

Abstract
Yersinia pestis is the etiologic agent of the plague. Reports of Y. pestis strains that are resistant to each of the currently approved first-line and prophylactic treatments point to the urgent need to develop novel antibiotics with activity against the pathogen. We previously reported that Y. pestis strain KIM6+, unlike most Enterobacteriaceae, is susceptible to the arylomycins, a novel class of natural-product lipopeptide antibiotics that inhibit signal peptidase I (SPase). In this study, we show that the arylomycin activity is conserved against a broad range of Y. pestis strains and confirm that it results from the inhibition of SPase. We next investigated the origins of this unique arylomycin sensitivity and found that it does not result from an increased affinity of the Y. pestis SPase for the antibiotic and that alterations to each component of the Y. pestis lipopolysaccharide—O antigen, core, and lipid A—make at most only a small contribution. Instead, the origins of the sensitivity can be traced to an increased dependence on SPase activity that results from high levels of protein secretion under physiological conditions. These results highlight the potential of targeting protein secretion in cases where there is a heavy reliance on this process and also have implications for the development of the arylomycins as an antibiotic with activity against Y. pestis and potentially other Gram-negative pathogens.
INTRODUCTION
The emergence of multidrug-resistant bacteria, especially Gram-negative pathogens, is a major health concern that can be combated only by the continued development of new antibiotics, particularly ones that act via novel mechanisms of action to limit the potential for cross-resistance. Yersinia pestis, a Gram-negative bacterium and the causative agent of plague, is of particular historical significance due to the mortality and social havoc wreaked by at least three major pandemics (1, 2). Today, plague continues to pose a threat, even in developed countries (3), and while Y. pestis infections are now treatable with available antibiotics, delays in the initiation of effective therapy, as can be caused by resistance to the employed antibiotic, results in significantly increased mortality (4). Thus, reports of Y. pestis strains that are resistant to first-line antibiotic therapies (5–7) are troubling, and the all but certain continued evolution of these strains toward resistance to all available antibiotics threatens to return the plague agent to its historical position as an important pathogen. This threat is unlikely to abate until new, effective antibiotics are discovered and developed; however, no new class of antibiotics with activity against Gram-negative bacteria has been approved in over 40 years (8). Moreover, a recent report by the Infectious Diseases Society of America (IDSA) identified only seven new candidate antibiotics that have progressed into clinical development for the treatment of multidrug-resistant Gram-negative bacilli since 2010 (9).
A general challenge in developing antibiotics against Gram-negative bacteria is the barrier to penetration provided by the lipopolysaccharide (LPS) leaflet of the outer membrane (OM) (10–12). Generally, LPS consists of three parts—the O antigen, the core oligosaccharide, and lipid A—and metal-mediated cross-linking results in the formation of a tightly packed, extended structure. While small hydrophilic molecules may pass through the outer membrane via porins, larger and more hydrophobic molecules must traverse it passively through exposed membrane phospholipids, which are largely precluded by the tight packing of the LPS (10, 11).
The arylomycins are a promising family of natural-product antibiotics that inhibit type I signal peptidase (SPase) (13–15). SPase is highly conserved and essential in both Gram-negative and Gram-positive bacteria because it is required to release proteins from their membrane-bound N-terminal leader sequences after translocation across the cytoplasmic membrane via the Sec or Tat translocation pathway (16, 17). Thus, it was somewhat surprising that the arylomycins were initially reported to possess activity against only a narrow spectrum of Gram-positive bacteria (18, 19). However, a more extensive characterization, made possible by the total synthesis of several arylomycin variants (20–24), including arylomycin A-C16 (Fig. 1), revealed that they are active against a broad range of Gram-positive and Gram-negative bacteria and that the majority of the observed resistance was associated with the idiosyncratic presence of a specific proline residue in SPase (14, 25). While this proline appears to universally reduce sensitivity, the absolute values of the sensitivities are species and sometimes even strain specific (14, 25), suggesting that other factors also contribute. Of particular note, despite encoding an SPase with the resistance-conferring proline, Y. pestis KIM6+ is sensitive to arylomycin A-C16, with a MIC of 4 μg/ml (14).
FIG 1.
Structure of arylomycin A-C16.
Here, we show that the arylomycins have activity against a broad range of Y. pestis strains and that the activity indeed results from the potent inhibition of SPase. To investigate why Y. pestis is sensitive while related Enterobacteriaceae, such as Escherichia coli, are resistant, we determined the contributions of different factors that are unique to Y. pestis. We found that the sensitivity does not result from a greater affinity of the arylomycins for Y. pestis SPase and that its unique LPS is insufficient on its own to explain the sensitivity. In contrast, we found that deletion of the gene encoding the highly expressed cell adhesion protein Ail (locus tag y1324; UniProt entry Q8D0Z7) significantly alleviates sensitivity and that overexpression of Ail or E. coli maltose-binding protein then restores sensitivity, suggesting that it results from a high secretion burden at physiological temperatures. These results highlight the potential of the arylomycins as antibiotics for the treatment of Y. pestis, and potentially other Gram-negative bacteria, especially those that rely on high levels of protein secretion for viability or virulence.
MATERIALS AND METHODS
Medium and antibiotics.
Bacteria were routinely grown at 28°C or 37°C, as appropriate, on Mueller-Hinton II agar (MHIIA) or in cation-adjusted Mueller-Hinton II broth (CAMHB). Antibiotic stock solutions were prepared at the following concentrations: arylomycin A-C16, 10 mg/ml (in dimethyl sulfoxide [DMSO]); actinomycin D (in DMSO), 10 mg/ml; deoxycholic acid (in 1:1 CAMHB-H2O), 250 mg/ml; polymyxin B nonapeptide (PMBN) (in H2O), 10 mg/ml; l-arabinose (in CAMHB), 50% (wt/vol); tetracycline (in ethyl alcohol [EtOH]), 1 mg/ml; and gentamicin (in H2O), 1 mg/ml. Arylomycin A-C16 was synthesized as described previously (20). PMBN and deoxycholic acid were obtained from Sigma-Aldrich (St. Louis, MO), actinomycin D was obtained from Fisher Scientific (Pittsburgh, PA), l-arabinose and gentamicin sulfate were obtained from MP Biomedicals (Solon, OH), and tetracycline hydrochloride was obtained from Fisher Bioreagents (Fairlawn, NJ).
Bacterial strains and plasmids.
The bacterial strains and plasmids used in this study are listed in Table 1 (also see Table 3). E. coli BAS901 is a hyperpermeable strain harboring the lptD mutation, which results in defective LPS assembly. In strains PAS0260 and DBS600, the Pro residue in the SPase responsible for arylomycin resistance was replaced by a residue that confers arylomycin sensitivity; the resulting SPases are LepB(P84L) in E. coli strain PAS0260 and LepB(P91S) in Y. pestis strain DBS600. Strain Y. pestis KIM5-pLpxL (which expresses a hexa-acylated lipid A) and its parental strain were kindly provided by Egil Lien (University of Massachusetts Medical School). Strain RS 058, a Y. pestis strain that harbors the E. coli lepB gene in the pBAD vector, was kindly provided by RQX Pharmaceuticals (La Jolla, CA). The KIM6+ phoP knockout strain was kindly provided by NIAID Rocky Mountain Laboratories (Crested Butte, CO).
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| Strains | ||
| BW27343 | E. coli DH5α pir+ | CGSC |
| MG1655 | E. coli K-12 F− λ− ilvG rbf-50 rph-1 | 75–77 |
| PAS0260 | MG1655 LepB(P84L)::Kanr | 14 |
| KIM6+ | Y. pestis pCD1− pMT1+ pPCP1+ pgm+ | 78 |
| DBS600 | KIM6+ expressing LepB(P91S) | This study |
| ATCC 23715 | Y. enterocolitica serotype 8 | 79 |
| BAS901 | E. coli lptD4213) mutant | 80 |
| RS 058 | KIM6+ containing pBAD (E. coli K-12 lepB) | RQX Pharmaceuticals |
| D21 | E. coli F− proA23 lac-28 tsx-81 trp-30 his-51 rpsL173(strR) ampCp-1 | 81, CGSC |
| D21e7 | E. coli F− proA23 lac-28 tsx-81 trp-30 his-51 rpsL173(strR) rfa-1 ampCp-1 | 82, CGSC |
| D21e19 | E. coli F− proA23 lac-28 tsx-81 trp-30 his-51 rpsL173(strR) rfa-11 ampCp-1 | 82, CGSC |
| D21f1 | E. coli F− proA23 lac-28 tsx-81 trp-30 his-51 rpsL173(strR) rfa-21 rfa-1 ampCp-1 | 81, CGSC |
| D21f2 | E. coli F− proA23 lac-28 tsx-81 trp-30 his-51 rpsL173(strR) rfa-31 rfa-1 ampCp-1 | 81, CGSC |
| KIM5 | Y. pestis KIM6 pCD1Ap pMT1+ pPCP1+ pgm mutant Apr | 83 |
| KIM5-pLpxL | KIM5 containing pBR322(E. coli K-12 lpxL) | E. Lien (University of Massachusetts Medical School) (53) |
| KIM6+ ΔphoP | KIM6+ lacking phoP | Rocky Mountain Laboratories |
| DBS601 | KIM6+ expressing LepB(P91S)::Kanr lacking ail | This study |
| DBS601- pBAD/His B | DBS601 complemented with pBAD/HisB (empty) | This study |
| DBS601- pBAD/His B-ail | DBS601 complemented with pBAD/HisB vector (Y. pestis KIM6+ ail) | This study |
| DBS601- pBAD/His B-malE | DBS601 complemented with pBAD/HisB vector (E. coli K-12 malE) | This study |
| Plasmids | ||
| pET23b | Expression vector; T7 promoter; His6 tag | EMD Millipore |
| pET23b-EclepB | pET23b harboring E. coli K-12 lepB | 14 |
| pET23B-YplepB | pET23b harboring a chimeric E. coli K-12/Y. pestis KIM6+ lepB gene | This study |
| pET23b- YplepB(P91S) | pET23b harboring a mutated, chimeric E. coli K-12/Y. pestis KIM6+ lepB gene that encodes the P91S substitution | This study |
| pKNG101 | Suicide vector; sacB | 28 |
| pKNG101- YplepB(P91S) | pKNG101 harboring a mutated Y. pestis KIM6+ lepB gene that encodes the P91S substitution | This study |
| pKD4 | 30; Addgene plasmid no. 45605 | |
| pKD46 | 30; CGSC | |
| pCP20 | 30; CGSC | |
| pBAD/His B | Expression vector; araBAD promoter | Invitrogen |
| pBAD/His B-ail | pBAD/HisB harboring Y. pestis KIM6+ ail | This study |
| pBAD/His B-malE | pBAD/HisB harboring E. coli K-12 malE | This study |
TABLE 3.
