

Abstract
The aim of this study was to examine the relationships between N-acetyltransferase genotypes, pharmacokinetics, and tolerability of granular slow-release para-aminosalicylic acid (GSR-PAS) in tuberculosis patients. The study was a randomized, two-period, open-label, crossover design wherein each patient received 4 g GSR-PAS twice daily or 8 g once daily alternately. The PAS concentration-time profiles were modeled by a one-compartment disposition model with three transit compartments in series to describe its absorption. Patients' NAT1 and NAT2 genotypes were determined by sequencing and restriction enzyme analysis, respectively. The number of daily vomits was modeled by a Poisson probability mass function. Comparisons of other tolerability measures by regimens, gender, and genotypes were evaluated by a linear mixed-effects model. The covariate effects associated with efavirenz, gender, and NAT1*3, NAT1*14, and NAT2*5 alleles corresponded to 25, 37, −17, −48, and −27% changes, respectively, in oral clearance of PAS. The NAT1*10 allele did not influence drug clearance. The time above the MIC of 1 mg/liter was significantly different between the two regimens but not influenced by the NAT1 or NAT2 genotypes. The occurrence and intensity of intolerance differed little between regimens. Four grams of GSR-PAS twice daily but not 8 g once daily ensured concentrations exceeding the MIC (1 mg/liter) throughout the dosing interval; PAS intolerance was not related to maximum PAS concentrations over the doses studied and was not more frequent after once-daily dosing. We confirm that the slow phenotype conferred by the NAT1*14 and NAT1*3 alleles resulted in higher PAS exposure but found no evidence of increased activity of the NAT1*10 allele.
INTRODUCTION
para-Aminosalicylic acid (PAS) was the first effective antituberculosis agent used to treat pulmonary tuberculosis (1); for a duration of 20 to 25 years, it was part of the standard “first-line” tuberculosis treatment (2). Valued for preventing resistance in companion drugs, it was nonetheless notorious for gastrointestinal intolerance, causing frequent nausea, vomiting, and abdominal discomfort. The replacement of PAS with rifampin and ethambutol was greeted with relief by patients, but with widespread multidrug-resistant (MDR) and extensively drug-resistant (XDR) tuberculosis in a number of countries, particularly in the developing world, PAS is again being used to treat drug-resistant tuberculosis.
The PAS preparation most commonly used in many countries is the granular slow-release (delayed-release) formulation (GSR-PAS), which prevents premature drug release in the stomach, avoiding the high PAS concentrations considered prone to cause intolerance. Several studies (3–7) have reported PAS concentrations associated with use of GSR-PAS in dosages from 4 g daily to 8 to 12 g daily in divided doses, as recommended by the World Health Organization (8). As PAS is usually considered bacteriostatic, divided dosing aims to provide concentrations consistently exceeding the PAS MIC of approximately 1 mg/liter (9–11). However, there is evidence from earlier studies that higher PAS concentrations may improve resistance prevention in relatively weak companion drugs and assist supervised programmatic drug administration (12).
At low concentrations, PAS is metabolized mainly by acetylation by N-acetyltransferase-1 (NAT1) to form acetyl-PAS (12); although at first considered monomorphic (13), it is now clear that PAS-acetylation is polymorphic (14). Although PAS has been utilized in several studies as a probe investigating the phenotype of NAT1 alleles (15, 16), particularly with a view to elucidate the role of NAT1 alleles in carcinogenesis, the potential pharmacokinetic consequences of different NAT1 genotypes in tuberculosis patients are uncertain.
In earlier papers, we described the pharmacokinetics of GSR-PAS in 10 children in comparison to a similar group of adults (6) and then the population pharmacokinetics of GSR-PAS in 73 adult MDR or XDR patients (17). In the latter paper, we reported GSR-PAS pharmacokinetics following 4 g GSR-PAS twice daily in 41 patients and also in 32 patients randomized in a crossover study to receive 4 g GSR-PAS twice daily or 8 g once daily and showed that HIV-infected patients were exposed to lower PAS concentrations due to an interaction with efavirenz. We have since completed analysis of the NAT1 and NAT2 genes of the 32 patients receiving 4 g GSR-PAS twice daily or 8 g once daily and now describe the relationships between NAT1 and NAT2 genotypes, PAS concentrations, and tolerability of GSR-PAS following the two regimens.
MATERIALS AND METHODS
This was a randomized, two-period, open-label, crossover study with each patient receiving the two PAS regimens for 8 days per regimen. Patients aged ≥18 years with MDR or XDR tuberculosis admitted to the Brooklyn Hospital for Chest Diseases, Cape Town, South Africa, were prospectively enrolled if in a stable condition, on a clinically indicated antituberculosis regimen, including GSR-PAS, for at least 2 weeks, and had no concurrent condition making their participation unsuitable. These studies were approved by the Committee for Human Research, Stellenbosch University (no. 9/08/212). All patients provided written informed consent for the study and its associated genetic study. Exclusion criteria were severe anemia, diarrhea, or dehydration. The patient characteristics recorded were age, race, sex, weight, and concomitant medications. Of necessity, companion drugs varied according to the susceptibility patterns of the Mycobacterium tuberculosis isolates.
Drug administration.
GSR-PAS was prescribed by attending physicians for all subjects as part of their multidrug regimens. Concomitant medication was allowed according to the resistance pattern of each subject and taking into account concurrent illnesses. HIV-infected patients received stavudine, lamivudine, and efavirenz.
All dose administrations were directly observed and were accompanied by either 100 g yoghurt or 125 ml amasi, an African curdled milk. These acidic beverages prevent early PAS release in the stomach (5). GSR-PAS was given 1 to 2 h before breakfast and also before other medications. All drug doses on study days were recorded, and patients were monitored for vomiting. Thirty-two patients were randomized to receive one of the two regimens. Blood samples were drawn at 0, 1, 2, 3, 4, 6, 8, 12, and 24 h postdose on days 8 and 16 to ensure that steady state was achieved before samples were drawn. On day 9, the regimens were crossed over. No washout period was implemented to avoid the further development of drug resistance in these patients.
For twice-daily dosing, patients received 4 g GSR-PAS every 12 h with other antituberculosis drugs and antiretroviral agents if patients were HIV infected. The second dose for the twice-daily regimen was given immediately after the 12-h blood sampling and 1 1/2 to 2 h after the evening meal.
For once-daily dosing, patients received 8 g oral GSR-PAS once daily with other antituberculosis drugs and antiretroviral agents if HIV infected.
Three-milliliter blood specimens were collected in EDTA-containing tubes through a catheter inserted into a forearm vein; samples were collected on ice and centrifuged for 10 min at 3,500 × g, and plasma was promptly harvested and frozen at −80°C awaiting analysis. The remainder was used for genetic analysis.
One patient was discharged from the hospital after receiving 4 g GSR-PAS twice daily, and two discharged patients similarly received only the 8-g once-daily dose.
Genotyping.
