

Abstract
The increasing incidence and severity of infection by Clostridium difficile have stimulated attempts to develop new antimicrobial therapies. We report here the relative abilities of two antibiotics (metronidazole and vancomycin) in current use for treating C. difficile infection and of a third antimicrobial, surotomycin, to kill C. difficile cells at various stages of development and to inhibit the production of the toxin proteins that are the major virulence factors. The results indicate that none of the drugs affects the viability of spores at 8× MIC or 80× MIC and that all of the drugs kill exponential-phase cells when provided at 8× MIC. In contrast, none of the drugs killed stationary-phase cells or inhibited toxin production when provided at 8× MIC and neither vancomycin nor metronidazole killed stationary-phase cells when provided at 80× MIC. Surotomycin, on the other hand, did kill stationary-phase cells when provided at 80× MIC but did so without inducing lysis.
INTRODUCTION
In the United States, the incidence of Clostridium difficile infection (CDI) has been steadily rising over the last 15 to 20 years (1–3). There has been a significant increase in primary, recurrent, and untreatable CDI on the global scale as well (3, 4). Treatment with oral vancomycin or metronidazole, the current standard practice, leads to recurrence of CDI in up to 30% of patients treated for an initial episode. For patients who have already suffered multiple recurrences, the future recurrence rate can be as high as 60% (3, 5–7). In addition, many cases of CDI in the United States are now linked to a family of more-virulent strains (NAP1/BI/027) that emerged first in the United Kingdom and then in Canada (8, 9).
C. difficile spores, the infectious form of the organism, are metabolically dormant but germinate in response to a combination of host-derived bile acids and glycine (10). Because spores are metabolically dormant, they are insensitive to the action of most antibiotics. Thus, after antibiotics are prescribed to treat the active infection, the spores that remain in the colon or that are acquired from the environment are able to germinate and reinitiate active infection. The process of spore germination is largely enzymatic and, once initiated, cannot be stopped. Once stimulated to germinate, the spore releases a large depot of dipicolinic acid (DPA), chelated 1:1 with Ca++ (Ca++-DPA), in exchange for water (11). Also, the cortex layer, a specialized peptidoglycan, is degraded (12, 13). After these changes, the spore is no longer dormant and does not have the resistances typically characteristic of a spore. Metabolism then resumes within the spore core, and a vegetative cell grows out from the germinated spore. Such outgrowth is susceptible to the action of several antibiotics (14). Moreover, the host intestine contains an assortment of antimicrobial compounds that are produced by the innate immune system and the host microbiota. To combat these defenses, C. difficile can modify its cell wall to resist antimicrobials or efflux a variety of antimicrobial peptides from the cell (15–17). These resistance mechanisms allow C. difficile to grow in the presence of antimicrobial peptides and may also contribute to resistance to therapeutic antimicrobials.
Given the difficulties in treating C. difficile infections, the identification of narrow-spectrum antibiotics that are potent inhibitors of C. difficile growth would fulfill a critical need. Also needed are compounds or methods useful for reducing spore abundance or viability in order to limit the onset of infection and to prevent relapsing disease. Here we investigated the activities of a newly developed antibiotic, surotomycin (18, 19), and compared its activity to that of metronidazole and vancomycin, two antibiotics that are currently used for treatment of C. difficile infections. Surotomycin is a lipopeptide antibiotic that acts through depolarization of the membrane, leading to the loss of a proton gradient and cell death (19). The mechanism of action of surotomycin differs significantly from that of vancomycin (cell wall synthesis inhibition) and metronidazole (inhibition of cellular enzymatic functions), suggesting that surotomycin may affect C. difficile physiology differently from those other antibiotics. We report here the relative sensitivities of C. difficile strains to surotomycin, metronidazole, and vancomycin at multiple stages of the life cycle, i.e., as dormant (phase-bright) spores, as germinated (phase-dark) spores, as exponential-phase cells, and as stationary-phase cells. In addition, we assessed the effects of the antibiotics on toxin gene expression and measured the effects of various mutations that affect the sensitivity of exponential-phase cells to nisin (an antibiotic that creates holes in membranes and disrupts cell wall synthesis) on susceptibility to the three tested compounds.
MATERIALS AND METHODS
Strains used and growth conditions.
