

Abstract
The tolerability, safety, and disposition of dihydroartemisinin (DHA) and piperaquine (PQ) were assessed in 32 pregnant (second/third trimester) and 33 nonpregnant Papua New Guinean women randomized to adult treatment courses of DHA-PQ (three daily doses) or sulfadoxine-pyrimethamine (SP)-PQ (three daily PQ doses, single dose of SP). All dose adminstrations were observed, and subjects fasted for 2 h postdose. Plasma PQ was assayed by using high-performance liquid chromatography, and DHA was assessed by using liquid chromatography-mass spectrometry. Compartmental pharmacokinetic models were developed using a population-based approach. Both regimens were well tolerated. There was an expected increase in the rate-corrected electrocardiographic QT interval which was independent of pregnancy and treatment. Two pregnant and two nonpregnant women had Plasmodium falciparum parasitemia which cleared within 48 h, and no other subject became slide positive for malaria during 42 days of follow-up. Of 30 pregnant women followed to delivery, 27 (90%) delivered healthy babies and 3 (10%) had stillbirths; these obstetric outcomes are consistent with those in the general population. The area under the plasma PQ concentration-time curve (AUC0–∞) was lower in the pregnant patients (median [interquartile range], 23,721 μg · h/liter [21,481 to 27,951 μg · h/liter] versus 35,644 μg · h/liter [29,546 to 39,541 μg · h/liter]; P < 0.001) in association with a greater clearance relative to bioavailability (73.5 liters/h [69.4 to 78.4] versus 53.8 liters/h [49.7 to 58.2]; P < 0.001), but pregnancy did not influence the pharmacokinetics of DHA. The apparent pharmacokinetic differences between the present study and results from other studies of women with uncomplicated malaria that showed no effect of pregnancy on the AUC0–∞ of PQ and greater bioavailability may reflect differences in postdose fat intake, proportions of women with malaria, and/or racial differences in drug disposition.
INTRODUCTION
Malaria in pregnancy is associated with preterm birth, low birth weight, intrauterine growth retardation, and maternal anemia (1, 2) and may attenuate the development of antimalarial immunity in infants (3). Various strategies have been employed in countries where malaria is endemic to improve obstetric outcome, including prompt treatment of symptomatic infections and intermittent presumptive treatment in pregnancy (IPTp) (4, 5). The World Health Organization (WHO) recommends sulfadoxine-pyrimethamine (SP) as IPTp in combination with the use of insecticide-treated bed nets (ITNs) (6, 7). However, as SP-resistant Plasmodium falciparum continues to spread, new IPTp regimens, including azithromycin-SP (AZI-SP), dihydroartemisinin-piperaquine (DHA-PQ), and AZI-PQ are being assessed (8–15).
In Papua New Guinea (PNG), IPTp originally comprised a treatment dose of chloroquine (CQ) plus SP at first antenatal presentation, followed by weekly CQ until delivery (16). More recently, and in line with WHO guidelines (7), this has changed to SP administered early in the second trimester and then at routine (at least monthly) antenatal visits. Women in PNG are usually first seen relatively late in pregnancy, and clinic attendance may be problematic for logistic and economic reasons (16). This can restrict IPTp to limited and/or irregular SP doses, which may diminish the potential benefits for mother and infant (17), in part because the two-component drugs have short elimination half-lives relative to 4-aminoquinolines and related compounds, such as PQ (18, 19). In addition, there is evidence that P. falciparum is developing resistance to SP in PNG (20), while SP has limited activity against Plasmodium vivax (21), which is a significant contributor to adverse obstetric outcomes in regions where it is transmitted (22).
Of novel chemoprophylactic regimens with potential use in pregnancy, DHA-PQ may be the most suitable for countries such as PNG, where there is intense transmission of multiple Plasmodium species. Artemisinin derivatives clear malaria parasites rapidly and are considered safe beyond the first trimester (5). Preclinical studies have provided reproductive toxicity data for PQ (23, 24), which has an elimination half-life of several weeks (24). A fixed-dose combination is in widespread use for treatment of uncomplicated falciparum and vivax malaria outside pregnancy (25, 26). Available pharmacokinetic and efficacy data from Southeast Asia (14, 27, 28) and Africa (12, 13) suggest that DHA-PQ is safe, well tolerated, and efficacious in pregnant women with uncomplicated malaria and that PQ exposure is similar in pregnant and nonpregnant women, even if DHA exposure may be lower in pregnancy.
There is, however, the view that artemisinin derivatives are not a logical choice as a component of IPT regimens, given that their very rapid elimination limits the benefit (apart from in individuals who are parasitemic when dosed) at additional cost and risk (29). In an African pediatric IPT efficacy study of monthly DHA-PQ or SP-PQ (30), incident malaria was lower in the SP-PQ-treated children. There is evidence that the elimination half-life of the pyrimethamine component of SP may be particularly long in pregnancy (19 days versus 10 days in nonpregnant women) (19) which, given that DHA is eliminated within hours of dosing (14, 27), might suggest that SP-PQ is a better choice for IPTp than DHA-PQ.
We have shown that there are significant ethno-geographic differences in the disposition of antimalarial drugs in pregnancy and these may have clinical implications (19, 31). In the case of DHA-PQ given as IPTp, the periodicity of administration should be determined primarily by the duration of postdose PQ prophylaxis, which reflects the elimination half-life (14). Since these factors may vary across populations, our primary aim was to conduct a pharmacokinetic study of DHA-PQ in pregnant and nonpregnant PNG women and to compare the data obtained to those from similar studies in women from other countries where malaria is endemic. In contrast to previous pharmacokinetic and efficacy studies which only enrolled women with malaria (12–14, 27, 28), we aimed to replicate real-life IPTp by recruiting patients from the antenatal clinic regardless of malaria status. In addition, we included SP-PQ treatment arms to provide parallel tolerability, safety, and preliminary efficacy data, as well as the opportunity to assess potential interactions between PQ, SP, and DHA.
MATERIALS AND METHODS
Study site, approval, and participants.
The present study was conducted at Alexishafen Health Center, Madang Province, which is located on the north coast of PNG, where there is transmission of P. falciparum and P. vivax (32). Pregnant women attending their first antenatal clinical visit at the Alexishafen Antenatal Clinic and who were at >14 weeks of gestation were invited to participate, together with one age-matched nonpregnant female (as confirmed on pregnancy testing) from the same community as each pregnant woman. Subjects were eligible if (i) they had not taken study drugs in the previous 28 days, (ii) they had no history of significant allergy to study drugs, (iii) there was no significant comorbidity or clinical evidence of severe malaria, and (iv) they were available for follow-up assessments. Witnessed informed consent was obtained from each participant. Approval for the study was obtained from the PNG Institute of Medical Research Institutional Review Board and the Medical Research Advisory Committee of the PNG Health Department.
Baseline assessment and treatment allocation.
A detailed medical history and symptom questionnaire (including full details of recent drug treatment) was completed and a physical examination (including body weight, height, axillary temperature, lying and standing blood pressure and pulse rate, respiratory rate, and estimation of gestational age by fundal height in those who were pregnant) was performed. Thick and thin blood films were taken for determination of parasite density and determination of species. An intravenous cannula was inserted, and a 2-ml baseline venous blood sample was collected for hemoglobin and blood glucose, while the remaining blood was centrifuged and the plasma was stored at −20°C for subsequent drug and biochemical assays. The red cell pellet was retained for molecular assays. Each woman had a baseline electrocardiogram (ECG) with the QT interval measured from lead II of the 12-lead trace and corrected for the heart rate using Bazett's method (QTc = QT/√RR interval).
Participants were randomized by a computer-generated schedule to receive either (i) three DHA-PQ tetraphosphate tablets (Eurartesim; Simga-Tau Industrie Farmaceutiche Riunite S.p.A., Italy) containing 40 mg DHA and 320 mg PQ tetraphosphate at a DHA dose of 7 mg/kg of body weight and a PQ dose of 58 mg/kg (equivalent to 33 mg/kg PQ base for a 50-kg woman) daily with water for 3 days (at 0, 24, and 48 h), or (ii) four PQ tablets containing 320 mg (Sigma-Tau Industrie Farmaceutiche Riunite S.p.A) daily with water for 3 days (at 0, 24, and 48 h) plus a single dose of SP (25 mg/kg S, 1.25 mg/kg P) as a single dose with the first PQ dose. All dose administrations were directly observed. Patients were fasted for 2 h before and after dosing, consistent with the recommendations of the manufacturer. Women who vomited within 30 min of drug administration were retreated. All pregnant women were provided with ITNs and educated on their use, with instructions to employ them for the duration of their pregnancy as an alternative to additional regular chemoprophylaxis.
Monitoring and follow-up.