The NAT1 genotype was successfully determined in 31 patients and the NAT2 genotype in all 32. A 1,492-bp domain of the NAT1 gene sequence (http://www.ncbi.nlm.nih.gov/nuccore/14018355) was amplified via PCR assay in the Gene Amp PCR system 9700 (Applied Biosystems). Briefly, each 25-μl reaction volume consisted of 15 ng of genomic DNA, 20 mM Tris buffer (pH 8.4), 50 mM KCl, 0.2 mM (each) dATP, dGTP, dCTP, and dTTP, 2.5 mM MgCl2, 0.25 U of Taq DNA polymerase 1, and 0.25 μM (each) the forward and reverse primers NAT1/117 (5′-117AGGATACCAGTTGGAATCTCTCT) and NAT1/1609 (5′-1609AACAATAAACCAACATTAAAAGCTT), respectively. The cycling conditions consisted of initial denaturation at 94°C for 5 min, followed by 35 cycles of alternating denaturation at 95°C for 30 s, annealing at 60°C for 40 s, and polymerization at 72°C for 80 s. This was followed by extended polymerization at 72°C for 10 min. Thereafter, sample tubes were cooled to 4°C.
The amplified domain is demarcated by nucleotides 117 to 1648 of the gene sequence and contained 22 NAT1 single nucleotide polymorphisms (SNPs), 19 exonic, and 3 located within the 3′-untranslated region of the gene. This PCR-amplified domain was analyzed via bidirectional Sanger sequencing at the Central Analytical Facility, Stellenbosch University. Variant analysis and SNP calling were performed using the software program Sequencher version 5.1 (Gene Codes).
NAT2 genotyping was done by analysis of the 191G→A, 282C→T, 341T→C, 481C→T, 590G→A, 803A→G, and 857G→A SNPs, which delineated the NAT2*14, -2*3, -2*5, -2*11, -2*6, -2*12, and -2*7 alleles, respectively. The NAT2 gene sequence (http://www.ncbi.nlm.nih.gov/nuccore/219871) analysis also proceeded via an initial PCR amplification, subsequent restriction enzyme digestion, and allele-specific-PCR analyses, as previously described (18).
PAS analysis.
PAS concentrations were quantified by the validated high-performance liquid chromatography-tandem mass spectrometry assay previously described (6). Analysis of quality control samples found good reproducibility and a coefficient of variation (CV) of ≤5%; between-sample differences did not exceed 2.5%. The lower limit of quantification was 0.01 mg/liter; all observed drug concentrations exceeded this limit.
Pharmacokinetic analysis.
The pharmacokinetic model for the PAS disposition in tuberculosis and tuberculosis-HIV patients was a one-compartment model with 3 transit compartments, as previously described (17). The covariates screened were treatment or dosing regimen, HIV status, gender, race, age, body weight, and the NAT1 and NAT2 genotypes. The hypothesis testing to discriminate among alternative hierarchical structural models was based on the P values for the forward inclusion of 0.05. The covariate model was based on the equation Pi = θ × exp(ηi + κij) × [1 + α1(X1/median1)] … [1 + αk(Xk/mediank)] × exp(β1Y1 + … + βlYl), where ηi and κij define the interindividual and interoccasion variabilities, respectively. Continuous covariates X1, … , Xk were incorporated as a proportional model centered on the median value of the covariate, and categorical or discrete covariates Y1, … Yl, wherein dummy variables 0 and 1 were used to represent absence and presence, respectively, were included in the parameter as exponential models.
Given the exploratory nature of the study, model selection was based on maximum likelihood statistics (defined as negative twice the log likelihood), goodness of fit, and visual predictive checks (VPC) (with 5,000 simulated profiles).
The time above the MIC (T>MIC) was computed by taking the log-concentration-time profile and utilizing the linear imputation function approxfun in R to estimate the time in the ascending phase and the time in the descending phase that intersect the MIC value; the difference between these two time points divided by the respective dosing interval and converted to percentage became the parameter %T>MIC.
Gastrointestinal intolerance.
Gastrointestinal intolerance was assessed using self-rating visual analogue scales (VAS) for diarrhea, nausea, bloating, and abdominal pain. The far left of the 10-point scale indicated no intolerance and the far right severe intolerance. Vomiting was scored by the number of times it occurred. Patients were individually interviewed daily, and the scales were thoroughly explained before completion; this took place 2 to 3 h after the last daily meal. Occurrence of intolerance refers to any reported experience of intolerance by a patient during any one of 8 observation days on each regimen. Intensity of intolerance refers to the single highest visual analog score out of a possible score of 10 allotted by a patient to a particular symptom on any of the 8 days of observation on each regimen.
Count model.
Daily vomit frequencies over 8 days were evaluated using a Poisson model, as previously described (19, 20). The suitability of the Poisson model was evaluated by plotting the individual mean frequency against variance to evaluate for equidispersion. The Poisson probability mass function has the form f(n; λ) = P(Y = n) = (λn × e−λ)/n!, where λ is the expected average number of vomits daily and n is the actual observed number of vomits daily. An exponential interindividual variability term was incorporated into the λ parameter; time, treatment, and covariate effects on λ were also evaluated: λi = θ × exp(ηi) × [1 − f(tj)] × exp[α1(X1/median) + … + αk(Xk/median) + β1Y1 + … +βlYl], where tj represents time and f(tj) = exp(γtj) is the form used to evaluate time effect. ηi describes the interindividual variability. X1, …, Xk refer to the continuous covariates 1 to k that were tested; α1, … ,αk are coefficients associated with continuous covariates. For discrete covariates Y1, … , Yl, the corresponding coefficients are β1, … , βl. Covariates that were evaluated for their potential effect on λ were the same as the ones evaluate for PAS pharmacokinetics, with the exception that the observed PAS maximum concentration (Cmax) and the observed area under the concentration-time curve (AUC) were also evaluated.
The predictability of the final model was evaluated using a visual predictive check wherein 1,000 Cmax values were simulated from a uniform distribution, υ(minimum [min] = 10 ng/ml, and maximum [max] = 135 ng/ml). The frequencies of individuals with 0 to 5 daily vomits were estimated and compared with the actual observed frequency, plotted as a histogram, to evaluate the predictive performance of the final model.
Statistical methods.
This was an observational study without a priori power calculation. Potential subjects were patients placed on PAS-containing regimens by their physicians. Based on experience, we estimated the period of time that would allow recruitment of at least 20 consenting subjects and proceeded to recruit all available subjects during this period.
Numerical data were summarized as median (range) as most quantitative variables did not have normal distributions. Genotype data were summarized as counts (frequency/proportion) for regimens and separately for race. For each individual, pairs of haplotypes were inferred, together with their probabilities; hence, haplotypes were summarized as frequencies (proportions). Other categorical traits were summarized as counts and percentages for the regimens separately. Logistic regression provided the P values for race differences in the genotype distributions and sex differences, adjusted for race. Genotype distributions were tested as categorical variables. Where genotype results were significant, a dose-response coding (additive allelic) was tested for each allele separately.
Both the pharmacokinetic and the intolerance results were obtained for the same patients at different times on separate regimens, which created pairs of correlated outcomes. We tested whether there was a difference in any outcome between those who had the once-daily regimen first and those who had it last. No differences were statistically significant, so we ignored this factor in further analyses. Linear and logistic mixed-effects models enabled the use of a patient-level random effect to adjust for the correlation between observations of the same patient and also allowed inclusion of observations of patients not completing both regimens. To obtain valid results, some numerical outcomes were transformed (log, square root) toward normality for modeling. All tests were adjusted for confounders, such as regimen, gender, and race, where possible, by including them in the models as fixed effects. The significance of effects (P values) and effect sizes (differences with 95% confidence intervals) are from the results of the confounder-adjusted models.
