

Abstract
KPC-2 is the most prevalent class A carbapenemase in the world. Previously, KPC-2 was shown to hydrolyze the β-lactamase inhibitors clavulanic acid, sulbactam, and tazobactam. In addition, substitutions at amino acid position R220 in the KPC-2 β-lactamase increased resistance to clavulanic acid. A novel bridged diazabicyclooctane (DBO) non-β-lactam β-lactamase inhibitor, avibactam, was shown to inactivate the KPC-2 β-lactamase. To better understand the mechanistic basis for inhibition of KPC-2 by avibactam, we tested the potency of ampicillin-avibactam and ceftazidime-avibactam against engineered variants of the KPC-2 β-lactamase that possessed single amino acid substitutions at important sites (i.e., Ambler positions 69, 130, 234, 220, and 276) that were previously shown to confer inhibitor resistance in TEM and SHV β-lactamases. To this end, we performed susceptibility testing, biochemical assays, and molecular modeling. Escherichia coli DH10B carrying KPC-2 β-lactamase variants with the substitutions S130G, K234R, and R220M demonstrated elevated MICs for only the ampicillin-avibactam combinations (e.g., 512, 64, and 32 mg/liter, respectively, versus the MICs for wild-type KPC-2 at 2 to 8 mg/liter). Steady-state kinetics revealed that the S130G variant of KPC-2 resisted inactivation by avibactam; the k2/K ratio was significantly lowered 4 logs for the S130G variant from the ratio for the wild-type enzyme (21,580 M−1 s−1 to 1.2 M−1 s−1). Molecular modeling and molecular dynamics simulations suggested that the mobility of K73 and its ability to activate S70 (i.e., function as a general base) may be impaired in the S130G variant of KPC-2, thereby explaining the slowed acylation. Moreover, we also advance the idea that the protonation of the sulfate nitrogen of avibactam may be slowed in the S130G variant, as S130 is the likely proton donor and another residue, possibly K234, must compensate. Our findings show that residues S130 as well as K234 and R220 contribute significantly to the mechanism of avibactam inactivation of KPC-2. Fortunately, the emergence of S130G, K234R, and R220M variants of KPC in the clinic should not result in failure of ceftazidime-avibactam, as the ceftazidime partner is potent against E. coli DH10B strains possessing all of these variants.
INTRODUCTION
In 2013, the Centers for Disease Control and Prevention (CDC) in the United States announced that carbapenem-resistant Enterobacteriaceae (CRE; e.g., Klebsiella pneumoniae possessing blaKPC-2) were one of the highest-priority target pathogens for the identification and development of novel antibacterials (1). CREs are often resistant to all antibiotics; thus, treatment options are limited to toxic agents, such as polymyxins B and E. Every year in the United States, 9,000 people become infected with CREs, and 600 of these individuals die (1). To combat these trends, the Infectious Diseases Society of America (IDSA) presented a “call to arms,” known as the 10 by '20 Initiative, which demands the development of 10 new antibiotics by 2020 (2). Regrettably, as of April 2013, a progress report revealed that only one such antibiotic (i.e., a β-lactam, ceftaroline-fosamil) had been approved by the Food and Drug Administration (FDA); importantly, ceftaroline-fosamil lacks activity against CREs (3).
Of the CREs, Klebsiella spp. possessing KPC-2 β-lactamases are the most prevalent in the United States as well as other regions of the world (e.g., Israel) (1). Resistance mediated by KPC-2 β-lactamase is 2-fold: not only does KPC-2 hydrolyze β-lactams of all classes (i.e., penicillins, cephalosporins, carbapenems, and monobactams), but KPC-2 also inactivates the commercially available β-lactamase inhibitors (i.e., clavulanic acid, sulbactam, and tazobactam) (4, 5).
Unlike the TEM and SHV β-lactamases, our understanding of structure-function relationships for KPC β-lactamase is still in its infancy. Site-directed mutagenesis has shown that several amino acid positions are critical for KPC-2's inhibitor-resistant phenotype as well as its carbapenemase and cephalosporinase activities (e.g., R220 and T237) (6, 7). A novel bridged diazabicyclooctane non-β-lactam β-lactamase inhibitor, avibactam, was shown to inactivate KPC-2 and restore susceptibility to certain β-lactams when given in combination with them (8). When avibactam's inhibition profile was compared among β-lactamases, avibactam was slowly hydrolyzed by KPC-2, suggesting differences in the reaction pathway and important differences in the catalytic machinery of this class A β-lactamase (8).
From the highest-resolution crystal structure of CTX-M-15 with acylated avibactam, stabilizing hydrogen bonds at key residues (e.g., S70, S130, and S237) in the active site were similar to those that formed with other β-lactamase inhibitors (Fig. 1) (9). Based upon this knowledge and results of previous studies on the mechanism of inactivation by clavulanic acid of other class A β-lactamases (e.g., SHV-1), we hypothesized that the structural and kinetic determinants that mediate resistance to clavulanic acid, sulbactam, and tazobactam in class A β-lactamases will also result in resistance to avibactam. In this work, we determined the amino acid sequence requirements of the KPC-2 β-lactamase as they affect β-lactam–avibactam resistance, focusing on residues (i.e., 69, 130, 234, 220, and 276) that were previously shown to be important for inhibitor resistance in TEM and SHV class A β-lactamases (6, 10–15).
FIG 1.
