PDB IDs 4OO9 and 4OR2 (Dore et al., 2014; Wu et al., 2014)]. Before this, researchers relied on family A 7TM templates to perform homology modeling and interpret site-directed mutagenesis data. Despite low sequence homology (<20%), these approaches proved fruitful, localizing allosteric binding pockets of mGlu1, mGlu2, mGlu4, and mGlu5 to a region analogous to that of the biogenic amine binding sites in family A GPCRs (Ott et al., 2000; Pagano et al., 2000; Malherbe et al., 2003a,b; Lundstrom et al., 2011; Molck et al., 2012; Gregory et al., 2013b, 2014; Rovira et al., 2015). In light of the new crystal structures, although alignments and secondary structure predictions of TMs 5–7 deviated from that observed in the mGlu1 and mGlu5 structures, the general localization of the pocket was consistent with earlier reports. Many residues that had previously been identified in mutagenesis-based studies were recapitulated in the crystal structures (discussed in detail below). Ongoing research continues to refine our understanding of binding pocket(s) within the 7TM bundle in particular in relation to determinants that govern selectivity, affinity, cooperativity, and efficacy of allosteric modulators.
The Common mGlu Allosteric Site
The concept for a common allosteric site between mGlu subtypes arises from three lines of evidence: (1) allosteric modulators that lack subtype selectivity; (2) competitive interactions between diverse chemotypes and allosteric radioligands; (3) overlapping binding pockets based on mutagenesis studies and observed in X-ray crystal structures. Although attaining selectivity with allosteric modulators is considerably more effective than for orthosteric ligands, there are a number of examples of allosteric modulators that have efficacy at multiple subtypes. MPEP is both a negative allosteric modulator of glutamate at mGlu5 and a positive allosteric modulator of L-AP4 at mGlu4 (Mathiesen et al., 2003). Conversely, DFB and CPPHA, which potentiate glutamate signaling at mGlu5 are weak negative allosteric modulators of glutamate at mGlu4 and mGlu8 (O'Brien et al., 2003, 2004). In addition, PHCCC, an mGlu4 PAM for glutamate and L-AP4, negatively modulates glutamate at mGlu1 (Annoura et al., 1996; Maj et al., 2003). The recent report of a molecular switch to mGlu3 NAM from an mGlu5 PAM scaffold (Sheffler et al., 2012) reveals that the subtleties of interactions within this common pocket that dictate subtype selectivity are not limited to crossover between group I and group III mGlu receptors alone. This lack of selectivity within the common binding pocket is also associated with differential cooperativity between subtypes, highlighting that the small molecule interactions within the common allosteric site can elicit either positive or negative cooperativity.
Identification of allosteric ligands that interact with this common mGlu allosteric site has been facilitated by the development of radiolabeled mGlu allosteric modulators. Multiple selective tritiated and positron emission tomography (PET) allosteric ligands have been developed for mGlu1, mGlu2, and mGlu5 (Anderson et al., 2002, 2003; Gasparini et al., 2002; Cosford et al., 2003b; Lavreysen et al., 2003; Kohara et al., 2005; Malherbe et al., 2006; Ametamey et al., 2007; Treyer et al., 2007; Baumann et al., 2010). Through the use of inhibition binding assays with these allosteric ligands a direct assessment of competitive versus noncompetitive binding modes can be performed (Lavreysen et al., 2003, 2004; Kinney et al., 2005; Malherbe et al., 2006; Hemstapat et al., 2006; Chen et al., 2007; Lundstrom et al., 2011). Such experiments are often the first step toward identifying allosteric modulators that bind to alternative allosteric sites [(Chen et al., 2008; Hammond et al., 2010) discussed in further detail later]. However, competition binding experiments may also be used to identify neutral allosteric ligands that interact with the common allosteric site but have neutral cooperativity with agonist(s) in functional screening assays (Rodriguez et al., 2005; Sams et al., 2011).
Early mutational studies investigating group I mGlu receptor allosteric binding pockets exploited nonconserved residues within the 7TMs. Substitution of mGlu1 nonconserved amino acids onto the equivalent positions in mGlu5, and vice versa, resulted in gain-of-function of selective allosteric modulators (Litschig et al., 1999; Pagano et al., 2000; Knoflach et al., 2001; Goudet et al., 2005; Surin et al., 2007). These mutagenesis studies pointed to a common location for allosteric binding pockets for group I mGlu receptors. Additional studies have shown that residues at positions 3.40 and 7.38 are important for allosteric modulation of both mGlu1 and mGlu5 [Ballesteros-Weinstein numbering (Ballesteros and Weinstein, 1995) adopted from mGlu1 structural alignment to family A GPCRs; Malherbe et al., 2003a,b, 2006; Muhlemann et al., 2006; Suzuki et al., 2007; Fukuda et al., 2009; Gregory et al., 2012, 2013b, 2014; Turlington et al., 2014; Wu et al., 2014]. Subsequently, key residues for the potency and/or binding of mGlu1, mGlu2, mGlu4, and mGlu5 allosteric modulators from diverse chemical scaffolds have been mapped to TMs 3–7. Across the four subtypes multiple residues have repeatedly been reported as contributing to allosteric modulation. For all four subtypes, residues in positions 3.36, 5.43, and 6.48 are crucial for both positive and negative allosteric modulators (Pagano et al., 2000; Malherbe et al., 2003a, 2006; Muhlemann et al., 2006; Fukuda et al., 2009; Lundstrom et al., 2011; Gregory et al., 2012, 2013b, 2014; Molck et al., 2012; Turlington et al., 2014; Wu et al., 2014; Rovira et al., 2015). Position 7.45 has also been implicated in mGlu4 and mGlu5 allosteric modulation (Molck et al., 2012; Gregory et al., 2013b, 2014; Turlington et al., 2014; Rovira et al., 2015); there are no reports investigating the influence of this amino acid on selective allosteric modulators of mGlu1 or mGlu2. Between group I mGlu receptors and mGlu2, three residues are common: 5.44, 5.47, and 6.55 (Knoflach et al., 2001; Malherbe et al., 2003b, 2006; Schaffhauser et al., 2003; Hemstapat et al., 2006; Muhlemann et al., 2006; Rowe et al., 2008; Fukuda et al., 2009; Lundstrom et al., 2011; Gregory et al., 2012, 2013b, 2014; Molck et al., 2012). Group 1 receptors and mGlu4 share a common determinant in F6.51 (Malherbe et al., 2003a,b, 2006; Muhlemann et al., 2006; Suzuki et al., 2007; Fukuda et al., 2009; Gregory et al., 2014; Rovira et al., 2015). Two residues at the top of TM3 (3.28 and 3.29) are important for allosteric modulation of both mGlu2 and mGlu4 (Lundstrom et al., 2011; Rovira et al., 2015), with position 3.29 also implicated in the activity of some mGlu5 allosteric modulators (Malherbe et al., 2003b, 2006; Molck et al., 2012). Therefore these mutagenesis data reinforce the notion of overlapping binding pockets across the mGlu family.
Comparison of the recently solved mGlu1 and mGlu5 crystal structures reveals a similar architecture for the backbone of the transmembrane-spanning helices (Fig. 2A). Overlay of the 7TM binding pockets for FITM [4-fluoro-N-(4-(6-(isopropylamino)pyrimidin-4-yl)thiazole-2-yl)-N-methylbenzamide] and mavoglurant in mGlu1 and mGlu5, respectively, confirmed that these subtypes have overlapping allosteric binding pockets within the 7TMs (Dore et al., 2014; Wu et al., 2014). Examination of binding pockets for mavoglurant and FITM reveals that mavoglurant binds deeper within the 7TM bundle (Fig. 2B). Side chain conformations of two key nonconserved residues in TM3 of mGlu5: P6553.36 and S6583.39 (mGlu1 equivalents are S668 and C671, respectively) create greater space within the pocket that allows mavoglurant to bind deeper within the 7TM bundle (Fig. 2C). These crystal structures of closely related mGlu receptors have yielded structural insights with respect to how selectivity can be achieved in the common mGlu allosteric site.
Fig. 2.

Comparison of allosteric pockets within the 7TM domains of mGlu1 and mGlu5. (A) X-ray crystal structures of 7TMs of mGlu5 (gray, PDB ID 4OO9) and mGlu1 (teal; PDB ID 4OR2) were aligned using MacPyMOL (The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC, New York, NY). (B) Inside surface rendering of the allosteric binding pockets of mavoglurant (black) in mGlu5 (gray) and FITM (blue) in mGlu1 (teal). (C) Zoomed in image of the bottom of the allosteric pockets with mavoglurant shown. Two amino acids differ between mGlu1 and mGlu5 in this region, the differences in the side chains (depicted as sticks) create more space in mGlu5, allowing mavoglurant to bind deeper within the 7TM bundle. All images created using MacPyMOL.
Delineating Affinity versus Cooperativity Determinants
The vast majority of research on allosteric modulator SAR and structure-function has relied on measures of allosteric modulator potency; however, potency values represent a composite of affinity and cooperativity. In recent years the application of rigorous analytical pharmacological methods has facilitated delineation of the impact of mutations on allosteric modulator affinity, cooperativity, and efficacy based on functional interaction data alone. By combining this research with earlier studies using radiolabeled allosteric modulators where affinity was determined or a loss of binding was observed we can start to map affinity determinants in the binding pocket. Residues known to be crucial affinity determinants are summarized in Table 4 (also see Supplemental Table 1) where the most well characterized representatives from individual allosteric modulator scaffolds were included. These affinity determinants are also mapped onto the mGlu1 and mGlu5 structures (Fig. 3, A and B). Significant overlap is observed between mGlu1 and mGlu5, although there is limited affinity data for mGlu1. For mGlu5, six residues have been implicated in the binding of most PAMs and NAMs: P6553.36, Y6593.40, T7816.44, W7856.48, S8097.45, and A8107.46 within the common allosteric site. Mapping the known affinity determinants for MPEP, the most well studied mGlu5 allosteric ligand with respect to structure-function, onto the mGlu5 crystal structure yields additional insights (Fig. 4; Pagano et al., 2000; Malherbe et al., 2003b; Gregory et al., 2012, 2013b, 2014). Coloring residues on the basis of the impact of mutations reveals that substitutions that cause a moderate (<10-fold) reduction in affinity are located on the fringes of the mavoglurant binding pocket or are immediately adjacent to key residues (Fig. 4; Supplemental Table 1).