Strain identifiers and MICs of arylomycin A-C16 and ciprofloxacin for the 30-member Y. pestis panela
| Y. pestis strain | MIC (μg/ml) |
|
|---|---|---|
| Arylomycin A-C16 (MIC50 = 8; MIC90 = 32) | Ciprofloxacin (MIC50 = 0.03; MIC90 = 0.03) | |
| CO92 | 8 | 0.03 |
| C12 | 4 | 0.06 |
| Antiqua | 8 | 0.015 |
| Pestoides B | 4 | 0.015 |
| Pestoides Fmp1 | 1 | 0.015 |
| Yeo154 | 8 | 0.03 |
| Angola | 8 | 0.015 |
| Java9 | 8 | 0.03 |
| M111(74) | 8 | 0.03 |
| LaPaz | 8 | 0.03 |
| 195P mp1 | 8 | 0.03 |
| T26 mp3 | 4 | 0.03 |
| KIM 10 | 32 | 0.03 |
| Pest E | 8 | 0.015 |
| RFPBM 19 | 8 | 0.03 |
| PeXu 429 | 2 | 0.03 |
| Yokohama | 8 | 0.03 |
| Nicholisk 41 | 32 | 0.03 |
| Nairobi | 16 | 0.015 |
| South Park | 16 | 0.03 |
| Cambodia | 16 | 0.03 |
| 27 | 8 | 0.03 |
| 31 | 8 | 0.03 |
| 390 | 1 | 0.015 |
| 590 | 8 | 0.03 |
| 25 | 32 | 0.06 |
| 316 | >64 | 0.06 |
| 366 | 8 | 0.015 |
| Harbin 35 | 4 | 0.008 |
| Pest C | 8 | 0.015 |
MIC50 and MIC90 are the drug concentrations that inhibit growth of 50% and 90% of the isolates tested in this panel, respectively.
Cloning and expression of SPase.
The primers used in this study are listed in Table 2. Plasmids for the expression of the wild-type Y. pestis SPase were constructed by amplifying the DNA corresponding to the soluble C-terminal fragment of the enzyme and cloning it into the previously constructed pET23b expression vector containing full-length E. coli SPase (14) using the polymerase incomplete primer extension method (26) with the primer pairs vP_EclepBpET-1/vP_EclepBpET-2 and iP_YplepB-3/ip_YplepB-4. This resulted in the replacement of the homologous E. coli SPase soluble C-terminal fragment and the generation of a chimeric construct consisting of an N-terminal His tag, several residues of E. coli SPase, and, finally, the soluble, active fragment of Y. pestis SPase (pET23b-YplepB). To express the P91S variant of this construct, the LepB(P91S) mutation was introduced into pET23b-YplepB using site-directed mutagenesis by inverse PCR with Phusion polymerase (New England BioLabs) and the primer pair 5′_QC_P91S_protein/3′_QC_P91S_protein.
TABLE 2.
Primers used in this study

His-tagged Y. pestis SPase proteins were expressed in BL21(DE3) containing pET23b-YplepB and pET23b-YplepB(P91S) and purified as described previously (14). Briefly, expression was induced with IPTG (isopropyl-β-d-thiogalactopyranoside), and the protein was recovered using Ni-nitrilotriacetic acid (NTA) Superflow resin (Qiagen) in the presence of 1% Elugent detergent (CalBioChem). Fractions were analyzed by SDS-PAGE to identify those with single-band purity, which were then combined, concentrated, and used in subsequent binding assays.
In vitro KD measurements.
Binding assays were done essentially as described previously (13, 14) using fluorescence quenching of arylomycin. Briefly, a 384-well plate was prepared, with wells containing a constant SPase concentration of 50 nM and variable concentrations of drug (ranging from 2 to 8,000 nM) and a control well containing 125 nM the inhibitor saturated with a 6-fold molar excess of enzyme. The assay buffer for both the E. coli and Y. pestis proteins was as follows: 100 mM NaCl, 20 mM Tris-HCl (pH 7.4), 1 mM EDTA, 1% n-octyl-β-glucopyranoside (Anatrace). The plates were incubated in the dark at room temperature for 240 (±30) min. The fluorescence intensity (excitation [λex], 280 nm; emission [λem], 405 nm) was corrected for the intrinsic fluorescence intensity of free protein and inhibitor, and the resulting amplitude as a function of the inhibitor concentration was fitted to the quadratic equation describing two-state equilibrium binding to yield equilibrium dissociation constant (KD) values. The KD values, as well as the total protein concentration, were determined by direct fitting of the data using a nonlinear least-squares fitting algorithm (27). The reported KD values are the averages of the results of at least three independent experiments and are reported with their standard deviations (SD). The precision with which the KD is determined using the nonlinear least-squares method is typically taken to be within a factor of 2 (27). The binding data are also presented graphically using semilog plots, where the data points and error bars represent the average values and standard deviations from the results of independent experiments.
Construction of mutant Y. pestis strains.
The Y. pestis LepB(P91S) mutant strain DBS600 was constructed via allelic exchange using previously described methods (28). Briefly, the point mutation was introduced using overlap PCR with the primer pairs 5′_lepB_flanking_ApaI/3′_P91S_mutant_strain and 5′_P91S_mutant_strain/3′_ lepB_flanking_XbaI, and the amplified DNA was cloned into the ApaI-XbaI site of the suicide vector pKNG101. The construct was propagated in and recovered from the E. coli pir+ DH5α strain BW27343. The recovered construct was transformed into electrocompetent Y. pestis KIM6+ prepared using a previously reported method (29). Colonies in which allelic exchange occurred were selected on 5% sucrose.
The full-length ail deletion in strain DBS600, which expresses LepB(P91S), and insertion of a kanamycin resistance cassette flanked by FLP recognition sequences to generate strain DBS601 were accomplished using lambda Red-mediated recombination as described by Datsenko and Wanner (30). The PCR products used to construct gene replacements were generated using the template plasmid pKD4 and the oligonucleotide primers 5′_ail_KAN and 3′_ail_KAN. The PCR product was purified using the DNA Clean and Concentrator kit (Zymo Research, Irvine, CA) and treated with DpnI. Electrocompetent DBS600 carrying plasmid pKD46, encoding the Red recombinase, were prepared using the same method described above (29) with the modification of induction with 0.2% l-arabinose for 2 h prior to harvest. Competent cells were electroporated with the purified and DpnI-treated PCR product, plated on LB agar containing 35 μg/ml of kanamycin, and grown at 30°C. To verify deletion of ail and replacement with the kanamycin cassette, colony PCR was conducted using the primers 5′_ail_KAN_conf and 3′_ail_KAN_conf. The pKD46 plasmid was cured by growth at 37°C.
Complementation plasmid construction.
Plasmids for the expression of the Y. pestis ail and E. coli malE genes were constructed by amplifying the corresponding DNA from Y. pestis KIM6+ or E. coli K-12 MG1655 using the primer set 5′_pBAD_ail_KpnI/3′_pBAD_ail_HindIII or 5′_pBAD_malE_KpnI/3′_pBAD_malE_HindIII and cloning into the KpnI and HindIII sites of pBAD-HisB to generate pBAD/His B-ail and pBAD/HisB-malE. These plasmids were electroporated into DBS601 [LepB(P91S) Δail] to yield DBS601-pBAD/HisB-malE and DBS601-pBAD/HisB-ail. As a control strain, the empty pBAD/His B vector was also electroporated into DBS601 to yield DBS601-pBAD/HisB.
Susceptibility determinations.
Antibiotic susceptibilities were determined for the strains listed in Table 1 by measuring MICs using the CLSI broth microdilution method (31). Briefly, 2-fold serial dilutions of antibiotics were prepared in 96-well plates containing 100 μl of CAMHB. Bacterial inocula were prepared by suspending colonies grown for 48 h (for Y. pestis) and 16 to 20 h (for all other strains) to a final density of 1 × 107 CFU per ml in CAMHB. Wells containing the antibiotic dilutions were inoculated to a final density of 5 × 105 CFU/ml, and the MICs were defined as the lowest drug concentration at which no visible growth occurred following 24 to 48 h (for Y. pestis) and 16 to 20 h (for all other strains) of incubation at 20°C, 28°C, or 37°C, as appropriate. All susceptibility determinations were performed at least in triplicate.
MICs were determined for the panel of virulent Y. pestis isolates listed in Table 3 by the broth microdilution method in 96-well plates according to CLSI guidelines. Bacterial inocula for Y. pestis strains were prepared by suspending in CAMHB colonies from isolates grown aerobically at 35°C on sheep blood agar (SBA) plates for 42 to 48 h. The suspended cultures were diluted with CAMHB to a bacterial cell density of 105 CFU/ml using 0.5 McFarland standard. Antibiotics were serially diluted 2-fold in 50 μl of CAMHB, and 50 μl of the adjusted bacterial dilution was added to each well of the 96-well plate. The plates were incubated at 35°C. MICs were determined visually at 42 to 48 h and also by optical density (OD) (600 nm; SpectroMax M2 plate reader; Molecular Devices). Susceptibility determinations were performed in triplicate.
