

Abstract
The emergence of influenza virus strains resistant to approved neuraminidase inhibitors and the time constrains after infection when these drugs can be effective constitute major drawbacks for this class of drugs. This highlights a critical need to discover new therapeutic agents that can be used for the treatment of influenza virus-infected patients. The use of broadly neutralizing anti-influenza monoclonal antibodies (MAbs) has been sought as an alternative immunotherapy against influenza infection. Here, we tested in mice previously characterized broadly neutralizing anti-hemagglutinin (HA) stalk MAbs prophylactically and therapeutically using different routes of administration. The efficacy of treatment against an influenza H1N1 pandemic virus challenge was compared between two systemic routes of administration, intraperitoneal (i.p.) and intravenous (i.v.), and two local routes, intranasal (i.n.) and aerosol (a.e.). The dose of MAb required for prophylactic protection was reduced by 10-fold in animals treated locally (i.n. or a.e.) compared with those treated systemically (i.p. or i.v.). Improved therapeutic protection was observed in animals treated i.n. on day 5 postinfection (60% survival) compared with those treated via the i.p. route (20% survival). An increase in therapeutic efficacy against other influenza virus subtypes (H5N1) was also observed when a local route of administration was used. Our findings demonstrate that local administration significantly decreases the amount of broadly neutralizing monoclonal antibody required for protection against influenza, which highlights the potential use of MAbs as a therapeutic agent for influenza-associated disease.
INTRODUCTION
Influenza virus is a highly infectious respiratory pathogen that remains a significant threat to public health. Historically, passive transfer of convalescent human sera has been a viable option as a functional therapy in situations of crisis (1, 2). According to reports, passive transfer techniques were implemented for influenza virus infection from as early as the 1918 pandemic to as recently as the H1N1 pandemic and exhibited good results (3–5). Immunotherapy with monoclonal antibodies (MAbs) is the only approved treatment for prophylactic use in children at risk of respiratory syncytial virus infection (6). Production of neutralizing antibodies generated through vaccination or viral infection is generally correlated with protection. Broadly neutralizing antibodies purified from patients, produced by immunization of mice, or recombinantly expressed in mammalian culture have been tested both prophylactically and therapeutically in animal models of influenza virus infection (7, 8). Monoclonal antibody therapies are currently in clinical trials to target influenza virus infection (https://clinicaltrials.gov/ct2/results?term=influenza+monoclonal+antibody&Search=Search). Most anti-influenza virus MAbs tested in animal studies are given using systemic routes, such as the intraperitoneal (i.p.) or intravenous (i.v.) route. In these studies, the amount of antibody required to protect against lethal challenge is usually quite high (9–12). Currently, both the manufacturing process and the amount of antibody needed for protection make monoclonal antibody therapy very expensive and unjustifiable for large-scale implementation.
During an influenza virus infection in mammals, the virus usually targets epithelial cells of the upper and lower respiratory tracts (13). Therefore, local administration of neutralizing monoclonal antibodies to the target tissue region may be a clinically relevant approach. Thus, we compared the efficacy of broadly neutralizing anti-hemagglutinin (HA) stalk antibodies to prevent or rescue influenza-challenged mice from clinical disease when administered systemically (intraperitoneal or intravenous route) or locally (intranasal [i.n.] route via droplets or by aerosol [a.e.]). Local administration of the monoclonal antibodies reduced the dose required for protection and improved survival in mice treated therapeutically.
MATERIALS AND METHODS
Animals.
All research studies involving the use of animals were reviewed and approved by the Institutional Animal Care and Use Committees (IACUC) at the Icahn School of Medicine at Mount Sinai. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Research Council (8th ed).
Female BALB/c mice (6 to 8 weeks old) purchased from The Jackson Laboratory (Bar Harbor, ME) were used for all experiments. For virus challenges, mice were anesthetized by intraperitoneal injection of a mixture of ketamine (100 mg/kg of body weight) and xylazine (5 mg/kg) before intranasal instillation of 5 mouse 50% lethal dose (mLD50) in a volume of 35 μl. The animals were monitored daily for clinical signs of illness, and body weights were recorded daily for 14 days. Upon reaching >75% of initial body weight, animals were humanely euthanized.
Antibodies and viruses.
The mouse monoclonal antibodies 6F12, GG3, and KB2 used in these experiments were previously characterized as broadly neutralizing anti-HA stalk-specific monoclonal antibodies (9–11). Influenza viruses A/Puerto Rico/8/1934 H1N1 (PR/8), A/Netherlands/602/2009 H1N1pdm (NL/09), and A/Vietnam/1203/2004 H5N1 (VN/04) with the polybasic cleavage site deleted (HALo) were used at 5 mLD50 to challenge mice. For the particular batches of viruses used in these experiments the mLD50s were as follows: PR/8, 50 PFU; NL/09, 80 PFU; and VN/04 HALo, 300 PFU.
Antibody bioavailability.
To determine the half-life of the MAb 6F12 in serum, mice were treated with 3 mg/kg of MAb 6F12 via the i.v. route, and blood samples were collected every other day for 21 days. Additionally, the antibody presence was measured in bronchoalveolar lavage (BAL) fluid or serum samples obtained from mice that received 3 mg/kg of MAb 6F12 via the i.p., i.n., or a.e. route. Terminal samples were collected at 24 or 72 h posttreatment with the MAbs. The concentrations of antibodies in BAL fluid and serum samples were evaluated by enzyme-linked immunosorbent assays (ELISAs) with the use of purified preparations of recombinant hemagglutinin of A/California/04/2009 at 2.5 μg/ml (BEI Resources catalog no. NR-13691) as the coating antigen (9). Convalescent-phase serum samples from mice infected with NL/09 were used as positive controls, and samples from mice that received phosphate-buffered saline (PBS) alone served as negative controls. A standard curve from a purified preparation of MAb 6F12 was used to generate a nonlinear regression curve with GraphPad Prism 6.0 (GraphPad Software, San Diego, CA), and then the amount of antibodies in each sample was calculated.
Antibody distribution.