Detailed clinical assessment, including a side effect questionnaire and blood smear, was carried out at each follow-up visit on days 1, 2, 3, 7, 14, 21, 28, and 42. Additional monitoring comprised (i) hemoglobin and blood glucose concentrations at 4, 8, and 12 h and then at each follow-up visit, (ii) lying and standing blood pressure measurements at 4, 8, and 12 h and on days 1, 2, 3, 7, 14, 28, and 42, (iii) ECG at 4, 12, and 24 h, (iv) assessment of fetal viability (including movements detected by mother and fetal heart beat on auscultation) daily for the first 4 days and then at each subsequent sampling time point (see below), and (v) ultrasound scans on days 28 and 42 to determine fetal lie/presentation and to confirm gestational age.
All blood smears from each patient were examined initially on site and then were reexamined with quantification of parasite density by two skilled independent microscopists in a central laboratory. Discrepancies in species determinations and density between the two microscopists were adjudicated by a third microscopist. Parasite densities were calculated from the number of parasites per 200 or 500 white cells (depending on parasitemia) and an assumed total peripheral white cell count of 8,000/μl, with the final density taken as the geometric mean of the two values (33).
Further 3-ml blood samples for drug assay were taken at 0.5, 1, 1.5, 2, 4, 8, 12, 18, 24, 32, and 48 h (the 24- and 48-h samples being taken immediately before the second and third doses, respectively), and by venipuncture on days 3, 4, 7, 14, 21, 28, and 42. The exact time of each sample was recorded. All blood samples were centrifuged promptly, with plasma separated and stored at or below −20°C prior to assay. There were a total of 19 samples for each study subject, including baseline in each case, equating to a total blood volume of 60 ml over 6 weeks.
After completion of study procedures, the pregnant participants were returned to usual antenatal care. At the time of delivery, thick and thin blood films were prepared, and the hemoglobin concentration was measured. A 3-ml sample of cord blood was taken for PQ assay, multiple thick and thin films were prepared from cord blood, a placental smear was prepared, and a section of placenta was collected into formal saline for evaluation if the cord or placental smears were positive for malaria parasites.
Drug assays.
Plasma PQ concentrations were measured by a validated high-performance liquid chromatography assay using liquid-liquid extraction followed by reversed-phase chromatography (34). In brief, extracted plasma samples were injected onto a Gemini C6-phenyl (150- by 4.6-mm internal diameter) 5-μm column coupled with a guard column (Phenomenex, Lane Cove, Australia). Analytes were detected at 346 nm and quantified using Chemstation software (version 9; Agilent Technology, Waldbronn, Germany). The intraday relative standard deviations (RSDs) of PQ in plasma were 8.1, 5.4, 7.4, 5.2, and 2.5 at 5, 25, 50, 200, and 500 μg/liter, respectively (n = 5), while interday RSDs were 8.4, 9.6, 6.5, 7.8, and 3.6 at 5, 25, 50, 200, and 500 μg/liter, respectively (n = 25). The limit of quantification (LOQ) and limit of detection (LOD) were 2 μg/liter and 1 μg/liter, respectively, with a signal-to-noise ratio of 3.0.
Plasma DHA concentrations were measured by liquid chromatography-mass spectrometry as previously described using solid-phase extraction and reversed-phase chromatography (35). In brief, extracted plasma samples were injected onto a Synergy fusion-RP C18 (150- by 2.0-mm internal diameter) column coupled with a 4-mm by 3-mm internal diameter 5-μm C18 guard column (Phenomenex). Optimized mass spectra were acquired, and quantitation was performed by selected ion monitoring using the dual ionization source mode on a single quad mass spectrometer (Shimadzu, Kyoto, Japan). Interday accuracy deviations across clinically relevant concentration ranges were <15%. The LOQ and LOD for DHA were 2 and 1 μg/liter, respectively.
Pharmacokinetic modeling.
Loge plasma concentration-time data sets for PQ and DHA were analyzed by nonlinear mixed effects modeling using NONMEM (v 7.2.0; ICON Development Solutions, Ellicott City, MD, USA) (36) with an Intel Visual Fortran 10.0 compiler. The Laplacian with interaction estimation method was used for PQ, and the first order conditional estimation (FOCE) with interaction estimation method was used for DHA. The minimum value of the objective function (OFV), goodness-of-fit plots, and condition number (<1,000) were used to arrive at suitable models during the model-building process. Allometric scaling was employed a priori, with volume terms multiplied by a factor of (BW/70)1.0 and clearance terms by a factor of (BW/70)0.75, where BW is body weight (37). Two structures for residual variability (RV), equivalent to proportional and combined RV structures on the normal scale, were used for the log-transformed data. Secondary pharmacokinetic parameters, including area under the curve (AUC0–∞) and elimination half-lives (t1/2) for the participants, were obtained from post hoc Bayesian predictions in NONMEM using the final model parameters. Previously published equations for calculation of half-lives were used (38). Base models were parameterized using ka (absorption rate constant), VC/F (central volume of distribution), CL/F (clearance), VP/F and Q/F (peripheral volumes of distribution[s] and their respective intercompartmental clearance[s]), where F represents bioavailability.
For PQ, two- and three-compartment models (ADVAN 4 and 12) with zero-, first-, or mixed-order absorption with and without lag time were tested. As found previously (39), there was significant variability in the absorption phase of PQ. Therefore, a transit compartment model, previously shown to be effective in characterizing the absorption phase of PQ (39), was also tested. In this model, the dose passes through a series of transit compartments before entering the absorption compartment, in order to model the delay often associated with drug absorption. A single rate constant (ktr) describes the entry and exit for all transit compartments. Using a previously described implementation of the transit compartment model in NONMEM (40), the number of transit compartments (NN) and the mean transit time [MTT = (1 + NN)/ktr] were estimated as continuous variables. For the DHA data set, 1- and 2-compartment models (ADVAN 2 and 4) with first-order absorption with and without lag time were evaluated. Once the structures of the models were established, interindividual variability (IIV), interoccasion variability (IOV), and correlations between IIV terms were estimated, where supported by the data. In particular, IOV of FPQ (relative bioavailability) was estimated to examine the potential differences in the relative bioavailability of PQ between doses, as noted previously for PQ and other lipophilic antimalarial drugs (31, 35, 39).
Finally, relationships between model parameters and the covariates pregnancy, dose group, and gestational age were identified through inspection of scatter plots and box plots of eta versus covariate and subsequently evaluated within NONMEM. The effect size (as a percentage) of pregnancy and dose group was assessed, while both linear and power relationships were evaluated for gestational age. For effect size, the individual parameter value was equal to the (population parameter value) × {1 + [effect parameter × covariate value (0 or 1)]}. For linear relationships, the individual parameter value was equal to the (population parameter value) × {1 + [(effect parameter) × (covariate value for individual)/(average value of covariate)]}. For power relationships, the individual parameter value was equal to the (population parameter value) × {(covariate value for individual)/[(average value of covariate)effect parameter]}. A stepwise forward inclusion and backward elimination method was used, with a significance of P < 0.05 required for inclusion of a covariate relationship and P < 0.01 to retain a covariate relationship.
Model evaluation.
A bootstrap using Perl speaks NONMEM (PSN) with 1,000 samples was performed, and the parameters derived from this analysis were summarized as the median and 2.5th and 97.5th percentiles (95% empirical confidence interval [CI]) to facilitate evaluation of final model parameter estimates. In addition, prediction corrected visual predictive checks (pcVPCs) were performed with 1,000 data sets simulated from the final models, and these were stratified according to pregnancy for PQ. The observed 10th, 50th, and 90th percentiles were plotted with their respective simulated 95% CIs. Numerical predictive checks (NPCs) were performed to complement the pcVPCs in assessing the predictive performance of the model.
Statistical analysis.
Statistical analysis was performed using IBM SPSS Statistics version 20 (IBM Corporation, Somers, NY, USA). Data are summarized as means ± standard deviations (SD) or medians and interquartile ranges [IQRs], as appropriate. Two-sample comparisons for normally distributed variables were performed using Student's t test, for nonnormally distributed variables the Mann-Whitney U test was used, and for proportions the Fisher's exact test was used. For multiple samples, analysis of variance (ANOVA) or the Kruskal-Wallis test was used. Differences between baseline parameters by pregnancy status and treatment group were assessed using Bonferroni-adjusted pairwise comparisons, and Fisher's two-by-four exact test was used to determine differences in frequencies of side effects by pregnancy status and treatment group. A two-tailed level of significance of 0.05 was used throughout.
RESULTS
Subject characteristics.
A total of 32 pregnant and 34 nonpregnant women were recruited. However, one nonpregnant woman withdrew consent prior to the administration of antimalarial drugs, and her data were not included in the analyses. The baseline characteristics of the participants by pregnancy status and treatment allocation are shown in Table 1. The groups were well matched except that, consistent with normal physiological changes of pregnancy, the pregnant participants had a higher pulse rate and a lower hemoglobin and blood glucose level than the nonpregnant subjects in each treatment group.