Arabinose-induced SPase overexpression.
The contribution of increased SPase expression to arylomycin susceptibility was determined by inoculating strain RS 058 into 96-well plates containing a checkerboard of 2-fold dilutions of arylomycin A-C16 and l-arabinose. To induce overexpression of the E. coli SPase in this Y. pestis strain, colonies were first inoculated into CAMHB supplemented with 0.2% l-arabinose and 5 mM CaCl2 and incubated at 37°C with shaking for 2 to 3 h. The cultures were diluted to a final density of 1 × 107 CFU per ml in fresh CAMHB and inoculated into test wells to a final density of 5 × 105 CFU/ml. The plate was read for growth following 24 h of incubation at 37°C. KIM6+ transfected with an empty pBAD vector was used as a control. The checkerboard analyses between arylomycin A-C16 and l-arabinose were performed at least in triplicate.
Arabinose-induced complementation.
To determine the effect of ectopic expression of E. coli MalE and Y. pestis Ail on the MICs of DBS601 [LepB(P91S) Δail], colonies of DBS601, DBS601-pBAD/HisB, DBS601-pBAD/HisB-malE, and DBS601-pBAD/HisB-ail were inoculated into CAMHB supplemented with 4 mM CaCl2 (plus 50 μg/ml ampicillin for the pBAD/HisB-containing strains) and incubated at 37°C with shaking overnight. The cultures were diluted 1:40 in corresponding fresh medium and incubated at 37°C with shaking for 2 to 3 h. The cultures were diluted to a final density of 1 × 107 CFU per ml in fresh CAMHB and inoculated into test wells to a final density of 5 × 105 CFU/ml. Testing was conducted in 96-well plates containing serial 2-fold dilutions of arylomycin A-C16 and an l-arabinose concentration of 0, 0.2, or 2% and performed at least in triplicate. The plates were read for growth following 24 h of incubation at 37°C.
Arylomycin A-C16–polymyxin B nonapeptide checkerboard analysis.
The contribution of increased membrane permeability to arylomycin susceptibility was determined by inoculating 96-well plates containing a checkerboard of 2-fold dilutions of arylomycin A-C16 and PMBN with E. coli strain PAS0260 harboring LepB(P84L), E. coli strain BAS901, Y. pestis KIM6+, Y. pestis strain DBS600 harboring LepB(P91S), Y. pestis KIM5, and Y. pestis KIM5-pLpxL. Bacterial inocula were prepared by suspending colonies grown for 48 h (for Y. pestis) and 16 to 20 h (for all other strains) to a final density of 1 × 107 CFU per ml in CAMHB. All test wells were inoculated to a final density of 5 × 105 CFU/ml, and the plate was read for growth following 24 h of incubation at either 28°C or 37°C, as appropriate. The checkerboard analyses between arylomycin A-C16 and PMBN were performed at least in triplicate.
Synergy studies with protein synthesis inhibitors.
The interaction of arylomycin and tetracycline or gentamicin was determined by inoculating the strains to be tested into 96-well plates containing a checkerboard of 2-fold dilutions of arylomycin A-C16 and either tetracycline or gentamicin. Bacterial inocula were prepared by suspending colonies grown for 48 h to a final density of 1 × 107 CFU per ml in CAMHB. All test wells were inoculated to a final density of 5 × 105 CFU/ml, and the plate was read for growth following 24 h of incubation at 37°C, as appropriate. The checkerboard analyses were performed at least in triplicate. Antibiotic interactions were determined by measuring the fractional inhibitory concentration (FIC) index of the microdilution checkerboard consisting of two drugs (for simplicity, drugs A and B). The FIC is the MIC of drug A or B in combination divided by the MIC of that drug alone, and the FIC index is defined as follows: ΣFICdrug A + FICdrug B. A FIC index of ≤0.5 denotes synergy, whereas a FIC index of ≥4 represents antagonism (32). The minimum and maximum FIC indexes are reported for each drug combination.
Determination of protein localization.
The levels of membrane and periplasmic proteins were determined by isolating the cold-shocked cell and periplasmic fractions from cells grown with and without arylomycin A-C16 using a previously reported protocol with some modification (33). Briefly, cells were grown with shaking at 37°C overnight in tryptic soy broth (TSB) supplemented with 4 mM CaCl2 to an OD of ∼0.3. The cells were pelleted, resuspended in fresh medium, grown with or without arylomycin A-C16 (0.5× MIC) with shaking at 37°C for an additional 3 h, and then pelleted at 1,300 × g for 10 min at 4°C. The supernatant was discarded, and the pellet was gently resuspended in 1/4 the original culture volume of ice-cold sucrose buffer (50 mM Tris, pH 7.4, 1 mM EDTA, 20% [wt/vol] sucrose) and incubated on ice for 10 min. The resulting suspension was centrifuged at 5,200 × g for 10 min at 4°C. The supernatant was discarded, and the pellets were resuspended in 1/10 the original culture volume of ice-cold 5 mM MgCl2. The incubation and centrifugation steps were repeated to yield the cold osmotic shock solution that contained the periplasmic proteins (supernatant) and the shocked-cell fraction that contained the membrane-bound proteins (pellet). The periplasmic fraction was precipitated with 10% trichloroacetic acid overnight and washed with cold 90% acetone, and the resulting protein pellet was resuspended in 50 mM NH4HCO3, 1% SDS. The shocked-cell fraction was treated with DNase and lysozyme and lysed by three cycles of freezing-thawing. The protein concentrations of the samples were determined using the Micro BCA Protein Assay kit (Life Technologies, Grand Island, NY) and were normalized by OD.
RESULTS
Susceptibility of Y. pestis to arylomycin A-C16.
We first characterized the susceptibility of Y. pestis to arylomycin A-C16 as a function of SPase activity by determining the sensitivity of a KIM6+ derivative strain (RS 058) that overexpresses E. coli SPase in trans under arabinose induction. A mock-transfected KIM6+ strain was assessed in parallel for comparison. The addition of arabinose alone did not significantly affect growth for either strain, and the addition of arabinose did not affect the arylomycin MIC of the mock-transfected strain. However, the arylomycin MIC values showed a clear concentration dependence for RS 058, with complete resistance (>64 μg/ml) observed at high arabinose concentrations (Fig. 2). These results confirm that the activity of the arylomycins against Y. pestis results from the inhibition of SPase.
FIG 2.
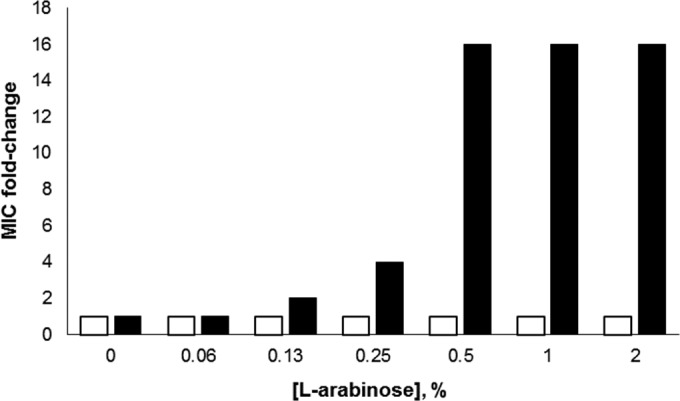
Arylomycin A-C16 MIC change for Y. pestis RS 058 (black bars), which contains pBAD-EcLepB, and a mock-transfected control Y. pestis containing pBAD-empty (white bars) as a function of increasing l-arabinose. The MICs for RS 058 and the mock-transfected Y. pestis in the absence of l-arabinose were 8 and 2 μg/ml, respectively. For RS 058, the arylomycin A-C16 MICs at 0.5 to 2% l-arabinose were >64 μg/ml. To calculate the fold change, the MICs were taken to be 128 μg/ml.
Having verified the on-target activity of arylomycin with KIM6+, we sought to determine its spectrum of activity against a broad range of other strains using a panel of 30 natural isolates representing all three biovars and including strains CO92, Antiqua, KIM10, and Angola (Table 3). As a control, MIC values for ciprofloxacin were determined and found to range between 0.008 and 0.06 μg/ml, as expected. The arylomycin A-C16 MIC50 and MIC90 values for the panel were 8 and 32 μg/ml, respectively, demonstrating the conservation of arylomycin susceptibility across this broad range of Y. pestis strains.
To quantitatively compare the sensitivity of Y. pestis to that of E. coli, which is naturally resistant (thus precluding a quantitative measurement of sensitivity), we constructed a Y. pestis strain harboring the sensitivity-conferring SPase point mutation P91S (strain DBS600) for comparison with the analogously sensitized E. coli strain (P84L; strain PAS0260), which has an MIC of 1 to 2 μg/ml (14). The mutant Y. pestis strain has an arylomycin A-C16 MIC of 0.03 μg/ml, which is reduced 128-fold compared to the parental Y. pestis strain. Comparison with PAS0260 reveals that Y. pestis is 30- to 60-fold more sensitive than E. coli to arylomycin A-C16.
The activity of the arylomycins against Y. pestis does not result from increased affinity for SPase.
To investigate whether the sensitivity of Y. pestis relative to E. coli is caused by a greater affinity of its SPase for the arylomycins, we determined equilibrium binding constants using micelle-reconstituted recombinant SPase. Wild-type Y. pestis SPase and the arylomycin-sensitized P91S mutant were expressed in E. coli as chimeric constructs with the N-terminal 66 residues of the E. coli protein. These constructs preserve the entire catalytic domain of the Y. pestis enzyme but incorporate the E. coli signal sequence and membrane anchor, which our experience suggested would facilitate expression in E. coli.