MAb 6F12 was labeled with XenoFluor750 using the fluorescent dye kit for in vivo imaging (PerkinElmer, Waltham, MA) following the conditions recommended by the manufacturer. A dose of 3 mg/kg of labeled MAb 6F12 was delivered systemically (i.p.) or locally (i.n. or a.e.). Levels of fluorescently labeled antibody were determined daily for 3 days. On day 4, mice were euthanized, and their lungs were also assessed for a fluorescence signal. Mice were imaged for 45 s in an IVIS Spectrum in vivo imaging system (PerkinElmer), and the images were processed with the system software using the same threshold applied to images across experimental groups.
Treatment regimens.
Monoclonal antibodies were diluted to the appropriate concentration in PBS and administered via i.p. or i.v. injection for systemic delivery or via i.n. instillation (under ketamine-xylazine anesthesia) or a.e. for local administration. We administered the MAbs in a prophylactic or a therapeutic approach using different doses of the antibody in a single application. To deliver MAbs via the a.e. route, mice were awake; therefore, they were trained for several sessions (using sterile PBS) to get them used to the procedure at least 1 week in advance. We used a nose-only aerosol delivery system (inExpose system; SCIREQ, Montreal, QC, Canada) with an Aerogen Lab nebulizer (Aerogen, Galway, Ireland) that delivers an average particle size of <3 μm. Briefly, we first determined the aerosolized drug delivery (ADD) value using the volume of nebulized drug (milliliters per liter) and multiplied it by the concentration of the drug solution (milligrams per milliliter). The amount of MAb delivered per mouse per minute (milligrams per minute per mouse) was calculated by multiplying the ADD value (milligrams per liter) by the minute volume of the mouse (liters per minute). We used this value to determine how long the mice needed to be in the aerosolization equipment based on the target dose (i.e., 3 or 15 mg/kg). An isotype of the immunoglobulin subtype G (IgG) antibody was used as a negative control for all the experiments (9). The readouts to determine protection were body weight loss and survival.
Lung viral titers.
Mice received MAb 6F12 via the i.p., i.n., or a.e. route prophylactically (3 mg/kg 2 h prior to infection with 3 mLD50 of NL/09 virus) or via the i.p. or i.n. route therapeutically (15 mg/kg 24 h after infection). The control groups received 15 mg/kg of the isotype IgG via the i.n. route 2 h before infection. On days 3 and 6 postinfection, groups of mice were euthanized, and the lungs were collected and homogenized (BeadBlaster 24; Benchmark Scientific) in 1 ml of sterile PBS. The lung homogenates were spun at 16,000 × g for 10 min to pellet tissue debris, and the supernatants were collected. Samples were stored at −80°C until titration by the standard plaque assay on Madin-Darby canine kidney cells. GraphPad Prism was used to calculate the statistical significance between groups using a two-way analysis of variance. A P value of <0.05 was considered statistically significant.
Histopathology.
Mice were administered 3 mg/kg of MAb 6F12 via the i.p., i.n., or a.e. route and challenged 2 h posttreatment with 5 mLD50 of NL/09 virus. Two days postinfection, mice were euthanized, and their lungs were perfused in situ with 1 ml of 10% buffered formalin. For the therapeutic analysis, mice were infected with 5 mLD50 of NL/09 virus and treated with 15 mg/kg of MAb (or PBS for the control group) 3 days postinfection. On day 4 (i.n. and i.p.) or day 5 (i.n., i.p., and PBS) postinfection, mice were euthanized, and their lungs were perfused and collected. Lung sections were stained with hematoxylin-eosin and evaluated by a board-certified veterinary pathologist from the Center of Comparative Medicine and Surgery in the Icahn School of Medicine at Mount Sinai. The following score system was used to analyze the histopathology of the lung sections (on a scale of 0 to 4): 0, no lesions; 1, mild changes with scattered cell necrosis/vacuolation and few/scattered inflammatory cells in the bronchiolar epithelium with minimal perivascular inflammation; 2, moderate multifocal vacuolation with peribronchiolar inflammation (<5 cell layers thick); 3, marked, multifocal/segmental necrosis, epithelial loss/effacement, and peribronchiolar inflammation (>5 cell layers thick); and 4, severe, coalescing areas of necrosis, parenchymal effacement with confluent areas of inflammation. The overall pathology scores per group were used for the statistical analysis of the results.
Statistical analysis.
Statistical differences were analyzed using Student's t test followed by the Holm-Sidak test. For the analysis of the survival rates, Kaplan-Meier curves were compared using the log rank (Mantel-Cox) test. All the analyses were performed using GraphPad software, and P values of <0.05 were considered significant.
RESULTS
In vivo bioavailability and distribution of administered MAb 6F12.
Influenza virus replication is typically limited to the respiratory tract; therefore, the amount of systemically administered antibody reaching the site of infection is critically important for achieving protection. To determine bioavailability, we first calculated the serum half-life in mice that received 3 mg/kg of MAb 6F12 (60 μg) via an intravenous bolus injection. We used MAb 6F12 due to its cross-reactivity within the H1 subtype (9, 10). We detected the antibody in sera up to 19 days posttreatment by ELISA, with a calculated half-life of 5.5 days (data not shown). We then quantified the amount of MAb 6F12 in bronchoalveolar lavage (BAL) fluids and sera of mice that received 3 mg/kg of MAb 6F12 via the i.p. (systemic), i.n. (local), or a.e. (local) route of administration. The amounts of antibody in BAL fluids were significantly higher (P < 0.05) in mice that received the MAb via the i.n. route than for the a.e. or i.p. route of administration at 24 h (Fig. 1A). In contrast, the amount of antibody in serum samples was significantly higher (P < 0.05) in mice that received the MAb via the i.p. route at 24 h (Fig. 1B). Very small amounts of MAb 6F12 were observed in BAL fluid or serum samples of mice that received the antibody via the a.e. route. These data suggest that local administration via the i.n. route delivers a higher MAb dose at the site of infection.
FIG 1.

Distribution of monoclonal antibody 6F12 in mice. (A) Concentration of MAb 6F12 in bronchoalveolar lavage (BAL) fluid of mice that received 3 mg/kg of the antibody via a.e., i.n., or i.p. administration. A group of mice received phosphate-buffered saline (PBS) via intranasal administration as a negative control. (B) Serum concentration of MAb 6F12 after administration of 3 mg/kg using the same routes as in panel A. Values represent means ± SEM (n = 3 mice per group). Significant differences are shown: *, P < 0.05. (C) Antibody detection using a fluorescently labeled MAb. Mice received labeled MAb 6F12 via aerosol, intranasal, or intraperitoneal administration, and live images of whole mice were collected at 24 and 72 h or of only lungs at 96 h posttreatment. Mice and lungs on the left of each picture only received PBS.