Software.
The nonlinear mixed-effects modeling of PAS disposition was performed using NONMEM (version VII.2) and NM-TRAN preprocessor with first-order conditional maximum likelihood estimation and η-interaction, running G-fortran 95. The subroutine was ADVAN6 with a tolerance value of 9. The visual predictive check was performed with PerlspeaksNONMEM 3.5.5 running ActivePerl 5.12. The Poisson probability mass function model was coded to run on NONMEM with first-order conditional estimation and Laplacian procedure.
Statistical analyses were done with functions from base R (version 3.1.0) and R packages: genetics for genotype and allelic counts and haplo.stats for inferring and modeling NAT2 haplotypes, lme4 (linear mixed-effects models), and MASS for logistic mixed-effects models.
RESULTS
Thirty-two patients, 18 male (56%), with resistance to at least isoniazid and rifampin and a clinical indication for PAS use were enrolled. Twenty-seven patients (84%) were of mixed race, and 5 (16%) were black. Nine patients (28%) were HIV infected. Patients had a median age of 36 years (range, 18 to 54 years); neither the weight of female patients (median, 49 kg; range, 42 to 86 kg) nor body mass index (median, 19 kg/m2; range, 16 to 25 kg/m2) differed significantly from those of males (median weight, 60 kg; range, 40 to 73 kg; median body mass index, 20 kg/m2; range, 15 to 26 kg/m2, respectively). Serum creatinine values on enrollment (median, 73 μmol/liter; range, 44 to 152 μmol/liter) and glomerular filtration rate (median, 88 ml/min; range, 37 to 206 ml/min) gave no indication of serious renal pathology; the patients' median alanine transaminase value was 16 U/liter (range, 5 to 57 U/liter).
Table 1 provides details of NAT1 and NAT2 alleles and genotypes and inferred haplotypes. The most common NAT1 genotype was *4/*10, found in 14 patients (44%); the putative NAT1 slow-acetylator genotype *14A (21, 22) was present in only 2 (6%) patients; 66% of patients were either heterozygous or homozygous NAT2 rapid acetylators (*4). The NAT2 genotype distributions differed between races (P = 0.046): specifically, black patients had a much higher NAT2*6 frequency (80%) than in the mixed-race patients (26%; P = 0.001), and this was reflected in much higher 1*10/2*6 (60% versus 22%) and 1*4/2*6 (80% versus 15%) haplotype frequencies in black than mixed-race patients.
TABLE 1.
Genotype, allele, and haplotype distributions of NAT1 and NAT2 in patients with tuberculosis randomized in a crossover study to receive either 4 g granular slow-release para-aminosalicylic acid twice daily or 8 g once daily
| Allele, genotype, or haplotype | Count (frequency) by race |
Count (frequency) by gender |
HWEa |
|||||
|---|---|---|---|---|---|---|---|---|
| All | Mixed | Black | P value | Male | Female | P value | P value | |
| NAT1 | ||||||||
| Typed | 32 | 27 | 5 | 18 | 14 | |||
| Allele | 0.92 | |||||||
| 1*4 | 23 (0.71) | 19 (0.70) | 4 (0.80) | 13 (0.72) | 10 (0.71) | |||
| 1*10 | 22 (0.68) | 18 (0.66) | 4 (0.80) | 10 (0.56) | 12 (0.86) | |||
| 1*14A | 2 (0.06) | 2 (0.07) | 2 (0.11) | |||||
| 1*3 | 1 (0.03) | 1 (0.04) | 1 (0.07) | |||||
| Unknown | 1 (0.03) | 1 (0.03) | 1 (0.06) | |||||
| Genotype | 0.770 | 0.172 | ||||||
| 1*4/1*4 | 7 (0.22) | 6 (0.22) | 1 (0.20) | 5 (0.28) | 2 (0.14) | |||
| 1*4/1*10 | 14 (0.44) | 11 (0.41) | 3 (0.60) | 6 (0.33) | 8 (0.57) | |||
| 1*10/1*10 | 7 (0.22) | 6 (0.22) | 1 (0.20) | 4 (0.22) | 3 (0.21) | |||
| 1*4/1*14A | 2 (0.06) | 2 (0.07) | 2 (0.11) | |||||
| 1*10/1*3 | 1 (0.03) | 1 (0.04) | 1 (0.07) | |||||
| NAT2 | ||||||||
| Typed | 32 | 27 | 5 | 18 | 14 | |||
| Allele | 0.76 | |||||||
| 2*4 | 21 (0.66) | 18 (0.67) | 3 (0.60) | 12 (0.67) | 9 (0.64) | |||
| 2*6 | 11 (0.34) | 7 (0.26) | 4 (0.80) | 7 (0.39) | 4 (0.29) | |||
| 2*5 | 12 (0.38) | 10 (0.37) | 2 (0.40) | 7 (0.39) | 5 (0.36) | |||
| 2*3 | 4 (0.13) | 4 (0.15) | 1 (0.06) | 3 (0.21) | ||||
| 2*14 | 2 (0.6) | 2 (0.07) | 2 (0.14) | |||||
| Genotype | 0.046 | 0.169 | ||||||
| 2*4/2*4 | 7 (0.22) | 7 (0.26) | 4 (0.22) | 3 (0.21) | ||||
| 2*4/2*6 | 5 (0.16) | 3 (0.11) | 2 (0.40) | 3 (0.17) | 2 (0.14) | |||
| 2*4/2*5 | 7 (0.22) | 6 (0.22) | 1 (0.2) | 4 (0.22) | 3 (0.21) | |||
| 2*4/2*3 | 1 (0.03) | 1 (0.04) | 1 (0.06) | |||||
| 2*4/2*14 | 1 (0.03) | 1 (0.04) | 1 (0.07) | |||||
| 2*6/2*6 | 3 (0.09) | 2 (0.07) | 1 (0.20) | 3 (0.17) | ||||
| 2*6/2*5 | 1 (0.03) | 1 (0.20) | 1 (0.06) | |||||
| 2*6/2*3 | 3 (0.09) | 3 (0.11) | 3 (0.21) | |||||
| 2*5/2*5 | 3 (0.09) | 3 (0.11) | 2 (0.11) | 1 (0.07) | ||||
| 2*5/2*14 | 1 (0.03) | 1 (0.04) | 1 (0.07) | |||||
| NAT1/NAT2 haplotype | 0.022 | 0.436 | ||||||
| 1*10/2*6 | 9 (0.28) | 6 (0.22) | 3 (0.60) | 5 (0.28) | 4 (0.29) | |||
| 1*4/2*6 | 8 (0.25) | 4 (0.15) | 4 (0.80) | 5 (0.28) | 3 (0.21) | |||
| 1*10/2*14 | 2 (0.06) | 1 (0.04) | 1 (0.20) | 2 (0.14) | ||||
| 1*14A/2*5 | 1 (0.03) | 1 (0.04) | 1 (0.06) | |||||
| 1*14A/2*3 | 1 (0.03) | 1 (0.04) | 1 (0.06) | |||||
| 1*3/2*14 | 1 (0.03) | 1 (0.04) | 1 (0.07) | |||||
| 1*4/2*5 | 7 (0.22) | 6 (0.22) | 1 (0.20) | 4 (0.22) | 3 (0.21) | |||
| 1*10/2*5 | 8 (0.25) | 7 (0.26) | 1 (0.20) | 5 (0.28) | 3 (0.21) | |||
| 1*10/2*3 | 3 (0.09) | 3 (0.11) | 3 (0.21) | |||||
| 1*4/2*4 | 16 (0.50) | 14 (0.52) | 2 (0.20) | 9 (0.50) | 7 (0.50) | |||
| 1*10/2*4 | 13 (0.41) | 11 (0.41) | 2 (0.20) | 5 (0.28) | 8 (0.57) | |||
| 1*4/2*3 | 3 (0.09) | 3 (0.11) | 1 (0.06) | 2 (0.14) | ||||
| 1*4/2*14 | 1 (0.03) | 1 (0.04) | 1 (0.07) | |||||
HWE, Hardy-Weinberg equilibrium.
