

Abstract
The human and bacterial A site rRNA binding as well as the aminoglycoside-modifying enzyme (AME) activity against a series of neomycin B (NEO) dimers is presented. The data indicate that by simple modifications of linker length and composition, substantial differences in rRNA selectivity and AME activity can be obtained. We tested five different AMEs with dimeric NEO dimers that were tethered via triazole, urea, and thiourea linkages. We show that triazole-linked dimers were the worst substrates for most AMEs, with those containing the longer linkers showing the largest decrease in activity. Thiourea-linked dimers that showed a decrease in activity by AMEs also showed increased bacterial A site binding, with one compound (compound 14) even showing substantially reduced human A site binding. The urea-linked dimers showed a substantial decrease in activity by AMEs when a conformationally restrictive phenyl linker was introduced. The information learned herein advances our understanding of the importance of the linker length and composition for the generation of dimeric aminoglycoside antibiotics capable of avoiding the action of AMEs and selective binding to the bacterial rRNA over binding to the human rRNA.
INTRODUCTION
Although aminoglycosides (AGs) have recently been explored as antiviral, antiprotozoal, and antifungal agents and a potential treatment for genetic disorders associated with premature termination codons, they remain best known for their use as broad-spectrum antibiotics (1, 2). To elicit their antibacterial response, AGs bind to a highly conserved set of nucleotides on helix 44 (h44) of the bacterial 16S rRNA (3–5). To a lesser extent, AGs have also been shown to bind to the mammalian ribosomes, and efforts have been devoted to achieve higher selectivity of these drugs toward their bacterial target (6–8). Three main groups of AGs have been structurally defined based on the substitution pattern of their common 2-deoxystreptamine (2-DOS) ring: (i) the monosubstituted 2-DOS AGs (e.g., apramycin and hygromycin), (ii) the 4,5-disubstituted 2-DOS AGs (e.g., butirosin, neomycin B [NEO], paromomycin, and ribostamycin), and (iii) the 4,6-disubstituted 2-DOS AGs (e.g., amikacin, kanamycin, tobramycin, etc.). Over the last 70 years of use, the emergence of resistance to AGs has greatly limited their effectiveness as antibiotics. Among the resistance mechanisms known to affect AGs, namely, decreased uptake, efflux pumps, ribosomal mutation or modification by methyltransferases, and the acquisition of aminoglycoside-modifying enzymes (AMEs), the latter is the most prominent. Three families of AMEs that chemically modify amine and hydroxyl moieties have evolved to help bacteria evade the action of these drugs: the AG N-acetyltransferases (AACs), O-phosphotransferases (APHs), and O-nucleotidyltransferases (ANTs) (9, 10). NEO (compound 1; Fig. 1), a potent antibacterial, is mainly sold for topical use due to its significant ototoxicity. A number of purified AMEs, such as AAC(6′)-Ie/APH(2″)-Ia (11), AAC(3)-IV (12), ANT(4′)-Ia (13), and APH(3′)-IIIa (14, 15), among others, have been found to modify NEO in vitro and to limit its effectiveness in vivo (7, 16). NEO dimers have been previously reported to bind the bacterial A site with differing affinities and showed large differences in bacterial inhibition (17).
FIG 1.

Structures of NEO (compound 1) and its dimers (compounds 2 to 18) used in this study.
Here, we report our studies on the activities of a number of AMEs with NEO dimers (Fig. 1). Additionally, a screening assay for studying the selectivity of these dimers for binding to the bacterial A site versus human A site rRNA is presented. Since any modification to an existing AG is expected to affect its binding, transport, and resistance mechanisms, work is needed to map the effect of modifications on such factors. In this report, we investigate how linker modification in NEO dimers influences binding of the drug to its ribosomal target, its selectivity in binding to bacterial versus human ribosome, and its influence on resistance enzymes that modify NEO.
MATERIALS AND METHODS
Materials and instrumentation.
The Eis (18), AAC(6′)-Ie/APH(2″)-Ia (11) (Note: AAC(6′)-Ie/APH(2″)-Ia was used only for its acetyltransferase activity and is referred to as AAC(6′)-Ie from here on), AAC(6′)-Ib′ (19), AAC(3)-IV (12), AAC(2′)-Ic (20), and APH(2″)-Ia (19) enzymes were expressed and purified as previously described. Acetyl-coenzyme A (AcCoA), 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), GTP, NADH, phosphoenolpyruvate (PEP), and a pyruvate kinase lactic dehydrogenase (PK-LDH) mixture were purchased from Sigma-Aldrich (Milwaukee, WI). The AG neomycin B (NEO) (compound 1) and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) were purchased from AK Scientific (Mountain View, CA). The NEO dimers with triazole linkers (compounds 2 to 8), urea linkers (compounds 9 to 12), and thiourea linkers (compounds 13 to 18) were synthesized as previously described (Fig. 1; note that the DPA numbers previously reported for these NEO dimers appear in parentheses in this figure) (21–23). The methicillin-resistant Staphylococcus aureus (MRSA) strain was a gift from David H. Sherman (University of Michigan, MI), and all other strains were donated by Paul J. Hergenrother (University of Illinois at Urbana-Champaign, IL). The pET28a plasmid containing the APH(3′)-Ia gene (24) was a generous gift from Gerald D. Wright (McMaster University, Canada). The 96-well plates were purchased from Thermo Fisher Scientific (Waltham, MA). UV-visible light (UV-Vis) assays were monitored on a SpectraMax M5 plate reader.
Determination of aminoglycoside-modifying enzyme activity against novel NEO dimers by UV-Vis assays.
To determine if various AMEs chemically added functionalities to our NEO dimers, we used previously developed assays to visualize the transformation of these compounds (Fig. 2) (11). All reactions were monitored at 25°C [with the exception of those with AAC(6′)-Ie and APH(2″)-Ia, which were monitored at 37°C] on a SpectraMax M5 microplate reader and performed at least in duplicate. All rates were normalized to that of NEO (compound 1) (Fig. 2).