Using a fluorescence-quenching assay, the binding constants (KD ± SD) for arylomycin A-C16 were found to be 31 ± 14 nM for the wild-type Y. pestis protein and 6 ± 2 nM for the P91S variant (Fig. 3). These dissociation constants are similar to those determined for the E. coli protein and its corresponding P84S mutant (60 ± 16 nM and 5.7 ± 1.0 nM, respectively) (14; see Fig. S1 in the supplemental material). These results demonstrate that the difference in susceptibility between Y. pestis and E. coli does not result from altered SPase affinity for the arylomycins.
FIG 3.
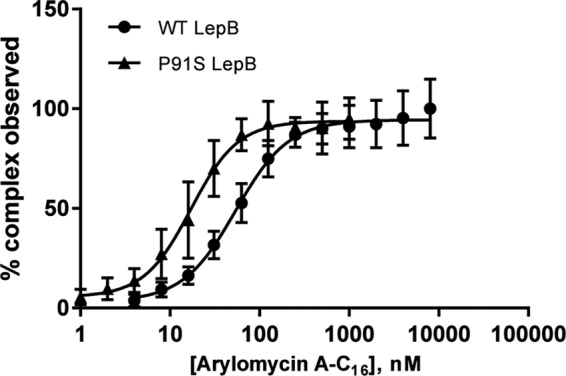
Equilibrium binding curves for wild-type and sensitized P91S Y. pestis LepB. The error bars represent SD.
The arylomycin susceptibility of Y. pestis is temperature dependent.
Y. pestis is able to survive and propagate in insect and mammalian hosts due to its ability to regulate its membrane characteristics and proteome expression in response to environmental cues, such as temperature and cation (i.e., calcium or iron) concentration (34–37). To test whether such factors are important for arylomycin sensitivity, we determined arylomycin A-C16 MICs with Y. pestis KIM6+ at 20, 28, and 37°C (Table 4). We observed an inverse correlation between the MIC and the temperature, with the lowest MIC (4 μg/ml) at 37°C and the highest (>64 μg/ml) at 20°C. Supplementation of the medium with calcium or iron did not have a significant effect on the arylomycin MIC at any of the temperatures examined (data not shown). A similar trend was observed with the hydrophobic antibiotic actinomycin D and the hydrophobic bile salt deoxycholic acid. These data suggest that the sensitivity of Y. pestis to the arylomycins may result from temperature-dependent alterations of the pathogen's membrane, as is thought to be the case with hydrophobic agents (38), or from changes in secreted proteins.
TABLE 4.
MICs of actinomycin D, arylomycin A-C16, and deoxycholic acid for Gram-negative strains determined at 37, 28, and 20°C
| Temp (°C) | Strain | MIC (μg/ml) |
||
|---|---|---|---|---|
| Actinomycin D | Arylomycin A-C16 | Deoxycholic acid | ||
| 37 | E. coli MG1655 | >64 | >64 | >50,000 |
| Y. enterocolitica ATCC 23715 | 64 | >64 | >50,000 | |
| Y. pestis KIM6+ | 4 | 4 | 6,250 | |
| Y. pestis KIM6+ LepB(P91S) (DBS600) | 4 | 0.03 | 6,250 | |
| E. coli lptD4213 (BAS901) | 0.06 | 16 | 500 | |
| E. coli LepB(P84L) (PAS0260) | 64 | 2 | >50,000 | |
| Y. pestis KIM6+ ΔphoP | 8 | 16 | 25,000 | |
| Y. pestis KIM5 | 8 | 4 | 3,100 | |
| Y. pestis KIM5-pLpxL | 16 | 8 | 25,000 | |
| Y. pestis KIM6+ LepB(P91S) Δail (DBS601) | 16 | 1 | 12,500 | |
| 28 | Y. enterocolitica ATCC 23715 | 64 | >128 | >50,000 |
| Y. pestis KIM6+ | 32 | 16 | 50,000 | |
| Y. pestis KIM6+ LepB(P91S) (DBS600) | 32 | 0.06 | 50,000 | |
| E. coli lptD4213 (BAS901) | −a | 16 | − | |
| E. coli LepB(P84L) (PAS0260) | >64 | 2 | >50,000 | |
| Y. pestis KIM6+ ΔphoP | 32 | >64 | 50,000 | |
| Y. pestis KIM5 | >64 | 16 | 50,000 | |
| Y. pestis KIM5-pLpxL | >64 | 16 | 50,000 | |
| Y. pestis KIM6+ LepB(P91S)Δail (DBS601) | 32 | 1 | 50,000 | |
| 20 | Y. pestis KIM6+ | >64 | >64 | 50,000 |
| Y. pestis KIM6+ LepB(P91S) (DBS600) | − | 1 | − | |
| E. coli lptD4213 (BAS901) | − | 16 | − | |
| E. coli LepB(P84L) (PAS0260) | >64 | 2 | >50,000 | |
| Y. pestis KIM6+ ΔphoP | >64 | >64 | 50,000 | |
| Y. pestis KIM5 | 32 | >64 | 50,000 | |
| Y. pestis KIM5-pLpxL | − | >64 | − | |
| Y. pestis KIM6+ LepB(P91S)Δail (DBS601) | 64 | 1 | 50,000 | |
−, not determined.
The unique LPS of Y. pestis provides at most a small contribution to arylomycin sensitivity.
In general, the LPS barrier of Gram-negative bacteria is thought to provide an effective barrier that prevents large and hydrophobic molecules from passing through the outer membrane. Interestingly, Y. pestis is unique in that it possesses “rough,” or R-type, LPS that lacks the O antigen (39), and it has been shown to be more susceptible to hydrophobic agents than other Yersinia species (38). Moreover, Y. pestis has been shown to modify its LPS as a function of temperature, and such modifications have been shown to impart to Y. pestis a temperature-dependent susceptibility to several agents that is similar to that observed with arylomycin A-C16 (38, 40).
To begin to explore the potential contribution of the LPS barrier to arylomycin sensitivity, we first examined a panel of Gram-negative bacteria with different LPS characteristics. We also examined the responses of the panel to the effects of added PMBN, which selectively permeabilizes LPS (41), using a checkerboard analysis with serial 2-fold dilutions of arylomycin A-C16 and PMBN (Fig. 4) (the PMBN MICs for all strains tested were >256 μg/ml, and thus, PMBN has no activity on its own at the concentrations employed). E. coli BAS901, which lacks LPS altogether due to an lptD mutation, is hypersensitive to arylomycin A-C16, and PMBN coadministration does not increase its sensitivity further, consistent with the complete absence of an LPS barrier. Conversely, PMBN coadministration dramatically sensitizes E. coli PAS0260 to arylomycin A-C16 (as well as E. coli MG1655 [20]), consistent with the intact E. coli LPS acting as a barrier to arylomycin penetration. The effect of PMBN on E. coli PAS0260 is independent of temperature. At 37°C, PMBN had a modest effect on the sensitivity of Y. pestis KIM6+ and its arylomycin-sensitized P91S SPase strain, but the effect was more pronounced at 28°C. These data suggest that the Y. pestis LPS is a somewhat less efficient barrier to arylomycin penetration than is that of E. coli, or possibly that PMBN is less efficient at disrupting the LPS of Y. pestis.
FIG 4.
Median fractional arylomycin A-C16 MICs as a function of increasing PMBN concentrations at 37°C and 28°C. The fractional arylomycin A-C16 MIC is equal to the MIC of arylomycin A-C16 in the presence of the tested PMBN concentration divided by the MIC of arylomycin A-C16 alone. The shading of the bars indicates the PMBN concentration tested (1 to 64 μg/ml in a 2-fold dilution series). The strains tested (the MICs in the absence of PMBN are shown in parentheses) were Y. pestis KIM6+ (4 μg/ml at 37°C; 16 μg/ml at 28°C), Y. pestis expressing P91S LepB (DBS600) (0.03 μg/ml at 37°C; 0.06 μg/ml at 28°C), E. coli expressing P84L LepB (PAS0260) (2 μg/ml at 37°C and 28°C), and the E. coli lptD4213 mutant (BAS901) (16 μg/ml). To standardize the MIC values among the different strains, the changes in the MICs are reported as fractional arylomycin A-C16 MICs, which are defined as the MIC of arylomycin A-C16 in the presence of the tested PMBN concentration divided by the MIC of arylomycin A-C16 alone.
Like Y. pestis in general, E. coli MG1655 (and its P84L SPase derivative, PAS0260) does not express the LPS O antigen (in this case due to a defect in the rfb gene cluster [42]); thus, the resistance of these strains suggests that the loss of the O antigen is unlikely to be the cause of Y. pestis sensitivity. To further support this conclusion, we examined Yersinia enterocolitica, which downregulates O antigen expression at higher temperatures (43). Consistent with the unimportance of the O antigen, this strain was resistant to arylomycin A-C16 at both 28 and 37°C (Table 4).
While there is typically one major variant of the LPS core oligosaccharide when Y. pestis is cultured at 37°C, modifications are made when Y. pestis is cultured at lower temperatures, resulting in the production of up to four different core isoforms (44–48; reviewed in reference 39). However, different subspecies of Y. pestis have been shown to have different core structures (46), and the general susceptibility of Y. pestis spp. to arylomycin (see above) suggests that core modifications do not substantially contribute to the observed sensitivity. To more systematically address the contribution of the LPS core to arylomycin permeability, we examined the sensitivities of a panel of isogenic E. coli mutants with well-defined core truncations: strain D21 lacks the O antigen but retains the entire LPS core, strain D21e7 contains only the heptose portion of the core plus a glucose residue, strain D21e19 is similar to strain D21e7 but contains an additional galactose residue, strain D21f1 contains only the Kdo and heptose portions of the core, and strain D21f2 contains only the Kdo residue. Despite the varying LPS truncations, each of these strains is resistant to arylomycin A-C16 (MIC > 128 μg/ml). Thus, as with the loss of the O antigen, it is unlikely that the LPS core of Y. pestis is itself sufficient to account for sensitivity to the arylomycins.