TABLE 4.
Known affinity determinants for mGlu allosteric modulators interacting with the common allosteric site.
Most well characterized representatives from individual allosteric modulator scaffolds were selected for inclusion. Alignment of amino acids and assignment of Ballesteros-Weinstein numbering was adopted from the structural alignment to class A GPCRs in Wu et al., 2014. A colorized version of this table (Supplemental Table 1) is included in the online supplemental materials.
| Ligand/Subtype | Amino Acid Residue Position |
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3.29 | 3.36 | 3.39 | 3.40 | 5.43 | 5.44 | 5.47 | 5.48 | 6.44 | 6.48 | 6.51 | 6.52 | 6.55 | 6.56 | 7.38 | 7.45 | 7.46 | |||
| EM-TBPC | 1 | Y | V/L | W+ | F | – | Y | T | |||||||||||
| FITM | 1 | s | P | – | T | – | |||||||||||||
| MPEP | 5 | R | P | S | Y | P | L | – | G | T | W | F | V | Y | f+ | – | S | A | |
| Fenobam | 5 | R | P | S | Y | T | W | F | Y | A | |||||||||
| VU0366248 | 5 | P | S | Y | N | – | t | W | f# | – | Y | – | S | S+ | A | ||||
| VU0285683 | 5 | p# | Y | – | N | – | T | W | F | – | Y | S | A | ||||||
| VU0366058 | 5 | p# | Y | – | g+ | W | F+ | v+ | Y+ | s | A | ||||||||
| VU0409106 | 5 | p# | Y | N | – | T | W | F | – | Y | f | S | A | ||||||
| DPFE | 5 | – | P | Y | P | L+ | – | T | W | – | V+ | y+ | f+ | s | A | ||||
| VU29 | 5 | – | p# | – | p | l | G | T | w+ | f | v | y | f+ | – | A | ||||
| VU0403602 | 5 | – | P | s | Y# | p | – | – | T | W | – | S | A | ||||||
DPFE, 1-(4-(2,4-difluorophenyl)piperazin-1-yl)-2-((4-fluorobenzyl)oxy)ethanone; EM-TBPC, 1-ethyl-2-methyl-6-oxo-4-(2,3,4,5-tetrahydro-1H-3-benzazepin-3-yl)-1,6-dihydropyrimidine-5-carbonitrile; VU0285683, 3-fluoro-5-(3-pyridin-2-yl-1,2,4-oxadiazol-5-yl)benzonitrile; VU0366058, 2-[(1,3-benzoxazol-2-yl)amino]-4-(4-fluorophenyl)pyrimidine-5-carbonitrile; VU0403602, N-cyclobutyl-5-((3-fluorophenyl)ethynyl)picolinamide.
Lower case, mutations caused a change (2- to 10-fold) in affinity estimates that did not reach significance (determined from either radioligand binding or from functional interactions with orthosteric agonists); upper case, mutations caused a significant change in affinity (determined from either radioligand binding or from functional interactions with orthosteric agonists; not included: T6.43 for MPEP, C7.52 for VU0403602). Grayscale: white, 3- to 10-fold change; light gray, 10- to 30-fold change; mid gray, 30- to 100-fold decrease; dark gray, >100-fold decrease or complete loss of radiolabeled allosteric modulator binding (data summarized from: Pagano et al., 2000; Malherbe et al., 2003a,b, 2006; Gregory et al., 2012, 2013b, 2014; Turlington et al., 2014; Wu et al., 2014).
Mutation caused a loss in allosteric modulation of orthosteric agonist functional response; impact on affinity inferred from effects across other scaffolds.
–Mutation(s) had no effect on modulator affinity (determined from either radioligand binding or from functional interactions with orthosteric agonists).
+Mutation increases modulator affinity, in all other cases mutations cause a decrease (determined from either radioligand binding or from functional interactions with orthosteric agonists).
Fig. 3.

Affinity and cooperativity determinants for allosteric modulation of metabotropic glutamate receptors. (A) X-ray crystal structure of mGlu1 7TMs with FITM (rendered in blue sticks) bound (PDB ID 4OR2) with known mGlu1 allosteric ligand affinity determinants highlighted in dark blue. (B) X-ray crystal structure of mGlu5 7TMs with mavoglurant (rendered in black sticks) bound (PDB ID 4OO9), affinity determinants for mGlu5 allosteric ligands are highlighted in red. (C) X-ray crystal structure of mGlu5 and mGlu1 7TM domains were aligned; only the mGlu5 structure is represented in cartoon. The binding poses of FITM and mavoglurant show the overlapping allosteric pockets of mGlu1 and mGlu5. Highlighted are residues that when mutated change allosteric ligand cooperativity: dark red, reduce mGlu5 NAM cooperativity; light red, increase NAM cooperativity; orange, differential effects on NAM cooperativity; dark blue, increase PAM cooperativity; light blue, increase cooperativity of NAMs and PAMs; purple, differential effects on cooperativity of NAMs versus PAMs. (D) Structures of mGlu1 and mGlu5 were aligned as per (C); highlighted are residues that when mutated switch allosteric modulator cooperativity: blue, PAM to either NAM or neutral switch; red, NAM to PAM switch; purple, PAM to NAM and vice versa for NAMs. All images created using MacPyMOL.
Fig. 4.

Relative contribution of 7TM residues to the binding affinity of prototypical mGlu5 allosteric modulator, MPEP. Known affinity determinants for MPEP are mapped onto the mGlu5-7TM X-ray crystal structure with mavoglurant bound (PDB ID 4OO9). The relative contribution of pocket residues are color-coded with side chains rendered as sticks based on mutational data as indicated in the key. All images created using MacPyMOL
In addition to effects on affinity, molecular determinants for a large set of allosteric modulators spanning multiple scaffolds with both positive and negative cooperativity have been explored at mGlu5. Some residues that are important for modulator affinity also contribute to cooperativity; however, there are select amino acids that only influence cooperativity (Fig. 3). Negative cooperativity with glutamate of multiple allosteric modulators at mGlu5 is decreased at P6553.36 mutants (Gregory et al., 2013b, 2014). Additionally mutations in TMs 3–7 [Y6593.40, N7475.47, T7816.44, W7856.48, S8097.45 (Molck et al., 2012; Gregory et al., 2014)] reduced cooperativity for negative modulators of glutamate at mGlu5. Notably, in mGlu1 mutations of T7946.44 and S8227.45 had no effect on the interaction between glutamate and FITM (Wu et al., 2014), suggesting that the structural determinants that govern cooperativity can be subtype selective even for conserved amino acids.
Negative cooperativity with glutamate at mGlu5 was increased for M-5MPEP [2-(2-(3-methoxyphenyl)ethynyl)-5-methylpyridine] and/or VU0366248 [N-(3-chloro-2-fluorophenyl)-3-cyano-5-fluorobenzamide] when mutations were introduced to S6583.39, P7435.43, G7485.48, and V7896.52 (Gregory et al., 2014). Both M-5MPEP and VU0366248 have limited negative cooperativity at the wild-type receptor, with saturating concentrations unable to completely abolish the glutamate response (Rodriguez et al., 2005; Felts et al., 2010). The cooperativity of other mGlu5-negative allosteric modulators tested at these mutations was not discernibly different to wild type (Gregory et al., 2014). However, given that these all have strong negative cooperativity at wild type, fully abolishing the maximal glutamate response, further increases in negative cooperativity were likely beyond the limits of detection. For P7435.43 this increase is opposite to that observed for the equivalent mGlu1 mutation (Wu et al., 2014), again highlighting the subtype selective nature of cooperativity. Interestingly, two point mutations (N7475.47 and S8097.45) had differential effects on allosteric ligands, actually increasing negative cooperativity of M-5MPEP and VU0366248, respectively (Fig. 3C; Gregory et al., 2014). It is clear that the amino acids within the common allosteric site can differentially contribute to affinity and the manifestation of negative cooperativity in a manner that is very much ligand dependent.
With respect to positive allosteric modulators, no mutations have yet been demonstrated to unequivocally reduce cooperativity. However, there are multiple examples of mutations that increase the magnitude of positive cooperativity with glutamate (Fig. 3C). Shared with the low cooperativity negative modulators, mutation of P7435.43 and V7896.52 also increased positive cooperativity of diverse allosteric modulators with glutamate at mGlu5 (Gregory et al., 2013b, 2014). Four residues (L7445.44, Y7926.55, A8107.46, and C8167.52) are implicated in positive but not negative cooperativity (Gregory et al., 2013b, 2014). Interestingly, compared with negative allosteric modulators, mutation of T7816.44 and W7856.48 had differential effects, increasing positive cooperativity with glutamate at mGlu5 (Gregory et al., 2013b, 2014). The complicated nature of cooperativity determinants is further highlighted through evidence of mutations that engender “molecular switches” in allosteric modulator cooperativity (Fig. 3D). Substitution of Y6.51 at mGlu5 switches DFB from a PAM of glutamate to a NAM, whereas the reverse is true for YM298198 at mGlu1 (Muhlemann et al., 2006; Fukuda et al., 2009). Multiple additional point mutations in TMs 3, 6, and 7 can also switch acetylenic PAMs to have either neutral or negative cooperativity with glutamate at mGlu5 (Gregory et al., 2013b; Turlington et al., 2014). Mutation of conserved W7856.48 in mGlu5 can switch the cooperativity of select NAMs of glutamate to positive (Gregory et al., 2014). Collectively, these data highlight the subtleties and complexities of interactions within the common allosteric site that dictate whether the binding of a ligand will result in enhancement or inhibition of mGlu receptor function. Furthermore, developing a better understanding of the dynamic ligand-receptor interactions that give rise to cooperativity will likely improve interpretation of allosteric modulator SAR and perhaps inform as to why “molecular switches” are prevalent for some chemical scaffolds.