FIG 2.
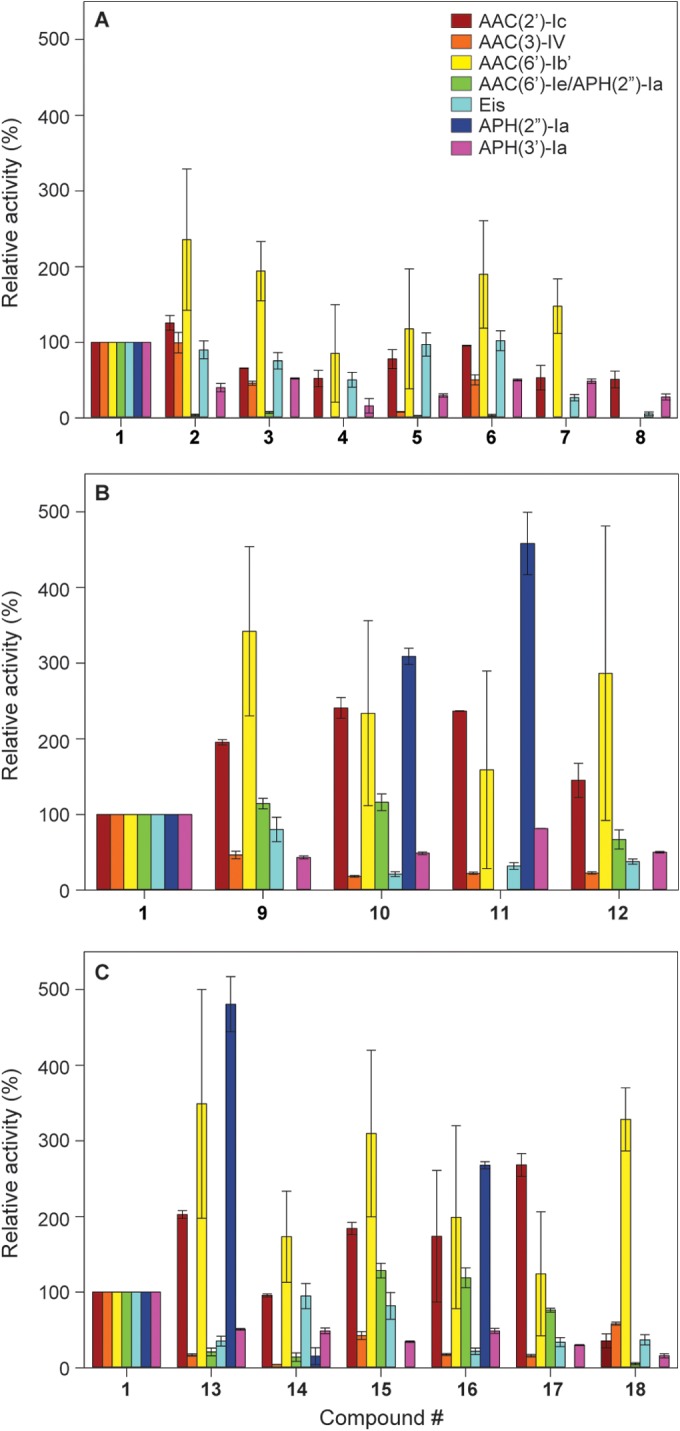
Bar graphs showing the relative enzymatic modification initial rates of the listed AACs and APHs with NEO (compound 1) and its dimers 2 to 18. All rates were normalized to that of NEO (compound 1), which was used for the synthesis of the dimers. Graphs are presented for (A) the triazole-linked NEO dimers, (B) the urea-linked NEO dimers, and (C) the thiourea-linked NEO dimers.
(i) Acetylation.
The activities of several AG acetyltransferases [AAC(6′)-Ie, AAC(6′)-Ib′, AAC(3)-IV, AAC(2′)-Ic, and Eis] were determined by monitoring the coupling of the released free sulfhydryl group of CoA with DTNB at 412 nm (ε 14,150 cm−1 M−1). Briefly, reaction mixtures (200 μl) containing antibiotic compound (100 μM) and AcCoA (500 μM for Eis and 150 μM for all other AG acetyltransferases) were incubated with the AG acetyltransferases [0.125 μM for AAC(3)-IV and AAC(2′)-Ic and 0.5 μM for all remaining AG acetyltransferases] in the presence of DTNB (2 mM) and the appropriate buffer [50 mM morpholineethanesulfonic acid, pH 6.6, for AAC(6′)-Ie and AAC(3)-IV; 50 mM Tris-HCl, pH 7.5, for AAC(6′)-Ib; 100 mM sodium phosphate, pH 7.4, for AAC(2′)-Ic; and 50 mM Tris-HCl, pH 8.0, for Eis]. Reactions were monitored by taking measurements every 30 s for 30 min. Initial rates of the reactions were calculated using the first 5 min of the reaction.
(ii) Phosphorylation.
The phosphorylation activities of APH(2″)-Ia and APH(3′)-Ia [purified identically to APH(2″)-Ia] (19) were monitored at 340 nm through the consumption of NADH in the enzyme-coupled response to the production of ADP. Reaction mixtures (200 μl) containing antibiotic compound (100 μM), Tris-HCl [50 mM, pH 8.0, for APH(2″) or HEPES, pH 7.5, for APH(3′)-Ia], MgCl2 (10 mM), KCl (40 mM), NADH (0.5 mg/ml), PEP (2.5 mM), GTP (2 mM), and PK-LDH (4 μl) were initiated by the addition of the AG phosphotransferase (1 μM). Reactions were monitored by taking measurements every 30 s for 30 min. Initial rates were determined using the first 5 min of the reaction.
rRNA A site binding screening.