TABLE 1.
Baseline characteristics of the women included in each treatment groupa
| Characteristic | Pregnant |
Nonpregnant |
P value | ||
|---|---|---|---|---|---|
| DHA-PQ (n = 16) | SP-PQ (n = 16) | DHA-PQ (n = 16) | SP-PQ (n = 17) | ||
| Age (yrs) | 27 (21 to 37) | 25 (20 to 32) | 21 (18 to 30) | 25 (22 to 37) | 0.35 |
| Weight (kg) | 51.6 ± 5.3 | 53.9 ± 7.5 | 54.7 ± 7.2 | 52.5 ± 5.8 | 0.53 |
| Height (cm) | 155 ± 4 | 153 ± 5 | 157 ± 5 | 155 ± 5 | 0.19 |
| Axillary temp (°C) | 36.8 (36.5 to 37.0) | 36.8 (36.6 to 37.0) | 36.7 (36.5 to 36.9) | 36.7 (36.2 to 37.0) | 0.68 |
| P. falciparum parasitemia (%) | 12.5 | 6.2 | 0 | 11.8 | 0.74 |
| Gestational age (wk) | 26 (20 to 31) | 29 (24 to 31) | 0.35 | ||
| Gravidity | 3 (1 to 5) | 2 (1 to 4) | 1 (0 to 3.8) | 0 (0 to 3) | 0.71 |
| Parity | 2 (0 to 4) | 1 (0 to 3) | 0.5 (0 to 2.8) | 0 (0 to 3) | 0.98 |
| Respiratory rate (min−1) | 18 (18 to 20) | 18 (18 to 20) | 20 (18 to 21) | 20 (18 to 20) | 0.38 |
| Pulse rate (min−1) | 83 ± 13 | 79 ± 10 | 73 ± 10 | 68 ± 9**,ǂ | 0.001 |
| Systolic blood pressure (mm Hg) | 99 ± 11 | 99 ± 8 | 102 ± 12 | 101 ± 11 | 0.79 |
| Diastolic blood pressure (mm Hg) | 59 ± 8 | 60 ± 8 | 65 ± 14 | 64 ± 10 | 0.43 |
| Systolic fall on standing (mm Hg) | 0 (−4 to 0) | −2 (−5 to 0) | 0 (−10 to 8) | −2 (−5 to 0) | 0.57 |
| Diastolic fall standing (mm Hg) | −2 (−4 to −2) | −4 (−4 to 0) | 0 (−6 to 0) | −3 (−10 to 0) | 0.78 |
| Hemoglobin (g/liter) | 91 ± 13 | 94 ± 19 | 116 ± 17**,ǂǂ | 120 ± 19***,ǂǂǂ | <0.001 |
| Blood glucose (mmol/liter) | 5.4 ± 0.7 | 5.0 ± 0.9 | 5.9 ± 1.0ǂ | 5.9 ± 0.9ǂ | 0.010 |
| QTc (ms0.5) | 423 ± 16 | 435 ± 16 | 433 ± 20 | 424 ± 17 | 0.19 |
Data are percentages, means ± SD, or medians (with IQRs indicated in parentheses). For comparisons with pregnant women receiving DHA-PQ: *, P < 0.05; **, P < 0.01; ***, P < 0.001. For comparisons with pregnant women receiving SP-PQ: ǂ, P < 0.05; ǂǂ, P < 0.01; ǂǂǂ P < 0.001.
Efficacy, tolerability, and safety.
Both treatments were well tolerated by pregnant and nonpregnant participants. Table 2 summarizes the self-reported side effects which were experienced by at least 3% of the sample during the first 7 days after start of treatment. All were mild (they did not interfere with daily activities) and self-limiting (resolution of symptoms within 2 days). There were no significant between-group differences in the frequency of symptoms (P ≥ 0.09 by Fisher's exact test). No participant developed hypoglycemia (blood glucose, <2.5 mmol/liter) or severe anemia (hemoglobin, <50 g/liter) during the 42-day follow-up period. There were no significant between-group differences in hemoglobin concentrations at any time point during follow-up (data not shown).
TABLE 2.
Side effects reported by participants during the first week after treatment
| Side effect | No. (%) of participants with symptom in group |
P value | |||
|---|---|---|---|---|---|
| Pregnant |
Nonpregnant |
||||
| DHA-PQ (n = 16) | SP-PQ (n = 16) | DHA-PQ (n = 16) | SP-PQ (n = 17) | ||
| Fever | 4 (27) | 1 (7) | 2 (13) | 5 (29) | 0.29 |
| Chills | 1 (7) | 0 (0) | 0 (0) | 3 (18) | 0.15 |
| Headache | 5 (33) | 2 (13) | 5 (33) | 8 (47) | 0.20 |
| Nausea | 1 (7) | 3 (20) | 3 (20) | 4 (24) | 0.63 |
| Vomiting | 0 (0) | 2 (13) | 0 (0) | 1 (6) | 0.37 |
| Abdominal pain | 0 (0) | 1 (7) | 1 (7) | 2 (12) | 0.80 |
| Anorexia | 1 (7) | 2 (13) | 0 (0) | 1 (6) | 0.80 |
| Insomnia | 1 (7) | 0 (0) | 0 (0) | 1 (6) | 0.74 |
| Dizziness | 1 (7) | 0 (0) | 1 (7) | 2 (12) | 0.80 |
| Bone or joint pain | 5 (33) | 0 (0) | 2 (13) | 2 (12) | 0.09 |
| Rash | 2 (13) | 0 (0) | 0 (0) | 1 (6) | 0.56 |
The QTc increased from baseline to 4 h after the first dose by a median (IQR) of 13.5 ms0.5 (5 to 26 ms0.5) in pregnant DHA-PQ-treated women, 14.5 ms0.5 (9 to 30 ms0.5) in pregnant SP-PQ-treated women, 3.5 ms0.5 (−7 to 19 ms0.5) in nonpregnant DHA-PQ-treated women, and 16 ms0.5 (8 to 27 ms0.5) in nonpregnant SP-PQ-treated women (P = 0.17 by Kruskal-Wallis test). Values for most participants had returned toward baseline at 12 h (Fig. 1). Nineteen of 32 pregnant women (59%) and 12 of 33 nonpregnant women (36%) developed a QTc of >440 ms0.5 at 4 h postdose, with the longest QTc recorded being 512 ms0.5. None of the women experienced episodic palpitations, breathlessness, or light-headedness during follow-up. Postural hypotension (>20 mm Hg systolic fall after standing) occurred in nine pregnant women from the DHA-PQ group (60%) and three from the SP-PQ group (30%) and in three nonpregnant women from the DHA-PQ group (30%) and four from the SP-PQ group (27%) (P = 0.053 by Fisher's exact test), but there were no associated symptoms in any of these subjects.
FIG 1.

Changes in the electrocardiographic QTc interval during the first 24 h in the pregnant groups (upper panels) and nonpregnant groups (lower panels) for dihydroartemisinin-piperaquine (left panels) and sulfadoxine-pyrimethamine-piperaquine (right panels). Data are medians (■) and ranges (vertical bars).
There were four cases of asymptomatic P. falciparum infections detected by microscopy at baseline. Two of these were in pregnant women randomized to DHA-PQ and the other two were in nonpregnant women randomized to SP-PQ. In all four cases, parasites had cleared by day 2 and no recurrent parasitemias were detected by microscopy during follow-up. Another nonpregnant woman randomized to SP-PQ had P. falciparum gametocytes by microscopy at enrollment but no asexual forms were seen. Gametocytemia persisted for 14 days in this woman. No subject was found to be slide positive for malaria during the 42-day follow-up period or at delivery in the cases of pregnant participants.
Obstetric outcomes.
Of the 30 pregnant women who completed study procedures, 10 successfully delivered their baby in their home village without seeking medical assistance, and 17 women delivered healthy babies at the Alexishafen Health Center (mean ± SD birth weight of 2.9 ± 0.6 kg; 57% males). Three participants had stillbirths. One of these women was high risk due to advanced maternal age (39 years) and a history of two previous stillbirths out of nine pregnancies. She delivered in her village at 22 weeks' gestation (26 days after DHA-PQ). The placenta and fetus were not available for examination. The remaining two stillbirths were intrauterine deaths during labor attributed to asphyxia. Both mothers presented with cervical effacement and footling breech and buttock presentations, respectively, and a nonpulsating cord on view. Heavy meconium staining was present in both cases. Review by a panel of three independent physicians concluded that all three stillbirths were unlikely to be a result of study medication, which had been administered 26, 70, and 137 days beforehand, respectively.
Pharmacokinetic modeling.