The distribution of the NAT1 and NAT2 alleles and haplotypes in our mixed-race patients and a small number of black patients is similar to that reported in previous studies of southern African populations (23, 24). The allele frequencies adhere to Hardy-Weinberg equilibrium. The occurrence of different NAT1 alleles and genotypes did not differ between the mixed-race and black patients (P = 0.77). The putative low-activity allele NAT1*14 occurred with a low frequency in our population similar to that reported for other populations (22). However, in comparison to mixed-race patients, the small number of black patients had significantly more slow NAT2 alleles (P = 0.046) and slow genotypes (P = 0.022).
The PAS concentrations and exposure following 4 g twice daily and 8 g once daily are summarized in Table 2. As expected, the once-daily regimen produced higher maximum concentrations (P < 0.001) and areas under the concentration-time curve from 0 to 12 h (AUC0–12) than the twice-daily regimen (P < 0.001); the time to maximum concentration (Tmax) occurred later in the once-daily regimen (P = 0.006). Following the twice-daily regimen, only one patient was exposed on one occasion to values of <1 mg/liter; following the once-daily regimen, however, 16 (52%) patients had a concentration at 0 h (C0) value of <1 mg/liter, and 12 (39%) patients had a C24 value of <1 mg/liter (all at P < 0.001). The percentages of time above MIC over the 24-h interval (%T>MIC) were significantly different between the regimens (P = 0.0003).
TABLE 2.
para-Aminosalicylic acid concentrations and pharmacokinetic characteristics in tuberculosis patients randomized in a crossover study to receive either 4 g granular slow-release para-aminosalicylic acid twice daily or 8 g once daily
| Pharmacokinetic parametera | Result for GSR-PAS dosing regimen |
|||
|---|---|---|---|---|
| 4 g twice daily |
8 g once daily |
|||
| n | Median (range) | n | Median (range) | |
| Cmax (μg/ml) | 30 | 61 (10–112) | 31 | 80 (21–135) |
| AUC0–12 (μg · h/ml) | 20 | 428 (119–934) | 31 | 652 (161–1055) |
| C0 (μg/ml) | 30 | 27.8 (1.5–105.0) | 31 | 1.0 (0.0–28.6) |
| C12 (μg/ml) | 30 | 21.4 (1.7–63.2) | 31 | 59.3 (20.6–121.0) |
| C24 (μg/ml) | 30 | 22.4 (2.4–81.9) | 31 | 1.4 (0.0–53.6) |
| Cmin (μg/ml) | 30 | 17.2 (0.8–63.2) | 31 | 0.9 (0.0–28.6) |
| Tmax (h) | 30 | 4.0 (0.0–12.0) | 31 | 8.0 (3.0–12.1) |
| %T>MIC (%) | 30 | 100 (100–100) | 31 | 94.6 (77.1–100) |
Cmax, C0, C12, C24, Cmin, AUC0–12, and %T>MIC values differed significantly between the regimens (P < 0.001).
There were considerable interindividual variations in PAS pharmacokinetics following both regimens as noted previously (3, 6). For several patients receiving both regimens, concentrations appeared to be still rising at the end of the 12 h of intensive sampling. For this reason, a previous pharmacokinetic model using a 1-compartment disposition model with its absorption described by a 3-transit compartment in series was used to evaluate the effect of polymorphisms in NAT1 and NAT2 on PAS pharmacokinetic (17). Efavirenz interaction as a function of PAS clearance had the greatest difference in the log-likelihood function, followed by NAT1*14, NAT1*3, NAT2*5, and gender. Due to the strong PAS-efavirenz drug-drug interaction, only the forward insertion without backward elimination was carried out to detect all likely effects of polymorphisms. Dosing regimen was not a significant covariate of oral clearance. The model simultaneously described both dosing regimens with a single set of parameters, as listed in Table 3. The corresponding visual predictive check of the final model stratified by dosing regimen is shown in Fig. 1. The structural parameters from this subset of data were very close to those previously reported (17). The final model that included all of the covariates resulted in a significant drop in objective function value of 92.633 U. The exponential coefficients for the covariate effects associated with efavirenz, gender (male), and NAT1*3, NAT1*14, and NAT2*5 alleles were 0.226, 0.315, −0.19, −0.664, and −0.32, which corresponded to 25, 37, −17, −48, and −27% changes in oral clearance of PAS, respectively. It is noted that HIV status is not a significant covariate of PAS clearance, further ruling out that the effect of efavirenz could be confounded by the disease state of the patients.
TABLE 3.
Population pharmacokinetic model parameters of para-aminosalicylic acid with covariate effects due to the NAT1*14, NAT1*3, and NAT2*5 alleles, gender, and concomitant efavirenz
| Parametera | Mean value (RSE %)b in final model |
|---|---|
| Structural model | |
| CL/F, liters/h | 8.14 (11.3) |
| Apparent V/F, liters | 48.9 (6.9) |
| Ktr, h−1 | 0.617 (10.2) |
| No. of transit compartments | 3 |
| Interindividual variability | |
| ωCL/F | 31.4 (36.9) |
| ωV/F | 25.2 (54.4) |
| ωKtr | 52.3 (35.1) |
| Interoccasion variability | |
| πCL/F | 32.2 (49.0) |
| πKtr | 44.7 (42.0) |
| Residual variability | |
| Proportional residual error | 0.142 (29.6) |
| Additive residual error, μg/ml | 10.2 (12.1) |
| Covariatesc | |
| Efavirenz on CL/F | 0.226 (65.5) |
| Gender on CL/F | 0.315 (40.3) |
| NAT1*3 allele on CL/F | −0.19 (56.3) |
| NAT1*14 allele on CL/F | −0.644 (26.2) |
| NAT2*5 allele on CL/F | −0.32 (35.0) |
| ΔOFV (from baseline model) | −92.633 |
Abbreviations: CL/F, oral clearance; V/F, volume of distribution; Ktr, transit rate constant; ωCL/F, interindividual % CV of CL/F; ωV/F, interindividual % CV of V/F; ωKtr, interindividual % CV of Ktr; πCL/F, interoccasion % CV of CL/F; πKtr, interoccasion % CV of Ktr; OFV, minimum objective function value.
RSE, relative standard error.
Covariate (Cov) relationships on CL/F are described by CL/Fi = CL/F × exp(θ1 × Cov1 + … + θj × Covj).