Additional mGlu Allosteric Sites
As mentioned earlier, the development of radiolabeled allosteric ligands has identified allosteric ligands that are noncompetitive. The first such ligand identified was CPPHA, a group I mGlu selective positive allosteric modulator of glutamate. CPPHA is unable to fully displace the mGlu5 radioligands [3H]mPEPy [3-methoxy-5-(2-pyridinylethynyl)pyridine] and [3H]MPEP or compete with [3H]R214127 [1-(3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-2-phenylethanone] for binding to mGlu1 (O'Brien et al., 2004; Chen et al., 2008; Bradley et al., 2011; Noetzel et al., 2013). Another structurally related selective mGlu5 PAM of glutamate, NCFP, is also thought to bind to this alternate allosteric site (Noetzel et al., 2013). Potentiation of glutamate by both NCFP and CPPHA is noncompetitively inhibited by the neutral allosteric ligand 5-methyl-2-(2-phenylethynyl)pyridine (Chen et al., 2008; Noetzel et al., 2013). This indicates that the second allosteric site is conformationally linked to the common allosteric site such that ligands binding to these sites can allosterically influence one another. Isoleucine substitution of F1.42 reduces potentiation by CPPHA (both mGlu1 and mGlu5) and NCFP (mGlu5 only) (Chen et al., 2008; Noetzel et al., 2013). Interestingly, NCFP also loses either affinity or the capacity to allosterically modulate [3H]mPEPy binding and has very little efficacy as an agonist at an N-terminal truncated mGlu5 (Noetzel et al., 2013). This suggests that the intact full-length receptor is required for this alternate allosteric site and/or receptor dimerization is also necessary. Also for mGlu5, VU0357121 [4-butoxy-N-(2,4-difluorophenyl)benzamide] and VU0400100 [2-[2-chloro-N-methylsulfonyl-5-(trifluoromethyl)anilino]-N-(cyclopropylmethyl)acetamide] do not completely displace [3H]mPEPy binding (Hammond et al., 2010; Gregory et al., 2012; Rodriguez et al., 2012). Multiple mGlu1 PAM scaffolds have also been flagged as binding to alternate allosteric site(s) based on no or incomplete displacement of the allosteric radioligand [3H]R214127 (Hemstapat et al., 2006). Overall, these data strongly support the hypothesis that multiple allosteric sites are present on the group I mGlu receptors.
In the absence of an allosteric radioligand, multiple allosteric sites have also been proposed for mGlu4. The concentration-response curve for VU0155041 as an allosteric agonist at mGlu4 was unaffected by coapplication of PHCCC, an mGlu4 PAM of glutamate and L-AP4 (Niswender et al., 2008). Recent mutational analysis of mGlu4 allosteric modulators suggests that these two allosteric ligands interact with the common allosteric pocket made up of TMs 3–7 but in a nonoverlapping manner (Rovira et al., 2015). Based on homology modeling and mutational data, PHCCC is proposed to bind deep within the allosteric site, analogous to the binding pose of mavoglurant in the mGlu5 structure (Rovira et al., 2015). On the other hand, VU0155041 binds higher in the pocket, interacting with residues at the very top of the TM domains as well as extracellular loops 1 and 2 (Rovira et al., 2015). This suggests that two allosteric ligands can be accommodated simultaneously by mGlu4; however, at least for PHCCC and VU0155041, there is neutral cooperativity between the binding of these ligands. Interestingly, VU0415374 appears to be bitopic, with homology modeling proposing it can span both binding sites. It is tempting to speculate that the reason VU0415374 has 10-fold (or more) higher apparent affinity than other mGlu4 positive allosteric modulators is due to its ability to engage more residues within the pocket. Furthermore, it is interesting to consider that CPPHA has weak activity at mGlu4 (Chen et al., 2008), as such there is the possibility that allosteric ligands may interact with mGlu4 in three distinct binding modes. The extensive literature surrounding orthosteric and allosteric binding pockets of class A GPCRs may offer some insights into the location of alternative allosteric sites (Christopoulos, 2014). As confirmed in the recent cocrystal structure of the M2 muscarinic acetylcholine receptor, class A GPCR allosteric modulators recognize a binding pocket defined by the top of the TMs and the second and third extracellular loops (Kruse et al., 2013). Given the parallels between the common allosteric site of class C GPCRs and the orthosteric site in class A GPCRs, there is the possibility that alternate allosteric sites in class C GPCRs may also involve the extracellular surfaces of the receptor. Allosteric modulators that interact with different binding pockets are likely to engender a different complement of receptor conformational states than those that interact with the common allosteric site. Therefore the functional consequences of allosteric modulation through different binding pockets are likely to be divergent. Indeed, CPPHA and related compounds have biased pharmacology at mGlu5 when compared with common site modulators (Zhang et al., 2005; Noetzel et al., 2013). We are just beginning to scratch the surface with respect to our understanding of what drives efficacy, cooperativity, and affinity through the different allosteric sites across the entire mGlu family.
Concluding Remarks
Since the discovery of the first mGlu receptor allosteric modulator in 1996, the mGlu allosteric modulator pharmacological toolbox has continued to expand with selective allosteric ligands now available for the majority of mGlu family members. This toolbox includes chemically diverse allosteric ligands that may possess positive, negative, or neutral cooperativity and/or positive or inverse agonism. At least two allosteric sites have been proposed, one of which is shared across mGlu receptor subtypes, but can still be selectively targeted. Which allosteric site is best targeted for therapeutic efficacy remains unknown. Given the complexity introduced by the phenomenon of biased modulation, the optimal allosteric ligand pharmacological profile and the relative contribution of affinity, efficacy, and cooperativity will likely be both pathophysiology and mGlu subtype dependent. With two X-ray crystal structures of the 7TMs for group I mGlu receptors now available, the field is now entering an exciting new era in our understanding of the dynamic interactions that govern allosteric ligand affinity, cooperativity, and efficacy.
Supplementary Material
Abbreviations
- AMN082
N,N′-dibenzhydrylethane-1,2-diamine dihydrochloride
- BAY36-7620
(3aS,6aS)-hexahydro-5-methylene-6a-(2-naphthalenylmethyl)-1H-cyclopenta[c]furan-1-one
- CDPPB
3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide
- CNS
central nervous system
- CPCCOEt
7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester
- CPPHA
N-(4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl)-2-hydroxybenzamide
- DCG-IV
(2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine
- DFB
((3-fluorophenyl)methylene)hydrazone-3-fluorobenzaldehyde
- DHPG
3,5-dihydroxyphenylglycine
- FITM
4-fluoro-N-(4-(6-(isopropylamino)pyrimidin-4-yl)thiazole-2-yl)-N-methylbenzamide
- GPCR
G protein–coupled receptor
- JNJ-40068782
3-cyano-1-cyclo-propylmethyl-4-(4-phenyl-piperidin-1-yl)-pyridine-2(1H)-one
- L-AP4
l-2-amino-4-phosphonobutyrate
- LSP4-2022
(2S)-2-amino-4-([[4-(carboxymethoxy)phenyl](hydroxy)methyl](hydroxy)phosphoryl)butanoic acid
- Lu AF21934
(1S,2R)-2-[(aminooxy)methyl]-N-(3,4-dichlorophenyl)cyclohexane-1-carboxamide
- LY341495
(2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid
- LY379268
(−)-2-oxa-4-minobicyclo[3.1.0]hexane-4,6-dicarboxylate
- LY541850
(1S,2S,4R,5R,6S)-2-amino-4-methylbicyclo[3.1.0]hexane2,6-dicarboxylic acid
- mGlu
metabotropic glutamate
- M-5MPEP
2-(2-(3-methoxyphenyl)ethynyl)-5-methylpyridine
- MPEP
2-methyl-6-(phenylethynyl)pyridine
- mPEPy
3-methoxy-5-(2-pyridinylethynyl)pyridine
- NAM
negative allosteric modulator
- NCFP
N-(4-chloro-2-[(1,3-dioxoisoindolin-2-yl)methyl)phenyl)picolinamide
- PAM
positive allosteric modulator
- pERK1/2
phosphorylated extracellular–signal regulated kinases 1 and 2
- PET
positron emission tomography
- PHCCC
N-phenyl-7-(hydroxyimino)cyclopropa[b] chromen-1a-carboxamide
- R214127
1-(3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-2-phenylethanone
- Ro67-4853
butyl (9H-xanthene-9-carbonyl)carbamate
- SAR
structure-activity relationships
- TM
transmembrane
- VFT
venus flytrap domain
- VU0092273
3′-(8-methyl-4-oxo-7-trifluoromethyl-4,5-dihydro-3H-benzo[b][1,4]diazepin-2-yl)-biphenyl-3-sulfonic acid
- VU0155041
(1R,2S)-2-[(3,5-dichlorophenyl)carbamoyl]cyclohexane-1-carboxylic acid
- VU0155094
methyl 4-(3-(2-((4-acetamidophenyl)thio)acetyl)-2,5-dimethyl-1H-pyrrol-1-yl)benzoate
- VU0357121
4-butoxy-N-(2,4-difluorophenyl)benzamide
- VU0366248
N-(3-chloro-2-fluorophenyl)-3-cyano-5-fluorobenzamide
- VU0400100
2-[2-chloro-N-methylsulfonyl-5-(trifluoromethyl)anilino]-N-(cyclopropylmethyl)acetamide
- VU0409551
5-[(4-fluorophenyl)carbonyl]-2-(phenoxymethyl)-4,5,6,7-tetrahydro[1,3]oxazolo[5,4-c]pyridine
- VU0422288
N-(3-chloro-4-((5-chloropyridin-2-yl)oxy)phenyl)picolinamide
- VU29
N-(1,3-diphenyl-1H-pyrazolo-5-yl)-4-nitrobenzamide
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Gregory, Conn.