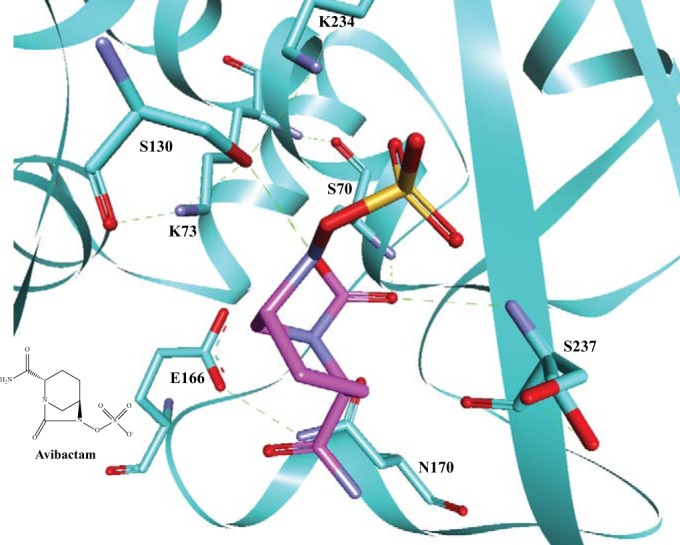
A “snapshot” of acylated avibactam (magenta) in the active site of CTX-M-15 (cyan), showing the interactions with active site residues. Potential hydrogen-bonding interactions are indicated by green dashed lines. Avibactam forms hydrogen-bonding interactions with S70, S130, and S237. The chemical structure of avibactam is presented in the bottom left corner of the figure.
MATERIALS AND METHODS
Bacterial strains and plasmids.
The methods for cloning of blaKPC-2 into the pBC SK(+), pBR322-catI-, and pET24a+ vectors were described (4, 5). The generation of the site-directed mutants of blaKPC-2 at amino acid positions R220 and E276 was also previously described (6). A similar approach was used to create the C69I, -L, and -V, S130A, - G, and -T, and K234A and -R variants in pBC SK(+) blaKPC-2 or pBR322-catI-blaKPC-2; these variants were expressed in Escherichia coli DH10B. Additionally, site-directed mutagenesis was conducted to construct the S130G variant in pET24a+ blaKPC-2 for protein expression in E. coli Origami 2 (DE3) cells.
Expression and purification of KPC-2 and variants.
The KPC-2 and S130G variant β-lactamases were purified as previously described (4). Briefly, cultures were grown in Super Optimal broth to an optical density at 600 nm of 0.6, at which point 1 mM isopropyl β-d-1-thiogalactopyranoside was added and then the cells were grown for 3 h under induction conditions. The cells were then pelleted, frozen, and subjected to stringent periplasmic fractionation. The β-lactamases were purified by preparative isoelectric focusing and fast protein liquid chromatography by using a HiTrap Q anion-exchange chromatography column (GE Life Sciences). The purity of the proteins was assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Protein concentrations were determined by measuring absorbance at a wavelength of 280 nm and using the protein's extinction coefficient (Δε, 39,545 M−1 cm−1), which was obtained using the ProtParam tool at http://us.expasy.org/tools.
Antimicrobial compounds.
Ampicillin and ceftazidime were purchased from Sigma-Aldrich. Imipenem, meropenem, ertapenem, and doripenem were obtained from their commercial sources. Avibactam, batches AFCH005151 and C565/5, was a kind gift from AstraZeneca. Nitrocefin was a kind gift made and tested by Shahriar Mobashery at the University of Notre Dame in Notre Dame, IN (16).
In vitro susceptibility testing methods.
MICs for various bacterial isolates were determined by the Mueller-Hinton agar dilution method according to the Clinical and Laboratory Standards Institute guidelines (17). The MIC measurements were performed using a Steers replicator that delivered 10 μl of a diluted overnight culture containing 104 CFU. Avibactam was tested at 4 mg/liter in combination with increasing concentrations of ampicillin or ceftazidime (18). MICs for imipenem, meropenem, ertapenem, and doripenem were obtained without avibactam.
Steady-state kinetic analysis.
Steady-state kinetic parameters were determined by using an Agilent 8453 diode array spectrophotometer as previously described (4, 8, 19). Briefly, each assay was performed in 10 mM phosphate-buffered saline (PBS) at pH 7.4 at room temperature.
To determine Vmax and Km values, the β-lactamases were maintained at 10 nM with nitrocefin at an excess molar concentration to establish pseudo-first-order kinetics (4). A nonlinear least square fit of the data (Henri Michaelis-Menten equation) using Enzfitter was employed to obtain the steady-state kinetic parameters Vmax and Km.
The interactions between KPC-2 and avibactam can be modeled according to the equation summarized in Fig. 2 (19, 20). Apparent Ki (Ki app) values can approximate the Ki of the inhibitor for β-lactamases that acylate very slowly; however, for β-lactamases that demonstrate a fast acylation rate, the Ki app approximates the Km of the enzyme for the inhibitor.
FIG 2.

Scheme representing the interactions of KPC-2 with avibactam (8). In this model, formation of the noncovalent complex, E:I, is represented by Ki, which is equivalent to k−1/k1. k2 is the first-order rate constant for the acylation step, or formation of E-I. k−2 is the first-order rate constant for the recyclization step, or reformation of E:I. The rate constant for the loss of the sulfate and imine hydrolysis, or formation of E-I′, corresponds to the rate constant k3. Finally, hydrolysis to the product, P, is represented by the rate constant k4.