FIG 1.

Visual predictive check showing the 5th, 50th, and 95th percentiles (lines) of the observed para-aminosalicylic acid concentration (dots) and 95% confidence intervals (shaded areas) of the percentiles based on the final population pharmacokinetic model in tuberculosis patients randomized in a crossover study to receive either 4 g granular slow-release para-aminosalicylic acid twice daily (left) or 8 g once daily (right). Horizontal lines indicate a MIC of 1 mg/liter.
To exclude body weight as being a confounding variable of gender, body weight was tested as a covariate of the model and was not a significant covariate of the pharmacokinetic model. The effect of efavirenz resulted in an increase of 25% in oral clearance, whereas all of the slow-acetylator phenotypes with NAT1*3, NAT1*14, and NAT2*5 alleles had a decrease in PAS oral clearance.
To evaluate how these changes affect the exposure of PAS, the observed Cmax, AUC, and %T>MIC were subjected to comparative statistical evaluation. The effect of efavirenz on PAS disposition was previously reported (17) and will not be discussed here. Women had a significantly higher Cmax (difference of 22.4 mg/liter; 95% confidence interval [CI], 7.4 to 22.4 mg/liter; P = 0.003) and AUC0–12 (difference of 216 mg · h/liter; 95% CI, 54 to 378 mg · h/liter; P < 0.007) than men following both regimens (Fig. 2). %T>MIC values were not significantly different between genders.
FIG 2.
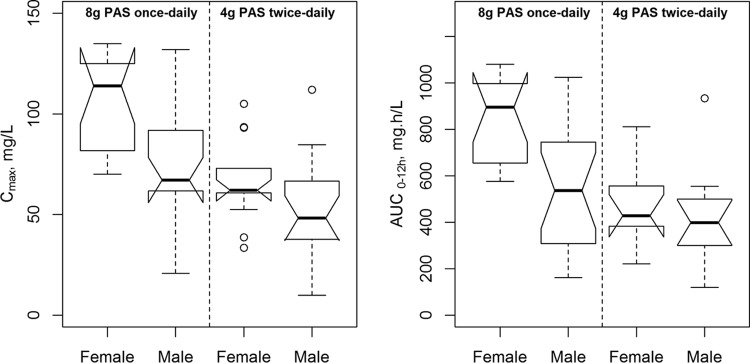
Notched box plots summarizing the maximum para-aminosalicylic acid serum concentrations (left) and the area under the concentration-time curve from 0 to 12 h (right) after dosing in male and female tuberculosis patients receiving either 4 g para-aminosalicylic acid twice daily or 8 g once daily.
The NAT1*14A allele was associated with a significantly higher PAS Cmax (difference of 51.6 mg/liter; 95% CI, 26.9 to 76.2 mg/liter; P < 0.001) and AUC0–12 exposure (difference of 458 mg · h/liter; 95% CI, 179 to 738 mg · h/liter; P = 0.002) irrespective of regimen, and these findings are illustrated in Fig. 3B. The NAT2*5 allele (Fig. 3C) was also associated with an increase in Cmax (difference of 14.3 mg/liter; 95% CI, 4.4 to 24.2 mg/liter; P = 0.006) and AUC0–12 (difference of 149 mg · h/liter; 95% CI, 42 to 257 mg · h/liter; P = 0.008). Individuals with two NAT2*6 alleles had significantly lower Cmax (Fig. 3D; P = 0.034). The NAT1*10/*3 genotype was encountered in only one patient, who was discharged after receiving 8 g once daily but had among the highest Cmax values (125 mg/liter) and the highest AUC0–12 (1,080.8 mg · h/liter); the C24 concentration was 33.2 mg/liter. We found no evidence of association of the NAT1*10 allele with increased enzymatic activity (Fig. 3A); neither Cmax (P = 0.460) nor AUC0–12 (P = 0.514) differed significantly in individuals with this allele. The estimated effect of each NAT1*10 allele was a reduction of 3.9 mg/liter (95% CI, −14.5 to 6.8 mg/liter) in Cmax and a reduction of 37 mg · h/liter (95% CI, −154 to 79 mg · h/liter) in AUC0–12.
FIG 3.
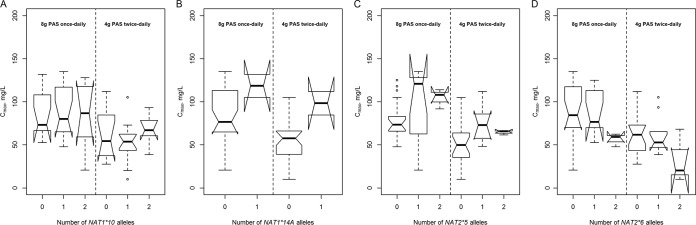
Maximum serum concentrations (Cmax) of para-aminosalicylic acid in patients with tuberculosis receiving either 4 g granular slow-release para-aminosalicylic acid twice daily or 8 g once daily and the influence of the NAT1*10 (A), NAT1*14A (B), NAT2*5 (C), and NAT2*6 (D) alleles during a randomized crossover study.
The occurrence and intensity of intolerance experienced by patients judged by visual analogue scales completed daily over each 8 days of PAS treatment are shown in Table 4. The daily scores by dosing regimens are shown in Fig. 4. Although a majority of patients experienced some form of intolerance on one or more days, the scores for intensity of the relevant symptom out of a possible score of 10 were generally low, and this is reflected in the low median value for the maximum intensity of each form of intolerance recorded over the 8 study days by each patient. The occurrence of symptoms and their intensity did not differ between regimens, with the exception of abdominal pain and cramps, which were more frequent following twice-daily GSR-PAS (P = 0.014). More than half of all scores recorded on each study day were 0, indicating no symptoms at all. Abdominal pain and cramps were experienced at least once—more frequently in men than women (63% versus 23%; P = 0.001)—as were nausea (63% versus 23%; P = 0.018) and diarrhea (86% versus 42%; P = 0.007).
TABLE 4.
Occurrence and severity of symptoms of intolerance assessed daily for 8 days by patients with tuberculosis randomized in a crossover study to receive either 4 g granular slow-release para-aminosalicylic acid twice daily or 8 g once daily in addition to a background regimen of other second-line antituberculosis agents
| Symptom | Result for GRS-PAS dosing regimena |
|||||
|---|---|---|---|---|---|---|
| 4 g twice daily |
8 g once daily |
|||||
| n | No. (%) with occurrence | Severity median (range) | n | No. (%) with occurrence (%) | Severity median (range) | |
| Abdominal pain and cramps | 30 | 18 (60) | 0.9 (0–10.0) | 31 | 10 (32) | 0 (0–8.1) |
| Nausea | 30 | 24 (80) | 1.3 (0–9.3) | 31 | 22 (71) | 1.4 (0–9.4) |
| Vomiting | 30 | 13 (43) | 0 (0–2.0) | 31 | 11 (35) | 0 (0–5.0) |
| Diarrhea | 30 | 21 (70) | 1.4 (0–10.0) | 31 | 21 (68) | 1.8 (0–9.4) |
| Bloated | 30 | 18 (60) | 0.8 (0–8.7) | 31 | 15 (48) | 0 (0–9.4) |
“Occurrence” refers to any reported experience of the symptom by a patient during any one of the 8 days of observation on each regimen. “Severity” (i.e., intensity) refers to the single highest visual analog score out of a possible score of 10 allotted by a patient to a particular symptom on any of the 8 days of observation on each regimen. For vomiting, intensity was counted as the number of times this occurred.