Footnotes
>Karen J. Gregory is a recipient of a National Health and Medical Research Council of Australia (NHMRC) Overseas Biomedical Postdoctoral Training Fellowship; work in her laboratory is supported by a NHMRC project grant [1084775]. Research on metabotropic glutamate receptors in the Conn laboratory is supported by the National Institutes of Health National Institute of Mental Health [Grant R01-MH062646-13], National Institute of Neurological Diseases and Stroke [Grant R01-NS031373-16A2], and National Institute of Drug Abuse [Grant R01-DA023947].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Ametamey SM, Treyer V, Streffer J, Wyss MT, Schmidt M, Blagoev M, Hintermann S, Auberson Y, Gasparini F, Fischer UC, et al. (2007) Human PET studies of metabotropic glutamate receptor subtype 5 with 11C-ABP688. J Nucl Med 48:247–252. [PubMed] [Google Scholar]
- Anderson JJ, Bradbury MJ, Giracello DR, Chapman DF, Holtz G, Roppe J, King C, Cosford ND, Varney MA. (2003) In vivo receptor occupancy of mGlu5 receptor antagonists using the novel radioligand [3H]3-methoxy-5-(pyridin-2-ylethynyl)pyridine). Eur J Pharmacol 473:35–40. [DOI] [PubMed] [Google Scholar]
- Anderson JJ, Rao SP, Rowe B, Giracello DR, Holtz G, Chapman DF, Tehrani L, Bradbury MJ, Cosford ND, Varney MA. (2002) [3H]Methoxymethyl-3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine binding to metabotropic glutamate receptor subtype 5 in rodent brain: in vitro and in vivo characterization. J Pharmacol Exp Ther 303:1044–1051. [DOI] [PubMed] [Google Scholar]
- Annoura H, Fukunaga A, Uesugi M, Tatsuoka T, Horikawa Y. (1996) A novel class of antagonists for metabotropic glutamate receptors, 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylates. Bioorg Med Chem Lett 6:763–766. [Google Scholar]
- Anwyl R. (1999) Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev 29:83–120. [DOI] [PubMed] [Google Scholar]
- Ayala JE, Chen Y, Banko JL, Sheffler DJ, Williams R, Telk AN, Watson NL, Xiang Z, Zhang Y, Jones PJ, et al. (2009) mGluR5 positive allosteric modulators facilitate both hippocampal LTP and LTD and enhance spatial learning. Neuropsychopharmacology 34:2057–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala JE, Niswender CM, Luo Q, Banko JL, Conn PJ. (2008) Group III mGluR regulation of synaptic transmission at the SC-CA1 synapse is developmentally regulated. Neuropharmacology 54:804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J, Weinstein H. (1995) Integrated methods for the construction of three dimensional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci 25:366–428. [Google Scholar]
- Baumann CA, Mu L, Wertli N, Krämer SD, Honer M, Schubiger PA, Ametamey SM. (2010) Syntheses and pharmacological characterization of novel thiazole derivatives as potential mGluR5 PET ligands. Bioorg Med Chem 18:6044–6054. [DOI] [PubMed] [Google Scholar]
- Becker JA, Clesse D, Spiegelhalter C, Schwab Y, Le Merrer J, Kieffer BL. (2014) Autistic-like syndrome in mu opioid receptor null mice is relieved by facilitated mGluR4 activity. Neuropsychopharmacology 39:2049–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone C, Lüscher C, Mameli M. (2008) Mechanisms of synaptic depression triggered by metabotropic glutamate receptors. Cell Mol Life Sci 65:2913–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belozertseva IV, Kos T, Popik P, Danysz W, Bespalov AY. (2007) Antidepressant-like effects of mGluR1 and mGluR5 antagonists in the rat forced swim and the mouse tail suspension tests. Eur Neuropsychopharmacol 17:172–179. [DOI] [PubMed] [Google Scholar]
- Bennett CE, Burnett DA, Greenlee WJ, Knutson CE, Korakas P, Li C, Tulshian D, Wu WL, Bertorelli R, Fredduzzi S, et al. (2012) Fused tricyclic mGluR1 antagonists for the treatment of neuropathic pain. Bioorg Med Chem Lett 22:1575–1578. [DOI] [PubMed] [Google Scholar]
- Beqollari D, Kammermeier PJ. (2008) The mGlu(4) receptor allosteric modulator N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxamide acts as a direct agonist at mGlu(6) receptors. Eur J Pharmacol 589:49–52. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Hessl D, Coffey S, Hervey C, Schneider A, Yuhas J, Hutchison J, Snape M, Tranfaglia M, Nguyen DV, et al. (2009) A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet 46:266–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley SJ, Langmead CJ, Watson JM, Challiss RA. (2011) Quantitative analysis reveals multiple mechanisms of allosteric modulation of the mGlu5 receptor in rat astroglia. Mol Pharmacol 79:874–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bräuner-Osborne H, Jensen AA, Krogsgaard-Larsen P. (1999) Interaction of CPCCOEt with a chimeric mGlu1b and calcium sensing receptor. Neuroreport 10:3923–3925. [DOI] [PubMed] [Google Scholar]
- Cabello N, Gandía J, Bertarelli DC, Watanabe M, Lluís C, Franco R, Ferré S, Luján R, Ciruela F. (2009) Metabotropic glutamate type 5, dopamine D2 and adenosine A2a receptors form higher-order oligomers in living cells. J Neurochem 109:1497–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campo B, Kalinichev M, Lambeng N, El Yacoubi M, Royer-Urios I, Schneider M, Legrand C, Parron D, Girard F, Bessif A, et al. (2011) Characterization of an mGluR2/3 negative allosteric modulator in rodent models of depression. J Neurogenet 25:152–166. [DOI] [PubMed] [Google Scholar]
- Carroll FY, Stolle A, Beart PM, Voerste A, Brabet I, Mauler F, Joly C, Antonicek H, Bockaert J, Müller T, et al. (2001) BAY36-7620: a potent non-competitive mGlu1 receptor antagonist with inverse agonist activity. Mol Pharmacol 59:965–973. [PMC free article] [PubMed] [Google Scholar]
- Chaki S, Ago Y, Palucha-Paniewiera A, Matrisciano F, Pilc A. (2013) mGlu2/3 and mGlu5 receptors: potential targets for novel antidepressants. Neuropharmacology 66:40–52. [DOI] [PubMed] [Google Scholar]
- Chen Y, Goudet C, Pin JP, Conn PJ. (2008) N-4-Chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl-2-hydroxybenzamide (CPPHA) acts through a novel site as a positive allosteric modulator of group 1 metabotropic glutamate receptors. Mol Pharmacol 73:909–918. [DOI] [PubMed] [Google Scholar]
- Chen Y, Nong Y, Goudet C, Hemstapat K, de Paulis T, Pin JP, Conn PJ. (2007) Interaction of novel positive allosteric modulators of metabotropic glutamate receptor 5 with the negative allosteric antagonist site is required for potentiation of receptor responses. Mol Pharmacol 71:1389–1398. [DOI] [PubMed] [Google Scholar]
- Christopoulos A. (2014) Advances in G protein-coupled receptor allostery: from function to structure. Mol Pharmacol 86:463–478. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Changeux JP, Catterall WA, Fabbro D, Burris TP, Cidlowski JA, Olsen RW, Peters JA, Neubig RR, Pin JP, et al. (2014) International union of basic and clinical pharmacology. XC. multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol Rev 66:918–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Lindsley CW, Meiler J, Niswender CM. (2014) Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating CNS disorders. Nat Rev Drug Discov 13:692–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. (1997) Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37:205–237. [DOI] [PubMed] [Google Scholar]
- Cosford ND, Roppe J, Tehrani L, Schweiger EJ, Seiders TJ, Chaudary A, Rao S, Varney MA. (2003b) [3H]-methoxymethyl-MTEP and [3H]-methoxy-PEPy: potent and selective radioligands for the metabotropic glutamate subtype 5 (mGlu5) receptor. Bioorg Med Chem Lett 13:351–354. [DOI] [PubMed] [Google Scholar]
- Coutinho V, Knöpfel T. (2002) Metabotropic glutamate receptors: electrical and chemical signaling properties. Neuroscientist 8:551–561. [DOI] [PubMed] [Google Scholar]
- Doré AS, Okrasa K, Patel JC, Serrano-Vega M, Bennett K, Cooke RM, Errey JC, Jazayeri A, Khan S, Tehan B, et al. (2014) Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 511:557–562. [DOI] [PubMed] [Google Scholar]
- Doria JG, Silva FR, de Souza JM, Vieira LB, Carvalho TG, Reis HJ, Pereira GS, Dobransky T, Ribeiro FM. (2013) Metabotropic glutamate receptor 5 positive allosteric modulators are neuroprotective in a mouse model of Huntington’s disease. Br J Pharmacol 169:909–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doumazane E, Scholler P, Zwier JM, Trinquet E, Rondard P, Pin JP. (2011) A new approach to analyze cell surface protein complexes reveals specific heterodimeric metabotropic glutamate receptors. FASEB J 25:66–77. [DOI] [PubMed] [Google Scholar]
- Dunayevich E, Erickson J, Levine L, Landbloom R, Schoepp DD, Tollefson GD. (2008) Efficacy and tolerability of an mGlu2/3 agonist in the treatment of generalized anxiety disorder. Neuropsychopharmacology 33:1603–1610. [DOI] [PubMed] [Google Scholar]