Clostridium difficile strains UK1 (ribotype 027; provided by D. Gerding) (20) and JIR8094 (ribotype 012; provided by J. Rood) were routinely grown at 37°C in brain heart infusion (BHI) medium supplemented with cysteine (0.1%) and yeast extract (0.5%), referred to here as BHIS medium, or in tryptose-yeast extract medium (3% tryptose, 2% yeast extract, 0.1% thioglycolate), referred to here as TY medium. All growth experiments were performed in Coy anaerobic chambers in an atmosphere containing 85% nitrogen, 10% hydrogen, and 5% carbon dioxide as previously described (21, 22). Susceptibility testing of surotomycin requires the presence of 50 mg (2.5 mEq) Ca++ per liter in the medium (23). The levels of Ca++ in the media used were determined by Laboratory Specialists Inc.; following their instructions, the Ca++ concentration in all media was raised to 50 mg/liter by addition of CaCl2·2H2O. Drugs were added where indicated. Surotomycin was provided by Cubist Pharmaceuticals. Metronidazole and vancomycin were purchased from Sigma-Aldrich. Nisin was obtained from MP Biomedicals.
The following mutant strains derived from JIR8094 were used in this study: MC112, a lytC (peptidoglycan hydrolase), nisin-tolerant mutant; MC119, a spontaneous cprK (nisin-resistant) mutant with increased resistance to lantibiotics and polymyxin B (16); MC120, a mutant with a TargeTron insertion in dltD resulting in increased susceptibility to nisin and polymyxin B (15); and MC141, a mutant with a TargeTron insertion in cprA causing increased susceptibility to lantibiotics (16).
MICs.
MICs were determined by growth of bacteria in culture tubes or in 96-well plates (that had been prereduced for at least 24 h prior to use) in medium (either BHIS or TY) supplemented with 2-fold serial dilutions of the test compounds. The initial bacterial titer was 5 × 105 CFU per ml. Each strain and each drug concentration were tested in duplicate in each MIC assay. The MIC was determined as the lowest concentration of drug at which no bacterial growth was detected after 18 to 24 h at 37°C. MIC assays were performed at least 3 times to ensure reproducibility of results.
Spore germination assay.
Spores of C. difficile strain UK1 were produced and purified as described previously (14, 24, 25). Purified spores (1 × 107) were suspended in 1 ml BHIS medium or germination salts [0.3 mM (NH4)2SO4, 6.6 mM KH2PO4, 15 mM NaCl, 59.5 mM NaHCO3, and 35.2 mM Na2HPO4] supplemented with 10 mM glycine. Cultures were further supplemented with 2 mM taurocholic acid (TA) and the test compounds at 8× or 80× MIC. The initiation of spore germination was detected as the loss of optical density at 600 nm (OD600) in a PerkinElmer Lambda 25 spectrophotometer.
Measurements of killing rates.
For experiments involving phase-dark spores, 1 × 107 phase-bright spores of strain UK1 were first incubated for 10 min in BHIS medium supplemented with 2 mM TA. The suspension was then centrifuged for 1 min at 14,000 × g. The resulting pellet of phase-dark spores was suspended in BHIS medium at a concentration of 1 × 105 per ml with or without the test compounds at 8× MIC or 80× MIC in Teflon-coated tubes to prevent the adherence of spores to the tubes. Spores were confirmed to be phase-dark by phase-contrast microscopy. For experiments using exponential-phase vegetative cells, cells of strain UK1 or JIR8094 (1 × 105 per ml) were suspended in BHIS medium supplemented with CaCl2 with or without addition of the test compounds at 8× MIC for strain JIR8094 or at 8× MIC and 80× MIC for strain UK1. For stationary-phase cell-killing assays, strain UK1 was grown in TY medium for ∼12 h and then diluted in 30 ml TY medium supplemented with CaCl2 to give an OD600 of ∼0.1. After 12 h of incubation at 37°C, at which point the cells had left the exponential-growth phase, the culture was subdivided and each of the test compounds was added to a separate culture tube at 8× or 80× MIC. For all assays, samples were withdrawn at the indicated time points thereafter and viability was scored by plating serial dilutions on BHIS agar without calcium or antibiotic. Plates were incubated in the anaerobic chamber overnight. Each killing-rate assay was performed at least twice to ensure the reproducibility of the results.
Measurements of toxin gene expression.