To evaluate overall antibody distribution, we administered 3 mg/kg of fluorescently labeled MAb 6F12 via different routes and measured the fluorescence signal over time using the IVIS Spectrum live imager. The fluorescence signal for mice that received the antibody via the a.e. route was observed only in the upper respiratory tract, while fluorescent antibody administered i.n. was well distributed in both the upper (data not shown) and lower respiratory tracts (Fig. 1C). When the antibody was administered via i.p. injection, distribution of the signal was observed throughout the body (Fig. 1C). On day 4 postadministration, lungs were harvested and the lung-specific fluorescence signal was also measured. Interestingly, in mice that received the MAb via the a.e. route, no fluorescent signal was detected (Fig. 1C). A strong signal was observed in the lungs of mice that received the antibody via the i.n. or i.p. route. Of note, mice were not flushed to get rid of the circulating blood before lungs were collected; therefore, the signal observed in the lungs of mice that received the labeled antibody via the i.p. route is coming partially from blood in the lung circulation.
Temporal bioavailability of locally administered MAb affords better protection.
To determine the relationship between the route of administration and pretreatment efficacy with MAb 6F12, we performed the following experiment. Mice were initially treated with 3 mg/kg of MAb 6F12 via the i.p. or i.n. route and then were infected with 5 mLD50 of NL/09 virus on day 2, 3, or 5 after treatment. When infection occurred on day 2, mice that received the MAb via the i.n. route did not lose weight, whereas mice that received it via the i.p. route lost almost 20% of their initial body weight (Fig. 2A). All mice treated with MAb 6F12 survived, irrespective of the route of administration. Mice that were infected on day 3 after i.n. administration of the MAb survived lethal challenge without transient weight loss, while only 60% of the mice that received the antibody via the i.p. route survived (Fig. 2B). All mice infected 5 days after i.n. MAb administration survived with a transient weight loss of only 5% of their initial body weight, while only 40% of the mice treated via the i.p. route survived and they had greater weight loss (Fig. 2C). This difference was statistically significant (P < 0.05).
FIG 2.

Window of opportunity for protection after administration of a broadly neutralizing antibody. Body weight and survival curves of mice that received 3 mg/kg of MAb 6F12 via the i.n. or i.p. route of administration and then were infected with influenza A/Netherlands/604/2009 H1N1pdm virus at 48 (A and B), 74 (C and D), or 120 (E and F) hours after MAb treatment. As a negative control, an irrelevant isotype IgG was used at 3 mg/kg and administered via the i.n. route 48 or 120 h before infection. Body weight curve values represent means ± SEM (n = 5 mice per group). Significant differences between the i.n. and i.p. routes are shown: *, P < 0.05.
Local administration is superior to systemic administration in a prophylaxis regimen.
One potential use of anti-influenza MAb therapy is the prophylactic administration of antibody to people who will be in contact with an infected individual to prevent infection. To evaluate the relationship between the routes of administration and prophylactic efficacy, groups of mice were treated with 3, 1, or 0.3 mg/kg of MAb 6F12 systemically (i.v. and i.p.) or locally (i.n. and a.e.) 2 h prior to infection with 5 mLD50 of NL/09 virus. All mice that received a dose of 3 mg/kg survived the virus challenge with no weight loss (Fig. 3A and B). When the dose was reduced to 1 mg/kg, all mice that received treatment via the i.n. or a.e. route of administration survived without transient weight loss. In contrast, 80% of mice administered the MAb via the i.v. or i.p. route succumbed to infection (Fig. 3C and D). Finally, mice that received 0.3 mg/kg of MAb 6F12, through either local administration route (i.n. or a.e.), were fully protected from lethal challenge and demonstrated no transient weight loss, whereas only 20% of mice that received i.v. or i.p. treatment survived (Fig. 3E and F). Overall, these results indicate that local administration of MAb 6F12 via the i.n. or a.e. route significantly (P < 0.05) reduces the amount of antibody required for protection in a prophylactic setting and is superior to systemic administration.
FIG 3.

Body weights and survival in mice treated with MAb 6F12 in a prophylactic experiment. Body weight and survival curves of mice treated with 3 mg/kg (A and B), 1 mg/kg (C and D), or 0.3 mg/kg (E and F) of MAb 6F12 via the a.e., i.n., i.p., or i.v. route before infection with 5 mLD50 of influenza A/Netherlands/604/2009 H1N1pdm virus. As a negative control, an isotype IgG was used at 3 mg/kg and administered via the i.n. route 2 h before infection (A and B). The same controls as in Fig. 2C to F were included as references for comparison. Body weight curve values represent means ± SEM (n = 5 mice per group). Significant differences between the i.n. and i.p. routes are shown: *, P < 0.05.
Local administration affords better protection against influenza disease in a therapeutic regimen.
After observing enhanced efficacy of MAb 6F12 in a prophylactic setting through i.n. or a.e. administration, a therapeutic regimen was tested to simulate a clinical scenario observed during an influenza virus infection. Mice were initially infected with 5 mLD50 of NL/09 virus and on day 3, 4, or 5 postinfection received 15 mg/kg of MAb 6F12 via either the i.p. or i.n. route. A control group received 15 mg/kg of an IgG isotype via the i.n. route 3 days postinfection. All mice treated 3 days postinfection exhibited similar transient weight losses and survived regardless of administration route (Fig. 4A and B). Mice treated 4 days postinfection exhibited similar transient body weight loss in both groups (Fig. 4C). All mice that received treatment via the i.p. route survived, while mice that received treatment via the i.n. route had a 90% survival (Fig. 4D), a difference that is not statistically significant. Significant differences, however, were observed when mice received treatment on day 5 postinfection where the i.n. route group had better body weight recovery by day 14 postinfection than the i.p. group (P < 0.05 on days 13 and 14 postinfection) (Fig. 4E). In addition, the group that received treatment via the i.n. route had a 60% survival rate, while only 20% of the mice treated via the i.p. route survived (Fig. 4F) although these differences were not statistically significant (P = 0.1). These data demonstrate the enhanced efficacy of locally administered MAb 6F12 when it is given therapeutically.