- Duvoisin RM, Pfankuch T, Wilson JM, Grabell J, Chhajlani V, Brown DG, Johnson E, Raber J. (2010) Acute pharmacological modulation of mGluR8 reduces measures of anxiety. Behav Brain Res 212:168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Moustaine D, Granier S, Doumazane E, Scholler P, Rahmeh R, Bron P, Mouillac B, Banères JL, Rondard P, Pin JP. (2012) Distinct roles of metabotropic glutamate receptor dimerization in agonist activation and G-protein coupling. Proc Natl Acad Sci USA 109:16342–16347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery AC, Diraddo JO, Miller E, Hathaway HA, Pshenichkin S, Takoudjou GR, Grajkowska E, Yasuda RP, Wolfe BB, Wroblewski J. (2012) Ligand Bias at Metabotropic Glutamate 1a Receptor: Molecular Determinants that Distinguish beta-arrestin from G Protein Mediated Signaling. Mol Pharmacol 82:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell MJ, Witkin JM, Falcone JF, Katner JS, Perry KW, Hart J, Rorick-Kehn L, Overshiner CD, Rasmussen K, Chaney SF, et al. (2011) N-(4-((2-(trifluoromethyl)-3-hydroxy-4-(isobutyryl)phenoxy)methyl)benzyl)-1-methyl-1H-imidazole-4-carboxamide (THIIC), a novel metabotropic glutamate 2 potentiator with potential anxiolytic/antidepressant properties: in vivo profiling suggests a link between behavioral and central nervous system neurochemical changes. J Pharmacol Exp Ther 336:165–177. [DOI] [PubMed] [Google Scholar]
- Felts AS, Lindsley SR, Lamb JP, Rodriguez AL, Menon UN, Jadhav S, Jones CK, Conn PJ, Lindsley CW, Emmitte KA. (2010) 3-Cyano-5-fluoro-N-arylbenzamides as negative allosteric modulators of mGlu(5): Identification of easily prepared tool compounds with CNS exposure in rats. Bioorg Med Chem Lett 20:4390–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda J, Suzuki G, Kimura T, Nagatomi Y, Ito S, Kawamoto H, Ozaki S, Ohta H. (2009) Identification of a novel transmembrane domain involved in the negative modulation of mGluR1 using a newly discovered allosteric mGluR1 antagonist, 3-cyclohexyl-5-fluoro-6-methyl-7-(2-morpholin-4-ylethoxy)-4H-chromen-4-one. Neuropharmacology 57:438–445. [DOI] [PubMed] [Google Scholar]
- Galici R, Jones CK, Hemstapat K, Nong Y, Echemendia NG, Williams LC, de Paulis T, Conn PJ. (2006) Biphenyl-indanone A, a positive allosteric modulator of the metabotropic glutamate receptor subtype 2, has antipsychotic- and anxiolytic-like effects in mice. J Pharmacol Exp Ther 318:173–185. [DOI] [PubMed] [Google Scholar]
- Gama L, Wilt SG, Breitwieser GE. (2001) Heterodimerization of calcium sensing receptors with metabotropic glutamate receptors in neurons. J Biol Chem 276:39053–39059. [DOI] [PubMed] [Google Scholar]
- Gasparini F, Andres H, Flor PJ, Heinrich M, Inderbitzin W, Lingenhöhl K, Müller H, Munk VC, Omilusik K, Stierlin C, et al. (2002) [(3)H]-M-MPEP, a potent, subtype-selective radioligand for the metabotropic glutamate receptor subtype 5. Bioorg Med Chem Lett 12:407–409. [DOI] [PubMed] [Google Scholar]
- Gasparini F, Floersheim P, Flor PJ, Heinrich M, Inderbitzin W, Ott D, Pagano A, Stierlin C, Stoehr N, Vranesic I, et al. (2001) Discovery and characterization of non-competitive antagonists of group I metabotropic glutamate receptors. Farmaco 56:95–99. [DOI] [PubMed] [Google Scholar]
- Gasparini F, Lingenhohl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, et al. (1999) 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology 38:1493–503. [DOI] [PubMed] [Google Scholar]
- González-Maeso J, Ang RL, Yuen T, Chan P, Weisstaub NV, López-Giménez JF, Zhou M, Okawa Y, Callado LF, Milligan G, et al. (2008) Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature 452:93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudet C, Gaven F, Kniazeff J, Vol C, Liu J, Cohen-Gonsaud M, Acher F, Prézeau L, Pin JP. (2004) Heptahelical domain of metabotropic glutamate receptor 5 behaves like rhodopsin-like receptors. Proc Natl Acad Sci USA 101:378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudet C, Kniazeff J, Hlavackova V, Malhaire F, Maurel D, Acher F, Blahos J, Prézeau L, Pin JP. (2005) Asymmetric functioning of dimeric metabotropic glutamate receptors disclosed by positive allosteric modulators. J Biol Chem 280:24380–24385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Herman EJ, Ramsey AJ, Hammond AS, Byun NE, Stauffer SR, Manka JT, Jadhav S, Bridges TM, Weaver CD, et al. (2013a) N-aryl piperazine metabotropic glutamate receptor 5 positive allosteric modulators possess efficacy in preclinical models of NMDA hypofunction and cognitive enhancement. J Pharmacol Exp Ther 347:438–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Nguyen ED, Malosh C, Mendenhall JL, Zic JZ, Bates BS, Noetzel MJ, Squire EF, Turner EM, Rook JM, et al. (2014) Identification of specific ligand-receptor interactions that govern binding and cooperativity of diverse modulators to a common metabotropic glutamate receptor 5 allosteric site. ACS Chem Neurosci 5:282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Nguyen ED, Reiff SD, Squire EF, Stauffer SR, Lindsley CW, Meiler J, Conn PJ. (2013b) Probing the metabotropic glutamate receptor 5 (mGlu₅) positive allosteric modulator (PAM) binding pocket: discovery of point mutations that engender a “molecular switch” in PAM pharmacology. Mol Pharmacol 83:991–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Noetzel MJ, Rook JM, Vinson PN, Stauffer SR, Rodriguez AL, Emmitte KA, Zhou Y, Chun AC, Felts AS, et al. (2012) Investigating metabotropic glutamate receptor 5 allosteric modulator cooperativity, affinity, and agonism: enriching structure-function studies and structure-activity relationships. Mol Pharmacol 82:860–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubellini P, Saulle E, Centonze D, Bonsi P, Pisani A, Bernardi G, Conquet F, Calabresi P. (2001) Selective involvement of mGlu1 receptors in corticostriatal LTD. Neuropharmacology 40:839–846. [DOI] [PubMed] [Google Scholar]
- Hammer R, Berrie CP, Birdsall NJ, Burgen AS, Hulme EC. (1980) Pirenzepine distinguishes between different subclasses of muscarinic receptors. Nature 283:90–92. [DOI] [PubMed] [Google Scholar]
- Hammond AS, Rodriguez AL, Townsend SD, Niswender CM, Gregory KJ, Lindsley CW, Conn PJ. (2010) Discovery of a Novel Chemical Class of mGlu(5) Allosteric Ligands with Distinct Modes of Pharmacology. ACS Chem Neurosci 1:702–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemstapat K, Da Costa H, Nong Y, Brady AE, Luo Q, Niswender CM, Tamagnan GD, Conn PJ. (2007) A novel family of potent negative allosteric modulators of group II metabotropic glutamate receptors. J Pharmacol Exp Ther 322:254–264. [DOI] [PubMed] [Google Scholar]
- Hemstapat K, de Paulis T, Chen Y, Brady AE, Grover VK, Alagille D, Tamagnan GD, Conn PJ. (2006) A novel class of positive allosteric modulators of metabotropic glutamate receptor subtype 1 interact with a site distinct from that of negative allosteric modulators. Mol Pharmacol 70:616–626. [DOI] [PubMed] [Google Scholar]
- Hughes ZA, Neal SJ, Smith DL, Sukoff Rizzo SJ, Pulicicchio CM, Lotarski S, Lu S, Dwyer JM, Brennan J, Olsen M, et al. (2013) Negative allosteric modulation of metabotropic glutamate receptor 5 results in broad spectrum activity relevant to treatment resistant depression. Neuropharmacology 66:202–214. [DOI] [PubMed] [Google Scholar]
- Iacovelli L, Felicioni M, Nisticò R, Nicoletti F, De Blasi A. (2014) Selective regulation of recombinantly expressed mGlu7 metabotropic glutamate receptors by G protein-coupled receptor kinases and arrestins. Neuropharmacology 77:303–312. [DOI] [PubMed] [Google Scholar]
- Jalan-Sakrikar N, Field JR, Klar R, Mattmann ME, Gregory KJ, Zamorano R, Engers DW, Bollinger SR, Weaver CD, Days EL, et al. (2014) Identification of positive allosteric modulators VU0155094 (ML397) and VU0422288 (ML396) reveals new insights into the biology of metabotropic glutamate receptor 7. ACS Chem Neurosci 5:1221–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang JY, Nagaraju M, Meyer RC, Zhang L, Hamelberg D, Hall RA, Brown EM, Conn PJ, Yang JJ. (2014) Extracellular calcium modulates actions of orthosteric and allosteric ligands on metabotropic glutamate receptor 1α. J Biol Chem 289:1649–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MP, Barda D, Britton TC, Emkey R, Hornback WJ, Jagdmann GE, McKinzie DL, Nisenbaum ES, Tizzano JP, Schoepp DD. (2005) Metabotropic glutamate 2 receptor potentiators: receptor modulation, frequency-dependent synaptic activity, and efficacy in preclinical anxiety and psychosis model(s). Psychopharmacology (Berl) 179:271–283. [DOI] [PubMed] [Google Scholar]