An overnight culture of strain UK1 was diluted in 30 ml TY-Ca++ to give an OD600 of ∼0.1. After 12 h of incubation at 37°C, the culture was split and each test compound was added to a separate subculture at 8× MIC. (Toxin gene expression was not tested in cells exposed to drugs at 80× MIC because such treatment with surotomycin at that concentration led to cell death and prevented the isolation of intact mRNA.) RNA was prepared from cells harvested at 0, 2, 4, 8, and 24 h after addition of the drugs as previously described (26). RNA was quantified by absorbance using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific). Primers for quantitative PCR (qPCR) were designed using the online PrimerQuest tool from Integrated DNA Technologies. Synthesis of cDNA was performed on 500 ng of RNA using random hexamer primers and a QuantiTect reverse transcription kit (Qiagen) according to the manufacturer's recommendations. To control for chromosomal DNA contamination, mock cDNA synthesis reaction mixtures containing no reverse transcriptase were used as negative controls in subsequent amplifications. cDNA samples were diluted 4-fold and used as the templates for qPCR of rpoC (primers oLB122 [CTAGCTGCTCCTATGTCTCACATC] and oLB123 [CCAGTCTCTCCTGGATCAACTA]) and tcdA (primers oLB131 [GTATGGATAGGTGGAGAAGTCA] and oLB132 [CTCTTCCTCTAGTAGCTGTAATGC]) using Roche SYBR green I PCR mix and a Roche LightCycler 480 II thermocycler. Reactions were performed in a final volume of 20 μl using 4 μl diluted cDNA and primers at 1 μM final concentration. Amplification included 45 cycles of the following steps: 10 s at 95°C, 10 s at 52°C, and 15 s at 72°C. Reactions were performed in triplicate using cDNA synthesized from each of a minimum of three biological replicates, and results are presented as the means and standard errors of the means (SEM) of the data obtained. Results were calculated using the threshold cycle (2−ΔΔCT) method, in which the amount of target mRNA is normalized to that of an internal control transcript (rpoC) (27).
Measurements of toxin protein accumulation by ELISA.
Culture supernatants of the samples used for RNA extraction (see above) were assayed for toxin A levels by enzyme-linked immunosorbent assay (ELISA) (28). In brief, 96-well plates were coated with antibody to toxin A (Novus Biologicals) overnight at 4°C. After being washed with phosphate-buffered saline (PBS) containing 0.1% Tween 20, the plates were incubated with blocking agent and, after the agent was discarded, incubated with culture fluid samples with or without dilution for 1 h at room temperature. After being washed with PBS-Tween, the plates were incubated with secondary antibodies fused to horseradish peroxidase (HRP) (Gallus Immunotech, Inc.). After 1 h at room temperature, the plates were washed again with PBS-Tween and mixed with peroxidase substrate for 40 min at room temperature. The reactions were stopped with H2SO4, and the OD450 was determined using a plate reader. Standard curves were generated using purified recombinant toxin A (29).
RESULTS
Determining the MICs of the test compounds.
The MICs for each of the test compounds were determined using exponential-phase cells as the target. The MIC values varied according to the strain being tested and the growth medium used (Table 1). For all further experimentation, the concentration of each antibiotic used was based on the MIC of that antibiotic in the strain to be tested growing in the medium being used. The growth medium chosen for each experiment was based on previous experimental conditions.
TABLE 1.
MICs of antimicrobial compoundsa
| Strain (growth medium) | MIC (μg/ml) |
||
|---|---|---|---|
| Surotomycin | Metronidazole | Vancomycin | |
| UK1 (BHIS + Ca++) | 1.5 | 0.5 | 0.3125 |
| JIR8094 (BHIS + Ca++) | 1 | 0.5 | 2 |
| UK1 (TY + Ca++) | 0.125 | 0.125 | 1 |
| ATCC 70057 (TY + Ca++) | 0.125–0.25 | 0.06–0.125 | 1 |
The various strains were grown in the indicated media and assayed for susceptibility to the indicated antibiotics provided in a series of 2-fold dilutions. The MIC was defined as the lowest concentration of antibiotic that prevented measurable growth.
MIC determination in mutant strains.
A panel of mutant strains derived from JIR8094 and altered in susceptibility to nisin and other small, cationic antimicrobial peptides (CAMPs) was tested for their susceptibility to surotomycin, metronidazole, and vancomycin. As shown in Table 2, the strains had identical metronidazole MIC values, but the surotomycin and vancomycin MIC values differed slightly. MC112 has a mutation in an autolysin that presumably alters the cell wall of the bacterium, resulting in increased tolerance of nisin though not a higher MIC value. (That is, MC112 survives exposure to higher concentrations of nisin than does the parent strain but does not grow in the presence of nisin.) MC112 had growth similar to that seen with the wild-type parent in all antibiotics tested, including surotomycin, in which the strain grew at a 2-fold-higher concentration.
TABLE 2.
MICs of antimicrobial compounds for mutant strainsa
| Strain | MIC (μg/ml) |
|||
|---|---|---|---|---|
| Nisin | Surotomycin | Metronidazole | Vancomycin | |
| JIR8094 | 180 | 1 | 0.5 | 2 |
| MC112 | 180 | 2 | 0.5 | 2 |
| MC119 | 1440 | 2 | 0.5 | 4 |
| MC120 | 90 | 1 | 0.5 | 2 |
| MC141 | 90 | 1 | 0.5 | 2 |
Each strain and each drug concentration were tested in cells growing in BHIS medium in duplicate for each assay. Assays were performed at least 3 times to ensure reproducibility of results.