FIG 4.

Body weight and survival curves of mice treated with MAb 6F12 in a therapeutic experiment. Mice received 15 mg/kg of MAb 6F12 via the i.n. or i.p. route on day 3 (A and B), 4 (C and D), or 5(E and F) postinfection with 5 mLD50 of the influenza virus A/Netherlands/604/2009 H1N1pdm. As a negative control, an isotype IgG was used at 15 mg/kg and administered via the i.n. route on day 3 postinfection (A and B). Body weight curve values represent means ± SEM (n = 10 mice per group). Significant differences between the i.n. and i.p. routes are shown: *, P < 0.05.
Local administration of MAb 6F12 significantly decreases lung viral titers and pathology in the lungs.
To determine if weight loss and/or disease correlated with the viral burden and lung pathology, mice were first administered MAb 6F12 via the i.p., i.n., or a.e. route 2 h prior to infection or 24 h postinfection (i.p. and i.n.) with 3 mLD50 of NL/09 virus. Mice were then euthanized on day 3 or day 6 postinfection to determine lung viral titers. When MAb 6F12 was administered prophylactically, significant reductions in lung viral titers (P < 0.05) were observed in the groups of mice that received the treatment via local administration compared to the results in the groups that received the MAb via the i.p. route (P < 0.05) (Fig. 5A) at both time points. The lung viral titers in the group that received the antibody via the i.n. route were below the limit of detection for the assay (Fig. 5A). For the groups that received MAb 6F12 therapeutically (24 h postinfection), a significant reduction (P < 0.05) in lung viral titers was observed on day 6 postinfection in the group that was treated via the i.n. route (Fig. 5B). Overall, the results showed that administration of MAb 6F12 prophylactically or therapeutically via local routes significantly reduces lung viral titers compared to those treated via systemic routes.
FIG 5.
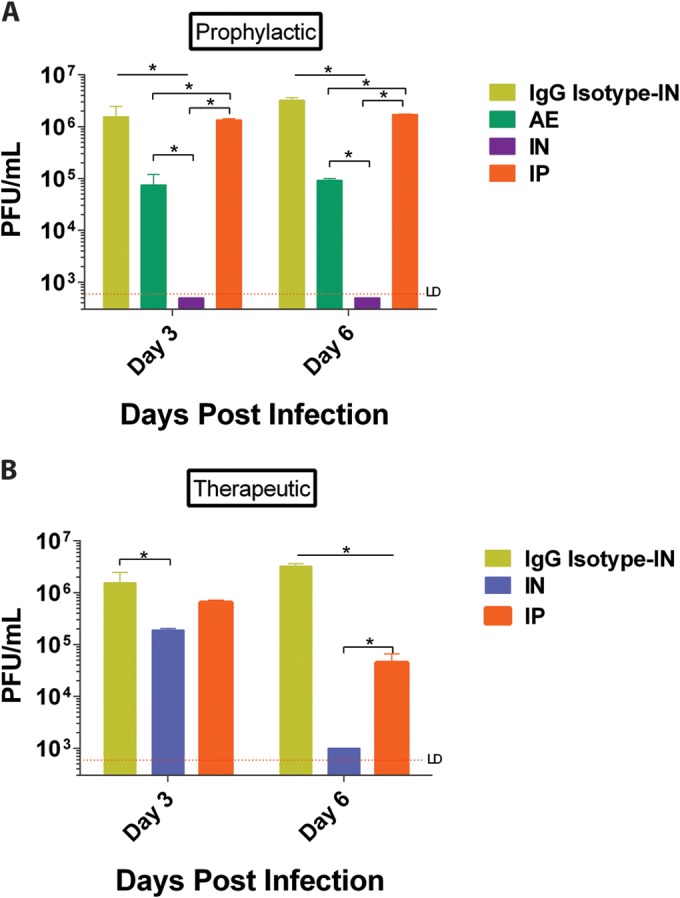
Lung viral titers of mice infected with influenza A/Netherlands/604/2009 H1N1pdm virus. Mice received 3 mg/kg of MAb 6F12 via the a.e., i.n., or i.p. route 2 h before infection (prophylactically) (A) or 15 mg/kg via the i.n. or i.p. route 24 h postinfection (therapeutically) (B). As a negative control an isotype IgG was used at 3 mg/kg and administered via the i.n. route 2 h before infection. Lungs were harvested from each group (n = 3) at 3 or 6 days postinfection, and lung viral titers were determined by a plaque assay (expressed in PFU per milliliter). Values represent means ± SEM. Significant differences are shown: *, P < 0.05. LD, limit of detection.
Histological analyses of lungs harvested 2 days postinfection from mice that received treatment prophylactically demonstrated either the absence of lesions or mild lesions (Table 1). Lungs from mice treated 3 days postinfection were harvested 24 (4 days postinfection) or 48 h (5 days postinfection) later. A higher degree of microscopic lesions were observed in lungs obtained on day 5 postinfection (i.e., 2 days after antibody treatment). Intranasally treated mice demonstrated moderate perivascular and mild alveolar inflammation, while i.p.-treated mice and the PBS-treated control mice demonstrated a wider range of pathologies. These included moderate perivascular inflammation and mild epithelial degeneration with scattered cell necrosis in the less affected animals. Some mice demonstrated more severe lesions, including marked epithelial degeneration with segmental necrosis, areas of epithelial loss, and moderate alveolar inflammation with interstitial damage. Overall, a significant reduction (P < 0.05) in pathology was observed in lungs from mice treated via the i.n. route on day 3 postinfection and harvested on day 5 postinfection compared with that for the groups that received treatment via the i.p. route or the control group.
TABLE 1.