Y. pestis modifies its LPS with 4-amino-4-deoxy-l-arabinose (Ara4N), which is mediated by the two-component response regulator PhoPQ (49). Moreover, the Ara4N content increases with decreased temperature, and these modifications have been linked to resistance to cationic antimicrobial peptides (49–51). To examine the contribution of Ara4N modification to arylomycin sensitivity, we examined the arylomycin A-C16 sensitivity of a Y. pestis phoP knockout strain (KIM6+ ΔphoP). The loss of phoP results in reduced susceptibility to arylomycin A-C16 (Table 4). The addition of 10 mM MgCl2 to the growth media, which is known to repress expression of phoPQ (52), also resulted in reduced arylomycin susceptibility of wild-type Y. pestis, but not the phoP deletion strain or E. coli strain BAS901 or PAS0260 (Table 5). Regardless of the specific mechanism, it is clear that the PhoP function actually sensitizes Y. pestis to the arylomycins, and because the Ara4N content and susceptibility in the context of the phoP deletion show opposite dependencies on temperature, LPS modification with Ara4N is unlikely to underlie the arylomycin sensitivity of Y. pestis.
TABLE 5.
Arylomycin A-C16 MICs in the presence of 10 mM Mg2+
| Strain | MIC (μg/ml) |
||
|---|---|---|---|
| 37°C | 28°C | 20°C | |
| Y. pestis | |||
| KIM6+ | 16 | >128 | >128 |
| KIM6+ ΔphoP | 16 | >128 | >128 |
| LepB(P91S) (DBS600) | 0.125 | 0.25 | 2 |
| KIM5 | 16 | >128 | >128 |
| KIM5 pLpxL | 16 | 16 | >128 |
| E. coli | |||
| lptD4213 (BAS901) | 16 | 16 | 8 |
| LepB(P84L) (PAS0260) | 2 | 4 | 4 |
At 37°C, lipid A of Y. pestis primarily exists in a tetra-acylated form, while at lower temperatures there is a shift toward the production of the hexa-acylated variant, which is more similar to that produced by other enterobacteria and is thought to result in lower permeability due to increased packing (39). To explore the contribution of lipid A acylation to arylomycin sensitivity, we extended our studies to Y. pestis KIM5-pLpxL, an engineered Y. pestis strain that expresses the E. coli acyltransferase LpxL and almost exclusively expresses hexa-acylated lipid A at all temperatures (53). At 37°C, Y. pestis KIM5-pLpxL was 2-fold less sensitive to arylomycin A-C16 than the KIM5 parental strain (Table 4). This is similar to the effect observed with actinomycin D but less than that observed with deoxycholic acid at 37°C. This suggests that, while the acylation state of Y. pestis lipid A is important for deoxycholic acid penetration through the LPS, it is less important for actinomycin D or arylomycin A-C16. In addition, coadministration of PMBN resulted in a very modest increase in arylomycin sensitivity (Fig. 5). While the effects on sensitivity and synergy with PMBN are small, they are reproducible, suggesting that the reduced acylation state of Y. pestis lipid A may underlie the slightly reduced efficiency of the Y. pestis LPS barrier to arylomycin penetration at 37°C. Nonetheless, the data demonstrate that core alterations in LPS structure on their own are not the dominant contributors to Y. pestis sensitivity to the arylomycins.
FIG 5.
Median fractional arylomycin A-C16 MICs as a function of increasing PMBN concentrations at 37°C and 28°C for Y. pestis KIM5 and KIM5-pLpxL. The arylomycin A-C16 MICs in the absence of PMBN were 4 μg/ml (37°C) and 16 μg/ml (28°C) for KIM5 and 8 μg/ml (37°C) and 16 μg/ml (28°C) for KIM5-pLpxL. The fractional arylomycin A-C16 MIC is equal to the MIC of arylomycin A-C16 in the presence of the tested PMBN concentration divided by the MIC of arylomycin A-C16 alone. The shading of the bars indicates the PMBN concentration tested (1 to 64 μg/ml in a 2-fold dilution series).
The arylomycin susceptibility of Y. pestis results from a temperature-dependent increase in protein secretion.
There are significant increases in the secretion of several proteins in Y. pestis at 37°C relative to lower temperatures (36, 37, 54, 55), and exacerbation of the associated secretion burden by inhibiting SPase could underlie the sensitivity to the arylomycins at physiological temperatures. Consistent with this possibility, reducing translation via mutation or the addition of translational inhibitors has been shown to rescue defects in the E. coli general secretory pathway (56). To begin to test this possibility, we first performed synergy studies with tetracycline, which inhibits translation without causing mistranslation, and with gentamicin, which causes mistranslation and the toxic accumulation of aberrant peptides in the cytoplasmic membrane (57) (Table 6). Antibiotic interactions were determined by measuring the FIC index using a microdilution checkerboard assay consisting of combinations of arylomycin A-C16 and either tetracycline or gentamicin. An FIC index of ≤0.5 denotes synergy, whereas an FIC index of ≥4 represents antagonism (32). The minimum (FICmin) and maximum (FICmax) FIC indexes measured are reported for each drug combination.
TABLE 6.
MICs of antibiotics alone and fractional inhibitory concentration indexes of antibiotics in combination with arylomycin A-C16 at 37°Ca
| Antibiotic |
E. coli PAS0260 |
Y. pestis |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KIM6+ |
DBS600 |
DBS601 |
||||||||||
| MIC | FICmin | FICmax | MIC | FICmin | FICmax | MIC | FICmin | FICmax | MIC | FICmin | FICmax | |
| Gentamicin | 1 | 0.58 ± 0.29 | 1.09 ± 0.05 | 2 | 0.60 ± 0.06 | 1.07 ± 0.06 | − | − | − | − | − | − |
| Tetracycline | 1 | 0.90 ± 0.23 | 2.35 ± 0.25 | 2 | 1.70 ± 0.59 | 8.42 ± 0.14 | 1 | 1.55 ± 0.59 | 5.50 ± 2.00 | 2 | 1.04 ± 0.02 | 2.25 ± 0.50 |
The FIC values are averages ± standard deviations. The MIC values are in micrograms per milliliter. −, not determined.
Based on the FIC indexes, the inhibition of protein synthesis effected by tetracycline cotreatment significantly rescues Y. pestis from the effects of arylomycin, and the mitigation of arylomycin's effects observed with Y. pestis is much greater than that observed for the sensitized PAS0260 E. coli strain expressing SPase with the P84L mutation (Table 6) (58). However, the FIC indexes for Y. pestis with gentamicin cotreatment do not differ significantly from those reported for PAS0260 (Table 6) (58). These data suggest that with the inhibition of SPase, Y. pestis is hypersensitive to the accumulation of normal proteins in the cytoplasmic membrane.
Y. pestis significantly increases the secretion of four proteins at 37°C relative to lower temperatures: the cell adhesion protein Ail (y1324) and the three hypothetical small β-barrel OM proteins (y1795, y2167, and y4083) (36). All four proteins are predicted (by SignalP version 3.0 [59]) to be translated with signal peptides, suggesting that their increased expression may create a high demand for SPase activity. Ail is a secreted protein, and at 37°C, it accounts for approximately 25% of the Y. pestis outer membrane proteome (60). Thus, to determine the extent to which this temperature-dependent secretion accounts for the increased sensitivity of Y. pestis relative to E. coli, we deleted ail in the arylomycin-sensitized strain DBS600, yielding strain DBS601 (Table 4). Deletion of ail resulted in 30- and 16-fold increases in the arylomycin MIC at 37°C and 28°C, respectively, and no change at 20°C. The increases in MICs that result from the deletion of ail result in the Y. pestis and E. coli strains (DBS600 and PAS0260, respectively) showing identical sensitivities. Moreover, synergy studies with tetracycline revealed that upon deletion of ail, Y. pestis and E. coli have indistinguishable FICs (Table 6). Finally, complementation with the plasmid-borne and l-arabinose-inducible ail or E. coli malE resulted in resensitization to arylomycin (Table 7). These data suggest that the increased secretion burden experienced by Y. pestis underlies virtually all of its increased sensitivity relative to E. coli.
TABLE 7.
MICs of arylomycin A-C16 as a function of added l-arabinose determined for Y. pestis P91S LepB Δail (DBS601) complemented with various constructs at 37°C
| Construct | MIC (μg/ml) at l-arabinose level (%): |
||
|---|---|---|---|
| 0 | 0.2 | 2 | |
| No vector | 1 | 1 | 1 |
| pBAD/His B (empty) | 1 | 1 | 0.5 |
| pBAD/His B-malE | 1 | 1 | 0.125 |
| pBAD/His B-ail | 1 | 0.5 | 0.03 |
To further support the conclusion that the altered sensitivities result from changes in the secretion burden, we assessed protein levels in a lysed cold-shocked cell fraction (which contained the cytoplasmic membrane) and the periplasmic fraction as a function of ail deletion and arylomycin addition (Fig. 6). As expected, the deletion of ail reduced the levels of proteins found in both fractions. Moreover, in the presence of Ail, the addition of arylomycin A-C16 increased the level of proteins in the shocked-cell fraction and decreased it in the periplasmic fraction, consistent with arylomycin sensitivity resulting from protein mislocalization. In contrast, in the absence of Ail, the addition of arylomycin had a diminished effect on the level of protein found in either fraction, consistent with resistance resulting from reduced protein mislocalization.
FIG 6.
Determination of protein content in the cold-shocked cell and periplasmic fractions of Y. pestis DBS600 and its Δail derivative, DBS601, with and without arylomycin A-C16 treatment (0.5× MIC).