Determination of Ki app values for KPC-2 and the S130G variant were obtained via a direct competition assay under steady-state conditions by measuring initial velocities. The velocity (V0) obtained after mixing corresponded to that derived using equation 1:
| (1) |
Concentrations of KPC-2 and the S130G variant were kept at 7.2 and 9.2 nM, respectively, while the avibactam concentration (I) was increased. Nitrocefin was used as the reporter substrate (S), at a final concentration of 100 μM. Data were linearized by plotting inverse initial steady-state velocities (1/V0) against the inhibitor concentration [I]. Ki app values were determined by dividing the value for the y-intercept by the slope of the line. Ki app values were corrected to account for the concentration and affinity of nitrocefin for the β-lactamases, according to equation 2:
| (2) |
To determine k2/K, progress curves were obtained by incubating β-lactamase (at 7.2 nM for KPC-2 and 9.3 nM for the S130G variant) with increasing concentrations of avibactam, using nitrocefin (100 μM) as the reporter substrate. Progress curves were subsequently fit to equation 3 to obtain kobs values. k2/K was determined using equation 4.
| (3) |
For equation 3, Vf is final velocity, V0 is initial velocity, and A0 is the initial absorbance at a wavelength of 482 nm.
| (4) |
For equation 4, [I] is the concentration of avibactam and [S] is the concentration of nitrocefin. The data for kobs versus the avibactam concentration were plotted. The k2/K value was obtained by correcting the value for the slope of the line for the concentration and affinity of nitrocefin (equation 5).
| (5) |
The ratio of inhibitor to enzyme (I:E) necessary to inhibit the hydrolysis of nitrocefin by greater than 90% was defined as the partition ratio (4). Partition ratios (kcat/kinact) at 5 min for KPC-2 and the S130G variant with avibactam were determined by incubating the β-lactamase with increasing concentrations of avibactam at room temperature in 10 mM PBS, pH 7.4. Partition ratios for longer time points could not be accurately determined for the S130G variant of KPC-2, as the enzyme lost activity at room temperature with time.
We attempted to determine the off-rate, koff, for the S130G variant of KPC-2, as previously described; the koff for wild-type KPC-2 has been published (8, 19). Unfortunately, the koff value for the S130G variant could not be accurately determined, due to the poor hydrolytic capacity of the enzyme for nitrocefin as well as the slow on-rate of avibactam for the β-lactamase.
ESI-MS.
To discern the nature of the intermediates of inactivation by avibactam in the reaction pathway with the KPC-2 S130G variant β-lactamase, electrospray ionization-mass spectrometry (ESI-MS) was performed on a Waters SynaptG2-Si quadrupole time of flight mass spectrometer equipped with a LockSpray dual electrospray ion source, using Glu-1–fibrinopeptide B as the lock mass. The Synapt G2-Si instrument was calibrated with sodium iodide based on a 50 to 2,000 m/z range. For the experiments, the S130G variant was incubated with avibactam for set times (i.e., 5 min, 1 h, and 3 h) at room temperature in 10 mM PBS, pH 7.4. Longer set times could not be used for the S130G variant of KPC-2, as the enzyme became inactive at room temperature with time. The inhibitor (avibactam)-to-enzyme (S130G variant) ratios (I:E) were 1:1, 10:1, 100:1, 1,000:1, and 10,000:1; the enzyme concentration was 20 μM. After the set time, reactions were terminated by the addition of 0.2% formic acid. All samples were desalted and concentrated using a C18 ZipTip (Millipore, Billerica, MA) according to the manufacturer's protocol. Eluted protein samples were diluted with 50% acetonitrile and 0.2% formic acid and directly infused at a rate of 50 μl per min, and data were collected for 1 min. Lock mass spectra were collected prior to each sample in a similar manner. The tune settings for each data run were as follows: capillary voltage at 3.2 kV, sampling cone at 30, source offset at 30, source temperature of 100°C, desolvation temperature of 450°C, cone gas at 50 liters/h, desolvation gas at 600 liters/h, and nebulizer bar at 6.0. Spectra were analyzed using MassLynx v4.1 and were modified for lock mass deviations by applying a gain factor and deconvoluted using the MaxEnt1 program.
Molecular modeling.
The crystal structure of KPC-2 (PDB ID 2OV5) was employed to construct Michaelis-Menten and acyl enzyme complexes with avibactam by using Discovery Studio 3.1 molecular modeling software (DS 3.1; Accelrys, Inc., San Diego, CA) (18). Avibactam was constructed using Fragment Builder tools and was minimized by a standard dynamics cascade protocol of DS 3.1. Avibactam was automatically docked into the active site of KPC-2 with the CDOCKER module of DS 3.1. This protocol uses a CHARMm-based molecular dynamics scheme to dock ligands into a receptor-binding site. The best conformations were automatically aligned to polar and nonpolar active site hot spots, and the best-scoring poses were reported. At this step, the hydrogen atoms were not maintained. To further optimize the docked poses (i.e., add hydrogens and prevent clashes between the receptor and ligand), a CHARMm minimization step was next used. For this step, the Smart Minimization algorithm was employed (i.e., 1,000 steps of steepest descent with a root mean square [RMS] gradient tolerance of 3 Å, followed by conjugate gradient minimization, with an RMS deviation [RMSD] minimization gradient of 0.001 Å). For the final minimization of the avibactam conformations into the active site of KPC-2, an RMSD cutoff of 1 Å was chosen.
The resulting conformations of KPC-2–avibactam complexes were analyzed, the most favorable positioning of avibactam was chosen (i.e., carbonyl oriented toward the oxyanion hole), and the complexes between the enzyme and inhibitor were created, as previously described (18). To check the stability of the complexes, an 8-ps molecular dynamics simulation (MDS) was conducted for the KPC-2–avibactam and S130G-avibactam Michaelis-Menten and acyl enzyme complexes, as previously described (6). During the heating/cooling, equilibration, and the production stages of the MDS, a temperature of 300 K and a constant pressure were maintained. The long-range electrostatics were treated with particle mesh Ewald and explicit solvation with the periodic boundary condition. The MDS and production step of MDS for KPC-2–avibactam and S130G-avibactam complexes were run without any constraints.
RESULTS
The S130G, K234R, and R220M variants demonstrated decreased susceptibility to ampicillin-avibactam.