Histopathology scores of lungs from mice treated with MAb 6F12 via intraperitoneal, intranasal, or aerosol administration and challenged with influenza A/Netherlands/602/2009
| Treatment regimena | Route of administrationb | Lung pathology scorec |
|||||
|---|---|---|---|---|---|---|---|
| Amount of tissue affected | Perivascular inflammation | Epithelial degeneration | Peribronchiolar inflammation | Intraluminal debris | Alveolar inflammation | ||
| Prophylactic | i.p. | 0.3 | 0.6 | 0 | 0 | 0 | 0.3 |
| i.n. | 1 | 0.6 | 0 | 0 | 0 | 1.3 | |
| a.e. | 0.6 | 1 | 0 | 0 | 0 | 1.3 | |
| Therapeutic D-4 | i.p. | 1 | 1.6 | 0 | 0 | 0 | 1 |
| i.n. | 0.6 | 0 | 0 | 0 | 0 | 0.6 | |
| Therapeutic D-5d | i.p. A | 1.7 | 2.3 | 3 | 2 | 2 | 2.3 |
| i.n. AB | 1.3 | 2 | 1.6 | 1 | 1.3 | 1.3 | |
| Control B | 1.5 | 2.5 | 3 | 2 | 2.5 | 3 | |
Prophylactic, treatment 2 h before infection; therapeutic, treatment on day 3 postinfection; D-4 and D-5, days postinfection when samples were collected.
i.p., intraperitoneal; i.n., intranasal; a.e., aerosol; control, mice received PBS via the i.n. route.
Lung pathology was scored on a scale of 0 to 4: 0, no lesions; 1, mild changes with scattered cell necrosis/vacuolation and few/scattered inflammatory cells in the bronchiolar epithelium with minimal perivascular inflammation; 2, moderate multifocal vacuolation with peribronchiolar inflammation (<5 cell layers thick); 3, marked, multifocal/segmental necrosis, epithelial loss/effacement and peribronchiolar inflammation (>5 cell layers thick); and 4, severe, coalescing areas of necrosis, parenchymal effacement with confluent areas of inflammation. Each score value represents the mean from 3 mice per group.
Significant differences (P < 0.05) in the overall pathology score between the i.n. and i.p. groups (A) and i.n. and control groups (B) are shown.
Enhanced efficacy of i.n. route of administration is not MAb or virus specific.
To determine if the improved efficacy observed upon local administration of MAb 6F12 was specific for this particular HA stalk antibody-virus combination, we tested various MAbs and influenza virus strains to demonstrate broader applicability of the monoclonal antibody therapy approach. For these experiments, mice received 3 or 0.3 mg/kg of antibody intraperitoneally (systemic administration) or intranasally (local administration) 1 day postinfection. First, we tested protection of mice treated with MAb 6F12 against infection with PR/8 (H1N1) virus. Mice that received treatment with 3 mg/kg of the MAb via the i.n. route survived influenza PR/8 infection and demonstrated minimal transient weight loss, while all mice that received treatment i.p. succumbed to infection (Fig. 6A and B). These trends continued in the groups that received 0.3 mg/kg of the MAb, in which we observed significant survival (P < 0.05) of the mice treated via the i.n. route compared to that of mice treated via the i.p. route (60% versus 0% survival, respectively). This demonstrated that the enhanced efficacy observed after local administration of MAb 6F12 was not virus strain specific. In order to ensure that this enhanced efficacy was not MAb specific, we tested MAb GG3, another HA stalk-specific broadly neutralizing antibody, in mice infected with 5 mLD50 of NL/09 or PR/8 virus. All mice treated with 3 mg/kg of GG3 survived against the NL/09 virus challenge, irrespective of the route of administration; however, mice treated i.p. had more pronounced weight loss (Fig. 6C and D). At a dose of 0.3 mg/kg, 40% of mice that received the MAb via the i.n. route survived the infection, while none of the mice that received treatment via the i.p. route survived (Fig. 6C and D), underscoring once again enhanced efficacy upon local MAb administration. In the groups that were infected with PR/8 virus, only mice that received 3 mg/kg of MAb GG3 via the i.n. route survived infection and demonstrated minimal transient weight loss, while mice treated i.p. or with the isotype IgG control succumbed to infection (Fig. 6E and F). These results confirm that the advantages observed by locally delivering MAb 6F12 also apply to other broadly neutralizing HA stalk monoclonal antibodies and different virus strains.
FIG 6.

Therapeutic administration of other broadly neutralizing anti-HA stalk monoclonal antibodies. Body weight loss (A) and survival (B) curves of mice treated with 3 mg/kg or 0.3 mg/kg of MAb 6F12 intranasally (i.n.) or intraperitoneally (i.p.) on day 1 postinfection with influenza A/Puerto Rico/08/1934 H1N1 virus (PR/8). Body weight loss (C) and survival (D) curves of mice treated with 3 mg/kg or 0.3 mg/kg of MAb GG3 via the i.n. or i.p. route on day 1 postinfection with influenza A/Netherlands/602/2009 H1N1pdm virus (NL/09). Body weight loss (E) and survival (F) curves of mice treated with 3 mg/kg or 0.3 mg/kg of MAb GG3 on day 1 postinfection with PR/8. As a negative control, an isotype IgG was used at 3 mg/kg and administered i.n. 1 day postinfection (B, D, and F). Body weight curve values represent means ± SEM (n = 5 mice per group). Significant differences between the i.n. and i.p. routes (within the same dose) are shown: *, P < 0.05.
Intranasal administration of monoclonal antibody therapy showed enhanced efficacy against H5N1.
Highly pathogenic avian influenza viruses belonging to the H5N1 subtype can cause severe disease when transmitted into humans. MAb KB2, another group 1 antistalk MAb, has neutralizing activity against the pathogenic H5N1 virus (10). Therefore, we tested if local administration improved the efficacy of this MAb for the treatment of H5N1 virus infection in a murine model. Mice were initially infected with the VN/04 (HALo) virus and then received 15 mg/kg of the MAb KB2 via i.n. or i.p. administration on day 1 or day 3 postinfection. As a control, a group of mice were infected and then treated with 15 mg/kg of an irrelevant IgG MAb via the i.n. route on day 1 postinfection. Mice that received treatment via the i.n. route exhibited only a transient weight loss of 3%, whereas mice that received treatment via the i.p. route had a more pronounced weight loss and lost almost 20% of their initial body weight (Fig. 7A). All mice in the groups that received treatment via the i.n. or i.p. route on day 1 postinfection survived (Fig. 7B). When the MAb was given on day 3 postinfection, all mice that received it via the i.n. route survived infection, while mice that were treated via the i.p. route had only a 60% survival rate (Fig. 7B). These data demonstrate an extension of our findings to one of the highly pathogenic avian origin influenza viruses circulating in the human population.