MC119 carries a mutation in a regulatory protein that is known to control an ABC-transporter system (cprABC). As a result, the MC119 mutant has an extraordinarily high drug MIC value for growth in nisin. In a previous study, we determined that MC119 also is less susceptible to polymyxin B (16). The MC119 strain had a 2-fold-higher drug MIC for both vancomycin and surotomycin. The strongly decreased susceptibility of MC119 to nisin and its modestly decreased susceptibility to surotomycin and to vancomycin are consistent with the hypothesis that this mutant is altered in its response to antimicrobials that target the cell surface.
Effects of antibiotic exposure on the initiation of C. difficile UK1 spore germination.
To test if surotomycin or vancomycin or metronidazole is able to affect C. difficile spore germination, purified spores of strain UK1 were incubated in BHIS-Ca++ medium with or without the germinant taurocholic acid (TA) (10) and with or without each of the antibiotics. The spores germinated, as measured by loss of OD600, when suspended in BHIS-Ca++ medium supplemented with 2 mM TA but not in the absence of TA (Fig. 1). (The loss of OD corresponds to the conversion of spores from a birefringent [phase-bright] form to a nonbirefringent [phase-dark] form.) The addition of surotomycin or metronidazole or vancomycin at 8× or 80× MIC had no effect, either positive or negative, on TA-dependent spore germination (Fig. 1). When similar experiments were performed in a buffer containing TA and glycine, a cogerminant, the same lack of effect of the antibiotics was seen (data not shown).
FIG 1.

Effect of antibiotic treatment on initiation of C. difficile spore germination. C. difficile UK1 spores were suspended in BHIS-Ca++ medium (black) or medium supplemented with surotomycin at 8× MIC (A) or 80× MIC (B) or vancomycin or metronidazole at 8× MIC (C) or 80× MIC (D). Where indicated, the medium was supplemented with taurocholic acid (TA), an activator of germination. The initiation of spore germination was detected by measuring the loss of OD600. Experiments were performed in duplicate; the presented data represent one such experiment. Germination plots from the two experiments were superimposable.
Effects of antibiotic exposure on germinated (phase-dark) spores.
To determine the time after spore germination at which outgrowing vegetative cells become sensitive to the antibiotics, we added antibiotics to germinated (phase-dark) spores in BHIS-Ca++ medium and followed bacterial viability at intervals thereafter. The outgrowing cells lost viability more rapidly after exposure to surotomycin than after exposure to the other drugs (Table 3). Note that this experiment did not distinguish between the time needed for killing and the growth stage at which outgrowing vegetative cells become susceptible to the drugs.
TABLE 3.
Time required to reduce viability of C. difficile UK1 phase-dark spores and exponential-phase cells by 90%
| Target | Time to 90% killing (h) |
|||||
|---|---|---|---|---|---|---|
| 8× MIC |
80× MIC |
|||||
| Surotomycin | Metronidazole | Vancomycin | Surotomycin | Metronidazole | Vancomycin | |
| UK1 phase-dark sporesa | 3.33 | >7 | >7 | 2 | 3 | >5 |
| UK1 exponential-phase cellsb | 0.33 | 1.5 | 1.5 | <0.33 | 0.66 | 1.33 |
Tubes containing BHIS medium supplemented with CaCl2 and the indicated antibiotics (at 8× MIC or 80× MIC) were inoculated with phase-dark spores of strain UK1 and tested, after various times of incubation, for survival by plating on BHIS medium.
Exponential-phase cells of strain UK1 growing in BHIS medium supplemented with CaCl2 were exposed to two different concentrations (8× MIC and 80× MIC) of the indicated antibiotics. Survival was assayed by plating serial dilutions on BHIS-Ca++ medium.
Effects of surotomycin, metronidazole, and vancomycin on exponential-phase cells.
Strains UK1 and JIR8094 were grown at 37°C to the early exponential phase in BHIS-Ca++ medium. After the initial titer was determined, the cultures were divided and inoculated with the test drugs at concentrations corresponding to 8× or 80× MIC. Samples of each culture were taken at multiple time points, and surviving cell titers were determined by serial dilution and plating on BHIS-Ca++ medium supplemented with taurocholic acid (TA). In exponential-phase cells of strain UK1, surotomycin reduced viability by 90% more rapidly than did the other drugs (Table 3). In exponential-phase cells of strain JIR8094, surotomycin, metronidazole, and nisin were all rapid killers, whereas vancomycin reduced viability more slowly (Fig. 2).