The results of our susceptibility testing are summarized in Table 1. Susceptibility testing with β-lactam–avibactam combinations against KPC-2 variants that possessed single amino acid substitutions at certain residues (i.e., Ambler positions 69, 130, 234, 220, and 276) revealed that several active site residues affected ampicillin-avibactam susceptibility. The S130G, K234R, and R220M variants expressed in E. coli DH10B demonstrated ampicillin resistance (MICs, ≥32 mg/liter), even when ampicillin was combined with 4 mg/liter avibactam. Conversely, ceftazidime-avibactam levels were below an MIC of 8 mg/liter for ceftazidime for all variants tested.
TABLE 1.
MICs of avibactama when combined with ampicillin or ceftazidime for E. coli strains containing KPC β-lactamases and variants
| Strains | MIC (mg/liter) |
|||
|---|---|---|---|---|
| Ampicillin | Ampicillin-avibactam | Ceftazidime | Ceftazidime-avibactam | |
| E. coli DH10B pBC SK(+) | 0.5 | 0.25 | 0.5 | 0.25 |
| E. coli DH10B pBC SK(+) blaKPC-2b | 2,048 | 2 | 16 | 0.5 |
| E. coli DH10B pBR322-catI-blaKPC-2c | 4,096 | 8 | 128 | 1 |
| E. coli DH10B pBR322-catI-blaKPC R220A | 2,048 | 8 | 2 | 1 |
| E. coli DH10B pBR322-catI-blaKPC R220K | 2,048 | 16 | 8 | 2 |
| E. coli DH10B pBR322-catI-blaKPC R220M | 4,096 | 32 | 4 | 2 |
| E. coli DH10B pBC SK(+) blaKPC C69I | 0.5 | 0.25 | 0.5 | 0.25 |
| E. coli DH10B pBC SK(+) blaKPC C69L | 1 | 0.25 | 0.5 | 0.5 |
| E. coli DH10B pBC SK(+) blaKPC C69V | 1 | 0.25 | 0.5 | 0.5 |
| E. coli DH10B pBC SK(+) blaKPC S130A | 16 | 8 | 0.5 | 0.5 |
| E. coli DH10B pBC SK(+) blaKPC S130G | 1,024 | 512 | 0.5 | 0.5 |
| E. coli DH10B pBC SK(+) blaKPC S130T | 8 | 1 | 2 | 1 |
| E. coli DH10B pBC SK(+) blaKPC E276A | 512 | 1 | 8 | 0.5 |
| E. coli DH10B pBC SK(+) blaKPC E276D | 2,048 | 1 | 8 | 0.25 |
| E. coli DH10B pBC SK(+) blaKPC E276N | 256 | 1 | 4 | 0.25 |
| E. coli DH10B pBR322-catI-blaKPC K234A | 0.5 | 0.25 | 0.5 | 0.5 |
| E. coli DH10B pBR322-catI-blaKPC K234R | 8,192 | 64 | 16 | 4 |
The avibactam concentration was kept at 4 mg/liter.
In the E. coli DH10B pBC SK(+) blaKPC-2 strain, blaKPC-2, including 124 bp upstream (i.e., this DNA span includes the −35, −10, and ribosomal binding sites) was cloned from the pBR322-catI-blaKPC-2 construct (7).
The pBR322-catI-blaKPC-2 construct contains ∼3,000 bp from the original Klebsiella oxytoca strain from which blaKPC-2 was first cloned (4, 5). The expression levels of blaKPC-2 in pBC SK(+) and pBR322-catI- are different; increased amounts of KPC-2 β-lactamase is produced at steady state from the pBR322-catI-blaKPC-2 expression vector than from pBC SK(+) blaKPC-2 in E. coli DH10B (26).
As KPC-2 is a carbapenemase, susceptibility testing was also conducted using carbapenems (imipenem, meropenem, ertapenem, and doripenem) with the selected KPC-2 variants. Fortunately, no significant increase in carbapenem MICs was observed for any variant compared to wild-type KPC-2 (Table 2).
TABLE 2.
MICs with carbapenems for E. coli strains containing KPC-2 variants
| Strain | MIC (mg/liter) |
|||
|---|---|---|---|---|
| Imipenem | Meropenem | Doripenem | Ertapenem | |
| E. coli DH10B pBC SK(+) | 0.12 | <0.06 | <0.06 | <0.06 |
| E. coli DH10B pBC SK(+) blaKPC-2 | 2 | 1 | 0.5 | 1 |
| E. coli DH10B pBR322-catI-blaKPC-2 | 16 | 8 | 4 | 16 |
| E. coli DH10B pBR322-catI-blaKPC R220A | 2 | 0.25 | 0.25 | <0.06 |
| E. coli DH10B pBR322-catI-blaKPC R220K | 4 | 2 | 0.5 | 2 |
| E. coli DH10B pBR322-catI-blaKPC R220M | 4 | 0.25 | 0.25 | <0.06 |
| E. coli DH10B pBC SK(+) blaKPC C69I | 0.12 | <0.06 | <0.06 | <0.06 |
| E. coli DH10B pBC SK(+) blaKPC C69L | 0.25 | <0.06 | <0.06 | <0.06 |
| E. coli DH10B pBC SK(+) blaKPC C69V | 0.25 | <0.06 | <0.06 | <0.06 |
| E. coli DH10B pBC SK(+) blaKPC S130A | 0.25 | <0.06 | <0.06 | <0.06 |
| E. coli DH10B pBC SK(+) blaKPC S130G | 0.12 | <0.06 | <0.06 | <0.06 |
| E. coli DH10B pBC SK(+) blaKPC S130T | 0.25 | <0.06 | <0.06 | <0.06 |
| E. coli DH10B pBC SK(+) blaKPC E276A | 1 | 1 | 0.5 | 2 |
| E. coli DH10B pBC SK(+) blaKPC E276D | 2 | 2 | 2 | 2 |
| E. coli DH10B pBC SK(+) blaKPC E276N | 2 | 1 | 0.5 | 2 |
| E. coli DH10B pBR322-catI-blaKPC K234A | 0.25 | <0.06 | <0.06 | <0.06 |
| E. coli DH10B pBR322-catI-blaKPC K234R | 4 | 2 | 2 | 4 |
The S130G variant of KPC-2 possesses decreased binding and acylation of avibactam.