FIG 4.

Daily visual analog scores (VAS) for abdominal pain and cramp, bloating, diarrhea, and nausea over 8 days in patients administered para-aminosalicylic acid in two dosing regimens.
Regarding the interaction of pharmacokinetics and intolerance, neither Cmax nor AUC0–12 was associated with intolerance. There was also little indication of the effects of the various alleles on intolerance: NAT1*14A was associated with the intensity of bloating (P = 0.029), NAT1*3 was associated with the intensity of abdominal pain and cramps (P = 0.004) and diarrhea (P = 0.037), and among the NAT2 alleles, only NAT2*14 was associated with the intensity of abdominal pain and cramps (P = 0.011), nausea (0.036), and diarrhea (P = 0.007). A higher degree of intolerance (abdominal pain and cramps and diarrhea) was associated with lower minimum concentration (Cmin) (P = 0.011). The estimates corresponding to the significant effects were extremely imprecise and are therefore not given.
The observed median frequency of vomiting was 0 for both dosing regimens (Table 4), whereas the mean frequencies were 0.12 and 0.1 day−1 in the 4-g twice-daily and 8-g once-daily regimens, respectively. The difference in frequency was not statistically significant between the two dosing regimens. The histogram of the number of episodes of vomiting by day and by dosing regimen did not show any time effect, with the majority of individuals having no vomiting for all 8 days of observation (data not shown). The appropriateness of the Poisson model was evaluated by visual inspection of the plot of the mean versus the variance of the frequencies, which indicates equidispersion (figure not shown). Table 5 shows the comparison between the Poisson model and the negative binomial model for the base model. The overdispersion parameter (OVDP) associated with the negative binomial was close to 0, indicating equidispersion (25). For these reasons, the vomit frequency data were modeled using Poisson probability mass function. The time effect on the λ parameter, as expected, did not improve the model, as judged by the log-likelihood ratio test. The treatment (or dosing regimen) effect also was not statistically significant, and neither were NAT1 or NAT2 alleles. The final model included the Cmax of PAS as a covariate of the model. Even though the difference in objective function value of 3.466 (P = 0.06) was borderline nonsignificant, we decided to include the Cmax as a covariate in the final model. The exponential covariate relationship was implemented after we realized that the relationship between vomit frequency and Cmax was not linear. The model-predicted baseline frequency was 0.0149 vomits/day (Table 5). The exponential coefficient for the covariate equation associated with the median-centered Cmax was 0.873. The exponentially distributed interindividual variability of the λ parameter was 160% in the CV.
TABLE 5.
Typical value of parameters, interindividual variability, and RSE percentage, comparing Poisson and negative binomial models of the vomit frequency data
| Parametera | Negative binomial estimate | Result by Poisson model |
|||
|---|---|---|---|---|---|
| Base |
Final |
||||
| Estimate | RSEb % | Estimate | RSE % | ||
| λ, day−1 | 0.0404 | 0.0425 | 52 | 0.0149 | 85 |
| OVDP | 0.0606 | ||||
| ω2λ,λ | 2.46 | 2.2 | 45 | 2.55 | 47 |
| % CV | 148 | 160 | |||
| ω2OVDP,OVDP | 10.3 | ||||
| ω2λ,OVDP | 0.135 | ||||
| Covariate Cmax effectc | 0.873 | 57 | |||
| OFV | 290.044 | 302.271 | 298.805 | ||
Abbreviations: OFV, minimum objective function value; OVDP, overdispersion.
RSE, relative standard error.
Covariate relationships on CL/F are described by CL/Fi = CL/F × exp[θ × (Cmax/median)].
The model was qualified by simulating 1,000 uniformly distributed Cmax values ranging from 10 to 135 mg/liter and estimating the distribution of vomiting by using the model parameters. Figure 5 shows that the Poisson model-simulated distribution of the number of daily vomits is similar to the observed distribution.
FIG 5.
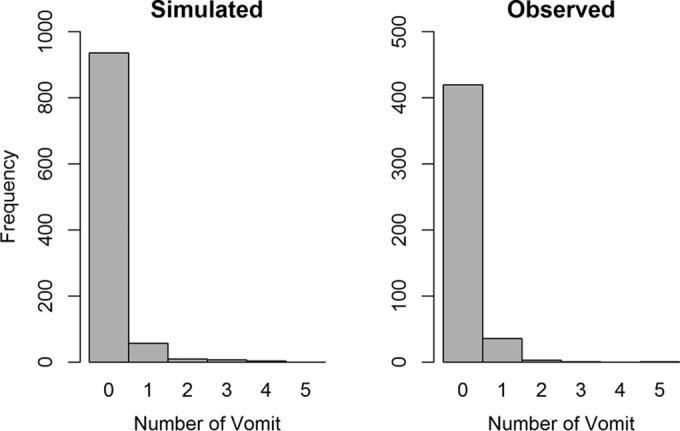
Visual predictive check comparing the Poisson model-simulated distribution of the number of daily vomits (left) and the distribution of the actual number of daily vomits (right) in para-aminosalicylic acid-treated patients.
DISCUSSION
PAS, for many years removed from consideration for tuberculosis treatment, has regained importance for management of extensively drug-resistant tuberculosis and might also be used following toxicity to first- or second-line drugs. Our study for the first time compares PAS concentrations achieved in the same patients following two regimens of GSR-PAS and the influence of NAT1 and NAT2 genetics on PAS concentrations and their influence on tolerability.
Accepting that PAS is a bacteriostatic drug; current recommendations for use in adults suggest 8 to 12 g PAS daily in two to three divided doses (8) and emphasize the maintenance of PAS concentrations above a MIC of approximately 1 mg/liter. Previous studies of GSR-PAS have found that 4-g doses twice daily were sufficient to maintain PAS concentrations above the MIC during a dosing interval but that a once-daily 4-g dose is insufficient for this purpose (3, 4). Our study found that although a minority of patients maintain concentrations above 1 mg/liter following once-daily dosing with 8 g GSR-PAS, half of the patients studied had C0 values of <1 mg/liter with a median %T>MIC of 94.6%. As we did not determine PAS concentrations for longer than 12 h after dosing, we cannot say with certainty at what point maximum concentrations were reached and when some concentrations fell below 1 mg/liter following the 8-g once-daily dosing regimen: although Fig. 1 suggests concentrations might have remained >1 mg/liter for the greater part of the dosing interval, PAS concentrations can fall rapidly once Cmax has passed (3). Whether slow release of PAS by the GSR-PAS formulation changes this paradigm at higher doses is uncertain.
Our analysis of NAT1 genetics and their relationship to PAS concentrations confirmed the loss of activity associated with the NAT1*14 allele; however, we found no evidence that NAT1*10 is associated with increased activity (26). Interestingly the single patient with a NAT1*10/1*3 genotype had particularly high PAS Cmax and AUC0–12, similar to two patients with a NAT1*14 allele, suggesting reduced enzyme function. A previous in vitro study among a Han Chinese population using para-aminobenzoic acid also found NAT1*3 associated with reduced enzyme activity (27). The influence of NAT2 genes on PAS metabolism found in our study is intriguing and reflects similar interactions between the NAT1 and NAT2 genes regarding aspects of carcinogenesis (28).