FIG 7.
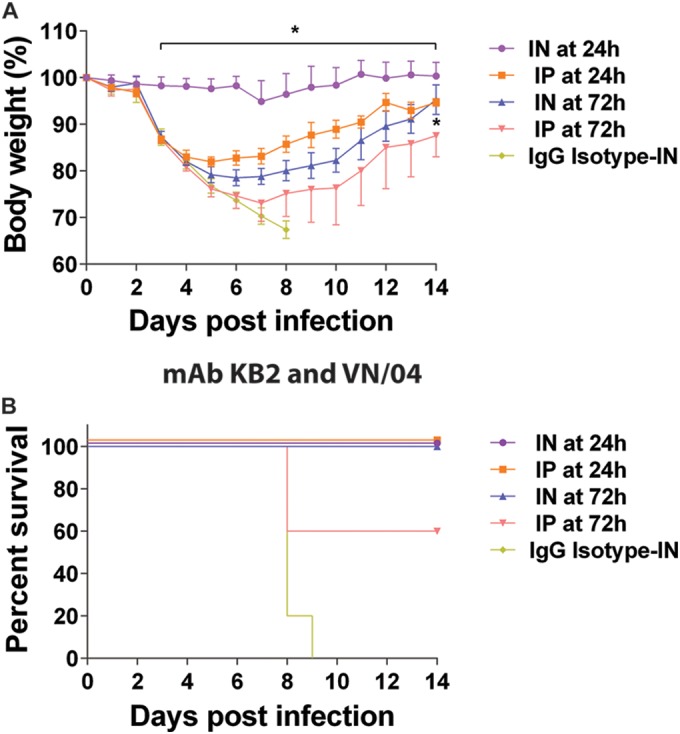
Therapeutic administration of broadly neutralizing anti-HA stalk antibody KB2. Body weight (A) and survival (B) curves of mice treated with 15 mg/kg of MAb KB2 via the i.n. or i.p. route at 24 h or 72 h postinfection with influenza A/Vietnam/1203/2004 H5N1 virus (VN/04). As a negative control, an isotype IgG was used at 15 mg/kg and administered via the i.n. route at 24 h postinfection. Body weight curve values represent means ± SEM (n = 5 mice per group). Significant differences between the i.n. and i.p. routes are shown: *, P < 0.05.
DISCUSSION
We previously characterized several broadly neutralizing monoclonal antibodies that target the hemagglutinin stalk region and demonstrated their ability to protect mice from developing overt clinical disease and succumbing to influenza virus infection (9, 10). Since influenza virus infection is mainly restricted to the respiratory tract in mammals, we tested previously characterized MAbs via the intranasal or aerosolized delivery route to determine if their efficacy can be improved when delivered locally rather than systemically. While some studies have used the intranasal route of administration to deliver anti-influenza antibodies (14–16), we have not found published studies comparing the efficacies of anti-influenza anti-HA stalk monoclonal antibodies via different routes of administration. We first characterized the bioavailability and distribution of MAb 6F12. The half-life of MAb 6F12 was 5.5 days, which is similar to that in previous reports for IgG mouse immunoglobulins in their natural host (17). Our bioavailability imaging studies revealed poor penetration into tissues, as has been observed for other monoclonal antibodies in mice (18). Therefore, we tested different systemic and local routes of delivery to quantify the amount of MAb that reached the respiratory epithelium. After systemic administration, higher levels of the antibody were observed in the serum, but low levels were present in the bronchoalveolar lavage fluid, demonstrating low penetration of the MAb in the lungs. Typically, a high concentration of IgGs (about 61 μg/ml) is necessary in the circulating plasma to enable transudation into the respiratory mucosa. The opposite is observed after local administration of a MAb via intranasal droplets, where the antibody maintains a higher concentration in the BAL fluid with low penetration into the bloodstream (19). The in vivo imaging of fluorescently labeled MAb 6F12 showed that after i.p. injection the MAb is primarily distributed in the bloodstream, while after i.n. delivery the MAb was distributed in the upper and lower respiratory tracts. Of note, aerosolization of MAbs resulted in distribution only in the upper respiratory tract, which we observed in our in vivo experiments. Therefore, it is not surprising that we did not detected aerosolized MAb in the BAL fluid and in sera by ELISAs, which reflect the lower respiratory tract and blood compartments of the mouse, respectively. The amount of MAb that goes to the lower respiratory tract, if any, may be too low to be detected with our approach. Ultimately, it should be noted that aerosolized MAb given prophylactically protects mice from disease, so although there is no detectable antibody in the lungs or in the BAL fluid of nebulized mice, this route of MAb administration is still effective. Similar distribution patterns have been observed in previous studies using fluorescently labeled anticancer monoclonal antibodies that compared systemic administration (i.v.) versus local administration via endotracheal aerosolization (20, 21).
Larger amounts of the MAb in the BAL fluid of mice that received the antibody intranasally may explain the extended protection observed in mice infected up to 5 days after antibody administration. This result has great clinical relevance in the event of a pandemic and suggests that a prolonged prophylactic treatment strategy may prevent infection in people at high risk, such as health care workers, the elderly, or immunocompromised patients.
In a prophylaxis regimen with the use of an escalating dose approach, we observed a 10-fold reduction (0.3 mg/kg local versus 3 mg/kg systemic) in the amount of antibody required for protection when the MAb was administered locally compared to that with systemic administration. Immunoglobulins are not secreted in the respiratory mucosa after influenza virus infection but reach the mucosa using a passive transudation process from serum subject to the prevailing concentration gradient (22). The amount of serum IgG needed to transude into the respiratory mucosa to confer protection in mice has been shown to be approximately 7 times the amount of serum IgG observed in a convalescent mouse (1, 19). This may explain why the efficacy of MAb 6F12 is lower when it is administered via systemic routes. In contrast, local administration provides the MAb direct access to the site of infection, which may facilitate virus neutralization, resulting in a dose-sparing effect.