The S130G variant of KPC-2 possessed the highest MIC for ampicillin-avibactam, at 512 mg/liter. In order to biochemically understand the basis for this phenotype, the S130G variant of KPC-2 was purified for steady-state kinetic analysis.
Significantly more avibactam was required to achieve the same level of inhibition for the S130G variant of KPC-2 than for the wild-type enzyme. As evidenced by the progress curves (Fig. 3A and B), complete inactivation occurred with 15 μM avibactam reacting with 7.2 nM wild-type KPC-2. However, to inactivate 9.2 nM of the S130G variant, the concentration of avibactam was increased to approximately 7.5 mM. Under similar conditions, the k2/K values for avibactam with KPC-2 compared to those for the S130G variant were also significantly reduced, from 21,580 M−1 s−1 to 1.2 M−1 s−1, respectively (Table 3). The k2/K determination suggested that acylation is significantly impaired in the S130G variant of KPC-2. Consistent with this observation, the Ki app value was ∼100-fold higher for the S130G variant of KPC-2 (Table 3).
FIG 3.

(A) Inhibition of nitrocefin hydrolysis (in absorbance units [a.u.]) by KPC-2 with increasing concentrations of avibactam. (B) Inhibition of nitrocefin hydrolysis by the S130G variant with increasing concentrations of avibactam.
TABLE 3.
Steady-state kinetics parameters for KPC-2 and the KPC-2 S130G variant with nitrocefin and avibactam
| Kinetic parameter | KPC-2 | KPC-2 S130G |
|---|---|---|
| NCF | ||
| Km (μM) | 9 ± 1 | 77 ± 9 |
| kcat/Km (μM−1 s−1) | 9 ± 1 | 2.1 ± 0.2 |
| AVI | ||
| Ki app (μM) | 1.0 ± 0.1 | 98 ± 9 |
| k2/K (M−1 s−1) | 21,580 | 1.2 |
| kcat/kinacta | 1 | 7,000 |
The ratio of inhibitor to enzyme required to inhibit the enzyme by 90%. (the tn after 5 min).
The partition ratio or ratio of the inhibitor versus enzyme required to inhibit the enzyme by 90% at 5 min for the S130G variant was 7,000-fold greater than that for KPC-2. However, we noted that the value of 7,000 was likely falsely high due to the impaired acylation of the S130G variant (see the mass spectrometry results for further evidence). Longer time points for partition ratios could not be accurately measured, as activity of the S130G variant was lost with time at room temperature.
The S130G variant of KPC-2 does not hydrolyze avibactam.
ESI-MS was used to identify the intermediates of inactivation of the S130G variant as well as to determine if hydrolysis of avibactam occurs with the S130G variant (8, 19). In previous studies, Ehmann et al. showed that KPC-2 hydrolyzes avibactam, which involves fragmentation of the acyl-enzyme complex (8). ESI-MS revealed that acylation is delayed in the S130G variant. At 5 min, a small amount of free enzyme (28,688 ± 3 atomic mass units [amu]) was observed even at a 1,000:1 avibactam-to-S130G ratio (Fig. 4). However, if the S130G variant was incubated with avibactam for longer time periods of 1 h or 3 h, more S130G-avibactam-acyl enzyme complex (28,953 ± 3 amu) was observed at lower I:E ratios (10:1 and 100:1). The mass spectrometry analysis suggested that hydrolytic turnover by the S130G variant did not occur during the time periods evaluated. Again, products from longer time points could not be accurately measured via mass spectrometry, as activity of the S130G variant was lost with an increased incubation time at room temperature.
FIG 4.
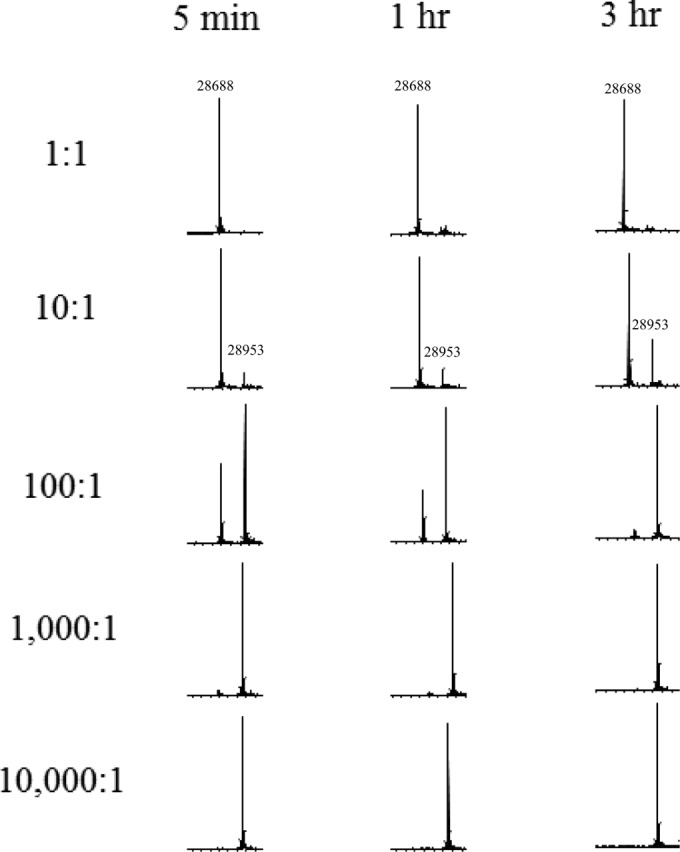
Mass spectrometry results for avibactam with the S130G variant of KPC-2 at different I:E ratios after incubation for 5 min, 1 h, or 3 h.