DISCUSSION
Gram-negative bacteria are emerging as a particularly troubling threat due to the evolution of resistance to available antibiotics and the challenge of developing new antibiotics. Protein secretion is an essential process in all bacteria, and the general secretory pathway is conserved across both Gram-positive and Gram-negative species (61–64). Interestingly, compared to Gram-positive organisms, a far greater number of proteins are processed by SPase in Gram-negative bacteria, which must maintain a protein-rich periplasm and outer membrane (65, 66), suggesting that targeting SPase may be a particularly effective strategy to combat these pathogens. Our previous demonstration that the arylomycin class of antibiotics, which act by inhibiting SPase, has the potential for broad-spectrum activity (14, 25, 58, 67), including against Gram-negative pathogens, suggests that they might be interesting for development as antibiotics. However, this will require an understanding of the varying susceptibilities of different bacteria and their physiological origins. Previously, we demonstrated that a specific SPase allele is responsible for much of the variations in sensitivity (14, 25), but variations among different species, or even within species, that possess the same SPase allele demonstrates that other factors must also contribute.
Among the Enterobacteriaceae tested that have SPases possessing the predicted arylomycin-resistant SPase allele, Y. pestis KIM6+ was unique in its susceptibility to the arylomycins (14). We have now shown that the arylomycins in fact show at least reasonable activity against a broad range of Y. pestis strains, including representative strains from all three classical biovars (antiqua, medievalis, and orientalis). This broad and at least relatively potent activity contrasts sharply with their activities against other Enterobacteriaceae, such as E. coli, which are typically resistant (14). While SPase is highly conserved, there are sequence differences among bacteria, and a trivial explanation for the higher sensitivity of Y. pestis is that arylomycins bind to its SPase with higher affinity. However, based on a comparison of the arylomycin dissociation constant with that determined previously for the E. coli protein (14), this is not the case.
A hint as to the factor(s) that might underlie the atypical arylomycin sensitivity of Y. pestis is its temperature dependence. We determined that the increased sensitivity of Y. pestis to the arylomycins is manifest only at higher (physiological) temperatures, with full resistance observed at 20°C. Interestingly, Y. pestis possesses an atypically truncated, or rough, LPS, and it has been shown to modify its LPS as a function of temperature. The ability of LPS to prevent large hydrophobic antibiotics from accessing periplasmic or cytoplasmic targets in many bacteria, the sensitization caused by chemical or genetic perturbations of the LPS with many of the same bacteria, and the fact that the observed temperature-dependent sensitivity of Y. pestis to arylomycin A-C16 appears to mirror changes in its LPS suggested that the atypical LPS of Y. pestis may be the cause of the arylomycin sensitivity. Indeed, the addition of PMBN, which disrupts LPS, had a limited effect on the susceptibility of Y. pestis to the arylomycin compared to E. coli, suggesting either that the Y. pestis LPS is a less efficient barrier to arylomycin penetration or that it is less efficiently disrupted by PMBN. However, little or no change in sensitivity to the arylomycin was observed upon alteration or deletion of the LPS O antigen or core or alteration of lipid A via acylation or with Ara4N. This suggests that while the unusual truncated, or rough, LPS of Y. pestis is somewhat less efficient at excluding the arylomycins, it is unlikely in and of itself to be the cause of the increased sensitivity of Y. pestis relative to E. coli.
A final possibility examined was that the known temperature-dependent increases in protein secretion (36) exacerbate the effects of arylomycin-mediated SPase inhibition at physiological temperatures. To test this hypothesis, we deleted the nonessential gene encoding the Ail protein, which is secreted and constitutes 25% of the outer membrane proteome. We found that deletion of ail reduced arylomycin sensitivity and synergy with tetracycline to levels indistinguishable from those of E. coli and that ectopic expression of ail or E. coli malE in the Δail strain restored sensitivity. In sensitized E. coli, we have previously shown that arylomycin A-C16 is bactericidal and that this activity appeared to result from protein mislocalization in the cytoplasmic membrane and either excessive accumulation of unprocessed preproteins, and perhaps membrane depolarization, or the misregulation of essential periplasmic and outer membrane processes (58). Similarly, it seems likely that the secretion-dependent sensitization of Y. pestis to arylomycin results from the accumulation of unprocessed proteins in the cytoplasmic membrane or from competition with the processing of essential proteins. This model is also consistent with the overexpression of SPase reducing the sensitivity of wild-type Y. pestis. However, we cannot exclude the possibility that the secretion burden and a slightly reduced LPS barrier act synergistically. Regardless of the detailed mechanism, the results demonstrate that the demands associated with the high levels of secretion induced at 37°C exacerbate the effects of SPase inhibition and account for most of the differences in arylomycin sensitivity between Y. pestis and E. coli.
Y. pestis has adapted to the different environments required for survival in its insect vector and in its mammalian host (68), in part by regulating the proteins secreted in response to different environmental cues (34–37). The increased secretion of Ail at 37°C is thought to facilitate survival in its mammalian host by mediating resistance to complement and adherence to epithelial cells (69, 70). However, reliance on this protein secretion appears to render Y. pestis more susceptible to SPase inhibition and thus to the arylomycins. Interestingly, the viability and/or virulence of many bacteria in different contexts depends on secretion in response to particular environmental cues (71–74), and the secretion of many of these proteins is mediated by the general secretory pathway and SPase. Because these cues are often not present during routine analysis of antibiotic susceptibility, the effect of SPase inhibition may be commonly underestimated. Thus, it seems likely that in the actual context of an infection, the arylomycins may possess a more potent and broad-spectrum activity than predicted based on MICs, especially against Gram-negative bacteria, which possess a single SPase and where high levels of secretion are typically required for the protein-rich periplasm and outer membrane of these organisms (65, 66). Along with the previously demonstrated activity of the arylomycins against many Gram-positive bacteria (25, 67), this reinforces the idea that they may be promising candidates for further development as therapeutics.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by Public Health Service grant AI-092182 from the National Institute of Allergy and Infectious Diseases.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00181-15.
REFERENCES
- 1.Perry RD, Fetherston JD. 1997. Yersinia pestis—etiologic agent of plague. Clin Microbiol Rev 10:35–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diamond J. 2002. Evolution, consequences and future of plant and animal domestication. Nature 418:700–707. doi: 10.1038/nature01019. [DOI] [PubMed] [Google Scholar]
- 3.Kugeler KJ, Staples JE, Hinckley AF, Gage KL, Mead PS. 2015. Epidemiology of human plague in the United States, 1900-2012. Emerg Infect Dis 21:16–22. doi: 10.3201/eid2101.140564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galimand M, Carniel E, Courvalin P. 2006. Resistance of Yersinia pestis to antimicrobial agents. Antimicrob Agents Chemother 50:3233–3236. doi: 10.1128/AAC.00306-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galimand M, Guiyoule A, Gerbaud G, Rasoamanana B, Chanteau S, Carniel E, Courvalin P. 1997. Multidrug resistance in Yersinia pestis mediated by a transferable plasmid. N Engl J Med 337:677–680. doi: 10.1056/NEJM199709043371004. [DOI] [PubMed] [Google Scholar]
- 6.Guiyoule A, Gerbaud G, Buchrieser C, Galimand M, Rahalison L, Chanteau S, Courvalin P, Carniel E. 2001. Transferable plasmid-mediated resistance to streptomycin in a clinical isolate of Yersinia pestis. Emerg Infect Dis 7:43–48. doi: 10.3201/eid0701.010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Welch TJ, Fricke WF, McDermott PF, White DG, Rosso ML, Rasko DA, Mammel MK, Eppinger M, Rosovitz MJ, Wagner D, Rahalison L, Leclerc JE, Hinshaw JM, Lindler LE, Cebula TA, Carniel E, Ravel J. 2007. Multiple antimicrobial resistance in plague: an emerging public health risk. PLoS One 2:e309. doi: 10.1371/journal.pone.0000309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Butler MS, Blaskovich MA, Cooper MA. 2013. Antibiotics in the clinical pipeline in 2013. J Antibiot 66:571–591. doi: 10.1038/ja.2013.86. [DOI] [PubMed] [Google Scholar]