FIG 2.
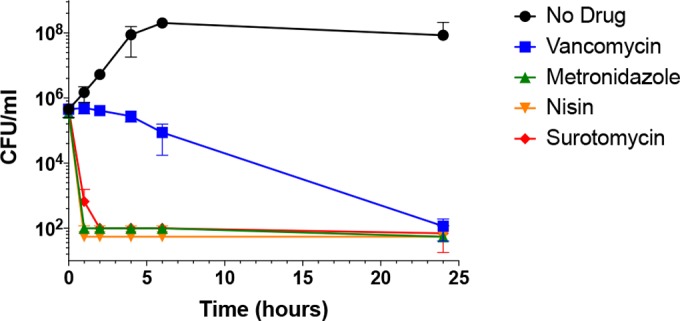
Effects of drugs at 8× MIC on survival of exponential-phase cells of C. difficile strain JIR8094. Exponential-phase cultures (OD600 = 0.45) in BHIS-Ca++ were exposed to the three antibiotics, and samples withdrawn at indicated times thereafter were assayed for viability (CFU/ml).
Effects of surotomycin, metronidazole, and vancomycin on stationary-phase cells.
After incubation of a culture of strain UK1 in TY plus CaCl2 at 37°C for 12 h, the culture was split and each drug was added to a separate culture tube at 8× or 80× MIC. Samples were harvested at various intervals for CFU counts and OD600 readings. At 8× MIC, none of the three drugs substantially reduced the turbidity of the culture (apparent cell mass) or viability (Fig. 3A and 4A). At 80× MIC, however, surotomycin greatly diminished the viability (but did not diminish cell mass significantly, indicating killing without cell lysis), whereas vancomycin and metronidazole had no detectable impact on cell mass or viability (Fig. 3B and 4B).
FIG 3.
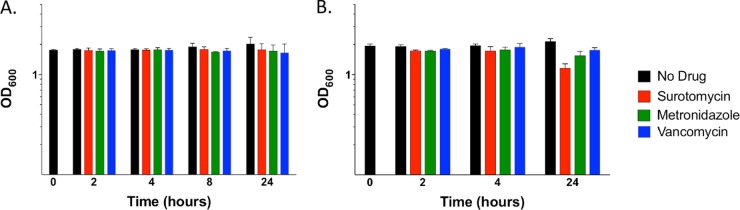
Effects of drugs on stationary-phase cells of strain UK1 as measured by cell mass. (A) Each drug was added at 8× MIC to the cultures at the early stationary phase. (B) Each drug was added at 80× MIC to the cultures at the early stationary phase. At least 3 biological replicates were performed for each drug at each concentration.
FIG 4.
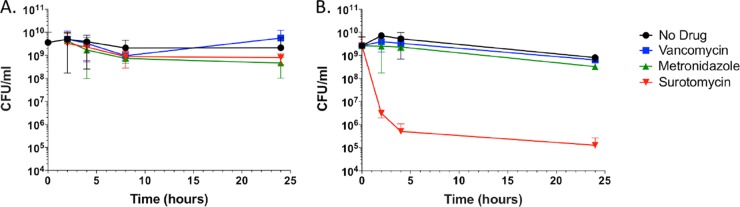
Effects of drugs at 8× and 80× MIC on the viability of strain UK1 stationary-phase cells. Early stationary-phase cells were exposed to antibiotics at 8× MIC (A) or 80× MIC (B). At the indicated time points thereafter, samples were removed and plated for viable counts (CFU).
Effects of surotomycin, metronidazole, and vancomycin on toxin gene expression and toxin release.
To test the effects of the various drugs on toxin production, cells at an early stage of the stationary phase were exposed to the individual compounds at 8× MIC. At intervals thereafter, samples were harvested and RNA was extracted for quantitation by quantitative reverse transcription-PCR (qRT-PCR). As shown in Fig. 5, expression of tcdA, the gene that encodes toxin A, increased with time to similar extents in untreated cells and cells treated with drugs.
FIG 5.