The Escherichia coli A site model (5′-GGCGUCACACCUUCGGGUAAGUCGCC-3′) and the human A site model (5′-GGCGUCGCUACUUCGGUAAAAGUCGCC-3′, with bold characters indicating where the human sequence differs from the E. coli sequence) (Fig. 3C) were synthesized using standard phosphoramidite solid-phase synthesis with a 2′-acetoxy ethyl orthoester (2′-ACE) protecting group (Thermo Scientific). All RNA oligonucleotides were deprotected before use according to the manufacturer's protocols, and the deprotection buffer was removed by evaporation using a SpeedVac (GeneVac). The RNA oligonucleotides were resuspended in diethyl pyrocarbonate-treated H2O (OmniPur) to the desired concentration.
FIG 3.
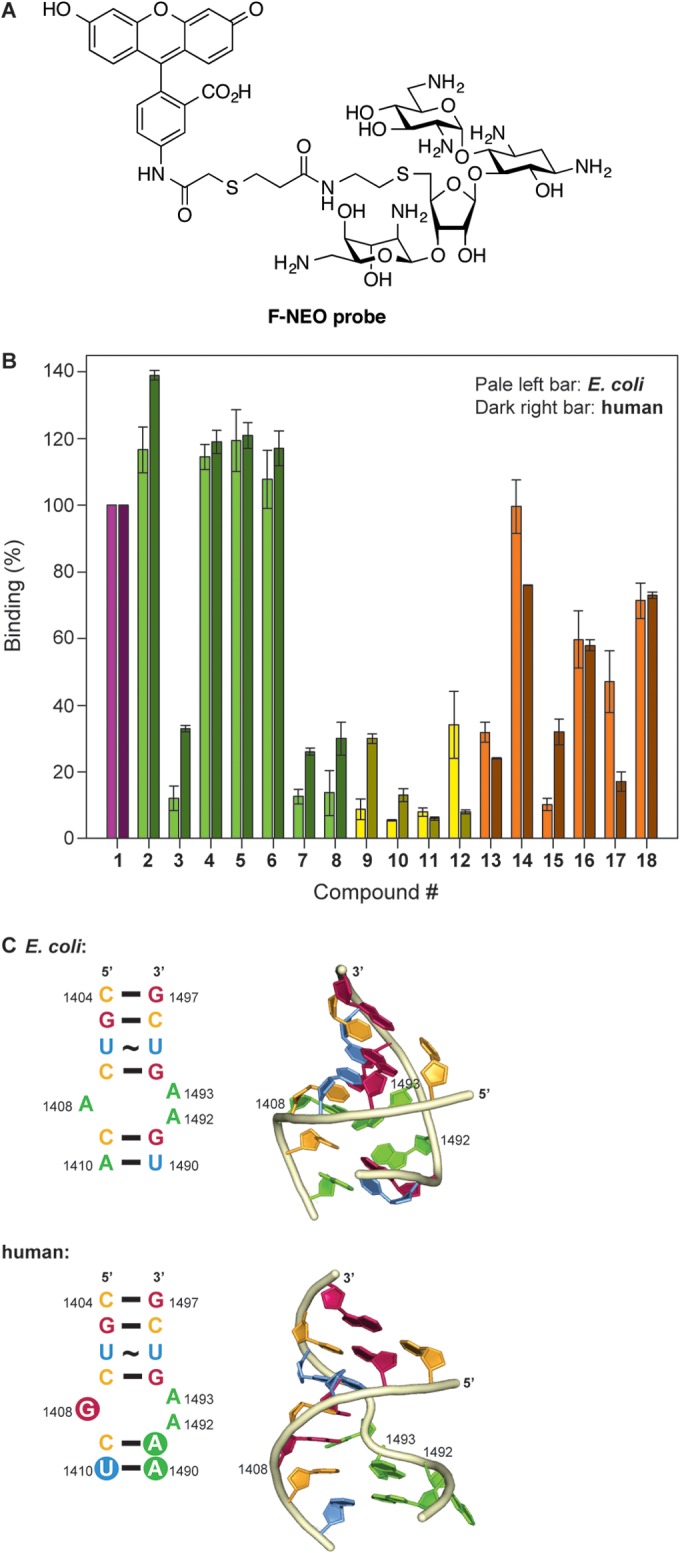
F-NEO competitive binding screen. (A) Structure of the F-NEO probe used in this study. (B) Bar graph showing the relative rRNA binding of NEO dimers 2 to 18 normalized to NEO (compound 1). NEO (compound 1) and the triazole-, urea-, and thiourea-linked NEO dimers are represented in purple, green, yellow, and orange, respectively. The competitive binding screen of E. coli and human model ribosomal A site are represented in pale color bars on the left and dark color bars on the right for each NEO dimer, respectively. (C) The E. coli A site rRNA (top) and mammalian A site rRNA (bottom) used for the screening assay using F-NEO. The differences between the E. coli and human sequence are highlighted by the filled circles in the secondary structure (left) of the human A site. Key residues of the binding pocket for AGs are labeled in the three-dimensional representation (right) of the E. coli and human A sites. The three dimensional structures were modeled from PDB ID: 1PBR for the E. coli A site and PDB ID: 2FQN for the human A site using only the residues present in the two structures. The bases of the representations are color coded red (guanine), pale orange (cytosine), blue (uracil), and green (adenine) and numbered according to the E. coli ribosome.
Compounds 1 to 18 were screened using an F-NEO (Fig. 3A) competitive binding assay, as previously described (17, 25). Briefly, the displacement assay was performed with each compound (0.3 μM) and F-NEO–A site complex (0.1 μM) using 100 reads per well at an excitation/emission of 485/535 nm. All experiments were performed in buffer containing MOPSO (10 mM, pH 7.0), NaCl (50 mM), and EDTA (0.4 mM). The displacements of F-NEO by compounds 1 to 18 were determined by the increase in fluorescence measured after the addition of the compound to wells of a 96-well plate containing F-NEO–A site complex compared to the fluorescence of F-NEO–A site complex alone (ΔF). The percentage of binding was normalized from the ratio of the change in fluorescence by addition of the test compound (ΔFDrug) with the change in fluorescence by addition of NEO (compound 1) (ΔFNEO), using the following equation: % binding = (ΔFDrug/ΔFNEO) × 100.