- 9.Boucher HW, Talbot GH, Benjamin DK Jr, Bradley J, Guidos RJ, Jones RN, Murray BE, Bonomo RA, Gilbert D, Infectious Diseases Society of America. 2013. 10 × '20 progress—development of new drugs active against gram-negative bacilli: an update from the Infectious Diseases Society of America. Clin Infect Dis 56:1685–1694. doi: 10.1093/cid/cit152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hancock RE. 1984. Alterations in outer membrane permeability. Annu Rev Microbiol 38:237–264. doi: 10.1146/annurev.mi.38.100184.001321. [DOI] [PubMed] [Google Scholar]
- 11.Nikaido H. 1979. Permeability of the outer membrane of bacteria. Angew Chem Int Ed Engl 18:337–350. doi: 10.1002/anie.197903373. [DOI] [PubMed] [Google Scholar]
- 12.Hitchcock PJ, Leive L, Makela PH, Rietschel ET, Strittmatter W, Morrison DC. 1986. Lipopolysaccharide nomenclature—past, present, and future. J Bacteriol 166:699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paetzel M, Goodall JJ, Kania M, Dalbey RE, Page MG. 2004. Crystallographic and biophysical analysis of a bacterial signal peptidase in complex with a lipopeptide-based inhibitor. J Biol Chem 279:30781–30790. doi: 10.1074/jbc.M401686200. [DOI] [PubMed] [Google Scholar]
- 14.Smith PA, Roberts TC, Romesberg FE. 2010. Broad-spectrum antibiotic activity of the arylomycin natural products is masked by natural target mutations. Chem Biol 17:1223–1231. doi: 10.1016/j.chembiol.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan YX, Romesberg FE. 2012. Latent antibiotics and the potential of the arylomycins for broad-spectrum antibacterial activity. Medchemcomm 3:916–925. doi: 10.1039/c2md20043k. [DOI] [Google Scholar]
- 16.Paetzel M, Dalbey RE, Strynadka NC. 2000. The structure and mechanism of bacterial type I signal peptidases. A novel antibiotic target. Pharmacol Ther 87:27–49. doi: 10.1016/S0163-7258(00)00064-4. [DOI] [PubMed] [Google Scholar]
- 17.Auclair SM, Bhanu MK, Kendall DA. 2012. Signal peptidase I: cleaving the way to mature proteins. Protein Sci 21:13–25. doi: 10.1002/pro.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schimana J, Gebhardt K, Höltzel A, Schmid DG, Sussmuth R, Muller J, Pukall R, Fiedler HP. 2002. Arylomycins A and B, new biaryl-bridged lipopeptide antibiotics produced by Streptomyces sp. Tu 6075. I. Taxonomy, fermentation, isolation and biological activities. J Antibiot 55:565–570. doi: 10.7164/antibiotics.55.565. [DOI] [PubMed] [Google Scholar]
- 19.Kulanthaivel P, Kreuzman AJ, Strege MA, Belvo MD, Smitka TA, Clemens M, Swartling JR, Minton KL, Zheng F, Angelton EL, Mullen D, Jungheim LN, Klimkowski VJ, Nicas TI, Thompson RC, Peng SB. 2004. Novel lipoglycopeptides as inhibitors of bacterial signal peptidase I. J Biol Chem 279:36250–36258. doi: 10.1074/jbc.M405884200. [DOI] [PubMed] [Google Scholar]
- 20.Roberts TC, Smith PA, Cirz RT, Romesberg FE. 2007. Structural and initial biological analysis of synthetic arylomycin A2. J Am Chem Soc 129:15830–15838. doi: 10.1021/ja073340u. [DOI] [PubMed] [Google Scholar]
- 21.Roberts TC, Smith PA, Romesberg FE. 2011. Synthesis and biological characterization of arylomycin B antibiotics. J Nat Prod 74:956–961. doi: 10.1021/np200163g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts TC, Schallenberger MA, Liu J, Smith PA, Romesberg FE. 2011. Initial efforts toward the optimization of arylomycins for antibiotic activity. J Med Chem 54:4954–4963. doi: 10.1021/jm1016126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J, Luo C, Smith PA, Chin JK, Page MG, Paetzel M, Romesberg FE. 2011. Synthesis and characterization of the arylomycin lipoglycopeptide antibiotics and the crystallographic analysis of their complex with signal peptidase. J Am Chem Soc 133:17869–17877. doi: 10.1021/ja207318n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Smith PA, Steed DB, Romesberg F. 2013. Efforts toward broadening the spectrum of arylomycin antibiotic activity. Bioorg Med Chem Lett 23:5654–5659. doi: 10.1016/j.bmcl.2013.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith PA, Powers ME, Roberts TC, Romesberg FE. 2011. In vitro activities of arylomycin natural-product antibiotics against Staphylococcus epidermidis and other coagulase-negative staphylococci. Antimicrob Agents Chemother 55:1130–1134. doi: 10.1128/AAC.01459-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klock HE, Lesley SA. 2009. The polymerase incomplete primer extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol Biol 498:91–103. doi: 10.1007/978-1-59745-196-3_6. [DOI] [PubMed] [Google Scholar]
- 27.Wilkinson K. 2004. Quantitative analysis of protein-protein interactions. Methods Mol Biol 261:15–32. doi: 10.1385/1-59259-762-9:015. [DOI] [PubMed] [Google Scholar]
- 28.Kaniga K, Delor I, Cornelis GR. 1991. A wide-host-range suicide vector for improving reverse genetics in gram-negative bacteria: inactivation of the blaA gene of Yersinia enterocolitica. Gene 109:137–141. doi: 10.1016/0378-1119(91)90599-7. [DOI] [PubMed] [Google Scholar]
- 29.Conchas RF, Carniel E. 1990. A highly efficient electroporation system for transformation of Yersinia. Gene 87:133–137. doi: 10.1016/0378-1119(90)90505-L. [DOI] [PubMed] [Google Scholar]
- 30.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.CLSI. 2009. Methods for dilution antimicrobial susceptibility. Tests for bacteria that grow aerobically. Approved standard, 9th ed. CLSI document M07-A9. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 32.Moody J. 2004. Synergism testing: broth microdilution checkerboard and broth macrodilution methods. In Isenberg H. D. (ed), Clinical microbiology procedures handbook, vol 2 American Society for Microbiology, Washington, DC. [Google Scholar]
- 33.Nilles ML, Williams AW, Skrzypek E, Straley SC. 1997. Yersinia pestis LcrV forms a stable complex with LcrG and may have a secretion-related regulatory role in the low-Ca2+ response. J Bacteriol 179:1307–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chromy BA, Choi MW, Murphy GA, Gonzales AD, Corzett CH, Chang BC, Fitch JP, McCutchen-Maloney SL. 2005. Proteomic characterization of Yersinia pestis virulence. J Bacteriol 187:8172–8180. doi: 10.1128/JB.187.23.8172-8180.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pieper R, Huang ST, Parmar PP, Clark DJ, Alami H, Fleischmann RD, Perry RD, Peterson SN. 2010. Proteomic analysis of iron acquisition, metabolic and regulatory responses of Yersinia pestis to iron starvation. BMC Microbiol 10:30. doi: 10.1186/1471-2180-10-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pieper R, Huang ST, Robinson JM, Clark DJ, Alami H, Parmar PP, Perry RD, Fleischmann RD, Peterson SN. 2009. Temperature and growth phase influence the outer-membrane proteome and the expression of a type VI secretion system in Yersinia pestis. Microbiology 155:498–512. doi: 10.1099/mic.0.022160-0. [DOI] [PubMed] [Google Scholar]
- 37.Pieper R, Huang ST, Clark DJ, Robinson JM, Parmar PP, Alami H, Bunai CL, Perry RD, Fleischmann RD, Peterson SN. 2008. Characterizing the dynamic nature of the Yersinia pestis periplasmic proteome in response to nutrient exhaustion and temperature change. Proteomics 8:1442–1458. doi: 10.1002/pmic.200700923. [DOI] [PubMed] [Google Scholar]
- 38.Bengoechea JA, Brandenburg K, Seydel U, Diaz R, Moriyon I. 1998. Yersinia pseudotuberculosis and Yersinia pestis show increased outer membrane permeability to hydrophobic agents which correlates with lipopolysaccharide acyl-chain fluidity. Microbiology 144:1517–1526. doi: 10.1099/00221287-144-6-1517. [DOI] [PubMed] [Google Scholar]
- 39.Knirel YA, Anisimov AP. 2012. Lipopolysaccharide of Yersinia pestis, the cause of plague: structure, genetics, biological properties. Acta Naturae 4:46–58. [PMC free article] [PubMed] [Google Scholar]
- 40.Anisimov AP, Dentovskaya SV, Titareva GM, Bakhteeva IV, Shaikhutdinova RZ, Balakhonov SV, Lindner B, Kocharova NA, Senchenkova SN, Holst O, Pier GB, Knirel YA. 2005. Intraspecies and temperature-dependent variations in susceptibility of Yersinia pestis to the bactericidal action of serum and to polymyxin B. Infect Immun 73:7324–7331. doi: 10.1128/IAI.73.11.7324-7331.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vaara M, Vaara T. 1983. Sensitization of Gram-negative bacteria to antibiotics and complement by a nontoxic oligopeptide. Nature 303:526–528. doi: 10.1038/303526a0. [DOI] [PubMed] [Google Scholar]
- 42.Liu D, Reeves PR. 1994. Escherichia coli K12 regains its O antigen. Microbiology 140:49–57. doi: 10.1099/13500872-140-1-49. [DOI] [PubMed] [Google Scholar]
- 43.al-Hendy A, Toivanen P, Skurnik M. 1991. The effect of growth temperature on the biosynthesis of Yersinia enterocolitica O:3 lipopolysaccharide: temperature regulates the transcription of the rfb but not of the rfa region. Microb Pathog 10:81–86. doi: 10.1016/0882-4010(91)90068-L. [DOI] [PubMed] [Google Scholar]
- 44.Gremyakova TA, Vinogradov EV, Lindner B, Kocharova NA, Senchenkova SN, Shashkov AS, Knirel YA, Holst O, Shaikhutdinova RZ, Anisimov AP. 2003. The core structure of the lipopolysaccharide of Yersinia pestis strain KM218. Influence of growth temperature. Adv Exp Med Biol 529:229–231, http://dx.doi.org/10.1007/0-306-48416-1_44. [DOI] [PubMed] [Google Scholar]