There were 1,078 PQ and 209 DHA individual plasma concentrations available for analysis. For PQ there were no concentrations below the LOQ, while for DHA there were 12 (6%), of which 10 (5%) were below the LOD. Given that <10% of data were below the LOQ, data points that were below the LOD were excluded from the analysis while those between the LOD and LOQ were included at their measured concentrations. For PQ, there were 17 observations in pregnant subjects that were made after delivery, and these were excluded from the analysis.
When 2-compartment models were tested for PQ plasma concentration-time data, a pattern in the CWRES was seen, suggesting model misspecification. With the exception of the absorption process, CWRES improved adequately with a 3-compartment model, and it was accompanied by a drop in the OFV of 68.415 (P < 0.001). Therefore, models with more distribution compartments were not tested. The absorption process remained poorly characterized with models that used a zero- and/or first-order absorption process with and without lag time. The use of a transit compartment model significantly improved the model fit (ΔOFV = −724.96; P < 0.001) and the characterization of the absorption process. As the ka was poorly estimated (high percent residual standard error [RSE] and high correlation with MMT) for models using transit compartments, it was set to be equal to the ktr {i.e., ka = ktr = [(1 + NN)/MTT]}.
The structural model parameters for the PQ model were, therefore, NN, MTT, VC/FPQ, VP1/FPQ, VP2/FPQ, CL/FPQ, Q1/FPQ, and Q2/FPQ. Of these, the IIV of NN, MTT, VC/FPQ, and CL/FPQ could be estimated. Correlation between IIV terms could be estimated for VC/FPQ and CL/FPQ, as this was estimated to be 0.99 and was fixed to 1 to help successful minimization of model estimation. The addition of IOV for FPQ resulted in a significant fall in the OFV (−485.338; P < 0.001). On examination of the initial pcVPC and NPC results, there appeared to be greater variability in the concentrations from pregnant women than from nonpregnant controls. To examine this potential difference, two separate IOVs were estimated, one for each of the two groups. The inclusion of this additional variable model parameter resulted in a significant improvement in the fit of the model (ΔOFV = −8.990; P < 0.002), and it improved pcVPC and NPC results. Finally, of the tested covariates, only pregnancy on CL/FPQ was significant (ΔOFV = −44.788; P < 0.001), with a 42.1% increase in the pregnancy group.
The final model parameter estimates and the bootstrap results for PQ are summarized in Table 3. All bootstrap runs produced estimates. Bias was <2% and <8% for all structural and random model parameters, respectively. Figures 2 and 3 show goodness-of-fit plots and pcVPCs (stratified according to pregnancy status), respectively. From the NPCs, 10% and 8% for nonpregnant women and 7% and 5% for pregnant women of the data points were above the 80% prediction interval, and all were within their respective 95% CI. Secondary pharmacokinetic parameters are shown in Table 4. Consistent with the results of modeling, CL/FPQ, t½α, t½ β, t½ γ, and AUC0–∞ were significantly different between the groups, with a 33% lower AUC0–∞ in the pregnant group. A trend to a significant difference in VC/FPQ (P = 0.054) was consistent with both a difference in BW between the two groups (53.5 versus 52.7 kg for pregnant and nonpregnant participants, respectively) and a correlation between CL/FPQ and VC/FPQ in the model.
TABLE 3.
Final population pharmacokinetic estimates and bootstrap results for piperaquine in pregnant and nonpregnant womena
| Parameter | Mean | % RSEb | Median bootstrap value (95% CI) |
|---|---|---|---|
| Objective function value | −691.189 | −702.788 (−875.573 to −536.245) | |
| Structural model parameters | |||
| MTT (h) | 1.02 | 8 | 1.02 (0.88 to 1.19) |
| NN | 1.91 | 24 | 1.94 (1.24 to 3.03) |
| CL/FPQ (liters/h/70 kg) | 65.6 | 7 | 65.3 (57.2 to 75.5) |
| VC/FPQ (liters/70 kg) | 3,410 | 11 | 3,386 (2,776 to 4,154) |
| Q1/FPQ (liters/h/70 kg) | 98.5 | 9 | 97.9 (81.3 to 117) |
| VP1/FPQ (liters/70 kg) | 24,200 | 8 | 24,157 (20,665 to 28,520) |
| Q2/FPQ (liters/h/70 kg) | 548 | 14 | 547 (417 to 718) |
| VP2/FPQ (liters/70 kg) | 9,883 | 15 | 9,841 (7,414 to 13,266) |
| Increase in CL with pregnancy (%) | 42.1 | 18 | 0.420 (0.283 to 0.563) |
| Variable model parameters (% shrinkage) | |||
| IOV in FPQ, pregnant women | 103 (44, 22, 54)c | 12 | 103 (80 to 127) |
| IOV in FPQ, nonpregnant women | 66 (36, 31, 44)c | 8 | 66 (55 to 77) |
| IIV in MTT | 52 (6) | 9 | 51 (42 to 61) |
| IIV in NN | 132 (22) | 18 | 131 (86 to 177) |
| IIV in CL/FPQ | 14 (36) | 28 | 13 (7 to 23) |
| IIV in VP1/FPQ | 31 (36) | 47 | 30 (38 to 44) |
| r(CL/FPQ,VP1/FPQ)d | 1 | ||
| RV for PQ | 30 (14) | 5 | 30 (27 to 32) |
Abbreviations: RSE, residual standard error; MTT, mean transit time; NN, number of transit compartments; CL/FPQ, clearance relative to bioavailability; VC/FPQ, central volume of distribution relative to bioavailability; Q1/FPQ, intercompartmental clearance for VP1/FPQ; VP1/FPQ, first peripheral volume of distribution relative to bioavailability; Q2/FPQ, intercompartmental clearance for VP2/FPQ; VP2/FPQ, second peripheral volume of distribution relative to bioavailability; IOV, interoccasion variability; IIV, interindividual variability; RV, residual variability. IOV and IIV are presented as 100% × √(variability estimate).
Obtained from bootstrap results.
Values in parentheses for IOV data represent represent shrinkage for the three doses of PQ given.
This value was fixed at unity (see text).
FIG 2.

Goodness-of-fit plots for piperaquine.
FIG 3.
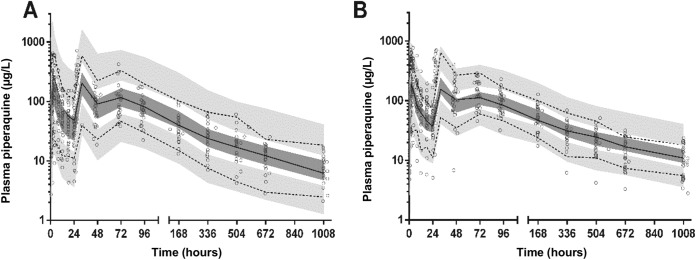
Prediction corrected visual predictive check with observed 50th (solid line) and 10th and 90th (dotted lines) percentiles within their simulated 95% CI (gray shaded areas) for piperaquine in pregnant women (A) and in nonpregnant women (B) (data are in micrograms per liter on a log10 scale) overlying the data points (○).
TABLE 4.
Post hoc Bayesian estimates of pharmacokinetic parameters and derived secondary parameters for piperaquine in pregnant and nonpregnant women
| Parametera | Median (IQR) value |
P value | |
|---|---|---|---|
| Nonpregnant (n = 33) | Pregnant (n = 32) | ||
| MTT (h) | 0.942 (0.813–1.17) | 0.977 (0.766–1.75) | 0.33 |
| NN | 2.57 (1.58–5.50) | 1.76 (1.11–5.44) | 0.28 |
| CL/FPQ (liters/h) | 53.8 (49.7–58.2) | 73.5 (69.4–78.4) | <0.001 |
| VC/FPQ (liters) | 2,601 (2,321–3,208) | 2,296 (2,117–2,720) | 0.054 |
| Q1/FPQ (liters/h) | 81.1 (73.6–84.4) | 77.7 (75.1–83.6) | 0.49 |
| VP1/FPQ (liters) | 18,639 (16,395–19,674) | 17,604 (16,827–19,415) | 0.49 |
| Q2/FPQ (liters/h) | 451 (410–470) | 432 (418–465) | 0.49 |
| VP2/FPQ (liters) | 7,623 (6,706–8,047) | 7,200 (6,882–7,941) | 0.49 |
| t½α (h) | 2.61 (2.33–2.85) | 2.33 (2.21–2.51) | 0.019 |
| t½β (h) | 50.7 (49.2–52.0) | 45.8 (45–46.8) | <0.001 |
| t½γ (h) | 488 (478–499) | 382 (374–388) | <0.001 |
| AUC0–∞ (μg · h/liter) | 35,644 (29,546–39,541) | 23,721 (21,481–27,951) | <0.001 |
Abbreviations: MTT, mean transit time; NN, number of transit compartments; CL/FPQ, clearance; VC/FPQ, central volume of distribution; Q1/FPQ, intercompartmental clearance for VP1/FPQ; VP1/FPQ, first peripheral volume of distribution; Q2/FPQ, intercompartmental clearance for VP2/FPQ; VP2/FPQ, second peripheral volume of distribution; t½α, t½β, and t½γ (first and second distribution and elimination half-lives, respectively; Cmax, maximum plasma concentration; AUC0–∞, area under the plasma drug concentration-time curve.