- Jones CK, Bubser M, Thompson AD, Dickerson JW, Turle-Lorenzo N, Amalric M, Blobaum AL, Bridges TM, Morrison RD, Jadhav S, et al. (2012) The metabotropic glutamate receptor 4-positive allosteric modulator VU0364770 produces efficacy alone and in combination with L-DOPA or an adenosine 2A antagonist in preclinical rodent models of Parkinson’s disease. J Pharmacol Exp Ther 340:404–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PJ, Xiang Z, Conn PJ. (2008) Metabotropic glutamate receptors mGluR4 and mGluR8 regulate transmission in the lateral olfactory tract-piriform cortex synapse. Neuropharmacology 55:440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong YJ, Kumar V, Kingston AE, Romano C, O’Malley KL. (2005) Functional metabotropic glutamate receptors on nuclei from brain and primary cultured striatal neurons. Role of transporters in delivering ligand. J Biol Chem 280:30469–30480. [DOI] [PubMed] [Google Scholar]
- Jong YJ, Kumar V, O’Malley KL. (2009) Intracellular metabotropic glutamate receptor 5 (mGluR5) activates signaling cascades distinct from cell surface counterparts. J Biol Chem 284:35827–35838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julio-Pieper M, Flor PJ, Dinan TG, Cryan JF. (2011) Exciting times beyond the brain: metabotropic glutamate receptors in peripheral and non-neural tissues. Pharmacol Rev 63:35–58. [DOI] [PubMed] [Google Scholar]
- Kalinichev M, Rouillier M, Girard F, Royer-Urios I, Bournique B, Finn T, Charvin D, Campo B, Le Poul E, Mutel V, et al. (2013) ADX71743, a potent and selective negative allosteric modulator of metabotropic glutamate receptor 7: in vitro and in vivo characterization. J Pharmacol Exp Ther 344:624–636. [DOI] [PubMed] [Google Scholar]
- Kammermeier PJ. (2012) Functional and pharmacological characteristics of metabotropic glutamate receptors 2/4 heterodimers. Mol Pharmacol 82:438–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keywood C, Wakefield M, Tack J. (2009) A proof-of-concept study evaluating the effect of ADX10059, a metabotropic glutamate receptor-5 negative allosteric modulator, on acid exposure and symptoms in gastro-oesophageal reflux disease. Gut 58:1192–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney GG, O’Brien JA, Lemaire W, Burno M, Bickel DJ, Clements MK, Chen TB, Wisnoski DD, Lindsley CW, Tiller PR, et al. (2005) A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J Pharmacol Exp Ther 313:199–206. [DOI] [PubMed] [Google Scholar]
- Kinon BJ, Zhang L, Millen BA, Osuntokun OO, Williams JE, Kollack-Walker S, Jackson K, Kryzhanovskaya L, Jarkova N, HBBI Study Group (2011) A multicenter, inpatient, phase 2, double-blind, placebo-controlled dose-ranging study of LY2140023 monohydrate in patients with DSM-IV schizophrenia. J Clin Psychopharmacol 31:349–355. [DOI] [PubMed] [Google Scholar]
- Knoflach F, Mutel V, Jolidon S, Kew JN, Malherbe P, Vieira E, Wichmann J, Kemp JA. (2001) Positive allosteric modulators of metabotropic glutamate 1 receptor: characterization, mechanism of action, and binding site. Proc Natl Acad Sci USA 98:13402–13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohara A, Toya T, Tamura S, Watabiki T, Nagakura Y, Shitaka Y, Hayashibe S, Kawabata S, Okada M. (2005) Radioligand binding properties and pharmacological characterization of 6-amino-N-cyclohexyl-N,3-dimethylthiazolo[3,2-a]benzimidazole-2-carboxamide (YM-298198), a high-affinity, selective, and noncompetitive antagonist of metabotropic glutamate receptor type 1. J Pharmacol Exp Ther 315:163–169. [DOI] [PubMed] [Google Scholar]
- Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hübner H, Pardon E, Valant C, Sexton PM, et al. (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504:101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Fahey PG, Jong YJ, Ramanan N, O’Malley KL. (2012) Activation of intracellular metabotropic glutamate receptor 5 in striatal neurons leads to up-regulation of genes associated with sustained synaptic transmission including Arc/Arg3.1 protein. J Biol Chem 287:5412–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Jong YJ, O’Malley KL. (2008) Activated nuclear metabotropic glutamate receptor mGlu5 couples to nuclear Gq/11 proteins to generate inositol 1,4,5-trisphosphate-mediated nuclear Ca2+ release. J Biol Chem 283:14072–14083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb JP, Engers DW, Niswender CM, Rodriguez AL, Venable DF, Conn PJ, Lindsley CW. (2011) Discovery of molecular switches within the ADX-47273 mGlu5 PAM scaffold that modulate modes of pharmacology to afford potent mGlu5 NAMs, PAMs and partial antagonists. Bioorg Med Chem Lett 21:2711–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavreysen H, Janssen C, Bischoff F, Langlois X, Leysen JE, Lesage AS. (2003) [3H]R214127: a novel high-affinity radioligand for the mGlu1 receptor reveals a common binding site shared by multiple allosteric antagonists. Mol Pharmacol 63:1082–1093. [DOI] [PubMed] [Google Scholar]
- Lavreysen H, Langlois X, Ahnaou A, Drinkenburg W, te Riele P, Biesmans I, Van der Linden I, Peeters L, Megens A, Wintmolders C, et al. (2013) Pharmacological characterization of JNJ-40068782, a new potent, selective, and systemically active positive allosteric modulator of the mGlu2 receptor and its radioligand [3H]JNJ-40068782. J Pharmacol Exp Ther 346:514–527. [DOI] [PubMed] [Google Scholar]
- Lavreysen H, Wouters R, Bischoff F, Nóbrega Pereira S, Langlois X, Blokland S, Somers M, Dillen L, Lesage AS. (2004) JNJ16259685, a highly potent, selective and systemically active mGlu1 receptor antagonist. Neuropharmacology 47:961–972. [DOI] [PubMed] [Google Scholar]
- Li X, Need AB, Baez M, Witkin JM. (2006) Metabotropic glutamate 5 receptor antagonism is associated with antidepressant-like effects in mice. J Pharmacol Exp Ther 319:254–259. [DOI] [PubMed] [Google Scholar]
- Lindsley CW. (2014) 2013 Philip S. Portoghese Medicinal Chemistry Lectureship: drug discovery targeting allosteric sites. J Med Chem 57:7485–7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litschig S, Gasparini F, Rueegg D, Stoehr N, Flor PJ, Vranesic I, Prézeau L, Pin JP, Thomsen C, Kuhn R. (1999) CPCCOEt, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits receptor signaling without affecting glutamate binding. Mol Pharmacol 55:453–461. [PubMed] [Google Scholar]
- Lundström L, Bissantz C, Beck J, Wettstein JG, Woltering TJ, Wichmann J, Gatti S. (2011) Structural determinants of allosteric antagonism at metabotropic glutamate receptor 2: mechanistic studies with new potent negative allosteric modulators. Br J Pharmacol 164 (2b):521–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma AWS, Redka DS, Pisterzi LF, Angers S, Wells JW. (2007) Recovery of oligomers and cooperativity when monomers of the M2 muscarinic cholinergic receptor are reconstituted into phospholipid vesicles. Biochemistry 46:7907–7927. [DOI] [PubMed] [Google Scholar]
- Mabire D, Coupa S, Adelinet C, Poncelet A, Simonnet Y, Venet M, Wouters R, Lesage AS, Van Beijsterveldt L, Bischoff F. (2005) Synthesis, structure-activity relationship, and receptor pharmacology of a new series of quinoline derivatives acting as selective, noncompetitive mGlu1 antagonists. J Med Chem 48:2134–2153. [DOI] [PubMed] [Google Scholar]
- Maj M, Bruno V, Dragic Z, Yamamoto R, Battaglia G, Inderbitzin W, Stoehr N, Stein T, Gasparini F, Vranesic I, et al. (2003) (-)-PHCCC, a positive allosteric modulator of mGluR4: characterization, mechanism of action, and neuroprotection. Neuropharmacology 45:895–906. [DOI] [PubMed] [Google Scholar]
- Malherbe P, Kratochwil N, Knoflach F, Zenner MT, Kew JN, Kratzeisen C, Maerki HP, Adam G, Mutel V. (2003a) Mutational analysis and molecular modeling of the allosteric binding site of a novel, selective, noncompetitive antagonist of the metabotropic glutamate 1 receptor. J Biol Chem 278:8340–8347. [DOI] [PubMed] [Google Scholar]
- Malherbe P, Kratochwil N, Mühlemann A, Zenner M-T, Fischer C, Stahl M, Gerber PR, Jaeschke G, Porter RHP. (2006) Comparison of the binding pockets of two chemically unrelated allosteric antagonists of the mGlu5 receptor and identification of crucial residues involved in the inverse agonism of MPEP. J Neurochem 98:601–615. [DOI] [PubMed] [Google Scholar]
- Malherbe P, Kratochwil N, Zenner MT, Piussi J, Diener C, Kratzeisen C, Fischer C, Porter RH. (2003b) Mutational analysis and molecular modeling of the binding pocket of the metabotropic glutamate 5 receptor negative modulator 2-methyl-6-(phenylethynyl)-pyridine. Mol Pharmacol 64:823–832. [DOI] [PubMed] [Google Scholar]