S130 is critical for binding and acylation of avibactam and KPC-2.
Avibactam was modeled as a Michaelis complex and acyl enzyme complex with KPC-2 and the S130G variant to begin to understand the basis of avibactam inhibition of KPC-2 and resistance to avibactam by the S130G variant.
The representation of the KPC-2:avibactam Michaelis complex revealed that E166 did not appear to be the general base for S70, as the catalytic water molecule was not present to initiate activation of S70. The model presented here suggests that steric interference from the carboxamide of avibactam may be a contributing factor to the absence of this water molecule. Instead, K73 is postulated to be within hydrogen-bonding distance of S70 (2.5 Å) and may play a role as a general base to activate S70 (Fig. 5A), as was alternatively proposed elsewhere for class A enzymes (21). In the context of this model, we advance that S130 may play a role in protonating the sulfate nitrogen during the acylation process.
FIG 5.

Molecular modeling of avibactam in the active sites of KPC-2 (gray) and the S130G variant (green). (A and B) Michaelis complexes. (C and D) Acyl enzyme complexes. In panel A, K73 is within hydrogen-bonding distance of S70 to activate the nucleophile; however, in panel B, we show that K73 is positioned >3.5 Å from S70 and is unlikely to activate S70. In panels C and D, we show that avibactam is positioned very similarly in both KPC-2 and the S130G variant. All potential hydrogen-bonding interactions are represented by dashed black lines.
Similarly, in the model of the S130G variant:avibactam Michaelis complex, the catalytic water molecule was absent. Here, K73 formed hydrogen-bonding interactions with S130G, E166, and N132. However, K73 was positioned more than 3.5 Å from S70 in all conformations, and thus was less likely to activate S70 (Fig. 5B). In addition, G130 does not have a proton to donate to the sulfate nitrogen during the acylation process.
The KPC-2–avibactam and S130G variant-avibactam acyl complexes were similar (Fig. 5C and D). Therefore, conclusions cannot be drawn about the recyclization and/or hydrolysis with KPC-2 or the S130G variant.
DISCUSSION
Susceptibility testing against β-lactam–avibactam combinations using KPC-2 variants that possess single amino acid substitutions at certain residues (i.e., Ambler positions 69, 130, 234, 220, and 276) that were shown previously to confer inhibitor (i.e., clavulanic acid, sulbactam, and tazobactam) resistance in TEM and SHV revealed that several active site residues (S130, K234, and R220) are particularly important for avibactam's inhibitory activity against KPC-2. Interestingly, we also showed that R220, C69, S130, E276, and K234 modulate KPC-2's ability to hydrolyze carbapenems. Previous work showed that R220, C69, and E276 are important for carbapenem resistance (6, 22). Our analysis defined the additional contribution of K234 (especially the R variant) in carbapenem resistance. The MIC profile was particularly interesting, as the ceftazidime-avibactam MIC was 4 mg/liter. Further studies are planned to explore these structure-function relationships.
To appreciate the importance of these findings, we must first revisit our understanding of the mechanism of inactivation by clavulanic acid (23). Resistance to clavulanic acid via an S130G substitution in SHV-1 and TEM-1 is attributed to a lack of terminal inactivation or cross-linking of the inhibitor within the active site, and not to an acylation deficiency (11, 15, 24). In addition, unlike in SHV-1, a water molecule resides adjacent to S130G in TEM-1, and this water molecule might assist in partially restoring some of the catalytic activity to the enzyme. As avibactam is not known to cross-link with S130, the mechanism of resistance to avibactam by the S130G variant of KPC-2 warranted further consideration. The K234R substitution in SHV-1 is hypothesized to cause clavulanic acid resistance as a result of K234R's interactions with S130 (14, 25). Resistance due to substitutions of R220M and R220K in KPC-2 were shown to alter the binding of clavulanic acid (6).
As the S130G variant demonstrated the highest ampicillin-avibactam MIC, we chose to investigate the mechanism conferring this phenotype. The biochemical analyses performed here indicated that k2/K is significantly impaired in the S130G variant of KPC. The MICs and kinetic studies supported each other regarding the impact of S130. The model of the KPC-2:avibactam Michaelis complex revealed that K73 may activate S70 for avibactam acylation (Fig. 6A). There is a possibility that a water molecule may be recruited into the active site to allow E166 to serve as a general base; however, this was not observed during our 8-ps simulation. We note here that crystallographic evidence from the only class A β-lactamase crystallized with avibactam, CTX-M-15, suggests that E166 is the general base for acylation in that β-lactamase (9). In addition, S130 most likely donates a proton to the sulfate nitrogen during acylation. The proton donor to S130 thereafter is in question: is it K73, as suggested by Ehmann et al. (8), that donates the proton back to S130, or is it another residue? We hypothesize that K234 may donate a proton to S130 during avibactam acylation, as the K234R variant demonstrated an elevated ampicillin-avibactam MIC as well. Additional experiments and calculations will need to be completed to verify this hypothesis and identify the correct proton donor for S130.
FIG 6.

Hypothetical mechanisms for acylation of KPC-2 (A) and the S130G variant (B) by avibactam. Potential hydrogen-bonding interactions are represented by dashed green lines. See the text for mechanism interpretations.