For DHA, a model with two compartments fitted the data significantly better than those with a single compartment (P < 0.001; df = 2). A three-compartment model was not tested, as two compartments described the data adequately, demonstrated by no bias in the CWRES plot. Absorption was best described by a first-order process and was significantly improved after the inclusion of a lag time (ΔOFV = −37.501; P < 0.001). Differences in disposition between doses could not be estimated for DHA, as the sampling regimen in the present study only allowed for analysis of DHA concentrations after the first dose. The structural model parameters for DHA were ka, lag, VC/FDHA, VP/FDHA,CL/FDHA, and Q/FDHA. IIV was estimated for ka, VC/FDHA, and CL/FDHA, as well as the correlation between the IIV terms for VC/FDHA and CL/FDHA. None of the tested covariates resulted in a significant improvement in the model.
The final model parameter estimates and the bootstrap results for DHA are summarized in Table 5. Almost all (99%) of the bootstrap runs produced estimates, and bias was <9% for all structural and random model parameters. Figures 4 and 5 show goodness-of-fit plots and pcVPCs, respectively. From the NPCs, 10% of the data points were above and below the 80% prediction interval. Secondary pharmacokinetic parameters are shown in Table 6. There were no significant differences between the two groups by pregnancy status.
TABLE 5.
Final population pharmacokinetic estimates and bootstrap results for dihydroartemisinin in pregnant and nonpregnant women
| Parametera | Mean | % RSE | Median bootstrap value (95% CI) |
|---|---|---|---|
| Objective function value | −716.816 | −736.766 (−893.437 to −579.301) | |
| Structural model parameters | |||
| Lag (h) | 0.733 | 1 | 0.736 (0.448 to 2.147) |
| ka (h−1) | 0.408 | 29 | 0.409 (0.372 to 0.472) |
| CL/FDHA (liters/h/70 kg) | 148 | 13 | 147 (109 to 184) |
| VC/FDHA (liters/70 kg) | 69.3 | 42 | 62.6 (20.5 to 223.6) |
| Q/FDHA (liters/h/70 kg) | 23.5 | 42 | 23.5 (2.07 to 66.6) |
| VP/FDHA (liters/70 kg) | 82.7 | 33 | 86.8 (17.0 to 1,379) |
| Variable model parameters (% shrinkage) | |||
| IIV in CL/FDHA | 54 (4) | 32 | 55 (37 to 81) |
| IIV in VC/FDHA | 152 (9) | 21 | 147 (83 to 236) |
| IIV in ka | 30 (38) | 31 | 28 (0 to 71) |
| r(CL/FDHA,VC/FDHA) | 0.838 | 37 | 0.818 (0.464 to 0.986) |
| RV for DHA | 51 | 16 | 48 (40 to 56) |
Abbreviations: lag, lag time; ka, absorption rate constant; CL/FDHA, clearance relative to bioavailability; VC/FDHA, central volume of distribution relative to bioavailability; Q/FDHA, intercompartmental clearance for VP/FDHA; VP/FDHA, peripheral volume of distribution relative to bioavailability; IIV, interindividual variability; r, correlation coefficient; RV, residual variability. IIV is presented as 100% × √(variability estimate).
FIG 4.

Goodness-of-fit plots for dihydroartemisinin.
FIG 5.
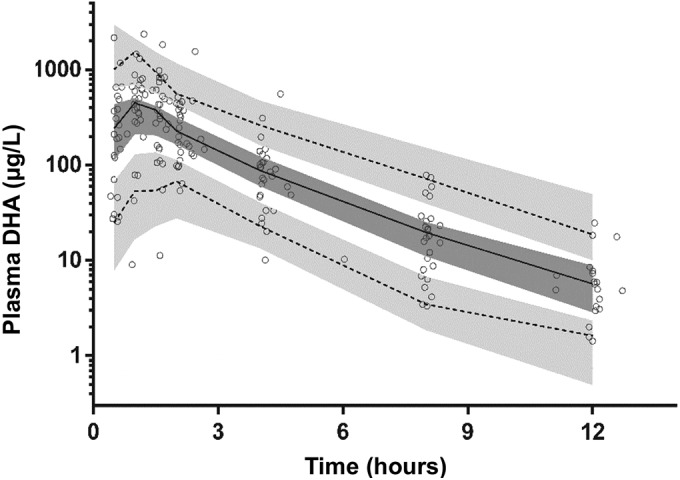
Prediction corrected visual predictive check with observed 50th (solid line) and 10th and 90th (dotted lines) percentiles within their simulated 95% CI (gray shaded areas) for dihydroartemisinin.
TABLE 6.
Post hoc Bayesian estimates of pharmacokinetic parameters and derived secondary parameters for dihydroartemisinin in pregnant and nonpregnant women
| Parametera | Median (IQR) |
P value | |
|---|---|---|---|
| Nonpregnant (n = 33) | Pregnant (n = 32) | ||
| Lag (h) | 0.408 (0.408–0.408) | 0.408 (0.408–0.408) | |
| ka (h−1) | 0.761 (0.708–0.801) | 0.738 (0.633–0.808) | 0.55 |
| CL/FDHA (liters/h) | 127 (78.1–144) | 134 (90.8–188) | 0.28 |
| VC/FDHA (liters) | 46.5 (23.4–79.2) | 45.0 (29.7–229) | 0.47 |
| Q/FDHA (liters/h) | 19.4 (17.9–20.8) | 18.3 (18.1–19.8) | 0.23 |
| VP/FDHA (liters) | 63.8 (57.6–70.3) | 59.1 (58.5–65.6) | 0.23 |
| t½α (h) | 0.235 (0.163–0.354) | 0.254 (0.143–0.671) | 0.52 |
| t½β (h) | 2.76 (2.71–2.87) | 2.69 (2.58–2.78) | 0.30 |
| AUC0–∞ (μg · h/liter) | 946 (834–1537) | 893 (656–1,325) | 0.28 |
Abbreviations: lag, lag time; ka, absorption rate constant; CL/FDHA, clearance relative to bioavailability; VC/FDHA, central volume of distribution relative to bioavailability; Q/FDHA, intercompartmental clearance for VP/FDHA; VP/FDHA, peripheral volume of distribution relative to bioavailability; t½α and t½β, distribution and elimination half-lives, respectively; AUC0–∞, area under the plasma drug concentration-time curve.
DISCUSSION
In the present study, the pharmacokinetic properties of PQ and DHA were examined in pregnant and nonpregnant PNG women randomized to either DHA-PQ or SP-PQ treatment. We previously characterized the disposition of SP in these groups of subjects (19), but there have been no published pharmacokinetic studies of PQ or DHA in women from the Oceania region. We included an SP-PQ arm because of the potential for this combination to be a more cost-effective IPTp than DHA-PQ (29, 30). In contrast to all previous pharmacokinetic and efficacy studies utilizing DHA-PQ (12–14, 27, 28), malaria was not an inclusion criterion, and a detailed safety assessment was performed on all patients. Safety monitoring was primarily to ensure that the cardiovascular effects of PQ were not augmented in pregnancy, especially since PQ is known to prolong the QTc (24) and pregnancy can be proarrhythmic (41, 42). Both drug regimens proved well tolerated, safe, and effective at both eliminating and, in association with ITNs, preventing parasitemia. While we found that there was no pregnancy-associated change in DHA disposition, there were significant differences in the pharmacokinetic properties of PQ between pregnant and nonpregnant women, including a reduction in PQ exposure that may have implications for IPTp regimens.
The conventional adult doses of DHA-PQ and SP-PQ utilized in the present study were generally well tolerated by both pregnant and nonpregnant women. Symptoms during the first week, including those related to postural hypotension, were mild and short-lived. There was the expected PQ-associated QTc prolongation, but this was not increased by pregnancy. The few women who presented with asymptomatic P. falciparum infections cleared their parasitemia promptly and without subsequent recurrence, while there were no new clinical or microscopically detected malaria infections during follow-up in any patient. Gametocyte carriage was identified in a single participant which, consistent with the known effects of SP and PQ (43, 44), persisted for 2 weeks after this form of treatment. The three serious adverse obstetric outcomes during the study were unlikely to be related to drug administration and were consistent with the background risks of such events in pregnant PNG women, even when they receive hospital care (45).