MIC value determination.
MIC values were determined using the microdilution method as outlined in the CLSI Antimicrobial Susceptibility Testing Standard. Several strains of S. aureus (USA 100, USA 200, USA 300, USA 600, ATCC 29213, and one MRSA strain) and one Bacillus cereus (ATCC 11778) strain were tested in Mueller-Hinton broth. Cultures were grown for 16 to 24 h before determination of the MIC values by visual inspection and confirmation with MTT staining. MIC values for these eight bacterial strains along with those of an additional S. aureus (ATCC 33591) strain previously reported are presented in Table 1.
TABLE 1.
MICs for NEO dimers 1 to 18 against B. cereus and various S. aureus strains
| Compound | MIC for strain: |
|||||||
|---|---|---|---|---|---|---|---|---|
| S. aureus USA 100 | S. aureus USA 200 | S. aureus USA 300 | S. aureus USA 600 | S. aureus ATCC 29213 | S. aureus ATCC 33591a | MRSA | B. cereus ATCC 11778 | |
| 1 (NEO) | >50 | >50 | >50 | >50 | 3.13 | 159.5 | >50 | 1.56 |
| 2 | >50 | >50 | >50 | >50 | 1.56 | >44.4 | >50 | 12.5 |
| 3 | >50 | >50 | >50 | >50 | 3.13 | 43.2 | >50 | 12.5 |
| 4 | >50 | >50 | >50 | >50 | 6.25 | >42.6 | >50 | 25 |
| 5 | >50 | >50 | >50 | >50 | 3.13 | 20.1 | >50 | 25 |
| 6 | >50 | >50 | >50 | >50 | 1.56 | >44.2 | >50 | 25 |
| 7 | >50 | >50 | >50 | >50 | 3.13 | >41.2 | >50 | >25 |
| 8 | 25 | 25 | 25 | 25 | 3.13 | 20.1 | 25 | 25 |
| 9 | >50 | 50 | >50 | >50 | 3.13 | >43.6 | >50 | 12.5 |
| 10 | >50 | >50 | >50 | >50 | >50 | >41.7 | >50 | >25 |
| 11 | >50 | >50 | >50 | >50 | >50 | >43.8 | >50 | >25 |
| 12 | >50 | >50 | >50 | >50 | 12.5 | >43.8 | >50 | >25 |
| 13 | >50 | >50 | >50 | >50 | 25 | >46.9 | >50 | >25 |
| 14 | >50 | >50 | >50 | >50 | 3.13 | 43.9 | >50 | 12.5 |
| 15 | >50 | 50 | >50 | >50 | 3.13 | >42.2 | 50 | 6.25 |
| 16 | >50 | >50 | >50 | >50 | >50 | 20 | >50 | >25 |
| 17 | >50 | 25 | >50 | >50 | 3.13 | >27.3 | >50 | 25 |
| 18 | 25 | 25 | 25 | 50 | 3.13 | >39.7 | 12.5 | >25 |
The data for these S. aureus isolates were previously reported in reference 17. They have been included as the resistance enzymes for this strain and were probed by PCR (see Fig. 4).
Identification of AMEs in eight bacterial strains.
To determine the AME genes present in each of the nine bacterial strains selected (three ATCC strains, four clinically isolated strains, and two additional MRSA strains), colonies were picked and subjected to PCR analysis. We used primer probes previously reported to detect the genes encoding AAC(6′)-Ib, AAC(3)-IV, ANT(2″)-Ia, and APH(3′)-Ia (26). An initial 5 min at 95°C was used to lyse the bacterial cell and denature the DNA. For 35 cycles, the samples were subjected to 2 min at 95°C, 1 min at 58°C, and 1 min at 72°C. After the 35 cycles, the reactions were held at 72°C for 10 min and then analyzed on a 1.5% agarose gel. Expected band sizes were 482 bp [aac(6′)-Ib], 230 bp [aac(3)-IV], 534 bp [ant(2″)-Ia], and 624 bp [aph(3′)-Ia] (Fig. 4).
FIG 4.
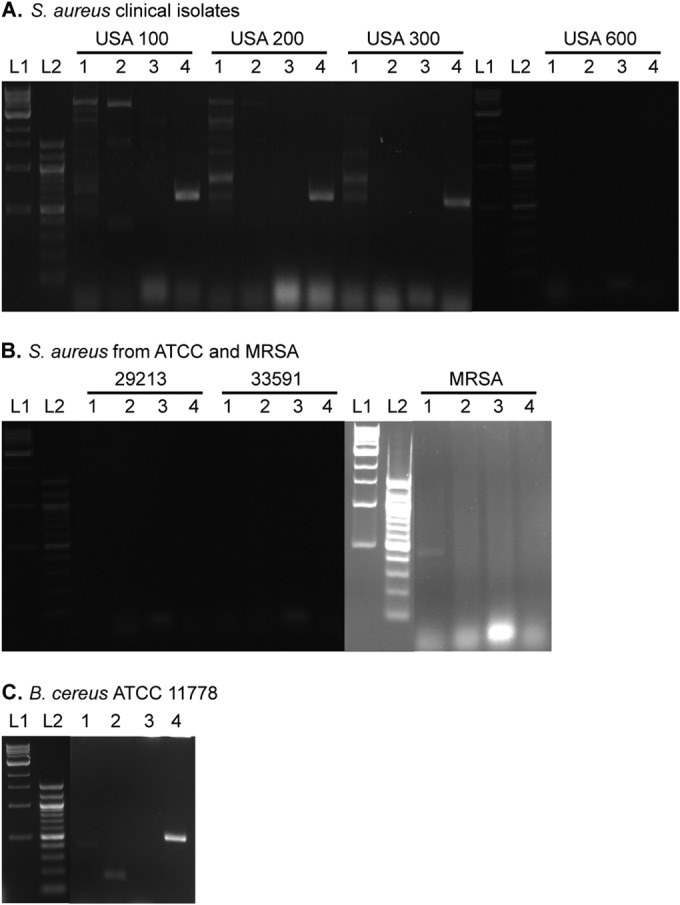
Agarose gels of PCRs probing for AME genes in various ATCC strains and clinical isolates. Strains of (A) clinically isolated MRSA USA 100, USA 200, USA 300, and USA 600, (B) S. aureus ATCC 29213, S. aureus ATCC 33591, and an uncategorized MRSA, and (C) B. cereus ATCC 11778 tested for aac(6′)-Ib (lane 1), aac(3)-IV (lane 2), ant(2″)-Ia (lane 3), and aph(3′)-Ia (lane 4). L1 and L2 represent 100-bp and 1-kb DNA ladders, respectively. Expected band sizes are as follows: aac(6′)-Ib, 482 bp; aac(3)-IV, 230 bp; ant(2″)-Ia, 534 bp; aph(3′)-Ia, 624 bp.