As influenza virus infection progresses, the inflammatory response recruits immune effector cells to the site of infection and also increases permeability for higher-molecular-weight molecules such as immunoglobulins. We previously demonstrated that mice receiving 30 mg/kg of MAb 6F12 via the i.p. route were protected, even when treated as late as 5 days postinfection (9). Therefore, we also examined if local administration of MAb 6F12 can improve the therapeutic efficacy of these antibodies. When treatment was administered 5 days postinfection, we observed a significant improvement in body weight recovery and better survival, although the improvement in survival was not statistically significant in the group that received treatment intranasally compared with that in the group that received treatment intraperitoneally. On day 5 postinfection, immunopathology development is still in progress; however, we observed significantly less histopathology in mice treated via the i.n. route than in mice treated via the i.p. route or the control group.
We hypothesize that to control infection at this advanced stage (5 days postinfection) a higher concentration of the protective MAb at the site of infection is required; therefore, only the i.n. administration of 15 mg/kg provided the amount of MAb necessary to improve protection. During an influenza virus infection in mice, the amount of virus replicating in the upper respiratory tract peaks on 1 day postinfection and then gradually declines over time and can be detected up to 5 days postinfection (23). This may explain why aerosolization of MAb 6F12 protected mice treated prophylactically despite its localization only to the upper respiratory tract, suggesting that the aerosolized MAb is able to neutralize the virus at the site of entry. In addition, the earliest point used to evaluate the distribution and concentration of the MAb was at 24 h after treatment, which may indicate that the concentration of the MAb delivered via aerosolization declines faster than when delivered intranasally but still maintained protective concentrations at the time when the challenge with the virus was done (2 h after treatment).
Increased efficacy upon local administration of the MAbs was observed with different virus strains. Previous studies using polyclonal or monoclonal anti-HA antibodies have shown some protection in mice treated intranasally against challenges with H1N1 viruses such as PR/8 and A/California/04/2009 H1N1pdm (14, 16). In these experiments, a prophylactic dose of 12.5 mg/kg (14) or 6.5 mg/kg (16) was required to protect mice from influenza. Studies previously published also investigated anti-head-specific MAbs or polyclonal serum being administered intranasally, but to date, anti-stalk-specific MAbs have not been tested for local administration. Broadly neutralizing anti-HA stalk antibodies confer heterosubtypic protection against highly divergent subtypes within group 1, which is the potential gold standard for broad-spectrum anti-influenza virus prophylactic and therapeutic regimens (24). These results demonstrated a potential broader applicability of our local administration treatment approach.
One of the main disadvantages of antibody therapy is the amount required for protection. For example, the recommended dose of palivizumab, a commercially available monoclonal antibody against respiratory syncytial virus infection in infants and young children, is 15 mg/kg once a month for 5 months (25). This means that 375 mg of the MAb is necessary to treat one 5-kg child. Currently, no monoclonal antibody therapy has been approved for the treatment of influenza; however, some monoclonal antibodies tested for protective efficacy in clinical trials have used 15 to 30 mg/kg of the antibody via the i.v. route (https://clinicaltrials.gov/ct2/results?term=influenza+monoclonal+antibody&Search=Search). Considering that production of large amounts of antibody is still a very expensive process, approaches that allow dose sparing are highly desirable. Here we demonstrate that treatment with a broadly neutralizing MAb via the intranasal or aerosol route can significantly decrease the amount of MAb required for protection by 1 log compared with administration via the intraperitoneal or intravenous route. The use of aerosol as a route of administration is also of interest because aerosols are routinely used to deliver drugs to treat both inflammatory and infectious conditions of the lung (20). Overall, our results demonstrate that local administration of broadly neutralizing antibodies, particularly anti-HA stalk antibodies, is a viable alternative in the prevention and treatment of influenza virus infections.
ACKNOWLEDGMENTS
We thank Virginia Gillespie from the Center of Comparative Medicine and Surgery at the Icahn School of Medicine for the pathology evaluation of the lung samples.
This work was supported in part by the Centers for Excellence for Influenza Research and Surveillance (CEIRS) grant (HHSN266200700010C) and NIH grants U19 AI109946 and P01 AI097092 (P.P.).
REFERENCES
- 1.Renegar KB, Small PA Jr. 1991. Passive transfer of local immunity to influenza virus infection by IgA antibody. J Immunol 146:1972–1978. [PubMed] [Google Scholar]
- 2.Saylor C, Dadachova E, Casadevall A. 2009. Monoclonal antibody-based therapies for microbial diseases. Vaccine 27(Suppl):G38–G46. doi: 10.1016/j.vaccine.2009.09.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luke T, Kilbane EM, Jackson JL, Hoffman SL. 2006. Meta-analysis: convalescent blood products for Spanish influenza pneumonia: a future H5N1 treatment? Ann Intern Med 145:599–609. doi: 10.7326/0003-4819-145-8-200610170-00139. [DOI] [PubMed] [Google Scholar]
- 4.Luke T, Casadevall A, Watowich SJ, Hoffman SL, Beigel JH, Burgess TH. 2010. Hark back: passive immunotherapy for influenza and other serious infections. Crit Care Med 38:e66–e73. doi: 10.1097/CCM.0b013e3181d44c1e. [DOI] [PubMed] [Google Scholar]