As in similar previous pharmacokinetic studies of PQ in pregnancy that have employed long-duration sampling schedules (13, 14), we found that a three-compartment model with the incorporation of a transit compartment provided the best fit. Whether DHA or SP was given with PQ did not influence PQ disposition. Pregnancy was associated with a 42% higher CL and a 33% lower AUC0–∞ in our subjects. In a study of Thai women that used similar population modeling as the present study (14), pregnancy also increased the PQ CL (by 45%) in uncomplicated malaria. However, the AUC0–92 days was not significantly different from that of the nonpregnant women with malaria, reflecting a 46.8% increased relative bioavailability (14). We did not find an effect of pregnancy on bioavailability and, although the IOV for FPQ was greater in the pregnant women, there was no evidence of the time-dependent increase in relative bioavailability observed in both groups in the study of Thai women with uncomplicated malaria treated with DHA-PQ (14). The differences between the present data and those of the Thai study may relate to the ingestion of fat around the time of PQ dosing, differences in the proportions of women with malaria, and/or racial differences in PQ disposition.
We kept our PNG women fasting for 2 h after dosing, and in a healthy volunteer population pharmacokinetic study in which Vietnamese subjects were also kept fasting for 2 h postdose, there was also no evidence of a time-dependent increase in bioavailability (46). Food intake was not controlled in the Thai study (14). Although a small amount of fat (<10 g) does not increase PQ bioavailability (47–49), greater fat intake, such as that associated with a normal meal, can as much as double relative bioavailability (50–52). It is possible that the women in the Thai study, whether pregnant or not, consumed increasing amounts of fat as they recovered from malaria, progressively increasing bioavailability as a result of the lipophilicity of PQ. It is also possible that the greater time-independent relative bioavailability in the pregnant versus nonpregnant women in the Thai study represented pregnancy-associated physiological changes favoring PQ absorption, including reduced small intestine motility, prolonged gastric emptying, and increased intestinal blood flow (14, 53, 54), which are of pharmacokinetic significance after consumption of food but not in the fasting state.
We had too few parasitemic women to examine whether malaria infection, as a covariate in the modeling, influenced PQ disposition, and there are no published studies which have examined this question in matched groups of patients with malaria and healthy volunteers. While the disposition of CQ does not appear to be influenced by malaria (55), recent population pharmacokinetic modeling involving mefloquine, another long-half-life antimalarial drug, suggests that the volume of distribution is more sensitive to acute malaria (with a 40% reduction) than CL/F, which is largely related to the terminal phase during convalescence after treatment (56). However, the median total volumes of distribution for PQ in pregnant and nonpregnant Thai women with uncomplicated malaria (529 and 829 liters/kg, respectively) (14) were larger rather than smaller than those in our pregnant and nonpregnant PNG women, most of whom did not have malaria (507 and 548 liters/kg, respectively). We cannot exclude an effect of malaria on PQ pharmacokinetics that underlies the differences between the present study and the Thai study but, if present, it would appear to differ from malaria-associated effects on other slowly eliminated partner drugs.
The metabolism of PQ has not been characterized in detail but, apart from the possibility of cytochrome P450 enzyme involvement (57), there are no major metabolites seen from chromatography of plasma from treated patients (24). Indeed, PQ has been regarded as a drug without suspected pharmacogenetic variability in absorption, distribution, metabolism, or excretion (57). At present, there is not a plausible reason why PQ disposition might differ in Melanesian populations but, as with the longer-than-expected elimination half-life of pyrimethamine in pregnant PNG women (19), this cannot be excluded.
Previous studies have shown that the pharmacokinetic properties of DHA after oral administration of artesunate may include lower drug exposure during treatment for malaria in pregnancy (58, 59), consistent with data from the Thai study involving DHA-PQ, which revealed a reduction of 37.5% in relative DHA bioavailability in pregnancy (14). In contrast, we did not find an effect of pregnancy on DHA pharmacokinetics in a sample of women with relatively few malaria cases. One reason for this apparent discrepancy is that we used a two-compartment rather than the one-compartment model employed in other studies (14). We had a total of 209 individual plasma DHA concentrations from relatively rich blood sampling after the first dose, of which only 12 (6%) fell below the LOQ and 10 (5%) below the LOD. This relatively low number, together with the model fit and bootstrapping, suggest that the structural model was appropriate. A one-compartmental model may miss late contributions to parameters such as the AUC, which could bias comparisons between groups. In any case, despite apparently diminished drug exposure, clearance of parasitemia was as prompt in the pregnant as nonpregnant patients in the Thai study (14), suggesting that there are no pharmacodynamic consequences.
The present study had some limitations. Few of the women were parasitemic at recruitment (6.7%), but this rate was similar to the 7.4% who were slide positive at baseline in a recent large trial of SP versus AZI-SP as IPTp that was conducted in pregnant PNG women at ≤26 weeks of gestation in Madang Province (15). The low numbers of baseline malaria infections meant that we could not assess the effect of malaria infection on pharmacokinetic parameters. Our study was not powered for assessment of prophylactic efficacy, especially since ITNs were issued to all participants.
An appropriate DHA-PQ or SP-PQ IPTp needs to be based on various factors. In countries such as PNG, in which monthly antenatal attendances are recommended, the present and other data (14) suggest that this may represent an appropriate time between PQ-based regimens given in conventional adult doses to pregnant women at risk of malaria. However, the faster elimination of PQ in pregnant patients combined with significant between-patient variability in plasma drug concentrations may lead to more breakthrough cases of malaria if attendance is sporadic or delayed. As well as having pharmacokinetic determinants, the duration of protection will depend on the degree of malarial immunity. Where there has been a reduction in exposure to malaria through strategies, including ITNs (including PNG), immunity may decline as a result (60), and pregnant women may therefore have a shorter period of protection after the same DHA-PQ dose. There is a clear need for monitoring the incidence of malaria in pregnancy where IPTp is used, as part of in vitro and other in vivo measures of local parasite resistance.
ACKNOWLEDGMENTS
We thank the mothers and other relatives for supporting this study and Simga-Tau Industrie Farmaceutiche Riunite S.p.A. for manufacture and provision of all antimalarial drugs. We are also most grateful to Valsi Kurian, Maria Christina, and the staff of the Alexishafen Health Center and labor ward for their kind assistance and cooperation during the study. We also acknowledge the staff of the Papua New Guinea Institute of Medical Research for clinical and logistic assistance.
This study was funded by the National Health and Medical Research Council (NHMRC) of Australia (grants 458555 and 634343). B.R.M. was supported by an NHMRC Early Career Fellowship (1036951), J.B. was supported by a Fogerty International Masters Scholarship, L.J.R. was supported by an NHMRC Early Career Fellowship (1016443), I.M. was supported by an NHMRC Senior Research Fellowship (1043345), and T.M.E.D. was supported by an NHMRC Practitioner Fellowship (572761).