In the S130G variant:avibactam Michaelis complex model, K73 is less able to participate in acylation readily because of an altered position in the active site. Movements of K73 and E166 as well as recruitment of a water molecule may occur allowing acylation to proceed in the S130G variant, although at a much lower rate. In addition, as the hydroxyl side chain is missing in the S130G variant, protonation of the sulfate nitrogen is hypothesized to be slowed and result in a decreased k2/K (Fig. 6B). We advance that another residue, such as protonated K234 (see above), may compensate for this. Taken together, the Michaelis complex models support that acylation is the rate-limiting step in acyl enzyme complex formation with the S130G variant. ESI-MS demonstrated that acylation does occur at a low rate in the S130G variant; thus, movement of active site residues K73 or E166 and recruitment of a water molecule must occur at some point to activate S70. Finally, we raise the possibility that K234 may donate the proton to the sulfate nitrogen for acylation.
In both acyl enzyme complexes, the proton donor for recyclization was unclear, as was the mechanism of hydrolysis by KPC-2. We posit that residues within the dynamic hydrogen-bonding network between the sulfate of avibactam, T237, R220, E276, and a water molecule may be responsible for the slow hydrolysis of avibactam by KPC-2 that was observed previously (8). The confirmation of these mechanistic considerations requires further testing.
Conclusions.
We have demonstrated here that susceptibility to ampicillin-avibactam is altered as a result of substitutions at S130, R220, and K234. However, using the combination of ceftazidime-avibactam against these variants of KPC-2 should not result in clinical failure of ceftazidime-avibactam, as the ceftazidime partner is potent against these strains in combination with avibactam (MIC range, 0.25 to 4 mg/liter). In addition, as KPC-2 variants identified to date, such as KPC-3, possess substitutions outside the active site, we advance that the results presented here apply to the current KPC family of enzymes. It is important to note that these susceptibility and kinetic studies of KPC-2 variants with avibactam also revealed important structure-function relationships for KPC that are needed for future studies. Important questions raised by our observations remain. Is the mechanism of resistance conferred by S130G the same as that conferred by R220M or K234R? In the past 2 decades, the analyses of inhibitor-resistant TEM and SHV enzymes have informed us of the details of catalysis (23). We now see that a similar approach can be applied to a more “versatile” enzyme, KPC with a novel DBO inhibitor, and can provide insights into which changes are essential for the next generation of β-lactams and β-lactamase inhibitors.
ACKNOWLEDGMENTS
This work was supported by a research grant from AstraZeneca (K.M.P.-W. and R.A.B.). This study was also supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, the Department of Veterans Affairs Career Development Program (K.M.P.-W.), Department of Veterans Affairs Merit Review Program award 1I01BX001974 (R.A.B.), and the Veterans Integrated Service Network 10 Geriatric Research, Education, and Clinical Center (VISN 10 GRECC) (R.A.B.). Research reported in this publication was also supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R01AI100560 and R01AI063517 to R.A.B. M.L.W. was supported by a Medical Scientist Training Program Training Grant, Case Western Reserve University (T32 GM07250).
The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Centers for Disease Control and Prevention. 2013. Antibiotic resistance threats in the United States, 2013. CDC, Atlanta, GA: http://www.cdc.gov/drugresistance/threat-report-2013/index.html. [Google Scholar]
- 2.Gilbert DN, Guidos RJ, Boucher HW, Talbot GH, Spellberg B, Edwards JE, Scheld WM, Bradley JS, Bartlett JG. 2010. The 10 x '20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin Infect Dis 50:1081–1083. doi: 10.1086/652237. [DOI] [PubMed] [Google Scholar]
- 3.Boucher HW, Talbot GH, Benjamin DK Jr, Bradley J, Guidos RJ, Jones RN, Murray BE, Bonomo RA, Gilbert D, Infectious Diseases Society of America. 2013. 10 x '20 progress. Development of new drugs active against Gram-negative bacilli: an update from the Infectious Diseases Society of America. Clin Infect Dis 56:1685–1694. doi: 10.1093/cid/cit152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papp-Wallace KM, Bethel CR, Distler AM, Kasuboski C, Taracila M, Bonomo RA. 2010. Inhibitor resistance in the KPC-2 β-lactamase, a preeminent property of this class A β-lactamase. Antimicrob Agents Chemother 54:890–897. doi: 10.1128/AAC.00693-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yigit H, Queenan AM, Rasheed JK, Biddle JW, Domenech-Sanchez A, Alberti S, Bush K, Tenover FC. 2003. Carbapenem-resistant strain of Klebsiella oxytoca harboring carbapenem-hydrolyzing β-lactamase KPC-2. Antimicrob Agents Chemother 47:3881–3889. doi: 10.1128/AAC.47.12.3881-3889.