- 5.Hung IFN, To KKW, Lee CK, Lee KL, Yan WW, Chan K, Chan WM, Ngai CW, Law KI, Chow FL, Liu R, Lai KY, Lau CCY, Liu SH, Chan KH, Lin CK, Yuen KY. 2013. Hyperimmune IV immunoglobulin treatment: a multicenter double-blind randomized controlled trial for patients with severe 2009 influenza A (H1N1) infection. Chest 144:464–473. doi: 10.1378/chest.12-2907. [DOI] [PubMed] [Google Scholar]
- 6.Null D Jr, Pollara B, Dennehy PH, Steichen J, Sánchez PJ, Givner LB, Carlin D, Landry B, Top FH Jr, Connor E. 2005. Safety and immunogenicity of palivizumab (Synagis) administered for two seasons. Pediatr Infect Dis J 24:1021–1023. doi: 10.1097/01.inf.0000183938.33484.bd. [DOI] [PubMed] [Google Scholar]
- 7.Dunand CJ, Leon PE, Kaur K, Tan GS, Zheng NY, Andrews S, Huang M, Qu X, Huang Y, Salgado-Ferrer M, Ho IY, Taylor W, Hai R, Wrammert J, Ahmed R, Garcia-Sastre A, Palese P, Krammer F, Wilson PC. 2015. Preexisting human antibodies neutralize recently emerged H7N9 influenza strains. J Clin Invest 125:1255–1268. doi: 10.1172/JCI74374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DiLillo DJ, Tan GS, Palese P, Ravetch JV. 2014. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcγR interactions for protection against influenza virus in vivo. Nat Med 20:143–151. doi: 10.1038/nm.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan GS, Krammer F, Eggink D, Kongchanagul A, Moran TM, Palese P. 2012. A pan-H1 anti-hemagglutinin monoclonal antibody with potent broad-spectrum efficacy in vivo. J Virol 86:6179–6188. doi: 10.1128/JVI.00469-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heaton NS, Leyva-Grado VH, Tan GS, Eggink D, Hai R, Palese P. 2013. In vivo bioluminescent imaging of influenza A virus infection and characterization of novel cross-protective monoclonal antibodies. J Virol 87:8272–8281. doi: 10.1128/JVI.00969-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krammer F, Hai R, Yondola M, Tan GS, Leyva-Grado VH, Ryder AB, Miller MS, Rose JK, Palese P, Garcia-Sastre A, Albrecht RA. 2014. Assessment of influenza virus hemagglutinin stalk-based immunity in ferrets. J Virol 88:3432–3442. doi: 10.1128/JVI.03004-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shao H, Ye J, Vincent AL, Edworthy N, Ferrero A, Qin A, Perez DR. 2011. A novel monoclonal antibody effective against lethal challenge with swine-lineage and 2009 pandemic H1N1 influenza viruses in mice. Virology 417:379–384. doi: 10.1016/j.virol.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Short KR, Veldhuis Kroeze EJB, Fouchier RAM, Kuiken T. 2014. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect Dis 14:57–69. doi: 10.1016/S1473-3099(13)70286-X. [DOI] [PubMed] [Google Scholar]
- 14.Ye J, Shao H, Hickman D, Angel M, Xu K, Cai Y, Song H, Fouchier RAM, Qin A, Perez DR. 2010. Intranasal delivery of an IgA monoclonal antibody effective against sublethal H5N1 influenza virus infection in mice. Clin Vaccine Immunol 17:1363–1370. doi: 10.1128/CVI.00002-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He F, Kumar SR, Khader SMS, Tan Y, Prabakaran M, Kwang J. 2013. Effective intranasal therapeutics and prophylactics with monoclonal antibody against lethal infection of H7N7 influenza virus. Antiviral Res 100:207–214. doi: 10.1016/j.antiviral.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 16.Rinaldi C, Penhale WJ, Stumbles PA, Tay G, Berry CM. 2014. Modulation of innate immune response by influenza-specific ovine polyclonal antibodies used for prophylaxis. PLoS One 9:e89674. doi: 10.1371/journal.pone.0089674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vieira P, Rajewsky K. 1988. The half-lives of serum immunoglobulins in adult mice. Eur J Immunol 18:313–316. doi: 10.1002/eji.1830180221. [DOI] [PubMed] [Google Scholar]
- 18.Sigounas G, Harindranath N, Donadel G, Notkins AL. 1994. Half-life of polyreactive antibodies. J Clin Immunol 14:134–138. doi: 10.1007/BF01541346. [DOI] [PubMed] [Google Scholar]
- 19.Renegar KB, Small PA Jr, Boykins LG, Wright PF. 2004. Role of IgA versus IgG in the control of influenza viral infection in the murine respiratory tract. J Immunol 173:1978–1986. doi: 10.4049/jimmunol.173.3.1978. [DOI] [PubMed] [Google Scholar]
- 20.Maillet A, Guilleminault L, Lemarie E, Lerondel S, Azzopardi N, Montharu J, Congy-Jovilet N, Reverdiau P, Legrain B, Parent C, Douvin DH, Hureaux J, Courty Y, De Monte M, Diot P, Paintaud G, Le Pape A, Watier H, Heunze-Vourch N. 2011. The airways, a novel route for delivering monoclonal antibodies to treat lung tumors. Pharm Res 28:2147–2156. doi: 10.1007/s11095-011-0442-5. [DOI] [PubMed] [Google Scholar]
- 21.Guilleminault L, Azzopardi N, Arnoult C, Sobilo J, Herve V, Montharu J, Guillon A, Andres C, Herault O, Le Pape A, Diot P, Lemarie E, Paintaud G, Gouilleux-Gruart V, Heunze-Vourch N. 2014. Fate of inhaled monoclonal antibodies after deposition of aerosolized particles in the respiratory system. J Control Release 196:344–354. doi: 10.1016/j.jconrel.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Wagner DK, Clements ML, Reimer CB, Snyder M, Nelson DL, Murphy BR. 1987. Analysis of immunoglobulin G antibody responses after administration of live and inactivated influenza A vaccine indicates that nasal wash immunoglobulin G is a transudate from serum. J Clin Microbiol 25:559–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Novak M, Moldoveanu Z, Schafer DP, Mestecky J, Compans RW. 1993. Murine model for evaluation of protective immunity to influenza virus. Vaccine 11:55–60. doi: 10.1016/0264-410X(93)90339-Y. [DOI] [PubMed] [Google Scholar]
- 24.Mancini N, Solforosi L, Clementi N, De Marco D, Clementi M, Burioni R. 2011. A potential role for monoclonal antibodies in prophylactic and therapeutic treatment of influenza. Antiviral Res 92:15–26. doi: 10.1016/j.antiviral.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 25.American Academy of Pediatrics Committee on Infectious Diseases, American Academy of Pediatrics Bronchiolitis Guidelines Committee. 2014. Updated guidance for palivizumab prophylaxis among infants and young children at increased risk of hospitalization for respiratory syncytial virus infection. Pediatrics 134:415–420. doi: 10.1542/peds.2014-1665. [DOI] [PubMed] [Google Scholar]