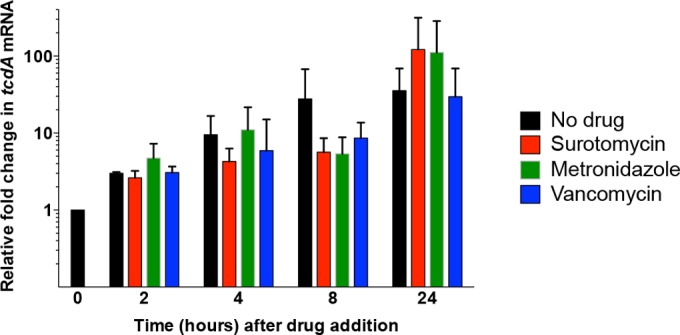
qRT-PCR analysis of tcdA expression following treatment with surotomycin, metronidazole, or vancomycin at 8× MIC. Strain UK1 was grown in CaCl2-supplemented TY medium for ∼12 h as described above, at which time the culture was split and drugs were added at 8× MIC. At 2, 4, 8, and 24 h after addition of drugs, cell samples were harvested, RNA was extracted, and cDNA was synthesized as described in Materials and Methods. cDNA corresponding to tcdA mRNA was quantified by real-time PCR. Reactions were performed in triplicate using cDNA synthesized from each of a minimum of three biological replicates, and results are presented as the means and SEM of the data obtained. Results were calculated using the threshold cycle (2−ΔΔCT) method, in which the amount of target mRNA was normalized to that of an internal control transcript (rpoC).
To measure toxin A release into the culture medium, the supernatant fluids of the samples used for RNA extraction as described above were assayed for toxin A levels by ELISA (see Materials and Methods). Standard curves were generated using purified toxin A. As shown in Fig. 6, the amount of toxin A in the culture fluid was not significantly altered compared to the amount seen with the no-drug control by exposure to surotomycin or metronidazole at 8× MIC at the early stationary phase. Addition of vancomycin, however, reduced toxin release at 24 h.
FIG 6.
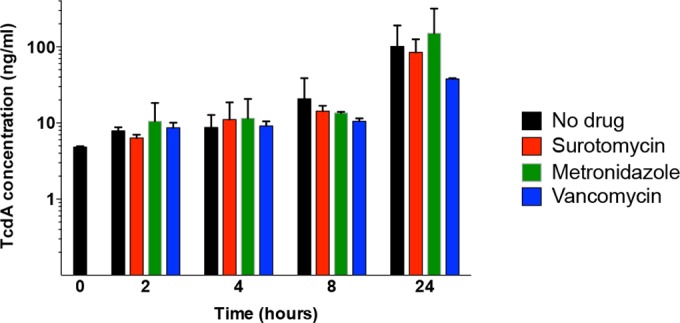
ELISAs of toxin A in cultures of strain UK1 treated with surotomycin, metronidazole, or vancomycin at 8× MIC. Culture fluids of cells exposed to antibiotics at the early stationary phase were collected at the indicated times and assayed for toxin A by ELISA.
DISCUSSION
The results presented here show that surotomycin kills cells that are growing out of germinated spores, exponential-phase cells, and stationary-phase cells (in the latter case at 80× MIC only) of strain UK1 more rapidly than does metronidazole or vancomycin. Surotomycin is also a more rapid killer of exponential-phase cells of strain JIR8094 than is vancomycin. None of the antibiotics tested was able to inhibit or activate C. difficile spore germination. This result seems to fit with the mechanisms of action of the tested antibiotics. The initiation of spore germination, a mostly enzymatic process, would be unaffected by antibiotics that need to be metabolized before becoming potent (metronidazole) or by antibiotics that inhibit new cell wall synthesis (vancomycin). Surotomycin leads to loss of the proton gradient but does so by depolarizing the membrane without causing permeability of molecules as large as 671 Da (19). Such a mechanism might explain its lack of effect on the initiation of spore germination if the pores created are also not large enough to allow the escape of Ca++-DPA from the spore core, a step necessary for the completion of germination (12, 13). Alternatively, surotomycin might be unable to penetrate the spore coat and cortex in order to gain access to the spore inner membrane.
Since outgrowing spores are metabolically active and synthesize new cell wall peptidoglycan, it is not surprising that such spores are susceptible to all of the antibiotics tested, although the heightened susceptibility of such cells to surotomycin was not predictable. The greater activity of surotomycin may be due to its ability to prevent the generation of the proton gradient needed for metabolic functions to resume. After the loss of dormancy, metabolism resumes in the core. Subsequently, a vegetative cell begins to grow from the germinated spore and this requires new cell wall synthesis. Metronidazole, which requires cellular metabolism for activity (30), inhibited growth in an intermediate time frame with respect to surotomycin and vancomycin. Because new cell wall synthesis occurs late during outgrowth, it is not surprising that vancomycin required more time to inhibit growth than the other two antibiotics tested.