RESULTS
Effect of AMEs on NEO dimers 2 to 18.
When tested against various AACs and APHs, the NEO dimers 2 to 18 displayed various levels of modification by these resistance enzymes (Fig. 2). In general, all AMEs displayed equal or reduced activity toward the triazole-linked NEO dimers (compounds 2 to 8) compared to the parent AG NEO (compound 1), with the exception of AAC(6′)-Ib′ for which the activity toward compounds 2 to 8 was increased by up to 235% that of standard 1 (Fig. 2A). Interestingly, contrary to the trend observed for AAC(6′)-Ib′, the NEO dimers 2 to 8 were all found to be extremely poor substrates of another AAC(6′) enzyme, the bifunctional AAC(6′)-Ie/APH(2″)-Ia (for which only the AAC(6′) activity was tested), with a maximum activity observed of 7%. The APH(2″)-Ia enzyme alone was also found to be completely inactive against compounds 2 to 8. Promising results were also observed against the AAC(3)-IV enzyme, which displayed from 0% to 50% activity against compounds 3 to 8 compared to that of compound 1. Compound 8 was found to be the most recalcitrant to the action of AMEs by being modified only by AAC(2′)-Ic (51% compared to compound 1) and basically no other AMEs.
As observed for the triazole-linked NEO dimers 2 to 8, the initial rate of acetylation of the urea-linked NEO dimers 9 to 12 was greatly increased (159% to 342%) by AAC(6′)-Ib′ (Fig. 2B). Similarly, the activity of AAC(2′)-Ic was increased by 145% to 241% for compounds 9 to 12 compared to that of the parent NEO (compound 1). Compounds 10 and 11 showed an increase in their initial rate of phosphorylation by APH(2″)-Ia (309% and 458%, respectively) while compounds 9 and 12 were not phosphorylated by this enzyme. All the urea-linked NEO dimers displayed slower initial rates of acetylation with AAC(3)-IV (18% to 46%) and Eis (21% to 80%). The AAC(6′)-Ie/APH(2″)-Ia enzyme was found to acetylate all urea-based linked dimers (compounds 9 to 12) with an efficiency similar to that of compound 1 (67% to 116%), with the exception of compound 11, which was not acetylated by this bifunctional enzyme.
Dimers with a thiourea linker (compounds 13 to 18) displayed mixed initial rates of activity compared to that of compound 1 (Fig. 2C). As observed for urea-linked NEO dimers 9 to 12, the initial rate of acetylation of thiourea-linked NEO dimers 13 to 18 was generally increased by AAC(2′)-Ic and AAC(6′)-Ib′. Compound 13 showed increased initial rates of modification by AAC(2′)-Ic (200%), AAC(6′)-Ib′ (349%), and APH(2″)-Ia (481%), and reduced rates of acetylation by AAC(6′)-Ie/APH(2″)-Ia (21%), AAC(3)-IV (17%), and Eis (35%). Compound 14 gave results similar to those observed for compound 13, with the exception of APH(2″)-Ia (15%) that displayed low phosphorylation activity and AAC(2′)-Ic (96%) that displayed activity similar to that of the enzyme against compound 1. Compounds 15 and 17 behaved similarly to compound 13 but were not phosphorylated by APH(2″)-Ia. Compound 16 had an activity profile similar to that of compound 13 but with a slightly elevated rate of acetylation by AAC(6′)-Ie/APH(2″)-Ia (119%). Compound 18 displayed an increase in its rate of acetylation by AAC(6′)-Ib′ (328%) but low to no modification by all other AMEs (0% to 58%) was observed.
Interestingly, all compounds displayed reduced phosphorylation rates by APH(3′)-Ia, which is an APH particularly adept at deactivating NEO. The fastest reaction rate was observed with compound 11 (52%), and all other compounds had a relative rate between 15% and 50% that of unmodified NEO. Despite the fact that all NEO dimers have two equivalents of the 3′-hydroxyl group, the reduced rates suggest that the resistance caused by this enzyme would generally not affect the compounds greatly.
F-NEO competitive binding screen.