REFERENCES
- 1.Desai M, ter Kuile FO, Nosten F, McGready R, Asamoa K, Brabin B, Newman RD. 2007. Epidemiology and burden of malaria in pregnancy. Lancet Infect Dis 7:93–104. doi: 10.1016/S1473-3099(07)70021-X. [DOI] [PubMed] [Google Scholar]
- 2.Steketee RW, Wirima JJ, Hightower AW, Slutsker L, Heymann DL, Breman JG. 1996. The effect of malaria and malaria prevention in pregnancy on offspring birthweight, prematurity, and intrauterine growth retardation in rural Malawi. Am J Trop Med Hyg 55:33–41. [DOI] [PubMed] [Google Scholar]
- 3.Mutabingwa TK, Bolla MC, Li JL, Domingo GJ, Li X, Fried M, Duffy PE. 2005. Maternal malaria and gravidity interact to modify infant susceptibility to malaria. PLoS Med 2:e407. doi: 10.1371/journal.pmed.0020407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis TM, Mueller I, Rogerson SJ. 2010. Prevention and treatment of malaria in pregnancy. Future Microbiol 5:1599–1613. doi: 10.2217/fmb.10.113. [DOI] [PubMed] [Google Scholar]
- 5.World Health Organization. 2010. Guidelines for the treatment of malaria, 2nd ed World Health Organization, Geneva, Switzerland. [Google Scholar]
- 6.World Health Organization. 2007. Technical expert group meeting on intermittent preventive treatment in pregnancy (IPTp). World Health Organization, Geneva, Switzerland. [Google Scholar]
- 7.World Health Organization Global Malaria Programme. 2012. Updated WHO policy recommendation (October 2012): intermittent preventive treatment of malaria in pregnancy using sulfadoxine-pyrimethamine (IPTp-SP). World Health Organization, Geneva, Switzerland. [Google Scholar]
- 8.Gosling RD. 2010. Intermittent preventive treatment against malaria: an update. Expert Rev Anti Infect Ther 8:589–606. doi: 10.1586/eri.10.36. [DOI] [PubMed] [Google Scholar]
- 9.van Vugt M, van Beest A, Sicuri E, van Tulder M, Grobusch MP. 2011. Malaria treatment and prophylaxis in endemic and nonendemic countries: evidence on strategies and their cost-effectiveness. Future Microbiol 6:1485–1500. doi: 10.2217/fmb.11.138. [DOI] [PubMed] [Google Scholar]
- 10.Moussiliou A, De Tove YS-S, Doritchamou J, Luty AJF, Massougbodji A, Alifrangis M, Deloron P, Ndam NT. 2013. High rates of parasite recrudescence following intermittent preventive treatment with sulphadoxine-pyrimethamine during pregnancy in Benin. Malar J 12:195. doi: 10.1186/1475-2875-12-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chico RM, Pittrof R, Greenwood B, Chandramohan D. 2008. Azithromycin-chloroquine and the intermittent preventive treatment of malaria in pregnancy. Malar J 7:255. doi: 10.1186/1475-2875-7-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adam I, Tarning J, Lindegardh N, Mahgoub H, McGready R, Nosten FH. 2012. Pharmacokinetics of piperaquine in pregnant women in Sudan with uncomplicated Plasmodium falciparum malaria. Am J Trop Med Hyg 87:35–40. doi: 10.4269/ajtmh.2012.11-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoglund RM, Adam I, Hanpithakpong W, Ashton M, Lindegardh N, Day NP, White NJ, Nosten F, Tarning J. 2012. A population pharmacokinetic model of piperaquine in pregnant and non-pregnant women with uncomplicated Plasmodium falciparum malaria in Sudan. Malar J 11:398. doi: 10.1186/1475-2875-11-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tarning J, Rijken MJ, McGready R, Phyo AP, Hanpithakpong W, Day NP, White NJ, Nosten F, Lindegardh N. 2012. Population pharmacokinetics of dihydroartemisinin and piperaquine in pregnant and nonpregnant women with uncomplicated malaria. Antimicrob Agents Chemother 56:1997–2007. doi: 10.1128/AAC.05756-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Unger HW, Ome-Kaius M, Wangnapi RA, Umbers AJ, Hanieh S, Suen CS, Robinson LJ, Rosanas-Urgell A, Wapling J, Lufele E, Kongs C, Samol P, Sui D, Singirok D, Bardaji A, Schofield L, Menendez C, Betuela I, Siba P, Mueller I, Rogerson SJ. 2015. Sulphadoxine-pyrimethamine plus azithromycin for the prevention of low birthweight in Papua New Guinea: a randomised controlled trial. BMC Med 13:9. doi: 10.1186/s12916-014-0258-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mueller I, Rogerson S, Mola GD, Reeder JC. 2008. A review of the current state of malaria among pregnant women in Papua New Guinea. P N G Med J 51:12–16. [PubMed] [Google Scholar]
- 17.Kayentao K, Garner P, van Eijk AM, Naidoo I, Roper C, Mulokozi A, MacArthur JR, Luntamo M, Ashorn P, Doumbo OK, ter Kuile FO. 2013. Intermittent preventive therapy for malaria during pregnancy using 2 vs 3 or more doses of sulfadoxine-pyrimethamine and risk of low birth weight in Africa: systematic review and meta-analysis. JAMA 309:594–604. doi: 10.1001/jama.2012.216231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nyunt MM, Adam I, Kayentao K, van Dijk J, Thuma P, Mauff K, Little F, Cassam Y, Guirou E, Traore B, Doumbo O, Sullivan D, Smith P, Barnes KI. 2010. Pharmacokinetics of sulfadoxine and pyrimethamine in intermittent preventive treatment of malaria in pregnancy. Clin Pharmacol Ther 87:226–234. doi: 10.1038/clpt.2009.177. [DOI] [PubMed] [Google Scholar]
- 19.Karunajeewa HA, Salman S, Mueller I, Baiwog F, Gomorrai S, Law I, Page-Sharp M, Rogerson S, Siba P, Ilett KF, Davis TM. 2009. Pharmacokinetic properties of sulfadoxine-pyrimethamine in pregnant women. Antimicrob Agents Chemother 53:4368–4376. doi: 10.1128/AAC.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koleala T, Karl S, Laman M, Moore BR, Benjamin J, Barnadas C, Robinson LJ, Kattenberg JH, Javati S, Wong RPM, Rosanas-Urgell A, Betuela I, Siba PM, Mueller I, Davis TME. 2015. Temporal changes in Plasmodium falciparum anti-malarial drug sensitivity in vitro and resistance-associated genetic mutations in isolates from Papua New Guinea. Malaria J 14:37. doi: 10.1186/s12936-015-0560-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barnadas C, Senn N, Iga J, Timinao L, Javati S, Malau E, Rarau P, Reeder JC, Siba P, Karunajeewa H, Zimmerman PA, Davis TM, Mueller I. 2014. Plasmodium falciparum and Plasmodium vivax genotypes and efficacy of intermittent preventive treatment in Papua New Guinea. Antimicrob Agents Chemother 58:6958–6961. doi: 10.1128/AAC.03323-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rijken MJ, McGready R, Boel ME, Poespoprodjo R, Singh N, Syafruddin D, Rogerson S, Nosten F. 2012. Malaria in pregnancy in the Asia-Pacific region. Lancet Infect Dis 12:75–88. doi: 10.1016/S1473-3099(11)70315-2. [DOI] [PubMed] [Google Scholar]
- 23.Batty KT, Moore BR, Stirling V, Ilett KF, Page-Sharp M, Shilkin KB, Mueller I, Rogerson SJ, Karunajeewa HA, Davis TM. 2010. Investigation of reproductive toxicity of piperaquine in mice. Reprod Toxicol 29:206–213. doi: 10.1016/j.reprotox.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 24.Davis TM, Hung TY, Sim IK, Karunajeewa HA, Ilett KF. 2005. Piperaquine: a resurgent antimalarial drug. Drugs 65:75–87. doi: 10.2165/00003495-200565010-00004. [DOI] [PubMed] [Google Scholar]
- 25.Gogtay N, Kannan S, Thatte UM, Olliaro PL, Sinclair D. 2013. Artemisinin-based combination therapy for treating uncomplicated Plasmodium vivax malaria. Cochrane Database Syst Rev 10:CD008492. doi: 10.1002/14651858.CDC008492.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zani B, Gathu M, Donegan S, Olliaro PL, Sinclair D. 2014. Dihydroartemisinin-piperaquine for treating uncomplicated Plasmodium falciparum malaria. Cochrane Database Syst Rev 1:CD010927. doi: 10.1002/14651858.CDD010927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rijken MJ, McGready R, Phyo AP, Lindegardh N, Tarning J, Laochan N, Than HH, Mu O, Win AK, Singhasivanon P, White N, Nosten F. 2011. Pharmacokinetics of dihydroartemisinin and piperaquine in pregnant and nonpregnant women with uncomplicated falciparum malaria. Antimicrob Agents Chemother 55:5500–5506. doi: 10.1128/AAC.05067-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poespoprodjo JR, Fobia W, Kenangalem E, Lampah DA, Sugiarto P, Tjitra E, Anstey NM, Price RN. 2014. Dihydroartemisinin-piperaquine treatment of multidrug resistant falciparum and vivax malaria in pregnancy. PLoS One 9:e84976. doi: 10.1371/journal.pone.0084976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.White NJ. 2005. Intermittent presumptive treatment for malaria. PLoS Med 2:e3. doi: 10.1371/journal.pmed.0020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cisse B, Cairns M, Faye E, O ND, Faye B, Cames C, Cheng Y, M ND, Lo AC, Simondon K, Trape JF, Faye O, JL ND, Gaye O, Greenwood B, Milligan P. 2009. Randomized trial of piperaquine with sulfadoxine-pyrimethamine or dihydroartemisinin for malaria intermittent preventive treatment in children. PLoS One 4:e7164. doi: 10.1371/journal.pone.0007164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karunajeewa HA, Salman S, Mueller I, Baiwog F, Gomorrai S, Law I, Page-Sharp M, Rogerson S, Siba P, Ilett KF, Davis TM. 2010. Pharmacokinetics of chloroquine and monodesethylchloroquine in pregnancy. Antimicrob Agents Chemother 54:1186–1192. doi: 10.1128/AAC.01269-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Michon P, Cole-Tobian JL, Dabod E, Schoepflin S, Igu J, Susapu M, Tarongka N, Zimmerman PA, Reeder JC, Beeson JG, Schofield L, King CL, Mueller I. 2007. The risk of malarial infections and disease in Papua New Guinean children. Am J Trop Med Hyg 76:997–1008. [PMC free article] [PubMed] [Google Scholar]