Although the ability of all three antibiotics to kill exponential-phase cells was expected, their differential effects on stationary-phase cells suggest that surotomycin may be more effective than vancomycin or metronidazole in reducing the severity and recurrence of infection. That is, at 80× MIC, a relatively low dose compared to those used clinically, surotomycin killed stationary-phase cells rapidly. Since stationary-phase cells are the ones that form spores and produce toxins, a drug that kills both exponential-phase and stationary-phase cells may be particularly effective in reducing the spore titer and toxin levels in the colon and in stool.
The appearance of C. difficile mutants that have decreased susceptibility to surotomycin is rare, and the MIC shift is not more than 8-fold to 16-fold (18). Therefore, it is interesting that certain mutant strains that were isolated on the basis of their increased tolerance or resistance to nisin also showed slightly reduced susceptibility to surotomycin (Table 2). The basis for this decreased susceptibility is not known but is likely to reflect changes in cell surface architecture. The decrease in susceptibility gained, however, would not be expected to influence the effectiveness of surotomycin in vivo.
ACKNOWLEDGMENTS
The research reported here was funded by a research contract jointly awarded to Tufts University, Texas A&M University, and Emory University by Cubist Pharmaceuticals, Inc. S.M. was also supported by the U.S. National Institutes of Health through research grants DK087763 and DK101870 and a Natalie V. Zucker research grant from Tufts University.
The content of the manuscript is solely our responsibility and does not necessarily reflect the official views of the National Institutes of Health.
REFERENCES
- 1.Bartlett JG. 2010. Clostridium difficile: progress and challenges. Ann N Y Acad Sci 1213:62–69. doi: 10.1111/j.1749-6632.2010.05863.x. [DOI] [PubMed] [Google Scholar]
- 2.Miller BA, Chen LF, Sexton DJ, Anderson DJ. 2011. Comparison of the burdens of hospital-onset, healthcare facility-associated Clostridium difficile infection and of healthcare-associated infection due to methicillin-resistant Staphylococcus aureus in community hospitals. Infect Control Hosp Epidemiol 32:387–390. doi: 10.1086/659156. [DOI] [PubMed] [Google Scholar]
- 3.Kelly CP, LaMont JT. 2008. Clostridium difficile—more difficult than ever. N Engl J Med 359:1932–1940. doi: 10.1056/NEJMra0707500. [DOI] [PubMed] [Google Scholar]
- 4.Jarvis WR, Schlosser J, Jarvis AA, Chinn RY. 2009. National point prevalence of Clostridium difficile in US health care facility inpatients, 2008. Am J Infect Control 37:263–270. doi: 10.1016/j.ajic.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Louie TJ, Miller MA, Mullane KM, Weiss K, Lentnek A, Golan Y, Gorbach S, Sears P, Shue YK. 2011. Fidaxomicin versus vancomycin for Clostridium difficile infection. N Engl J Med 364:422–431. doi: 10.1056/NEJMoa0910812. [DOI] [PubMed] [Google Scholar]
- 6.Pépin J, Valiquette L, Gagnon S, Routhier S, Brazeau I. 2007. Outcomes of Clostridium difficile-associated disease treated with metronidazole or vancomycin before and after the emergence of NAP1/027. Am J Gastroenterol 102:2781–2788. doi: 10.1111/j.1572-0241.2007.01539.x. [DOI] [PubMed] [Google Scholar]
- 7.Vardakas KZ, Polyzos KA, Patouni K, Rafailidis PI, Samonis G, Falagas ME. 2012. Treatment failure and recurrence of Clostridium difficile infection following treatment with vancomycin or metronidazole: a systematic review of the evidence. Int J Antimicrob Agents 40:1–8. doi: 10.1016/j.ijantimicag.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 8.Rupnik M, Wilcox MH, Gerding DN. 2009. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 9.Petrella LA, Sambol SP, Cheknis A, Nagaro K, Kean Y, Sears PS, Babakhani F, Johnson S, Gerding DN. 2012. Decreased cure and increased recurrence rates for Clostridium difficile infection caused by the epidemic C. difficile BI strain. Clin Infect Dis 55:351–357. doi: 10.1093/cid/cis430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sorg JA, Sonenshein AL. 2008. Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol 190:2505–2512. doi: 10.1128/JB.01765-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Setlow P. 2003. Spore germination. Curr Opin Microbiol 6:550–556. doi: 10.1016/j.mib.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Paredes-Sabja D, Shen A, Sorg JA. 2014. Clostridium difficile spore biology: sporulation, germination, and spore structural proteins. Trends Microbiol doi: 10.1016/j.tim.