One of our most interesting findings was the considerably higher PAS exposure in female patients compared to males. The weights of female patients did not differ from those of males, and our analysis taking weight into account found the difference in weight to be an insufficient explanation for the greater PAS exposure or lower PAS clearance in females. Two factors may offer an explanation. First it has been shown that NAT1 activity is androgen induced, and this may constitute a risk factor for cancer induction (29). Second it is known that NAT1 is widely expressed in muscle tissue, and males might have a greater muscle bulk than females (26). Early studies of the efficacy of regimens of PAS and streptomycin and PAS, streptomycin, and isoniazid did report better efficacy in females than males, but this was not statistically significant (30).
Regarding tolerance, several early studies of PAS pharmacokinetics found single daily doses better tolerated than divided doses (31, 32). Our study found little difference between once-daily and divided dosing but if anything suggests once-daily dosing was associated with less intolerance than twice-daily dosing. Regarding the influence of other aspects of pharmacokinetics, Cmin was associated with both abdominal pain and discomfort and diarrhea. Perhaps the more rapid absorption of PAS and its metabolism to a product such as acetyl-PAS might be responsible for increased abdominal pain. A previous study of GSR-PAS found it “well tolerated,” and our experience supports this opinion (3). Overall, although a majority of patients experienced one or more episodes of intolerance during 16 days of observation, the intensity of discomfort recorded was low, and the majority of responses were zero.
It should also be noted that other early studies also reported not only better tolerance of once-daily PAS doses but also improved efficacy (33). In early studies by the British Medical Research Council from 1952 (34), it was also shown that when administered with another relatively weak drug, such as streptomycin, prevention of resistance was considerably better with a daily dosage of 20 g in divided doses than after 10 g in divided doses, implying an advantage in reaching higher concentrations. John Crofton in a 1959 review paper emphasized that in the presence of any weakness in a regimen, a PAS dosage of 20 g daily should be preferred (2). Early laboratory studies (35, 36) and more recent studies have also demonstrated that PAS resistance occurs over a range of concentrations, and increasing the peak concentrations of PAS may well inhibit some of these bacilli from exceeding the mutant prevention concentration for PAS (10). Finally, although usually considered bacteriostatic, when evaluated in a study of early bactericidal activity in pulmonary tuberculosis patients, a single daily dosage of 15 g PAS had activity close to that of rifampin during the first 2 days of therapy (37).
The limitations of our study include the relatively low numbers of patients studied, especially the number of individuals with the slow genotypes NAT1*3 and -*14, and the fact that we did not determine concentrations of acetyl-PAS or other metabolites. A control group receiving second-line agents without PAS might clarify how much of the relatively low level of intolerance documented was in fact due to GSR-PAS and not accompanying second-line agents, such as ethionamide.
In conclusion, we show that when the total daily dose is the same in both regimens, the twice-daily 4-g GSR-PAS regimen but not the 8-g once-daily GSR-PAS regimen will ensure concentrations exceeding the PAS MIC of 1 mg/liter throughout dosing intervals, and PAS intolerance is not associated with serum PAS concentrations over the doses studied. We observed the slow phenotype of NAT1*14 and NAT1*3 alleles was associated with greater PAS exposure. However, the broader generalization may be limited, due to the small number of individuals belonging to these genotypes. No evidence of increased activity of the NAT1*10 allele was found in this study. Our literature review suggests that GSR-PAS once daily be explored as possibly being more efficacious in preventing resistance in companion drugs than intermittent dosing without necessarily causing greater intolerance.
ACKNOWLEDGMENT
P.R.D., A.H.D., and L.D.K. are supported by the South African National Research Foundation.
REFERENCES
- 1.Lehmann J. 1946. Para-aminosalicylic acid in the treatment of tuberculosis. Lancet i:15. [DOI] [PubMed] [Google Scholar]
- 2.Crofton J. 1959. Chemotherapy of pulmonary tuberculosis. Br Med J 1:1610–1614. doi: 10.1136/bmj.1.5138.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peloquin CA, Berning SE, Huitt GA, Childs JM, Singleton MD, James GT. 1999. Once-daily and twice-daily dosing of p-aminosalicylic acid granules. Am J Respir Crit Care Med 159:932–934. doi: 10.1164/ajrccm.159.3.9807131. [DOI] [PubMed] [Google Scholar]
- 4.Peloquin CA, Henshaw TL, Huitt GA, Berning SE, Nitta AT, James GT. 1994. Pharmacokinetic evaluation of para-aminosalicylic acid granules. Pharmacotherapy 14:40–46. [DOI] [PubMed] [Google Scholar]
- 5.Peloquin CA, Zhu M, Adam RD, Singleton MD, Nix DE. 2001. Pharmacokinetics of para-aminosalicylic acid granules under four dosing conditions. Ann Pharmacother 35:1332–1338. doi: 10.1345/aph.1A088. [DOI] [PubMed] [Google Scholar]
- 6.Liwa AC, Schaaf HS, Rosenkranz B, Seifart HI, Diacon AH, Donald PR. 2013. Para-aminosalicylic acid plasma concentrations in children in comparison with adults after receiving a granular slow-release preparation. J Trop Pediatr 59:90–94. doi: 10.1093/tropej/fms053. [DOI] [PubMed] [Google Scholar]
- 7.Kibleur Y, Brochart H, Schaaf HS, Diacon AH, Donald PR. 2014. Dose regimen of para-aminosalicylic acid gastro-resistant formulation (PAS-GR) in multidrug-resistant tuberculosis. Clin Drug Invest 34:269–276. doi: 10.1007/s40261-014-0172-7. [DOI] [PubMed] [Google Scholar]
- 8.World Health Organization. 20008. Guidelines for the programmatic management of drug-resistant tuberculosis. Emergency update 2008. WHO/HTM/TB/2008.402. World Health Organization, Geneva, Switzerland: http://www.who.int/tb/challenges/mdr/programmatic_guidelines_for_mdrtb/en/. [Google Scholar]
- 9.Heifets LB. 1991. Antituberculosis drugs: antimicrobial activity in vitro, p 13–58. In Heifets LB. (ed), Drug susceptibility in the chemotherapy of mycobacterial infections. CRC Press, Boca Raton, FL. [Google Scholar]