We have previously reported the binding of dimeric AGs to model bacterial A site rRNA using the F-NEO displacement assay (17). Here, we have expanded the utility of the assay to include the model human A site (Fig. 3B). Most NEO dimers in the library bound to E. coli and the human model of the ribosomal A site with a relative affinity that was similar to that of NEO (compound 1). Four of the triazole-linked NEO dimers (compounds 2 and 4 to 6) bound the two A site models with similar or greater affinity than NEO (compound 1). The remaining three triazole-linked NEO dimers (compounds 3, 7, and 8), which contain longer and/or more flexible linkers, bound with lower affinity to the human (<50% of the binding affinity of NEO [1]) and E. coli (<15% of the binding affinity of NEO [1]) binding sites. In general, the urea-linked NEO dimers bound weakly to the E. coli and human model A sites, with compounds 9 to 12 displacing F-NEO from the A site by less than 10% relative to NEO (compound 1). The only exceptions were the urea-linked NEO dimers 12, found to have a notable affinity for the E. coli A site (34% displacement of F-NEO compared to compound 1), and 9, found to have a notable affinity for the human A site (30% displacement of F-NEO compared to compound 1). As for the thiourea-linked NEO dimer series, compounds 14, 16, and 18 had a higher relative affinity for the E. coli and human A sites than did the urea-linked NEO dimers. Within the thiourea-linked NEO dimers, some interesting trends emerged. The compound with the shortest linkage, compound 13, showed weak binding relative to compound 1 and little differential binding to the two rRNAs. Going from dimer 15 to a longer linker in dimer 16 increased the overall affinities but eliminated the selectivity of 15 toward the human A site. Similarly, going from dimer 17 to a longer linker in dimer 18 increased the overall affinities but removed the bacterial rRNA selectivity of dimer 17. Overall, a few of the lower affinity dimers (compounds 12 and 17) showed a better selectivity for the bacterial rRNA model A site relative to that of the human version. In contrast, dimers 3, 9, and 15 showed a modest selectivity for the human A site rRNA. While the exact affinities of these compounds remain to be determined, the assay used (25), as shown before, is a reliable predictor of the effects of minor modifications to AG structure on rRNA binding. Our data suggest that such minor modifications in AG dimer linkages alone may lead to differential selectivity toward human versus bacterial model rRNAs and should be further explored.
MIC determination.
All compounds were tested against eight bacterial strains (Table 1). All MRSA strains were resistant to NEO and most of the derivatives with the exception of the untyped MRSA and S. aureus USA 100, which displayed MIC values of 12.5 μM and 3.13 μM, respectively, in the presence of compound 18. S. aureus USA 200, USA 300, and USA 600 also showed less resistance than NEO with the same compound. S. aureus ATCC 29213 showed the best susceptibility to the NEO dimers with all compounds showing a similar MIC value to that of NEO with the exception of compounds 13, 11, 10, and 16. B. cereus MIC values ranged from 6.25 μM to 25 μM, 4-fold to 16-fold higher values than that of NEO (1.56 μM).
The genetic probing provided insight into potential resistance mechanisms of the bacterial strains tested. Of the strains probed, S. aureus USA 100 had three AME genes, aac(6′)-Ib, aac(3)-IV, and aph(3′)-Ia; S. aureus USA 200 and USA 300 each had two AME genes, aac(6′)-Ib and aph(3′)-Ia; and the MRSA strain had aac(6′)-Ib in its genome. B. cereus also had three AME genes contained within its DNA, aac(6′)-Ib, aac(3)-IV, and aph(3′)-Ia .While there are AMEs contained within the strains tested herein and previously, this panel is far from complete and does not take into account other modes of resistance. S. aureus ATCC 29213 contains none of the four AMEs probed for, which agrees with the low MIC values for that strain. S. aureus USA 600 and S. aureus ATCC 33591 also show no AMEs in their respective genomes; however, these are methicillin-resistant phenotypes which are known to have many different mechanisms of resistance that can affect the efficacy of AGs.
DISCUSSION
Within the triazole-linked dimer series, compound 2, containing three methylene groups between the triazoles, showed the highest rate of modification. Conversely, compound 8, with the dodecanediol linker between the triazole groups, shows the least amount of modifications, suggesting that this linker would be the best to overcome modification by AMEs. The decrease in activity observed with compound 8 may arise if the long linker either (i) prevents the individual NEO molecules in the dimer from binding to the active site or (ii) prevents binding of AcCoA to the enzyme-substrate complex. With the exception of compound 4, the only linker that includes branching, and compound 5, containing a phenyl linker, as the linker length increases, fewer AMEs modify the NEO molecules in their respective dimers.
The dimers with the urea linkages (compounds 9 to 12) generally displayed greater activity than those in the triazole series (compounds 2 to 8). The most direct comparison can be drawn from compounds 3 and 9, which contain a 6-carbon linker between their defining functional groups. The difference in the enzymatic-modification profile of these compounds can be attributed to the method of linking the two NEO molecules and suggests that the triazole linkage is less susceptible to enzymatic modification than the urea linkage. From the data in the urea-linked NEO dimers, a long linker, such as that in compound 10, reduces the number of modifications by enzymes as well as the rate of modification that can occur on NEO (compound 1). Likewise, conformationally restricting the rotation of the dimer may also reduce the ability of enzymes to modify NEO (compound 1), as seen in compounds 11 and 12. The para-substitution of compound 12 shows reduced activity compared to that of compound 11 for all enzymes except AAC(6′)-Ie. From the urea-linked NEO dimers, compound 10, containing 10 methylene groups separating the urea linkages, showed the highest rates of modification while compound 12, linked via a para-phenyl linker, showed the least modification. Within the urea-linked NEO dimers, the best linker to minimize AME activity would be a conformationally restricted para-substituted phenyl group.