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Papp-Wallace KM, Taracila MA, Smith KM, Xu Y, Bonomo RA. 2012. Understanding the molecular determinants of substrate and inhibitor specificities in the carbapenemase KPC-2: exploring the roles of Arg220 and Glu276. Antimicrob Agents Chemother 56:4428–4438. doi: 10.1128/AAC.05769-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Papp-Wallace KM, Taracila M, Hornick JM, Hujer AM, Hujer KM, Distler AM, Endimiani A, Bonomo RA. 2010. Substrate selectivity and a novel role in inhibitor discrimination by residue 237 in the KPC-2 β-lactamase. Antimicrob Agents Chemother 54:2867–2877. doi: 10.1128/AAC.00197-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Durand-Reville TF, Lahiri S, Thresher J, Livchak S, Gao N, Palmer T, Walkup GK, Fisher SL. 2013. Kinetics of avibactam inhibition against class A, C, and D β-lactamases. J Biol Chem 288:27960–27971. doi: 10.1074/jbc.M113.485979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lahiri SD, Mangani S, Durand-Reville T, Benvenuti M, De Luca F, Sanyal G, Docquier JD. 2013. Structural insight into potent broad-spectrum inhibition with reversible recyclization mechanism: avibactam in complex with CTX-M-15 and Pseudomonas aeruginosa AmpC β-lactamases. Antimicrob Agents Chemother 57:2496–2505. doi: 10.1128/AAC.02247-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drawz SM, Bethel CR, Hujer KM, Hurless KN, Distler AM, Caselli E, Prati F, Bonomo RA. 2009. The role of a second-shell residue in modifying substrate and inhibitor interactions in the SHV β-lactamase: a study of ambler position Asn276. Biochemistry 48:4557–4566. doi: 10.1021/bi9003292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Helfand MS, Bethel CR, Hujer AM, Hujer KM, Anderson VE, Bonomo RA. 2003. Understanding resistance to β-lactams and β-lactamase inhibitors in the SHV β-lactamase: lessons from the mutagenesis of SER-130. J Biol Chem 278:52724–52729. doi: 10.1074/jbc.M306059200. [DOI] [PubMed] [Google Scholar]
- 12.Helfand MS, Hujer AM, Sonnichsen FD, Bonomo RA. 2002. Unexpected advanced generation cephalosporinase activity of the M69F variant of SHV β-lactamase. J Biol Chem 277:47719–47723. doi: 10.1074/jbc.M207271200. [DOI] [PubMed] [Google Scholar]
- 13.Thomson JM, Distler AM, Prati F, Bonomo RA. 2006. Probing active site chemistry in SHV β-lactamase variants at Ambler position 244. Understanding unique properties of inhibitor resistance. J Biol Chem 281:26734–26744. doi: 10.1074/jbc.M603222200. [DOI] [PubMed] [Google Scholar]
- 14.Winkler ML, Rodkey EA, Taracila MA, Drawz SM, Bethel CR, Papp-Wallace KM, Smith KM, Xu Y, Dwulit-Smith JR, Romagnoli C, Caselli E, Prati F, van den Akker F, Bonomo RA. 2013. Design and exploration of novel boronic acid inhibitors reveals important interactions with a clavulanic acid-resistant sulfhydryl-variable (SHV) β-lactamase. J Med Chem 56:1084–1097. doi: 10.1021/jm301490d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun T, Bethel CR, Bonomo RA, Knox JR. 2004. Inhibitor-resistant class A β-lactamases: consequences of the Ser130-to-Gly mutation seen in Apo and tazobactam structures of the SHV-1 variant. Biochemistry 43:14111–14117. doi: 10.1021/bi0487903. [DOI] [PubMed] [Google Scholar]
- 16.Lee M, Hesek D, Mobashery S. 2005. A practical synthesis of nitrocefin. J Org Chem 70:367–369. doi: 10.1021/jo0487395. [DOI] [PubMed] [Google Scholar]
- 17.Clinical and Laboratory Standards Institute. 2006. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 7th ed CLSI document M7-A7. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 18.Papp-Wallace KM, Winkler ML, Gatta JA, Taracila MA, Chilakala S, Xu Y, Johnson JK, Bonomo RA. 2014. Reclaiming the efficacy of β-lactam–β-lactamase inhibitor combinations: avibactam restores the susceptibility of ceftazidime against CMY-2-producing Escherichia coli. Antimicrob Agents Chemother 58:4290–4297. doi: 10.1128/AAC.02625-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Kern G, Walkup GK, Fisher SL. 2012. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc Natl Acad Sci U S A 109:11663–11668. doi: 10.1073/pnas.1205073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrison JF, Walsh CT. 1988. The behavior and significance of slow-binding enzyme inhibitors. Adv Enzymol Relat Areas Mol Biol 61:201–301. [DOI] [PubMed] [Google Scholar]
- 21.Meroueh SO, Fisher JF, Schlegel HB, Mobashery S. 2005. Ab initio QM/MM study of class A β-lactamase acylation: dual participation of Glu166 and Lys73 in a concerted base promotion of Ser70. J Am Chem Soc 127:15397–15407. doi: 10.1021/ja051592u. [DOI] [PubMed] [Google Scholar]
- 22.Majiduddin FK, Palzkill T. 2003. Amino acid sequence requirements at residues 69 and 238 for the SME-1 β-lactamase to confer resistance to β-lactam antibiotics. Antimicrob Agents Chemother 47:1062–1067. doi: 10.1128/AAC.47.3.1062-1067.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drawz SM, Bonomo RA. 2010. Three decades of β-lactamase inhibitors. Clin Microbiol Rev 23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas VL, Golemi-Kotra D, Kim C, Vakulenko SB, Mobashery S, Shoichet BK. 2005. Structural consequences of the inhibitor-resistant Ser130Gly substitution in TEM β-lactamase. Biochemistry 44:9330–9338. doi: 10.1021/bi0502700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubois V, Poirel L, Demarthe F, Arpin C, Coulange L, Minarini LA, Bezian MC, Nordmann P, Quentin C. 2008. Molecular and biochemical characterization of SHV-56, a novel inhibitor-resistant β-lactamase from Klebsiella pneumoniae. Antimicrob Agents Chemother 52:3792–3794. doi: 10.1128/AAC.00387-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Papp-Wallace KM, Taracila M, Wallace CJ, Hujer KM, Bethel CR, Hornick JM, Bonomo RA. 2010. Elucidating the role of Trp105 in the KPC-2 β-lactamase. Protein Sci 19:1714–1727. doi: 10.1002/pro.454. [DOI] [PMC free article] [PubMed] [Google Scholar]