- Marino MJ, Williams DL, Jr, O’Brien JA, Valenti O, McDonald TP, Clements MK, Wang R, DiLella AG, Hess JF, Kinney GG, et al. (2003) Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson’s disease treatment. Proc Natl Acad Sci USA 100:13668–13673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathiesen JM, Svendsen N, Bräuner-Osborne H, Thomsen C, Ramirez MT. (2003) Positive allosteric modulation of the human metabotropic glutamate receptor 4 (hmGluR4) by SIB-1893 and MPEP. Br J Pharmacol 138:1026–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsukawa K, Yamamoto R, Ofner S, Nozulak J, Pescott O, Lukic S, Stoehr N, Mombereau C, Kuhn R, McAllister KH, et al. (2005) A selective metabotropic glutamate receptor 7 agonist: activation of receptor signaling via an allosteric site modulates stress parameters in vivo. Proc Natl Acad Sci USA 102:18712–18717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B. (2004) Targeting metabotropic glutamate receptors for treatment of the cognitive symptoms of schizophrenia. Psychopharmacology (Berl) 174:39–44. [DOI] [PubMed] [Google Scholar]
- Mølck C, Harpsøe K, Gloriam DE, Clausen RP, Madsen U, Pedersen LO, Jimenez HN, Nielsen SM, Mathiesen JM, Bräuner-Osborne H. (2012) Pharmacological characterization and modeling of the binding sites of novel 1,3-bis(pyridinylethynyl)benzenes as metabotropic glutamate receptor 5-selective negative allosteric modulators. Mol Pharmacol 82:929–937. [DOI] [PubMed] [Google Scholar]
- Moussawi K, Kalivas PW. (2010) Group II metabotropic glutamate receptors (mGlu2/3) in drug addiction. Eur J Pharmacol 639:115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlemann A, Ward NA, Kratochwil N, Diener C, Fischer C, Stucki A, Jaeschke G, Malherbe P, Porter RH. (2006) Determination of key amino acids implicated in the actions of allosteric modulation by 3,3′-difluorobenzaldazine on rat mGlu5 receptors. Eur J Pharmacol 529:95–104. [DOI] [PubMed] [Google Scholar]
- Niswender CM, Johnson KA, Miller NR, Ayala JE, Luo Q, Williams R, Saleh S, Orton D, Weaver CD, Conn PJ. (2010) Context-dependent pharmacology exhibited by negative allosteric modulators of metabotropic glutamate receptor 7. Mol Pharmacol 77:459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Johnson KA, Weaver CD, Jones CK, Xiang Z, Luo Q, Rodriguez AL, Marlo JE, de Paulis T, Thompson AD, et al. (2008) Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol Pharmacol 74:1345–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noetzel MJ, Gregory KJ, Vinson PN, Manka JT, Stauffer SR, Lindsley CW, Niswender CM, Xiang Z, Conn PJ. (2013) A novel metabotropic glutamate receptor 5 positive allosteric modulator acts at a unique site and confers stimulus bias to mGlu5 signaling. Mol Pharmacol 83:835–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noetzel MJ, Jones CK, Conn PJ. (2012a) Emerging approaches for treatment of schizophrenia: modulation of glutamatergic signaling. Discov Med 14:335–343. [PMC free article] [PubMed] [Google Scholar]
- Noetzel MJ, Rook JM, Vinson PN, Cho HP, Days E, Zhou Y, Rodriguez AL, Lavreysen H, Stauffer SR, Niswender CM, et al. (2012b) Functional impact of allosteric agonist activity of selective positive allosteric modulators of metabotropic glutamate receptor subtype 5 in regulating central nervous system function. Mol Pharmacol 81:120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JA, Lemaire W, Chen TB, Chang RS, Jacobson MA, Ha SN, Lindsley CW, Schaffhauser HJ, Sur C, Pettibone DJ, et al. (2003) A family of highly selective allosteric modulators of the metabotropic glutamate receptor subtype 5. Mol Pharmacol 64:731–740. [DOI] [PubMed] [Google Scholar]
- O’Brien JA, Lemaire W, Wittmann M, Jacobson MA, Ha SN, Wisnoski DD, Lindsley CW, Schaffhauser HJ, Rowe B, Sur C, et al. (2004) A novel selective allosteric modulator potentiates the activity of native metabotropic glutamate receptor subtype 5 in rat forebrain. J Pharmacol Exp Ther 309:568–577. [DOI] [PubMed] [Google Scholar]
- Odagaki Y, Kinoshita M, Toyoshima R. (2011) Functional coupling between metabotropic glutamate receptors and G-proteins in rat cerebral cortex assessed by guanosine-5′-O-(3-[(35)S]thio)triphosphate binding assay. Basic Clin Pharmacol Toxicol 109:175–185. [DOI] [PubMed] [Google Scholar]
- Odagaki Y, Kinoshita M, Toyoshima R. (2013) Group II metabotropic glutamate receptor-mediated activation of G-proteins in rat hippocampal and striatal membranes. Neurosci Lett 539:22–26. [DOI] [PubMed] [Google Scholar]
- Ott D, Floersheim P, Inderbitzin W, Stoehr N, Francotte E, Lecis G, Richert P, Rihs G, Flor PJ, Kuhn R, et al. (2000) Chiral resolution, pharmacological characterization, and receptor docking of the noncompetitive mGlu1 receptor antagonist (+/-)-2-hydroxyimino- 1a, 2-dihydro-1H-7-oxacyclopropa[b]naphthalene-7a-carboxylic acid ethyl ester. J Med Chem 43:4428–4436. [DOI] [PubMed] [Google Scholar]
- Pagano A, Ruegg D, Litschig S, Stoehr N, Stierlin C, Heinrich M, Floersheim P, Prezèau L, Carroll F, Pin JP, et al. (2000) The non-competitive antagonists 2-methyl-6-(phenylethynyl)pyridine and 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester interact with overlapping binding pockets in the transmembrane region of group I metabotropic glutamate receptors. J Biol Chem 275:33750–33758. [DOI] [PubMed] [Google Scholar]
- Parmentier-Batteur S, Hutson PH, Menzel K, Uslaner JM, Mattson BA, O’Brien JA, Magliaro BC, Forest T, Stump CA, Tynebor RM, et al. (2014) Mechanism based neurotoxicity of mGlu5 positive allosteric modulators—development challenges for a promising novel antipsychotic target. Neuropharmacology 82:161–173. [DOI] [PubMed] [Google Scholar]
- Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, Avedisova AS, Bardenstein LM, Gurovich IY, Morozova MA, et al. (2007) Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med 13:1102–1107. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Yuan X, Lavezzari G, Roche KW, McBain CJ. (2007) mGluR7 undergoes rapid internalization in response to activation by the allosteric agonist AMN082. Neuropharmacology 52:108–117. [DOI] [PubMed] [Google Scholar]
- Pinheiro PS, Mulle C. (2008) Presynaptic glutamate receptors: physiological functions and mechanisms of action. Nat Rev Neurosci 9:423–436. [DOI] [PubMed] [Google Scholar]
- Pittolo S, Gómez-Santacana X, Eckelt K, Rovira X, Dalton J, Goudet C, Pin JP, Llobet A, Giraldo J, Llebaria A, et al. (2014) An allosteric modulator to control endogenous G protein-coupled receptors with light. Nat Chem Biol 10:813–815. [DOI] [PubMed] [Google Scholar]
- Porter RH, Jaeschke G, Spooren W, Ballard TM, Büttelmann B, Kolczewski S, Peters JU, Prinssen E, Wichmann J, Vieira E, et al. (2005) Fenobam: a clinically validated nonbenzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor antagonist with inverse agonist activity. J Pharmacol Exp Ther 315:711–721. [DOI] [PubMed] [Google Scholar]
- Purgert CA, Izumi Y, Jong YJ, Kumar V, Zorumski CF, O’Malley KL. (2014) Intracellular mGluR5 can mediate synaptic plasticity in the hippocampus. J Neurosci 34:4589–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days EL, et al. (2010) Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol Pharmacol 78:1105–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AL, Nong Y, Sekaran NK, Alagille D, Tamagnan GD, Conn PJ. (2005) A close structural analog of 2-methyl-6-(phenylethynyl)-pyridine acts as a neutral allosteric site ligand on metabotropic glutamate receptor subtype 5 and blocks the effects of multiple allosteric modulators. Mol Pharmacol 68:1793–1802. [DOI] [PubMed] [Google Scholar]
- Rodriguez AL, Zhou Y, Williams R, Weaver CD, Vinson PN, Dawson ES, Steckler T, Lavreysen H, Mackie C, Bartolomé JM, et al. (2012) Discovery and SAR of a novel series of non-MPEP site mGlu₅ PAMs based on an aryl glycine sulfonamide scaffold. Bioorg Med Chem Lett 22:7388–7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rook JM, Noetzel MJ, Pouliot WA, Bridges TM, Vinson PN, Cho HP, Zhou Y, Gogliotti RD, Manka JT, Gregory KJ, et al. (2013) Unique signaling profiles of positive allosteric modulators of metabotropic glutamate receptor subtype 5 determine differences in in vivo activity. Biol Psychiatry 73:501–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rook JM, Xiang Z, Lv X, Ghoshal A, Dickerson JW, Bridges TM, Johnson KA, Foster DJ, Gregory KJ, Vinson PN, et al. (2015) Biased mGlu5 positive allosteric modulators provide in vivo efficacy without potentiating mGlu5 modulation of NMDAR currents. Neuron, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovira X, Malhaire F, Scholler P, Rodrigo J, Gonzalez-Bulnes P, Llebaria A, Pin JP, Giraldo J, Goudet C. (2015) Overlapping binding sites drive allosteric agonism and positive cooperativity in type 4 metabotropic glutamate receptors. FASEB J 29:116–130. [DOI] [PubMed] [Google Scholar]