Compounds in the thiourea-linked dimers (compounds 13 to 18) showed similar enzymatic-modification profiles to those of the urea-linked dimers, suggesting that the oxygen-to-sulfur replacement does not greatly affect AME modification profiles. In this group, compound 13, containing two NEO molecules directly linked via a thiourea, showed the highest rates of modification. Compound 14, with three methylene units between two thiourea moieties, showed the lowest average rates of modifications, followed closely by compound 18. These data suggest that within the thiourea-linked NEO dimers, a relatively short 3-carbon chain would be an optimum choice. Additionally, the long polyethyleneglycol linker in compound 18 showed substantial activity only with AAC(6′)-Ib′, suggesting that it would be a good linker to avoid modification by AMEs. Directly comparing compounds 2 and 14 with the major difference being a triazole versus a thiourea, there is little difference in the rates of modification with the exception of AAC(3)-IV, where compound 2 displayed a 10-fold higher rate of conversion than that of compound 14. This result suggests that when choosing a short methylene linker length, a thiourea conjugation would be superior to a triazole.
While MIC values were previously determined with these compounds (17), eight additional strains were tested herein. Noting the excellent MIC values previously reported with S. aureus ATCC 25923 and the moderate activity with S. aureus ATCC 33591, a methicillin-resistant strain, we tested several more strains of S. aureus and the Gram-positive B. cereus (Table 1). While most of the compounds were as effective at killing S. aureus ATCC 29213 as they were at killing S. aureus ATCC 25923, only compound 18 stood out against the MRSA strains, showing significantly reduced MIC values. This structure should be the next stepping point for this series of NEO dimers, as it is effective against strains containing AG resistance enzymes and those that did not show the selected genes.
Other NEO dimers have been generated by using a disulfide approach, where each NEO molecule is attached to an aliphatic thiol and the thiols are oxidized, forming a disulfide bond in the linker (27), and using naphthalene diimide as the central linker (28, 29). Each type of dimer has improved binding to the tested ribosome compared to NEO; however, the effect of these dimers on bacteria has not been tested. These compounds reinforce the presented data showing that dimerization of NEO enhances binding to the ribosome, given the correct linker.
Overall, the longer linkers, containing a triazole linkage as the method of combining two NEO molecules, lead to a NEO dimer that most effectively avoids modification by AMEs. On the other hand, some of these long linkers (e.g., in compounds 7 and 8) lead to reduced binding to the model A site rRNA compared to that of NEO. Similar observations can be made from the urea-linked dimers, where reduction in AME activity also and a loss of bacterial rRNA binding (compounds 10 and 12) is observed. Compound 10, however, showed much reduced binding to the human A site rRNA, suggesting a binder more selective for bacteria. Finally, the best thiourea-linked dimers that avoid AME modification (compounds 14 and 18) also showed a high degree of binding to the bacterial rRNA, with dimer 14 also showing increased selectivity in binding, as evidenced by reduced binding to human A site. It should be noted here that all but two of the NEO dimers reduced the initial rate of acetylation for Eis. This is significant due to the multiacetylating ability this AME has. We have documented this enzyme to modify the 3″-amino, 2′-amino, 6′-amino, and 1-amino groups of certain AGs (18, 30); while the modified positions of NEO are as of yet unresolved, the fact that these compounds hinder Eis ability to transfer an acetyl group is significant. Further work to optimize the selectivity in ribosomal binding and AME activity within a modified AG will be necessary for the development of more potent antibacterials. This work is under way in our laboratories.
ACKNOWLEDGMENTS
This work was supported by start-up funds from the College of Pharmacy at the University of Kentucky (S.G.-T.) and by National Institutes of Health (NIH) grants AI090048 (S.G.-T.) and GM097917 (D.P.A).
REFERENCES
- 1.Houghton JL, Green KD, Chen W, Garneau-Tsodikova S. 2010. The future of aminoglycosides: the end or renaissance? Chembiochem 11:880–902. doi: 10.1002/cbic.200900779. [DOI] [PubMed] [Google Scholar]
- 2.Fosso MY, Li Y, Garneau-Tsodikova S. 2014. New trends in aminoglycosides use. Medchemcomm 5:1075–1091. doi: 10.1039/C4MD00163J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wimberly BT, Brodersen DE, Clemons WM Jr, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. 2000. Structure of the 30S ribosomal subunit. Nature 407:327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 4.Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. 2000. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 5.Francois B, Russell RJ, Murray JB, Aboul-ela F, Masquida B, Vicens Q, Westhof E. 2005. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res 33:5677–5690. doi: 10.1093/nar/gki862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perez-Fernandez D, Shcherbakov D, Matt T, Leong NC, Kudyba I, Duscha S, Boukari H, Patak R, Dubbaka SR, Lang K, Meyer M, Akbergenov R, Freihofer P, Vaddi S, Thommes P, Ramakrishnan V, Vasella A, Bottger EC. 2014. 4′-O-substitutions determine selectivity of aminoglycoside antibiotics. Nat Commun 5:3112. doi: 10.1038/ncomms4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maianti JP, Kanazawa H, Dozzo P, Matias RD, Feeney LA, Armstrong ES, Hildebrandt DJ, Kane TR, Gliedt MJ, Goldblum AA, Linsell MS, Aggen JB, Kondo J, Hanessian S. 2014. Toxicity modulation, resistance enzyme evasion, and A site X-ray structure of broad-spectrum antibacterial neomycin analogs. ACS Chem Biol 9:2067–2073. doi: 10.1021/cb5003416. [DOI] [PubMed] [Google Scholar]