- 45.Knirel YA, Lindner B, Vinogradov E, Shaikhutdinova RZ, Senchenkova SN, Kocharova NA, Holst O, Pier GB, Anisimov AP. 2005. Cold temperature-induced modifications to the composition and structure of the lipopolysaccharide of Yersinia pestis. Carbohydr Res 340:1625–1630. doi: 10.1016/j.carres.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 46.Knirel YA, Lindner B, Vinogradov EV, Kocharova NA, Senchenkova SN, Shaikhutdinova RZ, Dentovskaya SV, Fursova NK, Bakhteeva IV, Titareva GM, Balakhonov SV, Holst O, Gremyakova TA, Pier GB, Anisimov AP. 2005. Temperature-dependent variations and intraspecies diversity of the structure of the lipopolysaccharide of Yersinia pestis. Biochemistry 44:1731–1743. doi: 10.1021/bi048430f. [DOI] [PubMed] [Google Scholar]
- 47.Vinogradov EV, Lindner B, Kocharova NA, Senchenkova SN, Shashkov AS, Knirel YA, Holst O, Gremyakova TA, Shaikhutdinova RZ, Anisimov AP. 2002. The core structure of the lipopolysaccharide from the causative agent of plague, Yersinia pestis. Carbohydr Res 337:775–777. doi: 10.1016/S0008-6215(02)00074-5. [DOI] [PubMed] [Google Scholar]
- 48.Hartley JL, Adams GA, Tornabene TG. 1974. Chemical and physical properties of lipopolysaccharide of Yersinia pestis. J Bacteriol 118:848–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rebeil R, Ernst RK, Gowen BB, Miller SI, Hinnebusch BJ. 2004. Variation in lipid A structure in the pathogenic yersiniae. Mol Microbiol 52:1363–1373. doi: 10.1111/j.1365-2958.2004.04059.x. [DOI] [PubMed] [Google Scholar]
- 50.Gunn JS, Lim KB, Krueger J, Kim K, Guo L, Hackett M, Miller SI. 1998. PmrA-PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol Microbiol 27:1171–1182. doi: 10.1046/j.1365-2958.1998.00757.x. [DOI] [PubMed] [Google Scholar]
- 51.Hitchen PG, Prior JL, Oyston PC, Panico M, Wren BW, Titball RW, Morris HR, Dell A. 2002. Structural characterization of lipo-oligosaccharide (LOS) from Yersinia pestis: regulation of LOS structure by the PhoPQ system. Mol Microbiol 44:1637–1650. doi: 10.1046/j.1365-2958.2002.02990.x. [DOI] [PubMed] [Google Scholar]
- 52.Garcia Vescovi E, Soncini FC, Groisman EA. 1996. Mg2+ as an extracellular signal: environmental regulation of Salmonella virulence. Cell 84:165–174. doi: 10.1016/S0092-8674(00)81003-X. [DOI] [PubMed] [Google Scholar]
- 53.Montminy SW, Khan N, McGrath S, Walkowicz MJ, Sharp F, Conlon JE, Fukase K, Kusumoto S, Sweet C, Miyake K, Akira S, Cotter RJ, Goguen JD, Lien E. 2006. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat Immunol 7:1066–1073. doi: 10.1038/ni1386. [DOI] [PubMed] [Google Scholar]
- 54.Motin VL, Georgescu AM, Fitch JP, Gu PP, Nelson DO, Mabery SL, Garnham JB, Sokhansanj BA, Ott LL, Coleman MA, Elliott JM, Kegelmeyer LM, Wyrobek AJ, Slezak TR, Brubaker RR, Garcia E. 2004. Temporal global changes in gene expression during temperature transition in Yersinia pestis. J Bacteriol 186:6298–6305. doi: 10.1128/JB.186.18.6298-6305.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chauvaux S, Rosso ML, Frangeul L, Lacroix C, Labarre L, Schiavo A, Marceau M, Dillies MA, Foulon J, Coppee JY, Medigue C, Simonet M, Carniel E. 2007. Transcriptome analysis of Yersinia pestis in human plasma: an approach for discovering bacterial genes involved in septicaemic plague. Microbiology 153:3112–3124. doi: 10.1099/mic.0.2007/006213-0. [DOI] [PubMed] [Google Scholar]
- 56.Lee CA, Beckwith J. 1986. Suppression of growth and protein secretion defects in Escherichia coli secA mutants by decreasing protein synthesis. J Bacteriol 166:878–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ. 2008. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 135:679–690. doi: 10.1016/j.cell.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith PA, Romesberg FE. 2012. Mechanism of action of the arylomycin antibiotics and effects of signal peptidase I inhibition. Antimicrob Agents Chemother 56:5054–5060. doi: 10.1128/AAC.00785-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. 2004. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 60.Kolodziejek AM, Hovde CJ, Minnich SA. 2012. Yersinia pestis Ail: multiple roles of a single protein. Front Cell Infect Microbiol 2:103. doi: 10.3389/fcimb.2012.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.du Plessis DJ, Nouwen N, Driessen AJ. 2011. The Sec translocase. Biochim Biophys Acta 1808:851–865. doi: 10.1016/j.bbamem.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 62.Rusch SL, Kendall DA. 2007. Interactions that drive Sec-dependent bacterial protein transport. Biochemistry 46:9665–9673. doi: 10.1021/bi7010064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Driessen AJ, Nouwen N. 2008. Protein translocation across the bacterial cytoplasmic membrane. Annu Rev Biochem 77:643–667. doi: 10.1146/annurev.biochem.77.061606.160747. [DOI] [PubMed] [Google Scholar]
- 64.Desvaux M, Parham NJ, Scott-Tucker A, Henderson IR. 2004. The general secretory pathway: a general misnomer? Trends Microbiol 12:306–309. doi: 10.1016/j.tim.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 65.Beveridge TJ. 1999. Structures of gram-negative cell walls and their derived membrane vesicles. J Bacteriol 181:4725–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Song C, Kumar A, Saleh M. 2009. Bioinformatic comparison of bacterial secretomes. Genomics Proteomics Bioinformatics 7:37–46. doi: 10.1016/S1672-0229(08)60031-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Therien AG, Huber JL, Wilson KE, Beaulieu P, Caron A, Claveau D, Deschamps K, Donald RG, Galgoci AM, Gallant M, Gu X, Kevin NJ, Lafleur J, Leavitt PS, Lebeau-Jacob C, Lee SS, Lin MM, Michels AA, Ogawa AM, Painter RE, Parish CA, Park YW, Benton-Perdomo L, Petcu M, Phillips JW, Powles MA, Skorey KI, Tam J, Tan CM, Young K, Wong S, Waddell ST, Miesel L. 2012. Broadening the spectrum of beta-lactam antibiotics through inhibition of signal peptidase type I. Antimicrob Agents Chemother 56:4662–4670. doi: 10.1128/AAC.00726-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brubaker RR. 2002. Yersinia pestis, p 2033–2058. In Sussman M. (ed), Molecular medical microbiology, vol 3 Academic Press, London, United Kingdom. [Google Scholar]
- 69.Bartra SS, Styer KL, O'Bryant DM, Nilles ML, Hinnebusch BJ, Aballay A, Plano GV. 2008. Resistance of Yersinia pestis to complement-dependent killing is mediated by the Ail outer membrane protein. Infect Immun 76:612–622. doi: 10.1128/IAI.01125-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kolodziejek AM, Sinclair DJ, Seo KS, Schnider DR, Deobald CF, Rohde HN, Viall AK, Minnich SS, Hovde CJ, Minnich SA, Bohach GA. 2007. Phenotypic characterization of OmpX, an Ail homologue of Yersinia pestis KIM. Microbiology 153:2941–2951. doi: 10.1099/mic.0.2006/005694-0. [DOI] [PubMed] [Google Scholar]
- 71.Lee VT, Schneewind O. 2001. Protein secretion and the pathogenesis of bacterial infections. Genes Dev 15:1725–1752. doi: 10.1101/gad.896801. [DOI] [PubMed] [Google Scholar]
- 72.Rieder G, Fischer W, Haas R. 2005. Interaction of Helicobacter pylori with host cells: function of secreted and translocated molecules. Curr Opin Microbiol 8:67–73. doi: 10.1016/j.mib.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 73.Sandkvist M. 2001. Type II secretion and pathogenesis. Infect Immun 69:3523–3535. doi: 10.1128/IAI.69.6.3523-3535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stathopoulos C, Hendrixson DR, Thanassi DG, Hultgren SJ, St Geme JW III, Curtiss R III. 2000. Secretion of virulence determinants by the general secretory pathway in gram-negative pathogens: an evolving story. Microbes Infect 2:1061–1072. doi: 10.1016/S1286-4579(00)01260-0. [DOI] [PubMed] [Google Scholar]
- 75.Bachmann BJ. 1972. Pedigrees of some mutant strains of Escherichia coli K-12. Bacteriol Rev 36:525–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blattner FR, Plunkett G III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 77.Jensen KF. 1993. The Escherichia coli K-12 “wild types” W3110 and MG1655 have an rph frameshift mutation that leads to pyrimidine starvation due to low pyrE expression levels. J Bacteriol 175:3401–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fetherston JD, Perry RD. 1994. The pigmentation locus of Yersinia pestis KIM6+ is flanked by an insertion sequence and includes the structural genes for pesticin sensitivity and HMWP2. Mol Microbiol 13:697–708. doi: 10.1111/j.1365-2958.1994.tb00463.x. [DOI] [PubMed] [Google Scholar]
- 79.Sonnenwirth AC. 1970. Bacteremia with and without meningitis due to Yersinia enterocolitica, Edwardsiella tarda, Comamonas terrigena, and Pseudomonas maltophilia. Ann N Y Acad Sci 174:488–502. doi: 10.1111/j.1749-6632.1970.tb45575.x. [DOI] [PubMed] [Google Scholar]
- 80.Sampson BA, Misra R, Benson SA. 1989. Identification and characterization of a new gene of Escherichia coli K-12 involved in outer membrane permeability. Genetics 122:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Boman HG, Jonsson S, Monner D, Normark S, Bloom GD. 1971. Cell-surface alterations in Escherichia coli K-12 with chromosmal mutations changing ampicillin resistance. Ann N Y Acad Sci 182:342–357. doi: 10.1111/j.1749-6632.1971.tb30670.x. [DOI] [PubMed] [Google Scholar]
- 82.Eriksson-Grennberg KR, Nordstrom K, Englund P. 1971. Resistance of Escherichia coli to penicillins. IX. Genetics and physiology of class II ampicillin-resistant mutants that are galactose negative or sensitive to bacteriophage C21, or both. J Bacteriol 108:1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Goguen JD, Yother J, Straley SC. 1984. Genetic analysis of the low calcium response in Yersinia pestis mu d1(Ap lac) insertion mutants. J Bacteriol 160:842–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.