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Setlow P. 2014. Germination of spores of Bacillus species: what we know and do not know. J Bacteriol 196:1297–1305. doi: 10.1128/JB.01455-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allen CA, Babakhani F, Sears P, Nguyen L, Sorg JA. 2013. Both fidaxomicin and vancomycin inhibit outgrowth of Clostridium difficile spores. Antimicrob Agents Chemother 57:664–667. doi: 10.1128/AAC.01611-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McBride SM, Sonenshein AL. 2011. The dlt operon confers resistance to cationic antimicrobial peptides in Clostridium difficile. Microbiology 157:1457–1465. doi: 10.1099/mic.0.045997-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McBride SM, Sonenshein AL. 2011. Identification of a genetic locus responsible for antimicrobial peptide resistance in Clostridium difficile. Infect Immun 79:167–176. doi: 10.1128/IAI.00731-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suárez JM, Edwards AN, McBride SM. 2013. The Clostridium difficile cpr locus is regulated by a noncontiguous two-component system in response to type A and B lantibiotics. J Bacteriol 195:2621–2631. doi: 10.1128/JB.00166-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mascio CT, Chesnel L, Thorne G, Silverman JA. 2014. Surotomycin demonstrates low in vitro frequency of resistance and rapid bactericidal activity in Clostridium difficile, Enterococcus faecalis, and Enterococcus faecium. Antimicrob Agents Chemother 58:3976–3982. doi: 10.1128/AAC.00124-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mascio CT, Mortin LI, Howland KT, Van Praagh AD, Zhang S, Arya A, Chuong CL, Kang C, Li T, Silverman JA. 2012. In vitro and in vivo characterization of CB-183,315, a novel lipopeptide antibiotic for treatment of Clostridium difficile. Antimicrob Agents Chemother 56:5023–5030. doi: 10.1128/AAC.00057-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Killgore GTA, Johnson S, Brazier J, Kuijper E, Pepin J, Frost EH, Savelkoul P, Nicholson B, van den Berg RJ, Kato H, Sambol SP, Zukowski W, Woods C, Limbago B, Gerding DN, McDonald LC. 2008. Comparison of seven techniques for typing international epidemic strains of Clostridium difficile: restriction endonuclease analysis, pulsed-field gel electrophoresis, PCR-ribotyping, multilocus sequence typing, multilocus variable-number tandem-repeat analysis, amplified fragment length polymorphism, and surface layer protein A gene sequence typing. J Clin Microbiol 46:431–437. doi: 10.1128/JCM.01484-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sorg JA, Dineen SS. 2009. Laboratory maintenance of Clostridium difficile. Curr Protoc Microbiol Chapter 9:Unit9A.1. doi: 10.1002/9780471729259.mc09a01s12. [DOI] [PubMed] [Google Scholar]
- 22.Edwards AN, Suarez JM, McBride SM. 2013. Culturing and maintaining Clostridium difficile in an anaerobic environment. J Vis Exp 2013:e50787. doi: 10.3791/50787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.CLSI. 2015. Performance standards for antimicrobial susceptibility testing; twenty-fifth informational supplement. CLSI, Wayne, PA. [Google Scholar]
- 24.Sorg JA, Sonenshein AL. 2010. Inhibiting the initiation of Clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J Bacteriol 192:4983–4990. doi: 10.1128/JB.00610-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Francis MB, Allen CA, Shrestha R, Sorg JA. 2013. Bile acid recognition by the Clostridium difficile germinant receptor, CspC, is important for establishing infection. PLoS Pathog 9:e1003356. doi: 10.1371/journal.ppat.1003356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bouillaut L, McBride SM, Sorg JA. 2011. Genetic manipulation of Clostridium difficile. Curr Protoc Microbiol Chapter 9:Unit 9A.2. doi: 10.1002/9780471729259.mc09a02s20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 28.Mukherjee J, Tremblay JM, Leysath CE, Ofori K, Baldwin K, Feng X, Bedenice D, Webb RP, Wright PM, Smith LA, Tzipori S, Shoemaker CB. 2012. A novel strategy for development of recombinant antitoxin therapeutics tested in a mouse botulism model. PLoS One 7:e29941. doi: 10.1371/journal.pone.0029941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang G, Zhou B, Wang J, He X, Sun X, Nie W, Tzipori S, Feng H. 2008. Expression of recombinant Clostridium difficile toxin A and B in Bacillus megaterium. BMC Microbiol 8:192. doi: 10.1186/1471-2180-8-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Löfmark S, Edlund C, Nord CE. 2010. Metronidazole is still the drug of choice for treatment of anaerobic infections. Clin Infect Dis 50(Suppl 1):S16–S23. doi: 10.1086/647939. [DOI] [PubMed] [Google Scholar]