- 8.Blount KF, Tor Y. 2006. A tale of two targets: differential RNA selectivity of nucleobase-aminoglycoside conjugates. Chembiochem 7:1612–1621. doi: 10.1002/cbic.200600109. [DOI] [PubMed] [Google Scholar]
- 9.Ramirez MS, Tolmasky ME. 2010. Aminoglycoside modifying enzymes. Drug Resist Updat 13:151–171. doi: 10.1016/j.drup.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Labby KJ, Garneau-Tsodikova S. 2013. Strategies to overcome the action of aminoglycoside-modifying enzymes for treating resistant bacterial infections. Future Med Chem 5:1285–1309. doi: 10.4155/fmc.13.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Green KD, Chen W, Houghton JL, Fridman M, Garneau-Tsodikova S. 2010. Exploring the substrate promiscuity of drug-modifying enzymes for the chemoenzymatic generation of N-acylated aminoglycosides. Chembiochem 11:119–126. doi: 10.1002/cbic.200900584. [DOI] [PubMed] [Google Scholar]
- 12.Magalhaes ML, Blanchard JS. 2005. The kinetic mechanism of AAC3-IV aminoglycoside acetyltransferase from Escherichia coli. Biochemistry 44:16275–16283. doi: 10.1021/bi051777d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Porter VR, Green KD, Zolova OE, Houghton JL, Garneau-Tsodikova S. 2010. Dissecting the cosubstrate structure requirements of the Staphylococcus aureus aminoglycoside resistance enzyme ANT(4′). Biochem Biophys Res Commun 403:85–90. doi: 10.1016/j.bbrc.2010.10.119. [DOI] [PubMed] [Google Scholar]
- 14.McKay GA, Thompson PR, Wright GD. 1994. Broad spectrum aminoglycoside phosphotransferase type III from Enterococcus: overexpression, purification, and substrate specificity. Biochemistry 33:6936–6944. doi: 10.1021/bi00188a024. [DOI] [PubMed] [Google Scholar]
- 15.Thompson PR, Hughes DW, Wright GD. 1996. Regiospecificity of aminoglycoside phosphotransferase from Enterococci and Staphylococci (APH(3′)-IIIa). Biochemistry 35:8686–8695. doi: 10.1021/bi960389w. [DOI] [PubMed] [Google Scholar]
- 16.Wright GD, Thompson PR. 1999. Aminoglycoside phosphotransferases: proteins, structure, and mechanism. Front Biosci 4:D9–D21. [DOI] [PubMed] [Google Scholar]
- 17.King A, Watkins D, Kumar S, Ranjan N, Gong C, Whitlock J, Arya DP. 2013. Characterization of ribosomal binding and antibacterial activities using two orthogonal high-throughput screens. Antimicrob Agents Chemother 57:4717–4726. doi: 10.1128/AAC.00671-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen W, Biswas T, Porter VR, Tsodikov OV, Garneau-Tsodikova S. 2011. Unusual regioversatility of acetyltransferase Eis, a cause of drug resistance in XDR-TB. Proc Natl Acad Sci U S A 108:9804–9808. doi: 10.1073/pnas.1105379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green KD, Chen W, Garneau-Tsodikova S. 2011. Effects of altering aminoglycoside structures on bacterial resistance enzyme activities. Antimicrob Agents Chemother 55:3207–3213. doi: 10.1128/AAC.00312-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vetting MW, Hegde SS, Javid-Majd F, Blanchard JS, Roderick SL. 2002. Aminoglycoside 2′-N-acetyltransferase from Mycobacterium tuberculosis in complex with coenzyme A and aminoglycoside substrates. Nat Struct Biol 9:653–658. doi: 10.1038/nsb830. [DOI] [PubMed] [Google Scholar]
- 21.Kumar S, Kellish P, Robinson WE Jr, Wang D, Appella DH, Arya DP. 2012. Click dimers to target HIV TAR RNA conformation. Biochemistry 51:2331–2347. doi: 10.1021/bi201657k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar S, Arya DP. 2011. Recognition of HIV TAR RNA by triazole linked neomycin dimers. Bioorg Med Chem Lett 21:4788–4792. doi: 10.1016/j.bmcl.2011.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar S, Xue L, Arya DP. 2011. Neomycin-neomycin dimer: an all-carbohydrate scaffold with high affinity for AT-rich DNA duplexes. J Am Chem Soc 133:7361–7375. doi: 10.1021/ja108118v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stogios PJ, Spanogiannopoulos P, Evdokimova E, Egorova O, Shakya T, Todorovic N, Capretta A, Wright GD, Savchenko A. 2013. Structure-guided optimization of protein kinase inhibitors reverses aminoglycoside antibiotic resistance. Biochem J 454:191–200. doi: 10.1042/BJ20130317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watkins D, Norris FA, Kumar S, Arya DP. 2013. A fluorescence-based screen for ribosome binding antibiotics. Anal Biochem 434:300–307. doi: 10.1016/j.ab.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Almaghrabi R, Clancy CJ, Doi Y, Hao B, Chen L, Shields RK, Press EG, Iovine NM, Townsend BM, Wagener MM, Kreiswirth B, Nguyen MH. 2014. Carbapenem-resistant Klebsiella pneumoniae strains exhibit diversity in aminoglycoside-modifying enzymes, which exert differing effects on plazomicin and other agents. Antimicrob Agents Chemother 58:4443–4451. doi: 10.1128/AAC.00099-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, Tor Y. 1997. Dimeric aminoglycosides: design, synthesis and RNA binding. Bioorg Med Chem Lett 7:1951–1956. doi: 10.1016/S0960-894X(97)00339-9. [DOI] [Google Scholar]
- 28.Tok JB, Fenker J. 2001. Novel synthesis and RNA-binding properties of aminoglycoside dimers conjugated via a naphthalene diimide-based intercalator. Bioorg Med Chem Lett 11:2987–2991. doi: 10.1016/S0960-894X(01)00602-3. [DOI] [PubMed] [Google Scholar]
- 29.Tok JB, Dunn LJ, Des Jean RC. 2001. Binding of dimeric aminoglycosides to the HIV-1 rev responsive element (RRE) RNA construct. Bioorg Med Chem Lett 11:1127–1131. doi: 10.1016/S0960-894X(01)00149-4. [DOI] [PubMed] [Google Scholar]
- 30.Houghton JL, Biswas T, Chen W, Tsodikov OV, Garneau-Tsodikova S. 2013. Chemical and structural insights into the regioversatility of the aminoglycoside acetyltransferase Eis. Chembiochem 14:2127–2135. doi: 10.1002/cbic.201300359. [DOI] [PMC free article] [PubMed] [Google Scholar]