- Rowe BA, Schaffhauser H, Morales S, Lubbers LS, Bonnefous C, Kamenecka TM, McQuiston J, Daggett LP. (2008) Transposition of three amino acids transforms the human metabotropic glutamate receptor (mGluR)-3-positive allosteric modulation site to mGluR2, and additional characterization of the mGluR2-positive allosteric modulation site. J Pharmacol Exp Ther 326:240–251. [DOI] [PubMed] [Google Scholar]
- Rylander D, Iderberg H, Li Q, Dekundy A, Zhang J, Li H, Baishen R, Danysz W, Bezard E, Cenci MA. (2010) A mGluR5 antagonist under clinical development improves L-DOPA-induced dyskinesia in parkinsonian rats and monkeys. Neurobiol Dis 39:352–361. [DOI] [PubMed] [Google Scholar]
- Sams AG, Mikkelsen GK, Brodbeck RM, Pu X, Ritzén A. (2011) Efficacy switching SAR of mGluR5 allosteric modulators: highly potent positive and negative modulators from one chemotype. Bioorg Med Chem Lett 21:3407–3410. [DOI] [PubMed] [Google Scholar]
- Sanger H, Hanna L, Colvin EM, Grubisha O, Ursu D, Heinz BA, Findlay JD, Vivier RG, Sher E, Lodge D, et al. (2013) Pharmacological profiling of native group II metabotropic glutamate receptors in primary cortical neuronal cultures using a FLIPR. Neuropharmacology 66:264–273. [DOI] [PubMed] [Google Scholar]
- Scandroglio P, Brusa R, Lozza G, Mancini I, Petrò R, Reggiani A, Beltramo M. (2010) Evaluation of cannabinoid receptor 2 and metabotropic glutamate receptor 1 functional responses using a cell impedance-based technology. J Biomol Screen 15:1238–1247. [DOI] [PubMed] [Google Scholar]
- Schaffhauser H, Rowe BA, Morales S, Chavez-Noriega LE, Yin R, Jachec C, Rao SP, Bain G, Pinkerton AB, Vernier JM, et al. (2003) Pharmacological characterization and identification of amino acids involved in the positive modulation of metabotropic glutamate receptor subtype 2. Mol Pharmacol 64:798–810. [DOI] [PubMed] [Google Scholar]
- Schann S, Mayer S, Franchet C, Frauli M, Steinberg E, Thomas M, Baron L, Neuville P. (2010) Chemical switch of a metabotropic glutamate receptor 2 silent allosteric modulator into dual metabotropic glutamate receptor 2/3 negative/positive allosteric modulators. J Med Chem 53:8775–8779. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Wright RA, Levine LR, Gaydos B, Potter WZ. (2003) LY354740, an mGlu2/3 receptor agonist as a novel approach to treat anxiety/stress. Stress 6:189–197. [DOI] [PubMed] [Google Scholar]
- Sevastyanova TN, Kammermeier PJ. (2014) Cooperative signaling between homodimers of metabotropic glutamate receptors 1 and 5. Mol Pharmacol 86:492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Kedrowski J, Rook JM, Smith RL, Jones CK, Rodriguez AL, Conn PJ, Lindsley CW. (2009) Discovery of molecular switches that modulate modes of metabotropic glutamate receptor subtype 5 (mGlu5) pharmacology in vitro and in vivo within a series of functionalized, regioisomeric 2- and 5-(phenylethynyl)pyrimidines. J Med Chem 52:4103–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffler DJ, Conn PJ. (2008) Allosteric potentiators of metabotropic glutamate receptor subtype 1a differentially modulate independent signaling pathways in baby hamster kidney cells. Neuropharmacology 55:419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffler DJ, Wenthur CJ, Bruner JA, Carrington SJS, Vinson PN, Gogi KK, Blobaum AL, Morrison RD, Vamos M, Cosford NDP, et al. (2012) Development of a novel, CNS-penetrant, metabotropic glutamate receptor 3 (mGlu3) NAM probe (ML289) derived from a closely related mGlu5 PAM. Bioorg Med Chem Lett 22:3921–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukoff Rizzo SJ, Leonard SK, Gilbert A, Dollings P, Smith DL, Zhang MY, Di L, Platt BJ, Neal S, Dwyer JM, et al. (2011) The metabotropic glutamate receptor 7 allosteric modulator AMN082: a monoaminergic agent in disguise? J Pharmacol Exp Ther 338:345–352. [DOI] [PubMed] [Google Scholar]
- Surin A, Pshenichkin S, Grajkowska E, Surina E, Wroblewski JT. (2007) Cyclothiazide selectively inhibits mGluR1 receptors interacting with a common allosteric site for non-competitive antagonists. Neuropharmacology 52:744–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki G, Kimura T, Satow A, Kaneko N, Fukuda J, Hikichi H, Sakai N, Maehara S, Kawagoe-Takaki H, Hata M, et al. (2007) Pharmacological characterization of a new, orally active and potent allosteric metabotropic glutamate receptor 1 antagonist, 4-[1-(2-fluoropyridin-3-yl)-5-methyl-1H-1,2,3-triazol-4-yl]-N-isopropyl-N-methyl-3,6-dihydropyridine-1(2H)-carboxamide (FTIDC). J Pharmacol Exp Ther 321:1144–1153. [DOI] [PubMed] [Google Scholar]
- Treyer V, Streffer J, Wyss MT, Bettio A, Ametamey SM, Fischer U, Schmidt M, Gasparini F, Hock C, Buck A. (2007) Evaluation of the metabotropic glutamate receptor subtype 5 using PET and 11C-ABP688: assessment of methods. J Nucl Med 48:1207–1215. [DOI] [PubMed] [Google Scholar]
- Turlington M, Noetzel MJ, Bridges TM, Vinson PN, Steckler T, Lavreysen H, Mackie C, Bartolomé-Nebreda JM, Conde-Ceide S, Tong HM, et al. (2014) Discovery and SAR of a novel series of metabotropic glutamate receptor 5 positive allosteric modulators with high ligand efficiency. Bioorg Med Chem Lett 24:3641–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti O, Conn PJ, Marino MJ. (2002) Distinct physiological roles of the Gq-coupled metabotropic glutamate receptors Co-expressed in the same neuronal populations. J Cell Physiol 191:125–137. [DOI] [PubMed] [Google Scholar]
- Valenti O, Mannaioni G, Seabrook GR, Conn PJ, Marino MJ. (2005) Group III metabotropic glutamate-receptor-mediated modulation of excitatory transmission in rodent substantia nigra pars compacta dopamine neurons. J Pharmacol Exp Ther 313:1296–1304. [DOI] [PubMed] [Google Scholar]
- Vieira E, Huwyler J, Jolidon S, Knoflach F, Mutel V, Wichmann J. (2009) Fluorinated 9H-xanthene-9-carboxylic acid oxazol-2-yl-amides as potent, orally available mGlu1 receptor enhancers. Bioorg Med Chem Lett 19:1666–1669. [DOI] [PubMed] [Google Scholar]
- Vilar B, Busserolles J, Ling B, Laffray S, Ulmann L, Malhaire F, Chapuy E, Aissouni Y, Etienne M, Bourinet E, et al. (2013) Alleviating pain hypersensitivity through activation of type 4 metabotropic glutamate receptor. J Neurosci 33:18951–18965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woltering TJ, Wichmann J, Goetschi E, Knoflach F, Ballard TM, Huwyler J, Gatti S. (2010) Synthesis and characterization of 1,3-dihydro-benzo[b][1,4]diazepin-2-one derivatives: Part 4. In vivo active potent and selective non-competitive metabotropic glutamate receptor 2/3 antagonists. Bioorg Med Chem Lett 20:6969–6974. [DOI] [PubMed] [Google Scholar]
- Wood MR, Hopkins CR, Brogan JT, Conn PJ, Lindsley CW. (2011) “Molecular switches” on mGluR allosteric ligands that modulate modes of pharmacology. Biochemistry 50:2403–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wang C, Gregory KJ, Han GW, Cho HP, Xia Y, Niswender CM, Katritch V, Meiler J, Cherezov V, et al. (2014) Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science 344:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin S, Noetzel MJ, Johnson KA, Zamorano R, Jalan-Sakrikar N, Gregory KJ, Conn PJ, Niswender CM. (2014) Selective actions of novel allosteric modulators reveal functional heteromers of metabotropic glutamate receptors in the CNS. J Neurosci 34:79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin S, Zamorano R, Conn PJ, Niswender CM. (2013) Functional selectivity induced by mGlu(4) receptor positive allosteric modulation and concomitant activation of G(q) coupled receptors. Neuropharmacology 66:122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbib F, Keywood C, Strabach G. (2010) Efficacy, tolerability and pharmacokinetics of a modified release formulation of ADX10059, a negative allosteric modulator of metabotropic glutamate receptor 5: an esophageal pH-impedance study in healthy subjects. Neurogastroenterol Motil 22: 859–865. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Rodriguez AL, Conn PJ. (2005) Allosteric potentiators of metabotropic glutamate receptor subtype 5 have differential effects on different signaling pathways in cortical astrocytes. J Pharmacol Exp Ther 315:1212–1219. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Manka JT, Rodriguez AL, Weaver CD, Days EL, Vinson PN, Jadhav S, Hermann EJ, Jones CK, Conn PJ, et al. (2010) Discovery of N-Aryl Piperazines as Selective mGlu(5) Potentiators with Efficacy in a Rodent Model Predictive of Anti-Psychotic Activity. ACS Med Chem Lett 1:433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.