- 33.Laman M, Moore BR, Benjamin J, Padapu N, Tarongka N, Siba P, Betuela I, Mueller I, Robinson LJ, Davis TM. 2014. Comparison of an assumed versus measured leucocyte count in parasite density calculations in Papua New Guinean children with uncomplicated malaria. Malar J 13:145. doi: 10.1186/1475-2875-13-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karunajeewa HA, Ilett KF, Mueller I, Siba P, Law I, Page-Sharp M, Lin E, Lammey J, Batty KT, Davis TM. 2008. Pharmacokinetics and efficacy of piperaquine and chloroquine in Melanesian children with uncomplicated malaria. Antimicrob Agents Chemother 52:237–243. doi: 10.1128/AAC.00555-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salman S, Page-Sharp M, Griffin S, Kose K, Siba PM, Ilett KF, Mueller I, Davis TME. 2011. Population pharmacokinetics of artemether, lumefantrine, and their respective metabolites in Papua New Guinean children with uncomplicated malaria. Antimicrob Agents Chemother 55:5306–5313. doi: 10.1128/AAC.05136-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beal S, Sheiner LB. 1980. NONMEM users guide part I. Division of Clinical Pharmacology, University of California, San Fransisco, San Francisco, CA. [Google Scholar]
- 37.Anderson BJ, Holford NH. 2009. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet 24:25–36. doi: 10.2133/dmpk.24.25. [DOI] [PubMed] [Google Scholar]
- 38.Upton RN. 2004. Calculating the hybrid (macro) rate constants of a three-compartment mamillary pharmacokinetic model from known micro-rate constants. J Pharmacol Toxicol Methods 49:65–68. doi: 10.1016/j.vascn.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 39.Salman S, Page-Sharp M, Batty KT, Kose K, Griffin S, Siba P, Ilett KF, Mueller I, Davis TME. 2012. A pharmacokinetic comparison of two piperaquine-containing artemisinin combination therapies in Papua New Guinean children with uncomplicated malaria. Antimicrob Agents Chemother 56:3288–3297. doi: 10.1128/AAC.06232-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Savic RM, Jonker DM, Kerbusch T, Karlsson MO. 2007. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J Pharmacokinet Pharmacodyn 34:711–726. doi: 10.1007/s10928-007-9066-0. [DOI] [PubMed] [Google Scholar]
- 41.Lee SH, Chen SA, Wu TJ, Chiang CE, Cheng CC, Tai CT, Chiou CW, Ueng KC, Chang MS. 1995. Effects of pregnancy on first onset and symptoms of paroxysmal supraventricular tachycardia. Am J Cardiol 76:675–678. doi: 10.1016/S0002-9149(99)80195-7. [DOI] [PubMed] [Google Scholar]
- 42.Tawam M, Levine J, Mendelson M, Goldberger J, Dyer A, Kadish A. 1993. Effect of pregnancy on paroxysmal supraventricular tachycardia. Am J Cardiol 72:838–840. doi: 10.1016/0002-9149(93)91078-V. [DOI] [PubMed] [Google Scholar]
- 43.Baker DA. 2010. Malaria gametocytogenesis. Mol Biochem Parasitol 172:57–65. doi: 10.1016/j.molbiopara.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zwang J, Ashley EA, Karema C, D'Alessandro U, Smithuis F, Dorsey G, Janssens B, Mayxay M, Newton P, Singhasivanon P, Stepniewska K, White NJ, Nosten F. 2009. Safety and efficacy of dihydroartemisinin-piperaquine in falciparum malaria: A prospective multi-centre individual patient data analysis. PLoS One 4:e6358. doi: 10.1371/journal.pone.0006358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jimmy S, Kemiki AD, Vince JD. 2003. Neonatal outcome at Modilon Hospital, Madang: a 5-year review. P N G Med J 46:8–15. [PubMed] [Google Scholar]
- 46.Roshammar D, Hai TN, Hietala SF, Huong NV, Ashton M. 2006. Pharmacokinetics of piperaquine after repeated oral administration of the antimalarial combination CV8 in 12 healthy male subjects. Eur J Clin Pharmacol 62:335–341. doi: 10.1007/s00228-005-0084-9. [DOI] [PubMed] [Google Scholar]
- 47.Annerberg A, Lwin KM, Lindegardh N, Khrutsawadchai S, Ashley E, Day NJP, Singhasivanon P, Tarning J, White NJ, Nosten F. 2011. A small amount of fat does not affect piperaquine exposure in patients with malaria. Antimicrob Agents Chemother 55:3971–3976. doi: 10.1128/AAC.00279-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moore BR, Benjamin JM, Salman S, Griffin S, Ginny E, Page-Sharp M, Robinson LJ, Siba P, Batty KT, Mueller I, Davis TM. 2014. Effect of coadministered fat on the tolerability, safety, and pharmacokinetic properties of dihydroartemisinin-piperaquine in Papua New Guinean children with uncomplicated malaria. Antimicrob Agents Chemother 58:5784–5794. doi: 10.1128/AAC.03314-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tarning J, Lindegardh N, Lwin KM, Annerberg A, Kiricharoen L, Ashley E, White NJ, Nosten F, Day NP. 2014. Population pharmacokinetic assessment of the effect of food on piperaquine bioavailability in patients with uncomplicated malaria. Antimicrob Agents Chemother 58:2052–2058. doi: 10.1128/AAC.02318-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hai TN, Hietala SF, van Huong N, Ashton M. 2008. The influence of food on the pharmacokinetics of piperaquine in healthy Vietnamese volunteers. Acta Trop 107:145–149. doi: 10.1016/j.actatropica.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 51.Nguyen TC, Nguyen NQ, Nguyen XT, Bui D, Travers T, Edstein MD. 2008. Pharmacokinetics of the antimalarial drug piperaquine in healthy Vietnamese subjects. Am J Trop Med Hyg 79:620–623. [PubMed] [Google Scholar]
- 52.Sim I-K, Davis TME, Ilett KF. 2005. Effects of a high-fat meal on the relative oral bioavailability of piperaquine. Antimicrob Agents Chemother 49:2407–2411. doi: 10.1128/AAC.49.6.2407-2411.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anderson GD. 2005. Pregnancy-induced changes in pharmacokinetics: a mechanistic-based approach. Clin Pharmacokinet 44:989–1008. doi: 10.2165/00003088-200544100-00001. [DOI] [PubMed] [Google Scholar]
- 54.Little BB. 1999. Pharmacokinetics during pregnancy: evidence-based maternal dose formulation. Obstet Gynecol 93:858–868. doi: 10.1016/S0029-7844(98)00444-X. [DOI] [PubMed] [Google Scholar]
- 55.Edwards G, Looareesuwan S, Davies AJ, Wattanagoon Y, Phillips RE, Warrell DA. 1988. Pharmacokinetics of chloroquine in Thais: plasma and red-cell concentrations following an intravenous infusion to healthy subjects and patients with Plasmodium vivax malaria. Br J Clin Pharmacol 25:477–485. doi: 10.1111/j.1365-2125.1988.tb03332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reuter SE, Upton RN, Evans AM, Navaratnam V, Olliaro PL. 2015. Population pharmacokinetics of orally administered mefloquine in healthy volunteers and patients with uncomplicated Plasmodium falciparum malaria. J Antimicrob Chemother 70:868–876. doi: 10.1093/jac/dku430. [DOI] [PubMed] [Google Scholar]
- 57.Kerb R, Fux R, Morike K, Kremsner PG, Gil JP, Gleiter CH, Schwab M. 2009. Pharmacogenetics of antimalarial drugs: effect on metabolism and transport. Lancet Infect Dis 9:760–774. doi: 10.1016/S1473-3099(09)70320-2. [DOI] [PubMed] [Google Scholar]
- 58.McGready R, Stepniewska K, Ward SA, Cho T, Gilveray G, Looareesuwan S, White NJ, Nosten F. 2006. Pharmacokinetics of dihydroartemisinin following oral artesunate treatment of pregnant women with acute uncomplicated falciparum malaria. Eur J Clin Pharmacol 62:367–371. doi: 10.1007/s00228-006-0118-y. [DOI] [PubMed] [Google Scholar]
- 59.Morris CA, Onyamboko MA, Capparelli E, Koch MA, Atibu J, Lokomba V, Douoguih M, Hemingway-Foday J, Wesche D, Ryder RW, Bose C, Wright L, Tshefu AK, Meshnick S, Fleckenstein L. 2011. Population pharmacokinetics of artesunate and dihydroartemisinin in pregnant and non-pregnant women with malaria. Malar J 10:114. doi: 10.1186/1475-2875-10-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Teo A, Hasang W, Randall LM, Feng G, Bell L, Unger H, Langer C, Beeson JG, Siba PM, Mueller I, Molyneux ME, Brown GV, Rogerson SJ. 2014. Decreasing malaria prevalence and its potential consequences for immunity in pregnant women. J Infect Dis 210:1444–1455. doi: 10.1093/infdis/jiu264. [DOI] [PubMed] [Google Scholar]