- 10.Mathys V, Wintjens R, Lefevre P, Bertout J, Singhal A, Kiass M, Kurepina N, Wang XM, Mathema B, Baulard A, Kreiswirth BN, Bifani P. 2009. Molecular genetics of para-aminosalicylic acid resistance in clinical isolates and spontaneous mutants of Mycobacterium tuberculosis. Antimicrob Agents Chemother 53:2100–2109. doi: 10.1128/AAC.01197-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sy SK, Derendorf H. 2014. Pharmacometrics in bacterial infections, p 229–258. In Schmidt S, Derendorf H (ed), Applied pharmacometrics. Springer, New York, NY. [Google Scholar]
- 12.Lehmann J. 1969. The role of the metabolism of p-aminosalicylic acid (PAS) in the treatment of tuberculosis. Interaction with the metabolism of isonicotinic acid hydrazide (INH) and the synthesis of cholesterol. Scand J Respir Dis 50:169–185. [PubMed] [Google Scholar]
- 13.Motulsky AG, Steinmann L. 1962. Aryl amine acetylation in human red cells. J Clin Invest 41:1387. [Google Scholar]
- 14.Vatsis KP, Weber WW. 1993. Structural heterogeneity of Caucasian N-acetyltransferase at the NAT1 gene locus. Arch Biochem Biophys 301:71–76. doi: 10.1006/abbi.1993.1116. [DOI] [PubMed] [Google Scholar]
- 15.Grant D, Vohra P, Avis Y, Ima A. 1992. Detection of a new polymorphism of human arylamine N-acetyltransferase NAT1 using p-aminosalicylic acid as an in vivo probe. J Basic Clin Physiol Pharmacol 3:244. [Google Scholar]
- 16.Hughes NC, Janezic SA, McQueen KL, Jewett MA, Castranio T, Bell DA, Grant DM. 1998. Identification and characterization of variant alleles of human acetyltransferase NAT1 with defective function using p-aminosalicylate as an in-vivo and in-vitro probe. Pharmacogenetics 8:55–66. doi: 10.1097/00008571-199802000-00008. [DOI] [PubMed] [Google Scholar]
- 17.de Kock L, Sy SK, Rosenkranz B, Diacon AH, Prescott K, Hernandez KR, Yu M, Derendorf H, Donald PR. 2014. Pharmacokinetics of para-aminosalicylic acid in HIV-uninfected and HIV-coinfected tuberculosis patients receiving antiretroviral therapy, managed for multidrug-resistant and extensively drug-resistant tuberculosis. Antimicrob Agents Chemother 58:6242–6250. doi: 10.1128/AAC.03073-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schaaf HS, Parkin DP, Seifart HI, Werely CJ, Hesseling PB, van Helden PD, Maritz JS, Donald PR. 2005. Isoniazid pharmacokinetics in children treated for respiratory tuberculosis. Arch Dis Child 90:614–618. doi: 10.1136/adc.2004.052175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plan EL. 2014. Modeling and simulation of count data. CPT Pharmacometrics Syst Pharmacol 3:e129. doi: 10.1038/psp.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plan EL, Ma G, Nagard M, Jensen J, Karlsson MO. 2011. Transient lower esophageal sphincter relaxation pharmacokinetic-pharmacodynamic modeling: count model and repeated time-to-event model. J Pharmacol Exp Ther 339:878–885. doi: 10.1124/jpet.111.181636. [DOI] [PubMed] [Google Scholar]
- 21.Butcher NJ, Boukouvala S, Sim E, Minchin RF. 2002. Pharmacogenetics of the arylamine N-acetyltransferases. Pharmacogenomics J 2:30–42. doi: 10.1038/sj.tpj.6500053. [DOI] [PubMed] [Google Scholar]
- 22.Butcher NJ, Ilett KF, Minchin RF. 1998. Functional polymorphism of the human arylamine N-acetyltransferase type 1 gene caused by C190T and G560A mutations. Pharmacogenetics 8:67–72. doi: 10.1097/00008571-199802000-00009. [DOI] [PubMed] [Google Scholar]
- 23.Loktionov A, Moore W, Spencer SP, Vorster H, Nell T, O'Neill IK, Bingham SA, Cummings JH. 2002. Differences in N-acetylation genotypes between Caucasians and Black South Africans: implications for cancer prevention. Cancer Detect Prev 26:15–22. doi: 10.1016/S0361-090X(02)00010-7. [DOI] [PubMed] [Google Scholar]
- 24.Parkin DP, Vandenplas S, Botha FJ, Vandenplas ML, Seifart HI, van Helden PD, van der Walt BJ, Donald PR, van Jaarsveld PP. 1997. Trimodality of isoniazid elimination: phenotype and genotype in patients with tuberculosis. Am J Respir Crit Care Med 155:1717–1722. doi: 10.1164/ajrccm.155.5.9154882. [DOI] [PubMed] [Google Scholar]
- 25.Ahn JE, Plan EL, Karlsson MO, Miller R. 2012. Modeling longitudinal daily seizure frequency data from pregabalin add-on treatment. J Clin Pharmacol 52:880–892. doi: 10.1177/0091270011407193. [DOI] [PubMed] [Google Scholar]
- 26.Minchin RF, Hanna PE, Dupret JM, Wagner CR, Rodrigues-Lima F, Butcher NJ. 2007. Arylamine N-acetyltransferase I. Int J Biochem Cell Biol 39:1999–2005. doi: 10.1016/j.biocel.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 27.Zhangwei X, Jianming X, Qiao M, Xinhua X. 2006. N-Acetyltransferase-1 gene polymorphisms and correlation between genotype and its activity in a central Chinese Han population. Clin Chim Acta 371:85–91. doi: 10.1016/j.cca.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 28.Cascorbi I, Roots I, Brockmoller J. 2001. Association of NAT1 and NAT2 polymorphisms to urinary bladder cancer: significantly reduced risk in subjects with NAT1*10. Cancer Res 61:5051–5056. [PubMed] [Google Scholar]
- 29.Butcher NJ, Tetlow NL, Cheung C, Broadhurst GM, Minchin RF. 2007. Induction of human arylamine N-acetyltransferase type I by androgens in human prostate cancer cells. Cancer Res 67:85–92. doi: 10.1158/0008-5472.CAN-06-2635. [DOI] [PubMed] [Google Scholar]
- 30.Fox W, Sutherland I. 1956. A five-year assessment of patients in a controlled trial of streptomycin, para-aminosalicylic acid, and streptomycin plus para-aminosalicylic acid, in pulmonary tuberculosis. Q J Med 25:221–243. [PubMed] [Google Scholar]
- 31.Bang HO, Strandgaard E. 1960. Continued studies on the problems of PAS dosage. Acta Tuberc Scand 39:81–96. [PubMed] [Google Scholar]
- 32.Riska N, Tennberg C. 1962. Optimal PAS dosage. Am Rev Respir Dis 86:430–433. [DOI] [PubMed] [Google Scholar]
- 33.Bridge EV, Carr DT. 1958. Clinical use of a single daily dose of para-aminosalicylic acid in association with isoniazid. Am Rev Tuberc 78:749–752. [DOI] [PubMed] [Google Scholar]
- 34.Medical Research Council Investigation. 1952. Prevention of streptomycin resistance by combined chemotherapy. Br Med J 1:1157–1162. doi: 10.1136/bmj.1.4769.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsukamura M, Noda Y, Yamamoto M, Hayashi M. 1959. Genetic considerations of the mechanisms involved in PAS-resistant tubercle bacilli. Am Rev Tuberc 79:371–373. [DOI] [PubMed] [Google Scholar]
- 36.Singh B, Mitchison DA. 1955. Bactericidal activity of streptomycin and isoniazid in combination with p-aminosalicylic acid against Mycobacterium tuberculosis. J Gen Microbiol 12:76–84. doi: 10.1099/00221287-12-1-76. [DOI] [PubMed] [Google Scholar]
- 37.Jindani A, Aber VR, Edwards EA, Mitchison DA. 1980. The early bactericidal activity of drugs in patients with pulmonary tuberculosis. Am Rev Respir Dis 121:939–949. [DOI] [PubMed] [Google Scholar]